Week 4: Protein design I
PART A of HW: Answer any NINE of the following questions from Shuguang Zhang
1. How many molecules of amino acids do you take with a piece of 500 grams of meat?
Using Anthropic Clude I got the following answere and did some editions: There are more molecules of amino acids than the number of stars in the observable universe (~10²³). The calculation made by this IA modelis as follow:
Protein content of meat: ~25% by weight → 125 g of protein Average amino acid molecular weight: ~100 Da (100 g/mol) Moles of amino acids: $$\frac{125 \text{ g}}{100 \text{ g/mol}} = 1.25 \text{ mol}$$ Number of molecules (using Avogadro’s number, 6.022 × 10²³): $$1.25 \times 6.022 \times 10^{23} \approx \textbf{7.5 × 10}^{23} \textbf{ molecules}$$ Result: You consume roughly ~7.5 × 10²³ amino acid residues — about **1.25 times Avogadro’s number
2. Why are there only 20 natural amino acids?
Using Anthropic Clude I got the following answere and did some editions: We do not really know. However,there are some theories. Five of those are:
I. 🧬 The Genetic Code Constrains It: The standard genetic code uses 64 codons (4³) to encode only 20 amino acids + stop signals. The code is redundant but not expansive — evolution “froze” it early. Changing the code once life is established is catastrophically costly (one codon reassignment would corrupt thousands of proteins simultaneously). This is called the “frozen accident” hypothesis (Francis Crick, 1968).
II. ⚗️ Chemical Sufficiency: 20 amino acids already cover an enormous chemical diversity:
- Charged (+ and −)
- Hydrophobic / hydrophilic
- Tiny (Gly) to bulky (Trp)
- Flexible vs. rigid (Pro)
- Nucleophilic (Cys, Ser, His)
This diversity is apparently sufficient to fold proteins into almost any 3D shape and catalyze almost any reaction. More variety may simply not be needed.
III. 🌍 Prebiotic Availability: The amino acids found in the genetic code largely overlap with those found in meteorites and Miller-Urey-type experiments — suggesting the 20 were simply the ones available on early Earth when life was getting started. Notably, the “later” amino acids (Trp, His, Cys) are rarer in prebiotic chemistry — hinting at a gradual expansion from perhaps 10 → 20 over early evolution.
IV. 📐 Diminishing Returns: Theoretical and computational studies suggest that adding a 21st amino acid yields rapidly diminishing gains in protein foldability and function. The 20 already allow ~10¹³⁰ possible protein sequences — more than enough diversity.
V. 🔢 It’s Not Strictly 20 — Biology Cheats a Little (Nature has actually expanded the code in special cases):
Selenocysteine (#21) — encoded by a repurposed UGA stop codon, used in some enzymes
Pyrrolysine (#22) — found in some archaea
Synthetic biology labs have engineered organisms with expanded genetic codes (up to 23+ amino acids) This shows the limit isn’t absolute — but expanding it has a high evolutionary cost.
3. Can you make other non-natural amino acids? Design some new amino acids.
Using Anthropic Clude I got the following answere and did some editions: Yes - Synthetic biology labs have engineered organisms with expanded genetic codes (up to 23+ amino acids).
4. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? Using Anthropic Clude I got the following answere and did some editions: Left. The Logic: Normal L-amino acids form right-handed α-helices. This isn’t arbitrary — it’s a direct consequence of the stereochemistry at the Cα atom. D-amino acids are the mirror image of L-amino acids. Therefore, a helix built from D-amino acids is the mirror image of a normal α-helix → a left-handed helix.
5. Can you discover additional helices in proteins?
Using Anthropic Clude I got the following answere and did some editions: Yes — Several Avenues Exist. Some of those are:
I. 🤖 AI-Driven Structure Prediction
- AlphaFold2/3 and RoseTTAFold have predicted millions of structures, and researchers are data-mining them for unusual backbone geometries never seen in experimental structures. This is arguably the most promising current approach.
II. 🧪 Exotic Amino Acid Compositions
- β-peptides (backbone extended by one carbon) form helices with completely different geometries — 14-helix, 12-helix, etc. — that don’t exist in natural proteins
- D-amino acids form mirror-image helices
- Peptoids (N-substituted glycines) form their own helix families
These aren’t natural, but they reveal the chemical space of possible helices is much larger than biology uses.
III. 🔬 Cryo-EM Resolution Revolution Cryo-EM now routinely reaches 1.5–2 Å resolution, allowing detection of subtle backbone conformations that were invisible before. The π-helix was long thought to be a curiosity until cryo-EM showed it’s functionally important in ion channels and enzymes.
IV. 🧬 Extremophile & Novel Organism Proteomes Proteins from hyperthermophiles, deep-sea organisms, or newly sequenced microbial dark matter sometimes adopt unusual folds. Systematic structural surveys of these proteomes could reveal new helix types.
V. 📐 Computational Helix Hunting You can define a helix purely mathematically (repeating φ/ψ dihedral angles on the Ramachandran plot) and ask: which repeating geometries are sterically allowed but biologically unobserved? Some groups are doing exactly this.
6. Why are most molecular helices right-handed?
Using Anthropic Clude I got the following answere and did some editions: Most biological helices are right-handed because life uses L-amino acids, and the stereochemistry of L-amino acids energetically favors right-handed helical geometry.
7. Why do β-sheets tend to aggregate?
Using Anthropic Clude I got the following answere and did some editions: β-sheet aggregation it’s at the heart of some of the most devastating diseases known.
The core structure is a *ross-β spine:
- Strands run perpendicular to the fibril axis
- H-bonds run parallel to the fibril axis
- Two tightly mated β-sheets form a “steric zipper” — interdigitated side chains with almost no water inside
β-sheet aggregation is the molecular basis of a whole class of diseases:
| Disease | Protein |
|---|---|
| Alzheimer’s | Aβ peptide + Tau |
| Parkinson’s | α-synuclein |
| Type II Diabetes | IAPP (islet amyloid) |
| Prion diseases (CJD, BSE) | PrP |
| Huntington’s | Huntingtin (polyQ) |
| Systemic amyloidosis | Transthyretin, immunoglobulin light chain |
Spider silk: β-sheet aggregation may not be purely pathological — some organisms exploit it. Also, the same property that makes amyloid a disease culprit — extreme stability and cooperative assembly — makes it a remarkable material when controlled. Spider silk is a controlled amyloid-like β-sheet material with extraordinary mechanical properties
8. What is the driving force for β-sheet aggregation?
Using Anthropic Clude I got the following answere and did some editions:
The tendency to aggregate is essentially baked into β-sheet geometry:
Open edges + intermolecular H-bonding capacity + Hydrophobic stacking drive + Cooperative nucleation = Inevitable aggregation unless actively prevented
Life has spent billions of years evolving ways to suppress this tendency — and when those mechanisms fail, disease follows.
9. Why do many amyloid diseases form β-sheets?
Using Anthropic Clude I got the following answere and did some editions: This is actually a subtle question — it’s almost the inverse of the previous one. The question isn’t just why do β-sheets aggregate?" but"why do so many different, unrelated proteins converge on the same amyloid structure? Yet they all form the same cross-β amyloid architecture. Why?
In Zoom
Any polypeptide ↓ Partial unfolding (aging, mutation, stress, concentration) ↓ Exposed backbone + hydrophobic patches ↓ Nucleation (slow, rate-limiting) ↓ Cross-β amyloid — the thermodynamic sink ↓ Templated growth + cell-to-cell spreading ↓ Disease
Dobson’s view suggests that life is in a constant battle against amyloid. The entire proteostasis network — chaperones, the proteasome, autophagy, the unfolded protein response — exists largely to keep proteins away from their thermodynamic ground state. Amyloid disease is not a failure of a specific protein. It is a failure of the system that keeps all proteins from doing what they thermodynamically want to do. Aging is, in part, the slow losing of that battle.
10. Can you use amyloid β-sheets as materials?
Using Anthropic Clude I got the following answere and did some editions: Yes. The very properties that make amyloid dangerous in disease make it extraordinarily useful as a material. Some emarkable intrinsic properties are:
| Property | Value | Comparison |
|---|---|---|
| Young’s modulus | ~10–20 GPa | Similar to bone (~20 GPa) |
| Tensile strength | ~0.1–1 GPa | Comparable to steel |
| Width | 5–10 nm | Truly nanoscale |
| Persistence length | ~1–10 μm | Very stiff at nanoscale |
| Thermal stability | >100°C | Resists boiling |
| Chemical stability | Resists proteases, detergents | Extraordinary durability |
All of this self-assembles from cheap, abundant protein under mild aqueous conditions. No factory needed.
Nature already uses amyloid as a material, proving biocompatibility:
| Organism | Functional Amyloid | Purpose |
|---|---|---|
| Bacteria | Curli fibers (E. coli) | Biofilm scaffolding, surface adhesion |
| Fungi | Chaplins, hydrophobins | Waterproof spore coatings |
| Humans | Pmel17 | Melanin synthesis scaffold |
| Spiders | Silk β-sheet crystals | Structural fiber |
| Mussels | Adhesive plaques | Underwater adhesion |
| Sea cucumbers | Catch collagen | Variable-stiffness tissue |
Mussel adhesion is particularly interesting — they stick to rocks underwater using amyloid-like structures, inspiring wet adhesives for surgery and marine engineering.
🌱 Sustainability Angle
Amyloid materials are compelling from an environmental perspective:
Made from protein waste streams (whey, soy, egg white)
Self-assemble in water at mild temperatures — low energy process
Fully biodegradable under the right conditions
Could replace petroleum-based plastics in some applications
11. Design a β-sheet motif that forms a well-ordered structure
Using Copilot I got the following idea and did some editions to develop a biocuir with antimicrobial functiones:
- Turn the β‑sheet motif into an AMP-like sequence
Key AMP traits to build in: cationic, amphipathic, and sufficiently hydrophobic to interact with membranes. Frontiers Nature
A concrete β‑sheet–forming, antimicrobial‑oriented motif:
Ac–KFKFKFKF–NH2
(8‑mer, net charge +4 at neutral pH)
- Alternating pattern: Phe (F, hydrophobic/aromatic) and Lys (K, cationic) favor β‑sheet assembly and amphipathic surfaces.
- Cationic surface: Multiple Lys residues promote electrostatic attraction to negatively charged bacterial membranes and biofilms.
- End-capping: Ac-/–NH2 keeps the assembly more ordered and reduces terminal fraying.
You can use this as a “hard” antimicrobial layer interwoven with cellulose or with the more neutral structural peptide.
- Biocuir-specific hybrid motif
To keep β‑sheet order, add a short cellulose-contact and crosslinking segment:
Ac–KFKFKF–YGYG–K–NH2
- Segment 1 (AMP/β‑sheet core):
KFKFKF
Strong β‑sheet propensity, cationic, membrane-active. - Segment 2 (cellulose-contact):
YGYG
Aromatic + H‑bonding for interaction with glucan surfaces. - Segment 3 (handle):
K
Extra positive charge and a site for chemical/enzymatic crosslinking to oxidized cellulose or other polymers.
This should give you nanofibrillar β‑sheet assemblies that can both integrate into the cellulose network and present a cationic, antimicrobial surface.
- Design rules you can tune
- Net charge: Aim for overall +3 to +7 per peptide to keep strong antimicrobial character without extreme toxicity. Frontiers
- Hydrophobic fraction: Roughly 30–50% hydrophobic/aromatic residues (F, W, L, I, V) to ensure membrane insertion but avoid uncontrolled aggregation. MDPI
- β‑sheet bias: Use alternating hydrophobic/cationic pattern (e.g., K–F, K–L, R–W) to favor β‑sheets over α‑helices.
- How to test it in a biocuir context
- In solution:
- CD for β‑sheet signature (negative band ~218 nm).
- MIC/kill curves against E. coli and S. aureus to confirm antimicrobial activity.
- In the biocuir:
- Incorporate peptide during cellulose growth or post‑treat pellicles.
- Test zone of inhibition around biocuir disks and biofilm formation on the surface.
PART B of HW: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it: 🧠 Protein Arc — The Brain’s “Viral Memory”
Arc (Activity-Regulated Cytoskeleton-associated protein) Master regulator of synaptic plasticity that self-assembles into virion-like capsids that encapsulate RNAs and mediate intercellular RNA transfer in the nervous system. ARC protein is released from neurons in extracellular vesicles that mediate the transfer of ARC mRNA into new target cells, where ARC mRNA can undergo activity-dependent translation. ARC capsids are endocytosed and are able to transfer ARC mRNA into the cytoplasm of neurons. Acts as a key regulator of synaptic plasticity: required for protein synthesis-dependent forms of long-term potentiation (LTP) and depression (LTD) and for the formation of long-term memory. Regulates synaptic plasticity by promoting endocytosis of AMPA receptors (AMPARs) in response to synaptic activity: this endocytic pathway maintains levels of surface AMPARs in response to chronic changes in neuronal activity through synaptic scaling, thereby contributing to neuronal homeostasis. Acts as a postsynaptic mediator of activity-dependent synapse elimination in the developing cerebellum by mediating elimination of surplus climbing fiber synapses. Accumulates at weaker synapses, probably to prevent their undesired enhancement. This suggests that ARC-containing virion-like capsids may be required to eliminate synaptic material. Required to transduce experience into long-lasting changes in visual cortex plasticity and for long-term memory (By similarity). (https://www.uniprot.org/uniprotkb/Q7LC44/entry#sequences)
Identify the amino acid sequence of your protein:
sp|Q7LC44|ARC_HUMAN Activity-regulated cytoskeleton-associated protein OS=Homo sapiens OX=9606 GN=ARC PE=1 SV=1 MELDHRTSGGLHAYPGPRGGQVAKPNVILQIGKCRAEMLEHVRRTHRHLLAEVSKQVERE LKGLHRSVGKLESNLDGYVPTSDSQRWKKSIKACLCRCQETIANLERWVKREMHVWREVF YRLERWADRLESTGGKYPVGSESARHTVSVGVGGPESYCHEADGYDYTVSPYAITPPPAA GELPGQEPAEAQQYQPWVPGEDGQPSPGVDTQIFEDPREFLSHLEEYLRQVGGSEEYWLS QIQNHMNGPAKKWWEFKQGSVKNWVEFKKEFLQYSEGTLSREAIQRELDLPQKQGEPLDQ FLWRKRDLYQTLYVDADEEEIIQYVVGTLQPKLKRFLRHPLPKTLEQLIQRGMEVQDDLE QAAEPAGPHLPVEDEAETLTPAPNSESVASDRTQPE (https://rest.uniprot.org/uniprotkb/Q7LC44.fasta)
How long is it? 396
What is the most frequent amino acid? Glutamic Acid (E)
| Rank | Amino Acid | Count | Percentage |
|---|---|---|---|
| 🥇 1st | Glutamic Acid (E) | 46 | 11.6% |
| 2nd | Leucine (L) | 36 | 9.0% |
| 3rd | Glycine (G) | 30 | 7.5% |
| 4th | Proline (P) | 29 | 7.3% |
| 5th | Glutamine (Q) | 28 | 7.0% |
Why is Glutamic Acid (E) so abundant?
- Negatively charged at physiological pH — contributes to ARC’s electrostatic interactions
- Important for protein-protein interactions, which makes sense given ARC’s role as a synaptic scaffolding protein
- Involved in binding and signaling within the postsynaptic density
The high E content (~11.6%) is notably above its average abundance in typical human proteins (~6.4%), suggesting it plays a structural or functional role specific to ARC’s activity in synaptic plasticity and memory formation. (Anthropic Claude using the promt: What is the most frecuent Aminoacit in the ARC protein? MELDHRTSGGLHAYPGPRGGQVAKPNVILQIGKCRAEMLEHVRRTHRHLLAEVSKQVERE LKGLHRSVGKLESNLDGYVPTSDSQRWKKSIKACLCRCQETIANLERWVKREMHVWREVF YRLERWADRLESTGGKYPVGSESARHTVSVGVGGPESYCHEADGYDYTVSPYAITPPPAA)
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.: 243 (https://www.ebi.ac.uk/Tools/services/rest/ncbiblast/result/ncbiblast-R20260310-110450-0241-56195147-p2m/out)
Does your protein belong to any protein family? ARC belongs to the retroviral Gag protein superfamily (Anthroppc Calude promt: Does ARC protein belongs to any protein family?)
Structural Homology: ARC has a fold that resembles the proteins retroviruses use to form their protective protein shells (capsids).
Just like retroviral Gag proteins, ARC can: Self-assemble, Encapsulate RNA (including its own mRNA, Form extracellular vesicles, that transfer RNA between neurons — remarkably similar to how retroviruses package and deliver genetic material
Identify the structure page of your protein in RCSB: https://www.rcsb.org/structure/6YTU#entity-1
When was the structure solved? 2020-04-24
Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å): YES Resolution: 0.95 Å
Are there any other molecules in the solved structure apart from protein? YES, 1) Activity-regulated cytoskeleton-associated protein, 2) SMALL: (4S)-2-METHYL-2,4-PENTANEDIOL / C6 H14 O2 & SVTBMSDMJJWYQN-YFKPBYRVSA-N 3) SMALL (4S)-2-METHYL-2,4-PENTANEDIOL / C6 H14 O2 / SVTBMSDMJJWYQN-YFKPBYRVSA-N
Does your protein belong to any structure classification family?: Gag protein superfamily
Open the structure of your protein in any 3D molecule visualization software: (https://files.rcsb.org/header/6TN7.pdb)
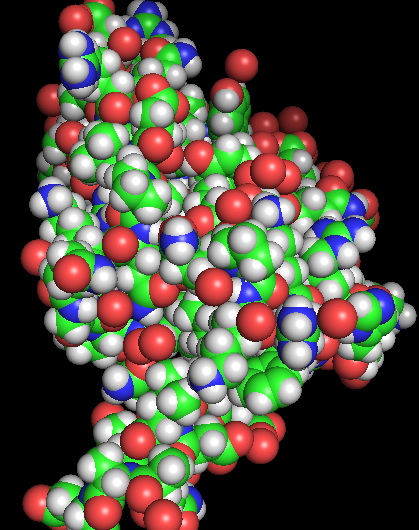
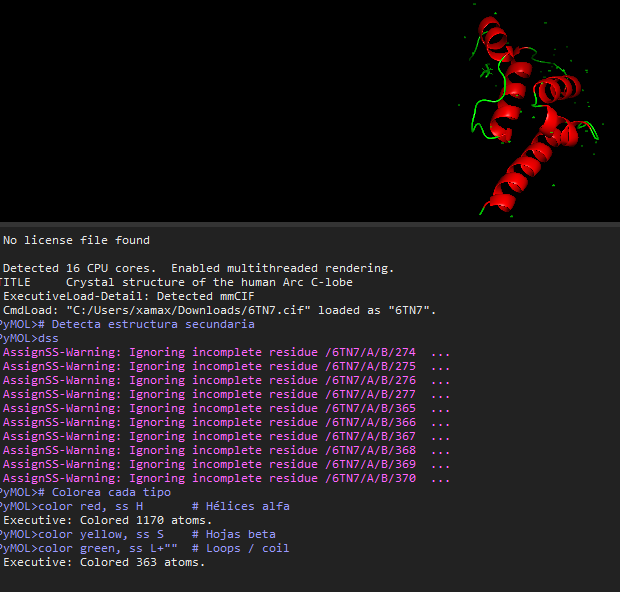
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
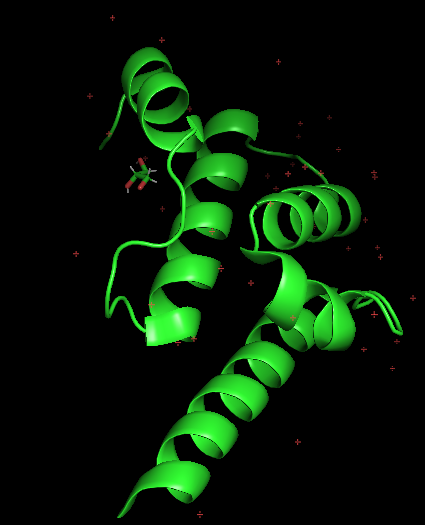

- Color the protein by secondary structure. Does it have more helices or sheets?
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)? YES (Copilot). The details about ARC’s binding cavity come directly from the structural description on the RCSB entry for 6TN7 rcsb.org.
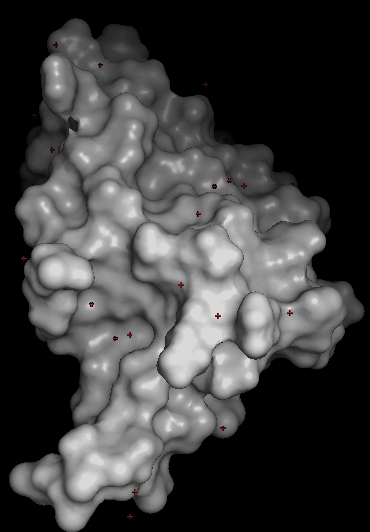
- Guide to visualizing the ARC surface in PyMOL. The surface view is the best way to inspect whether the protein has cavities or depressions (Copilot).
Basic surface visualization
This gives you a clean, semi‑transparent surface you can rotate to inspect for pockets.
🔍 Detecting cavities or “holes”
ARC does not have tunnels that pass through the protein, but it does have a binding cavity used for peptide recognition.
Automatic cavity detection in PyMOL
This highlights geometric depressions that PyMOL identifies as cavities.
Highlighting concave regions by curvature
- Blue = concave (potential pockets)
- Red = convex
🧬 Does ARC (6TN7) have holes or binding pockets? NO HOLES. The ARC’s C‑lobe is a compact capsid‑homology domain. It does not contain deep tunnels or internal cavities. YES BIDING CAVITY that recognizes short linear motifs (SLiMs) from proteins such as stargazin and GKAP. This means ARC has a functional pocket, but it is shallow and surface‑exposed, not a deep “hole.”
What the cavity is used for
The cavity binds:
- Stargazin peptides
- GKAP repeat peptides
- Other postsynaptic density motifs
This is central to ARC’s role in synaptic plasticity.
🧭 Practical PyMOL workflow to inspect ARC’s binding cavity
When you rotate the structure, the main cavity PyMOL highlights will correspond to the peptide‑binding cavity described in the RCSB entry.
PART C HOMEWORK. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling using ARC as example
Deep Mutational Scans
-Use ESM2 to generate an unsupervised deep mutational scan of your protein (ARC) based on language model likelihoods.
-Can you explain any particular pattern? (choose a residue and a mutation that stands out)
-(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
ESM2 result for ARC:
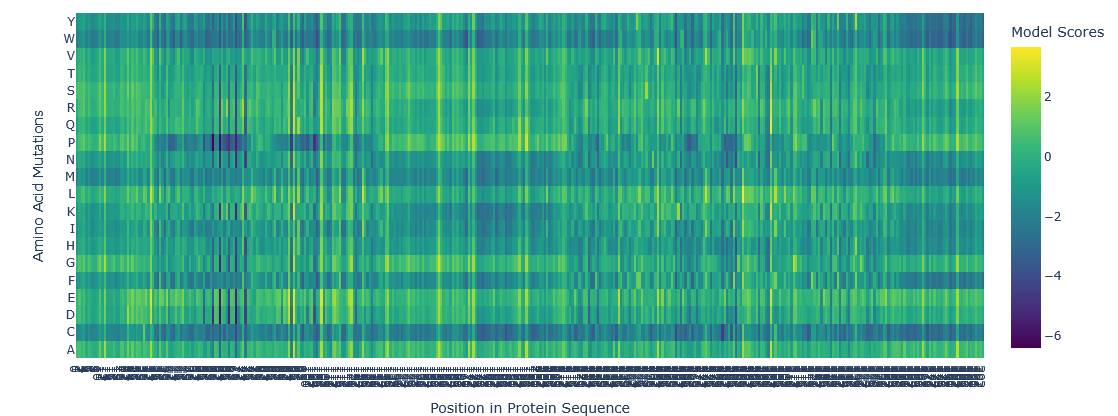
Asfar as I understand the A row in Y is more likely for the mutation to happen because is more yellow, meaning a more positive value. Whereas in the B row is less likely to happen because is blue, meaning a more negative value. This is a 2D representation of a 3D molecule, then is important to analize each position value to be sure about what is the best position for the mutation to occur."
Using the gemmini IA emmbeded in the notebook I got the following analysis about best and worst palces for mutations:
Most favorable mutation (highest LLR):
- LLR value: 3.6797
- Position in protein sequence: 290 (0-indexed, corresponding to 291 in 1-indexed view)
- Original amino acid: E
- Mutated to: L
Most unfavorable mutation (lowest LLR):
- LLR value: -6.4074
- Position in protein sequence: 60 (0-indexed, corresponding to 61 in 1-indexed view)
- Original amino acid:
- Mutated to: P
Latent Space Analysis
-Use the provided sequence dataset to embed proteins in reduced dimensionality.
-Analyze the different formed neighborhoods: do they approximate similar proteins?
-Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
 and a most unfavorable mutation with an LLR of -6.4074 (at position 60, changing an unknown original amino acid to ‘P’).
The protein exhibits a mixed resilience to mutations. While some positions show a degree of flexibility and can accommodate or even benefit from certain changes, other positions appear to be highly constrained, where specific mutations are strongly disfavored. The overall resilience would depend on the prevalence of such constrained versus flexible sites across the entire sequence. If many positions show low LLRs for most mutations, the protein would be considered less resilient.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
For the Inverse-Folding I got the following sequences:
Generating sequences…
5MBA, score=1.3477, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020 SLSAAEADLAGKSWAPVFANKNANGLDFLVALFEKFPDSANFFADFKGKSVADIKASPKLRDVSSRIFTRLNEFVNNAANAGKMSAMLSQFAKEHVGFGVGSAQFENVRSMFPGFVASVAAPPAGADAAWTKLFGLIIDALKAAGA T=0.1, sample=0, score=0.8171, seq_recovery=0.4658 ALTKEEAAKLAEAWKPFFADKEKNGKAFMLTLFAEYPELREKFPEFKGKTLEEIAASPKLTEHSMAFMDALEEFVKVADDAEKMKELLKAFAEKHVALGITAADFEKIRDIFPGFIASVAKPPEGADEAWKKLFDMVIAALKEAGG
I asked Gemini for hel to compare the predicted structure with the original and got the following answere:
Original Protein (Blue): ID=predicted_sequence_fold_552c4, PTM=0.321, pLDDT=73.364
Predicted Protein (Red): ID=predicted_sequence_fold_552c4, PTM=0.888, pLDDT=94.589
It appears that the predicted sequence, despite being significantly different from the original, yielded a structure with much higher confidence metrics (PTM and pLDDT).
pTM (predicted TM-score): The predicted protein has a significantly higher pTM (0.888 vs 0.321). A higher pTM indicates that the predicted structure of the new sequence is likely much closer to a true, stable structure than the original prediction of the sequence provided to ESMFold, or that the original sequence was a poor prediction. A pTM closer to 1 suggests a very good structural prediction, implying that the ProteinMPNN-designed sequence adopted a stable and well-defined fold. This is a very positive result, suggesting that the designed sequence is likely to fold well.
pLDDT (predicted Local Distance Difference Test): Similarly, the predicted protein has a much higher average pLDDT (94.589 vs 73.364). A pLDDT value close to 100 indicates very high confidence in the local structure of the protein. The significant increase in pLDDT suggests that the designed sequence leads to a more confidently predicted and likely more stable structure across its residues compared to the original. High pLDDT values generally correlate with well-folded and experimentally verifiable structures.
From a visual perspective, you can compare the blue (original) and red (predicted) structures. Given the large differences in pTM and pLDDT, you might observe that the red structure appears more compact or well-defined, while the blue structure might have regions of lower confidence that appear more flexible or disordered.
In summary, the ProteinMPNN-designed sequence, when folded by ESMFold, appears to result in a highly confident and presumably more stable protein structure, as indicated by the substantially improved pTM and pLDDT scores compared to the original sequence’s prediction.
Part D. Group Brainstorm on Bacteriophage Engineering
Find a group of ~3–4 students Read through the Phage Reading material listed under “Reading & Resources” below. Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why do you think those tools might help solve your chosen sub-problem?
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Include a schematic of your pipeline.
This resource may be useful: HTGAA Protein Engineering Tools
Each individually put your plan on your HTGAA website:
Proposal: Computational Stabilization of the L Protein by Preventing Protease Cleavage (With the advice of PhD Armando Garcia head of the Nanobiolab at UNAN and Copilot) Project Goal This project focuses on increasing the stability of the L protein by preventing its degradation by host proteases during recombinant expression. The working hypothesis, aligned with Armando García’s recommendations, is that a specific bacterial protease recognizes and cleaves a short motif in the L protein, leading to loss of function. By identifying this cleavage site and introducing targeted mutations that avoid protease recognition—while preserving the protein’s fold—we aim to computationally design a stabilized variant.
Computational Strategy
- Identify the protease and its cleavage motif The first step is to determine which bacterial protease is likely responsible for degrading the L protein. Sequence based protease site predictors (e.g., PROSPER, DeepProtease) will be used to scan the L protein for motifs recognized by common E. coli proteases such as Lon, ClpXP, OmpT, or DegP. This defines the minimal region where mutations should be introduced.
- Generate mutation candidates using Protein Language Models Protein Language Models (PLMs) such as ESM 2 or ProtT5 will be used to perform in silico mutagenesis at the predicted cleavage site. PLMs score each mutation by sequence likelihood, which correlates with foldability and evolutionary plausibility. Mutations that reduce protease recognition while maintaining high PLM likelihood will be prioritized. This step provides a computationally efficient filter before structural modeling.
- Predict structural effects of mutations with AlphaFold2 Following Armando García’s guidance, AlphaFold2 will be used to predict the structure of both the wild type L protein and the top PLM ranked mutants. Structural comparisons will evaluate: • RMSD between WT and mutant models • pLDDT and PAE confidence metrics • Local structural perturbations around the mutated region Only mutations that preserve the global fold and maintain high structural confidence will advance.
- Evaluate thermodynamic stability using ΔΔG calculations To quantify the stabilizing or destabilizing effect of each mutation, FoldX or Rosetta ΔΔG calculations will be performed. Mutations predicted to lower the folding free energy (ΔΔG < 0) while preserving structure will be considered strong candidates for increasing stability.
- Functional and biophysical sanity checks Final candidates will be evaluated for: • Solvent exposure and aggregation propensity • Disorder predictions • Potential disruption of functional or membrane associated regions This ensures that stabilization does not compromise biological activity.
Why These Tools Are Appropriate PLMs provide a rapid, sequence level exploration of mutational space; AlphaFold2 offers a structural safeguard to ensure mutations do not distort the fold; and ΔΔG tools quantify stability changes. Together, these methods directly address the mechanism described by García: mutate the protease sensitive region and verify that the structure remains intact.
Potential Pitfalls • AlphaFold2 limitations: If the L protein is highly novel or disordered, structural predictions may be unreliable. • Mutation induced misfolding: Some mutations may block protease recognition but still disrupt the fold, reducing stability rather than improving it. • Context dependence: Protease cleavage may depend on local flexibility or exposure, not just sequence, complicating prediction.
Pipeline Schematic WT L protein sequence | v [Protease-site Prediction]
- Identify likely bacterial protease cleavage motifs | v [Protein Language Models]
- Mutate cleavage site
- Rank mutations by sequence likelihood | v [AlphaFold2 Modeling]
- Predict WT and mutant structures
- Compare RMSD, pLDDT, PAE | v [ΔΔG Stability Scoring]
- FoldX/Rosetta evaluation
- Select stabilizing, structure-preserving variants | v [Biophysical Filters]
- Aggregation, disorder, solvent exposure | v Shortlist of stabilized L protein variants
Include your group’s short plan for engineering a bacteriophage (I was not able to work with a Nodo´s group due to timeshortage and probles for coordinating with my team)