Max Matus is a Zapotec indigenous scholar. He is the director of the Cultural Studies Department at El Colegio de la Frontera Norte where he also coordinates Xquenda_lab, a laboratory for the co-design of indigenous digital technologies to empower Pluriverses.
Project: Antimicrobial Biocuir Based on Grape Pomace Enhanced with Metabolites Produced Through Synthetic Biology Techniques
Xquenda_Lab, the Indigenous Digital Innovation Laboratory affiliated with El Colegio de la Frontera Norte (El Colef) in Tijuana, Mexico, has been developing since 2025 a series of biocuir recipes using grape pomace discarded from the wine industry at the Guadalupe Valley in Baja California. In coordination with the Autonomous University of Baja California (UABC), mechanical and bacteriological tests are being conducted on the material with the goal of achieving a recipe suitable for joint patenting by both institutions, intended for the exclusive use of the Kumiai Indigenous community. The aim is to support the creation of a social and solidarity-based enterprise led by Kumiai indigenous from the community of San Antonio Necua for the production and commercialization of grape-pomace biocuir. It is considered that the development of metabolites through synthetic biology techniques—later incorporated into the biocuir for antimicrobial purposes—would add value to the final product in the market while simultaneously improving community health.
Project: Antimicrobial Biocuir Based on Grape Pomace Enhanced with Metabolites Produced Through Synthetic Biology Techniques
Xquenda_Lab, the Indigenous Digital Innovation Laboratory affiliated with El Colegio de la Frontera Norte (El Colef) in Tijuana, Mexico, has been developing since 2025 a series of biocuir recipes using grape pomace discarded from the wine industry at the Guadalupe Valley in Baja California. In coordination with the Autonomous University of Baja California (UABC), mechanical and bacteriological tests are being conducted on the material with the goal of achieving a recipe suitable for joint patenting by both institutions, intended for the exclusive use of the Kumiai Indigenous community.
The aim is to support the creation of a social and solidarity-based enterprise led by Kumiai indigenous from the community of San Antonio Necua for the production and commercialization of grape-pomace biocuir. It is considered that the development of metabolites through synthetic biology techniques—later incorporated into the biocuir for antimicrobial purposes—would add value to the final product in the market while simultaneously improving community health.
Group Final Project
Homeworks
Subsections of Homeworks
Week 1: Principles and Practices
General Objective
To develop a grape-pomace biocuir with antimicrobial properties through the incorporation of metabolites produced using synthetic biology techniques, intended for the fabrication of tablecloths, kitchen coverings, and bathmats to improve community health among the Kumiai Indigenous population in San Antonio Necua, Baja California, Mexico.
Objective of Introducing Antimicrobial Metabolites
To incorporate antimicrobial metabolites into the grape-pomace biocuir developed by Xquenda_Lab at El Colef, in collaboration with the Autonomous University of Baja California (UABC) and the Kumiai community of San Antonio Necua.
Concept and expected outcome of the project (Copilot prompt: how to incorporate synthetic biology principles into the production of an antimicrobial biocuir based on grape pomace using the following biocuir recipes developed by Xqueda_lab)
The project develops a biocuir made from grape pomace, reinforced with natural biopolymers (gelatin, starch, glycerin, natural latex) and enriched with antimicrobial metabolites produced through synthetic biology principles in wet lab facilities at Autonomous University of Baja California (UABC).
These metabolites—purified and microencapsulated—are incorporated into the orujo based bioleather to create protective surfaces that reduce bacterial and fungal proliferation in rural domestic environments.
The antimicrobial Metabolites developed and introduced into the biocuir will be:
• For kitchen surfaces (gastrointestinal risks):
o Lactic acid
o Acetic acid
o Mild terpenes (limonene, low dose carvacrol)
• For bathroom and humid surfaces (respiratory and fungal risks):
o Biosurfactants (surfactin or sophorolipids)
o Antifungal terpenes (thymol, carvacrol)
These compounds will be produced using GRAS microorganisms genetically engineered in UABC wet laboratory. Only purified metabolites are transferred to Xquenda_lab and the Kumiai community to be incorporated into the biocuir -never living organisms.
Expected Outcome.
A community oriented antimicrobial orujo based biocuir, produced by Kumiai indigenous people in Xquenda_Lab – El Colef facilities at the Gadalupe Valley, in coordination with researchers from UABC. The biocuir will be used as:
• Antimicrobial tablecloths
• Kitchen surface covers
• Bathroom mats
• Protective domestic surfaces
The antimicrobial orujo based biocuir is:
• Microbially protective
• Biodegradable and safe
• Adaptable and manufacturable by the Kumiai community in Xquenda_lab - El Colef facilities at the Gaudalupe Valley.
• A vehicle for citizen science learning, community health improvement, and social entrepreneurship owned by the Kumiai community from San Antonio Necua and Xquenda_lab.
GOVERNANCE AND ETHICS PROPOSAL (Copilot prompt: how to devrlop a governance system including Xquenda_lab, El colef, the Kumiai community and UABC as stakeholders to make a safe and ethical production of the Antimicrobial Biocuir Based on Grape Pomace making use of Synthetic Biology techniques to produce Metabolites)
Synthetic Biology for Community Benefit in an Indigenous Context
A. Guiding Principles
• Kumiai autonomy and collective decision making
• Precaution and non harm
• Radical transparency
• Technology transfer
• Epistemic equity (Indigenous knowledge = scientific knowledge)
• Community benefit and shared outcomes
• Respect for cultural identity and territorial rights
B. Actors and Responsibilities
Kumiai Community of San Antonio Necua
• Collective decision making
• Defining needs, uses, and priorities
• Cultural and practical evaluation of prototypes
• Right to pause, modify, or redirect the project
Xquenda_Lab (Public and Citizen Laboratory)
• Space for technology transfer
• Training in biomaterials and basic synthetic biology principles
• Community based production of biocuir
• Accessible documentation
UABC (Biologists, Engineers, Biotechnologists)
• Genetic design and metabolite production
• Exclusive handling of genetically modified organisms
• Purification and characterization of metabolites
• Ensuring biosafety and regulatory compliance
Project Governance Council
• Kumiai representatives
• Xquenda_Lab researchers
• UABC researchers
• El Colef observer
Functions of the Governance council:
• Approve project directions
• Evaluate risks and benefits
• Oversee technology transfer
• Validate educational materials
• Resolve ethical questions
Biosecurity and Scientific Ethics Committee
• Review genetic constructs and metabolic pathways
• Supervise handling of organisms in wet lab settings
• Ensure safe disposal of biological waste
• Continuous risk assessment
C. Key Governance Processes
Collective Informed Consent
• Open assemblies
• Clear explanation of synthetic biology and metabolites
• Recorded community agreements
Technology Transfer
• Hands on training in Xquenda_Lab
• Accessible manuals
• Formation of local “technology stewards”
Transparency and Communication
• Open information on processes, risks, and results
• Pedagogical materials in clear language
Intellectual Property and Data
• Explicit recognition of Kumiai contributions
• Avoid exclusionary patents
• Explore communitarian, co ownership or open licenses
• Protection of sensitive cultural knowledge
Community Benefits
• Priority use in Kumiai comunities and households
• Potential Kumiai/Xquenda product line
• Fair distribution of income if commercialization occurs
Note: This is just a firt conceptual approach. I understand that might be very difficult to produce those metabolites. Therefore I might have to choose only one to produce it in large scale in order to encapsulated and include it into the biocuir.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
*
• By helping respond
Foster Lab Safety
• By preventing incident
*
• By helping respond
Protect the environment
• By preventing incidents
*
• By helping respond
Other considerations
• Minimizing costs and burdens to stakeholders
*
• Feasibility?
*
• Not impede research
*
• Promote constructive applications
*
Preparation for lecture 2:
Homework Questions from Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy? There are difere types of polomerase qualities and all of them have some kind of error. The leng of human genome is about 3.2 nillion pairs. Therefore corrections has to be made in any edition. Synthetic Biology deals with this big discrepancy by making corrections. For example a 10ns Base addition to the 1:10 error rate in the case of correcting Polimerase error in Biology synthesis as is shown in the slide 8 of the presentation.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest? There are two main different ways when it comes to code DNC: Chip based and non-chip based. In the case of the former many types of erros may happen depending of the choosen method; e.g. correction, reduction.
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently?
Direct synthesis using wall plate. 96 wale plate makes 1 gen. With a silicon platform it is possible to have 1M oligos in a chip to make 9.600 genes.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because chemical DNA synthesis is not perfectly efficient, and small errors accumulate with every added nucleotide. After ~200 cycles, the product becomes too impure and too heterogeneous to be useful (Direct answer from Copilot)
Why can’t you make a 2000bp gene via direct oligo synthesis?
Because chemical synthesis cannot reliably produce a single, accurate 2000 base molecule. The error rate and truncation rate make it chemically impossible (Direct answer from Copilot)
Homework Question from George Church:
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The “PVT TIM HALL” (Direct answer from Gemini to the question What are the 10 essential amino acids in all animals)
The “PVT” Group
• P – Phenylalanine: This is a precursor for signaling molecules like dopamine and adrenaline; it’s basically a building block for the “brain chemicals” that keep an animal alert.
• V – Valine: One of the three “branched-chain” amino acids (BCAAs). It focuses on muscle growth, tissue repair, and providing extra energy when the body is stressed.
• T – Threonine: This is a major component of structural proteins like collagen and elastin. It’s also vital for maintaining a healthy gut lining.
The “TIM” Group
• T – Tryptophan: Best known as the precursor to serotonin. It helps regulate mood, sleep, and even the “fullness” feeling after eating.
• I – Isoleucine: This BCAA is heavily concentrated in muscle tissue and is essential for producing hemoglobin (the stuff in blood that carries oxygen).
• M – Methionine: A sulfur-rich amino acid that is crucial for metabolism, detoxifying the liver, and helping the body absorb minerals like zinc.
The “HALL” Group
• H – Histidine: This is used to produce histamine, which is vital for the immune response, digestion, and maintaining the protective “sheaths” around nerve cells.
• A – Arginine: While adult humans can make some, most animals (and human infants) need it from food to help remove toxic ammonia from the body via the urea cycle.
• L – Leucine: Often called the “spark plug” for protein synthesis, this is the main amino acid that tells the body to start building and repairing muscle.
• L – Lysine: This one is critical for bone health, the production of antibodies to fight off sickness, and the overall creation of enzymes and hormones.
I believe the Lysine contingency hypothesis is very interesting if we think about it in relation to the Anthropocene and all the radical changes that are occurring in the world due to human activity. To think about the future where sources provide one of those 10 essential amino acids triggers some questions about how to synthetize and produce in a very efficient way some alternative source to get those amino acids. It also triggers some questions about the possibilities for humans to inhabit other planets and the importance of being sure about how to create an artificial environment able to produce in an efficient and sustainable way sources for those 10 essential amino acids.
[Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions?
Week 2: DNA Read Write and Edit
Part 0: Basics of Gel Electrophoresis
I watch the lecture on live and the recorded recitation video, However, when I wanted to review rhw recorded video ofr the lecture the link did not exist on the HTGAA 2026 page. However, we got via email a recording of the 2025 lecture. I also visited the Nanobiolab at UMAN in Mexico City were aI was able to follow an introduction to safe mesurement in wetlabs and the Electrophoresis machinery.
Part 1: Benchling & In-silico Gel Art
Intructions: See this week’s lab protocol “Gel Art: Restriction Digests and Gel Electrophoresis” for details. Overview:
Make a free account at benchling.com [Maxmatusbenchling](https://benchling.com/organizations/maxmatus)
Import the Lambda DNA (https://www.neb.com/en/-/media/nebus/page-images/tools-and-resources/interactive-tools/dna-sequences-and-maps/text-documents/lambdagbk.txt?rev=50c75f4579114750a9ad75d892d7d118&hash=EF15DDE468761F64D50E30418917B08D).
Simulate Restriction Enzyme Digestion with the following Enzymes:(https://benchling.com/xquenda_lab/enzyme-lists/22892)
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
You might find Ronan’s website a helpful tool for quickly iterating on designs!
**Part 3: DNA Design Challenge**
I choosed the protein P04439 · HLAA_HUMAN from the UniProt Database (https://www.uniprot.org/uniprotkb?query=Human)
I decide to choose this protein becouse it relates with human inmmunity and my finnal project is about how to develop a biomaterial to protect humans from some bacterias, so I found interesting this protein as starting point. Some of the characteristics assosiated to this protein according to UniProt are:
HLA class I histocompatibility antigen, A alpha chain · Gene: HLA-A (HLAA) · Homo sapiens (Human) · 365 amino acids · Evidence at protein level
The sequence of the P04439 · HLAA_HUMAN protein is:
>sp|O95905|ECD_HUMAN Protein ecdysoneless homolog OS=Homo sapiens OX=9606 GN=ECD PE=1 SV=1
Results for 644 residue sequence “sp|O95905|ECD_HUMAN Protein ecdysoneless homolog OS=Homo sapiens OX=9606 GN=ECD PE=1 SV=1” starting “MEETMKLATM”
Reverse translation of sp|O95905|ECD_HUMAN Protein ecdysoneless homolog OS=Homo sapiens OX=9606 GN=ECD PE=1 SV=1 to a 1932 base sequence of most likely codons.
atggaagaaaccatgaaactggcgaccatggaagataccgtggaatattgcctgtttctg
attccggatgaaagccgcgatagcgataaacataaagaaattctgcagaaatatattgaa
cgcattattacccgctttgcgccgatgctggtgccgtatatttggcagaaccagccgttt
aacctgaaatataaaccgggcaaaggcggcgtgccggcgcatatgtttggcgtgaccaaa
tttggcgataacattgaagatgaatggtttattgtgtatgtgattaaacagattaccaaa
gaatttccggaactggtggcgcgcattgaagataacgatggcgaatttctgctgattgaa
gcggcggattttctgccgaaatggctggatccggaaaacagcaccaaccgcgtgtttttt
tgccatggcgaactgtgcattattccggcgccgcgcaaaagcggcgcggaaagctggctg
ccgaccaccccgccgaccattccgcaggcgctgaacattattaccgcgcatagcgaaaaa
attctggcgagcgaaagcattcgcgcggcggtgaaccgccgcattcgcggctatccggaa
aaaattcaggcgagcctgcatcgcgcgcattgctttctgccggcgggcattgtggcggtg
ctgaaacagcgcccgcgcctggtggcggcggcggtgcaggcgttttatctgcgcgatccg
attgatctgcgcgcgtgccgcgtgtttaaaacctttctgccggaaacccgcattatgacc
agcgtgacctttaccaaatgcctgtatgcgcagctggtgcagcagcgctttgtgccggat
cgccgcagcggctatcgcctgccgccgccgagcgatccgcagtatcgcgcgcatgaactg
ggcatgaaactggcgcatggctttgaaattctgtgcagcaaatgcagcccgcattttagc
gattgcaaaaaaagcctggtgaccgcgagcccgctgtgggcgagctttctggaaagcctg
aaaaaaaacgattattttaaaggcctgattgaaggcagcgcgcagtatcgcgaacgcctg
gaaatggcggaaaactattttcagctgagcgtggattggccggaaagcagcctggcgatg
agcccgggcgaagaaattctgaccctgctgcagaccattccgtttgatattgaagatctg
aaaaaagaagcggcgaacctgccgccggaagatgatgatcagtggctggatctgagcccg
gatcagctggatcagctgctgcaggaagcggtgggcaaaaaagaaagcgaaagcgtgagc
aaagaagaaaaagaacagaactatgatctgaccgaagtgagcgaaagcatgaaagcgttt
attagcaaagtgagcacccataaaggcgcggaactgccgcgcgaaccgagcgaagcgccg
attacctttgatgcggatagctttctgaactattttgataaaattctgggcccgcgcccg
aacgaaagcgatagcgatgatctggatgatgaagattttgaatgcctggatagcgatgat
gatctggattttgaaacccatgaaccgggcgaagaagcgagcctgaaaggcaccctggat
aacctgaaaagctatatggcgcagatggatcaggaactggcgcatacctgcattagcaaa
agctttaccacccgcaaccaggtggaaccggtgagccagaccaccgataacaacagcgat
gaagaagatagcggcaccggcgaaagcgtgatggcgccggtggatgtggatctgaacctg
gtgagcaacattctggaaagctatagcagccaggcgggcctggcgggcccggcgagcaac
ctgctgcagagcatgggcgtgcagctgccggataacaccgatcatcgcccgaccagcaaa
ccgaccaaaaac
Reverse translation of sp|O95905|ECD_HUMAN Protein ecdysoneless homolog OS=Homo sapiens OX=9606 GN=ECD PE=1 SV=1 to a 1932 base sequence of consensus codons.
atggargaracnatgaarytngcnacnatggargayacngtngartaytgyytnttyytn
athccngaygarwsnmgngaywsngayaarcayaargarathytncaraartayathgar
mgnathathacnmgnttygcnccnatgytngtnccntayathtggcaraaycarccntty
aayytnaartayaarccnggnaarggnggngtnccngcncayatgttyggngtnacnaar
ttyggngayaayathgargaygartggttyathgtntaygtnathaarcarathacnaar
garttyccngarytngtngcnmgnathgargayaaygayggngarttyytnytnathgar
gcngcngayttyytnccnaartggytngayccngaraaywsnacnaaymgngtnttytty
tgycayggngarytntgyathathccngcnccnmgnaarwsnggngcngarwsntggytn
ccnacnacnccnccnacnathccncargcnytnaayathathacngcncaywsngaraar
athytngcnwsngarwsnathmgngcngcngtnaaymgnmgnathmgnggntayccngar
aarathcargcnwsnytncaymgngcncaytgyttyytnccngcnggnathgtngcngtn
ytnaarcarmgnccnmgnytngtngcngcngcngtncargcnttytayytnmgngayccn
athgayytnmgngcntgymgngtnttyaaracnttyytnccngaracnmgnathatgacn
wsngtnacnttyacnaartgyytntaygcncarytngtncarcarmgnttygtnccngay
mgnmgnwsnggntaymgnytnccnccnccnwsngayccncartaymgngcncaygarytn
ggnatgaarytngcncayggnttygarathytntgywsnaartgywsnccncayttywsn
gaytgyaaraarwsnytngtnacngcnwsnccnytntgggcnwsnttyytngarwsnytn
aaraaraaygaytayttyaarggnytnathgarggnwsngcncartaymgngarmgnytn
garatggcngaraaytayttycarytnwsngtngaytggccngarwsnwsnytngcnatg
wsnccnggngargarathytnacnytnytncaracnathccnttygayathgargayytn
aaraargargcngcnaayytnccnccngargaygaygaycartggytngayytnwsnccn
gaycarytngaycarytnytncargargcngtnggnaaraargarwsngarwsngtnwsn
aargargaraargarcaraaytaygayytnacngargtnwsngarwsnatgaargcntty
athwsnaargtnwsnacncayaarggngcngarytnccnmgngarccnwsngargcnccn
athacnttygaygcngaywsnttyytnaaytayttygayaarathytnggnccnmgnccn
aaygarwsngaywsngaygayytngaygaygargayttygartgyytngaywsngaygay
gayytngayttygaracncaygarccnggngargargcnwsnytnaarggnacnytngay
aayytnaarwsntayatggcncaratggaycargarytngcncayacntgyathwsnaar
wsnttyacnacnmgnaaycargtngarccngtnwsncaracnacngayaayaaywsngay
gargargaywsnggnacnggngarwsngtnatggcnccngtngaygtngayytnaayytn
gtnwsnaayathytngarwsntaywsnwsncargcnggnytngcnggnccngcnwsnaay
ytnytncarwsnatgggngtncarytnccngayaayacngaycaymgnccnacnwsnaar
ccnacnaaraay
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
To answere this question I used Copilot and privided the sequence that I got from the reverse translation of sp|O95905|ECD_HUMAN Protein. The reply was as follow:
Cell‑dependent systems:
Bacterial expression (e.g., E. coli) using a plasmid vector with a strong promoter (like T7).
Yeast or insect cells for eukaryotic expression.
Mammalian cells (e.g., HEK293, CHO) if proper human‑like folding or modifications are important.
Cell‑free systems:
E. coli lysate–based systems,
Wheat germ or rabbit reticulocyte lysates,
Or a PURE system with purified transcription/translation components.
In all cases, the same basic logic applies: the DNA encodes the amino‑acid sequence of ECD via triplet codons.
In my understanding the most easy and adfordable way to produce this protein would be a cell‑dependent systems in E. coli using a plasmid vector with a strong promoter. However, I still have to make a critic analyzis of this Copilot answere according to the most uptated literature.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
Maxmatusbenchling
I created a Twist account with my personal mail, however, when I am trying to login a window pop-up asking me to contact my local privider in Mexico. Nevertheless, it looks like the webpage of the local provider is not working, I tryed to charg it many times unsuscesfully, so I was not able to finish this secction of the homework: [Twist_Mexico] (https://ecommerce.twistdna.com/www.abalat.com.mx)
4.2. Build Your DNA Insert Sequence
4.3. On Twist, Select The “Genes” Option
4.4. Select “Clonal Genes” option
4.5. Import your sequence
4.6. Choose Your Vector
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would like to sequence the grape pomace DNA in order to understan if there are some antibacterial proteins in it.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
What is the output of your chosen sequencing technology?
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
If I find antibacterial proteins in grape pomenace I would like to mass produce it in order to enhance this property and then insert it into a biocuir made out of grape pomenace. I found this web page wich seems to be very usefull for my project: Grape_genome and also an interesting article related to the use of pomenace as antimicrobial in feed: Antimicrobial_pomenace
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
5.3 DNA Edit
(i) What DNA would you want to edit and why?
Not sure yet
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Week 3: Lab Automation
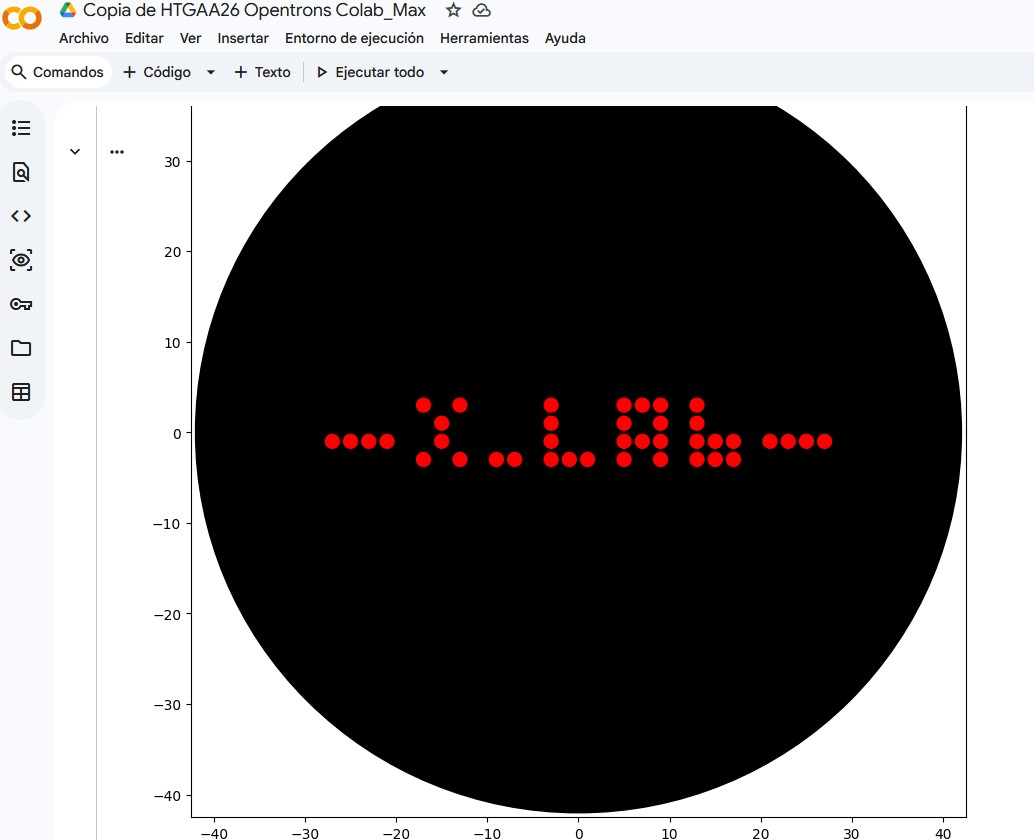
I. Assignment: Python Script for Opentrons Artwork: Max_XLAB_pattern
def location_of_color(color_string):
for well,color in well_colors.items():
if color.lower() == color_string.lower():
return color_plate[well]
raise ValueError(f"No well found with color {color_string}")
dispense_and_detach(pipette, 1, loc)
def dispense_and_detach(pipette, volume, location):
"""
Move laterally 5mm above the plate (to avoid smearing a drop); then drop down to the plate,
dispense, move back up 5mm to detach drop, and stay high to be ready for next lateral move.
5mm because a 4uL drop is 2mm diameter; and a 2deg tilt in the agar pour is >3mm difference across a plate.
"""
assert(isinstance(volume, (int, float)))
above_location = location.move(types.Point(z=location.point.z + 5)) # 5mm above
pipette.move_to(above_location) # Go to 5mm above the dispensing location
pipette.dispense(volume, location) # Go straight downwards and dispense
pipette.move_to(above_location) # Go straight up to detach drop and stay high
center_location = agar_plate[‘A1’].top()
cell_well = color_plate[‘A1’] # Changed ‘F1’ to ‘A1’ because F1 was not configured to have a color.
pipette_20ul.pick_up_tip()
X_LABpattern
X_LABpattern = [
# X
(0, 4), (2, 4), # Fila superior
(1, 3), # Diagonal bajando
(1, 2), # Centro
(0, 1), (2, 1), # Fila inferior
# First dot after X - moved from (3,1) to (4,1)
(4, 1),
# Second dot for separation - moved from (4,1) to (5,1)
(5, 1),
# L (shifted by 2 units from previous adjustment: 5+2=7)
(7, 4),
(7, 3),
(7, 2),
(7, 1), (8, 1), (9, 1),
# Espacio entre letras (col 10 vacía)
# A (shifted by 2 units from previous adjustment: 9+2=11)
(11, 1), (11, 2), (11, 3), (11, 4),
(12, 4),
(13, 4), (13, 3), (13, 2), (13, 1),
(12, 2),
# Espacio entre letras (col 14 vacía)
# b (minúscula) (shifted by 2 units from previous adjustment: 13+2=15)
(15, 1), (15, 2), (15, 3), (15, 4),
(16, 2),
(16, 1),
(17, 2),
(17, 1),
]
Determine pattern dimensions for scaling and centering
x_coords = [p[0] for p in X_LABpattern]
y_coords = [p[1] for p in X_LABpattern]
Scaling factor: Adjust this to control the size of your pattern on the plate
Each unit in X_LABpattern will represent ‘scale_factor’ mm on the plate.
For a 30mm wide pattern with the current 15 unit width: 30 / (15 - 0) = 2
scale_factor = 2.0 # mm per unit
Volume to dispense per dot
dispense_volume = 1 # uL
Aspiration tracking
aspirated_volume = 0
max_aspirate_volume = 20 # uL (matching the example’s approach)
for i, (px, py) in enumerate(X_LABpattern):
# If the pipette is empty or low on liquid, aspirate more
# We aspirate max_aspirate_volume or whatever is needed for the remaining dots
if aspirated_volume < dispense_volume:
volume_to_aspirate = min(max_aspirate_volume, (len(X_LABpattern) - i) * dispense_volume - aspirated_volume)
if volume_to_aspirate > 0:
pipette_20ul.aspirate(volume_to_aspirate, cell_well)
aspirated_volume += volume_to_aspirate
# Shift and scale the pattern coordinates
# The agar_plate['A1'].top() is the center, so we shift relative to that.
shifted_px = (px - center_pattern_x_unit) * scale_factor
shifted_py = (py - center_pattern_y_unit) * scale_factor
# Calculate the absolute position on the agar plate
adjusted_location = center_location.move(types.Point(x=shifted_px, y=shifted_py))
# Dispense the liquid using the helper function
dispense_and_detach(pipette_20ul, dispense_volume, adjusted_location)
aspirated_volume -= dispense_volume
Drop the tip at the end
pipette_20ul.drop_tip()
“”"### Protocolo Max_XLab_pattern
Este protocolo utiliza la plataforma Opentrons para patronar un diseño ‘XLab’ en una placa de agar. A continuación, se detalla el código completo:
Descripción general del funcionamiento:
Carga de laboratorio: Se configuran las puntas, la pipeta y el módulo de temperatura, junto con las placas de colores y agar.
Funciones auxiliares: Se definen location_of_color para obtener la ubicación de un color específico y dispense_and_detach para una dispensación limpia.
Definición del patrón X_LABpattern: Una lista de coordenadas (x, y) que forman el diseño ‘X LAb’. Este patrón incluye la separación solicitada entre ‘X’, los puntos y ‘L’.
Cálculo de escalado y centrado: El código calcula las dimensiones del patrón para escalarlo y centrarlo en la placa de agar.
Aspiración y dispensación: Itera sobre cada punto del patrón. La pipeta aspira líquido (‘Red’ de la posición ‘A1’) en bloques de 20 uL y dispensa 1 uL en cada coordenada ajustada en la placa de agar, utilizando la función dispense_and_detach para evitar manchas.
Descarte de punta: Al finalizar, la pipeta desecha la punta usada.
"""
pass this e.g. ‘Red’ and get back a Location which can be passed to aspirate()
def location_of_color(color_string):
for well,color in well_colors.items():
if color.lower() == color_string.lower():
return color_plate[well]
raise ValueError(f"No well found with color {color_string}")
For this lab, instead of calling pipette.dispense(1, loc) use this: dispense_and_detach(pipette, 1, loc)
def dispense_and_detach(pipette, volume, location):
"""
Move laterally 5mm above the plate (to avoid smearing a drop); then drop down to the plate,
dispense, move back up 5mm to detach drop, and stay high to be ready for next lateral move.
5mm because a 4uL drop is 2mm diameter; and a 2deg tilt in the agar pour is >3mm difference across a plate.
"""
assert(isinstance(volume, (int, float)))
above_location = location.move(types.Point(z=location.point.z + 5)) # 5mm above
pipette.move_to(above_location) # Go to 5mm above the dispensing location
pipette.dispense(volume, location) # Go straight downwards and dispense
pipette.move_to(above_location) # Go straight up to detach drop and stay high
YOUR CODE HERE to create your design
center_location = agar_plate[‘A1’].top()
cell_well = color_plate[‘A1’] # Changed ‘F1’ to ‘A1’ because F1 was not configured to have a color.
# X
(0, 4), (2, 4), # Fila superior
(1, 3), # Diagonal bajando
(1, 2), # Centro
(0, 1), (2, 1), # Fila inferior
# First dot after X - moved from (3,1) to (4,1)
(4, 1),
# Second dot for separation - moved from (4,1) to (5,1)
(5, 1),
# L (shifted by 2 units from previous adjustment: 5+2=7)
(7, 4),
(7, 3),
(7, 2),
(7, 1), (8, 1), (9, 1),
# Espacio entre letras (col 10 vacía)
# A (shifted by 2 units from previous adjustment: 9+2=11)
(11, 1), (11, 2), (11, 3), (11, 4),
(12, 4),
(13, 4), (13, 3), (13, 2), (13, 1),
(12, 2),
# Espacio entre letras (col 14 vacía)
# b (minúscula) (shifted by 2 units from previous adjustment: 13+2=15)
(15, 1), (15, 2), (15, 3), (15, 4),
(16, 2),
(16, 1),
(17, 2),
(17, 1),
# Línea horizontal derecha (puntos en fila central, columnas después de la b)
(19, 2), (20, 2), (21, 2), (22, 2),
]
Determine pattern dimensions for scaling and centering
x_coords = [p[0] for p in X_LABpattern]
y_coords = [p[1] for p in X_LABpattern]
Scaling factor: Adjust this to control the size of your pattern on the plate
Each unit in X_LABpattern will represent ‘scale_factor’ mm on the plate.
For a 30mm wide pattern with the current 15 unit width: 30 / (15 - 0) = 2
scale_factor = 2.0 # mm per unit
Volume to dispense per dot
dispense_volume = 1 # uL
Aspiration tracking
aspirated_volume = 0
max_aspirate_volume = 20 # uL (matching the example’s approach)
for i, (px, py) in enumerate(X_LABpattern):
# If the pipette is empty or low on liquid, aspirate more
# We aspirate max_aspirate_volume or whatever is needed for the remaining dots
if aspirated_volume < dispense_volume:
volume_to_aspirate = min(max_aspirate_volume, (len(X_LABpattern) - i) * dispense_volume - aspirated_volume)
if volume_to_aspirate > 0:
pipette_20ul.aspirate(volume_to_aspirate, cell_well)
aspirated_volume += volume_to_aspirate
# Shift and scale the pattern coordinates
# The agar_plate['A1'].top() is the center, so we shift relative to that.
shifted_px = (px - center_pattern_x_unit) * scale_factor
shifted_py = (py - center_pattern_y_unit) * scale_factor
# Calculate the absolute position on the agar plate
adjusted_location = center_location.move(types.Point(x=shifted_px, y=shifted_py))
# Dispense the liquid using the helper function
dispense_and_detach(pipette_20ul, dispense_volume, adjusted_location)
aspirated_volume -= dispense_volume
Drop the tip at the end
pipette_20ul.drop_tip()
Execute Simulation / Visualization – don’t change this code block
I choose this article because is very accesible and usefull for a non specialized public, udergrads or people that is starting to learn how to use Opentrons and automation toos for biological applicatios. This is the abstract:
Most members of the synthetic biology community, particularly plant scientists, lack access to
liquid handling robots to scale up experiments, enhance reproducibility, and accelerate the
Design, Build, Test, Learn cycle. Biofoundries enable high throughput data acquisition to train AI
models and to develop new bioproducts, but they are capital-intensive to set up and not widely
distributed. Entry-level, 3D-printed robots offer more affordable alternatives, but suffer from a
shortage of validated protocols that can be modified without prior coding experience. To
enhance access to biological automation, we developed a collection of modular BOTany
Methods using Opentrons OT-2 robots to streamline the most common methods for molecular
biology research and education. Our comprehensive workflow offers automation for a variety of
procedures, ranging from simple but repetitive tasks (such as primer dilution and PCR setup) to
more complex operations, including Plant Modular Cloning (MoClo), bacterial transformation,
and plasmid extraction. Our BOTany Methods enable undergraduate students and other early
career researchers to run designer experiments using table-based inputs, without editing the
custom Python scripts. This pipeline enables end-to-end molecular cloning with minimal user
intervention, enhancing throughput and traceability for synthetic biology applications.
III. Write a description about what you intend to do with automation tools for your final project
To produce an antimicrobial biocuir using grape pomenace. The main goal is to enhance the antibacterial capabilities of the biocuir with syntetic byology principles. Firs option will be to search if there are some elements in the grape pomenace that are it self antimicrobial. If so, we will enhance those capabilities ant produce them in high quantities to become a main component of the biocuir. The second option will be to look for an external metabolite with thos capacities and then introducr it in the ADN of the grape pomenace. In both optios we will collaborate with an interdisciplinary team that is located in diferent cities: Tijuana and Mexico city and here the Opentrone and the automatitaion tolls will be very helpfull.
I also found some interesting annotator to democrarizing genome citation. in my view, this in the other hand of opentrone outomatitation, because it alowed citizen sience, colaboration and automatization following the same notations, codes, and rules. The name of this toll is Apollo Genome Annotation Editor: http://138.102.159.70:8080/apollo/about/ Dunn, N. A. et al. Apollo: Democratizing genome annotation. PLoS Comput. Biol. 15, e1006790 (2019)
The Apollo Genome Annotation Editor is used by the Grapepedia team, wich focuses on researching grepes´s genome: https://grapedia.org/genomes/. I belive that the research developed so far by the Grapepedia proyect will be very helpfull for my own proyect, since they have advenced so much on the grape´s genome research. Hence, my interes focuses especifically on the grape´s pomenance in order to develop a pomenance biocuir with enhanced antibacterial properties using some principles of synthetic biology.
3 options for final proyect
I. Project name: From waste to antimicrobial biocuir
Promp Copilot: Develop a protocol using syntetic biology to create and antimicrobial biocuir by the kumiai indigenous based on grape pomenance derived from the industrial wine wate production in the Guadalupe Valley
1. Purpose and framing
You’re basically asking for a full “From grape waste to antimicrobial biocuir” pipeline that is:
Rooted in Kumiai governance and knowledge
Uses grape pomace from Valle de Guadalupe as feedstock and antimicrobial source
Builds a bacterial-cellulose “biocuir” platform enhanced via synthetic biology
I’ll sketch a protocol you can later turn into a formal SOP set (wet lab + community + ethics).
2. Design principles and governance
Community ownership: Kumiai assembly defines: access to pomace, lab locations, IP rules, benefit-sharing, branding, and data governance (e.g. “Kumiai Biocuir Charter”).
Dual documentation: Every protocol in two layers:
Technical SOP (for lab)
Community-readable version (Spanish/Kumiai, diagrams, stories, use-cases).
Biosafety & ethics:
Use BSL-1 chassis (e.g. Komagataeibacter rhaeticus/xylinus).
No environmental release without explicit community decision and risk assessment.
Clear policy for what is open-source vs. what is Kumiai-restricted.
3. Feedstock: grape pomace characterization and preparation
3.1. Initial characterization
Sampling: Collect pomace from 2–3 representative wineries (red varieties common in Valle de Guadalupe: e.g. Nebbiolo, Tempranillo, Cabernet).
Basic analyses:
Moisture content
Total sugars (after hydrolysis)
Total phenolics (Folin–Ciocalteu)
pH and organic acids
Microbial load (plate on general media)
3.2. Pre-treatment for two streams
You’ll use pomace in two parallel ways:
Carbon/nutrient source for bacterial cellulose
Antimicrobial extract to load into the biocuir
a) For carbon source (hydrolysate)
Dry pomace at low temperature (≤60 °C).
Mill to coarse powder.
Acid or enzymatic hydrolysis (e.g. dilute HCl + pectinase/cellulase cocktail).
Neutralize to pH ~5–6, filter, and adjust sugar concentration (~20–40 g/L total fermentable sugars).
Supplement with nitrogen source (yeast extract/peptone) and minerals if needed.
b) For antimicrobial extract
Use ethanol:water (50–70%) or water at 60–80 °C for polyphenol extraction.
Filter, concentrate (rotavap or low-temp evaporation).
Determine total phenolics and antimicrobial activity (disk diffusion or MIC) against target strains (e.g. E. coli, S. aureus, Candida).
4. Microbial platform and synthetic biology strategy
4.1. Base strain for biocuir
Preferred chassis:Komagataeibacter xylinus or K. rhaeticus (robust BC producers).
Maintain a wild-type production strain as baseline (no genetic modifications) for regulatory flexibility.
Define minimum antimicrobial effect and mechanical thresholds with Kumiai assembly and artisans (fit-for-purpose).
SOP-08 – Data, communication, and community reporting
Purpose: Ensure all technical data are translated into accessible formats and returned to Kumiai governance.
Procedure:
Data recording:
Maintain lab notebooks and digital records for all SOPs.
Tag each dataset with batch ID and date.
Translation and visualization:
Convert key results into bilingual (Spanish/Kumiai) summaries with simple graphs, photos, and short narratives.
Assembly reporting:
Present results periodically to Kumiai assembly: performance, risks, opportunities, and proposed changes.
Record feedback and integrate into protocol revisions.
Version control:
Maintain versioned SOPs; log changes and reasons (including community decisions).
II. Name: Antiseptic and anti-inflammatory pomenace patch
Copilot prompt: How to develop and Antiseptic and anti-inflammatory patch using syntetic biology metodhs in parthnership with the the kumiai indigenous based on grape pomenance derived from the industrial wine production in the Guadalupe Valley
1. The Kummiai community. Why? Governance system.
2. Characterize the grape pomace from Guadalupe Valley
2.1. Source and preprocessing
Source definition:
Identify 1–2 wineries willing to provide consistent pomace (same variety, same process).
Stabilization:
Drying: low temperature (≤50 °C) or lyophilization to preserve polyphenols.
Milling: standardize particle size for extraction reproducibility.
2.2. Chemical and bioactivity profiling
Polyphenol profiling:
Use methanolic/ethanolic extracts; quantify total phenolics, flavonoids, anthocyanins.
Target compounds: gallic acid, catechin, quercetin, resveratrol, etc.—all linked to antioxidant, anti-inflammatory, and antimicrobial activity. SpringerMDPI
Bioactivity assays (in vitro):
Antimicrobial: MIC/MBC against Staphylococcus aureus, E. coli, and skin-relevant strains. Grape pomace extracts often show stronger activity against Gram-positive bacteria. SpringerFrontiers
Anti-inflammatory: protein denaturation inhibition, NO inhibition in macrophage cell lines, etc.—grape pomace extracts have shown promising anti-inflammatory effects. SpringerMDPI
Antioxidant: DPPH, ABTS, FRAP to correlate with anti-inflammatory potential.
Outcome of this phase: a data-backed argument that “Kumiai grape pomace X from Guadalupe Valley has antiseptic and anti-inflammatory potential at Y concentration.”
3. Synthetic biology strategy: from extract to engineered system
Engineered microbes producing key actives
Target molecules:
Choose 1–3 “hero” compounds from your profiling (e.g., resveratrol, quercetin derivatives, gallic acid–related pathways) that correlate strongly with antimicrobial/anti-inflammatory activity. MDPI
Host selection:
Yeast (e.g., Saccharomyces cerevisiae): natural fit for resveratrol and other phenolics; compatible with wine narrative.
GRAS bacteria (e.g., Lactobacillus spp.): interesting if you want a “living” probiotic patch, but more complex regulatory path.
Pathway engineering:
Introduce or optimize phenylpropanoid/flavonoid pathways for your target compounds.
Use promoters responsive to simple inducers (e.g., temperature, small molecules) to control production.
Coupling to grape pomace:
Use pomace hydrolysates as carbon/nutrient source for the engineered strain—closing the circular bioeconomy loop and keeping the story “Kumiai + Guadalupe Valley wine waste” coherent.
Living or semi-living patch
Concept: A patch containing immobilized or killed engineered cells that have pre-produced the actives.
Safer intermediate: Use cell-free extracts or purified compounds from engineered microbes, not live GMOs on skin, at least initially.
4. Designing the antiseptic/anti-inflammatory patch
Think of the patch as: [biomaterial matrix] + [grape pomace actives] + (optional) [engineered bioactives].
4.1. Choose the base material
Biocuir / bacterial cellulose:
Produced by Komagataeibacter spp.; high water-holding capacity, biocompatible, and already used in wound dressings.
Can be grown on grape pomace–derived media to embed the circularity narrative.
Alternative matrices:
Chitosan (intrinsic antimicrobial), alginate, gelatin, or blends—depending on local availability and Kumiai preferences for texture and feel.
4.2. Loading the actives
Approach A: Soaking/adsorption:
Grow the biomaterial (e.g., bacterial cellulose), then soak in standardized grape pomace extract; dry to desired moisture level.
Approach B: In situ production:
Co-culture cellulose-producing bacteria with engineered yeast/bacteria that secrete phenolics into the medium; the matrix forms already loaded with actives.
Release profile testing:
In vitro diffusion assays (Franz cells or simple diffusion setups) to measure release of key phenolics over time.
4.3. Functional testing
Antimicrobial performance:
Place patch discs on bacterial lawns (agar diffusion) to test inhibition zones.
Anti-inflammatory performance:
Extract from patch and test in cell-based inflammation models (e.g., LPS-stimulated macrophages).
Later: small, ethically approved human patch tests for irritation.
III. Proyect name: A Yumano grape variety to promote circular economy
Promp Copilot: Develop a grape variety that is resistant to water stress and has organoleptic qualities similar to the pinyon nut—a fruit that is very common in the region and a traditional food staple of Yuman Indigenous groups—requires a culturally grounded and environmentally adapted approach. The grape variety will be for the exclusive use of local Yuman Indigenous communities, who will cultivate it and manage it sustainably, while also transforming the grape pomace into antibacterial biocuir. This means the entire initiative will function as a circular‑economy project from beginning to end, with a strong cultural dimension and a direct contribution to community health. For this reason, the grape’s properties must include very high antimicrobial capacity from the outset. To achieve this, synthetic biology techniques can be used.
🌱 Project goal
Develop a new grape variety designed to:
Withstand extreme water stress and poor soils.
Express organoleptic qualities inspired by pinyon nuts (a traditional food of Yuman peoples).
Produce intrinsic antimicrobial compounds in its skin and pulp.
Be cultivated and used exclusively by local Yuman Indigenous communities.
Enable the transformation of grape pomace into antibacterial biocuir (biocuir leather).
Operate as a fully circular, culturally grounded, community‑health–oriented system.
🍇 Organoleptic traits inspired by pinyon nuts
Pinyon nuts have a distinctive sensory profile:
Resinous notes (terpenes such as α‑pinene and limonene).
Buttery aromas (lactones).
Mild sweetness and a fatty texture (specific fatty acids).
To translate this into a grape variety:
Select parent lines rich in terpenes (e.g., Muscat, Malvasia).
Enhance metabolic pathways that increase lactones and sesquiterpenes.
Modulate the synthesis of volatile fatty acids that contribute to nut‑like aromas.
💧 Drought resistance
The environmental conditions of the Valle de Guadalupe require:
Thick cuticles.
Low stomatal density.
Deep root systems.
Efficient metabolism under water deficit.
Useful genetic sources include:
Vitis berlandieri (drought and alkaline soil tolerance).
Vitis arizonica (desert‑adapted).
Mediterranean varieties such as Garnacha and Monastrell.
🧬 Intrinsic antimicrobial capacity
To ensure the grape naturally produces antimicrobial compounds:
Increase stilbenes (resveratrol and derivatives).
Enhance flavonoids with bactericidal activity.
Boost terpenes with antifungal properties.
Introduce or upregulate plant antimicrobial peptides (AMPs) from desert species.
These traits support:
Reduced agricultural inputs.
Improved vineyard health.
Antibacterial properties in the biocuir produced from pomace.
🔬 Biotechnological strategies
Marker‑assisted classical breeding
Controlled crosses between selected parent lines.
Early seedling selection for drought tolerance, terpene production, and stilbene pathways.
Gene editing (CRISPR/Cas)
Modulate terpene synthesis genes to achieve pinyon‑like aromas.
NAC and bZIP TFs associated with ABA and drought signaling.
Aquaporins (PIP, TIP families) and proline biosynthesis enzymes (e.g., P5CS).
Promoter choices:
Stress‑inducible promoters:
Grapevine homologs of RD29A‑like promoters (dehydration‑responsive).
Native drought‑responsive promoters identified from grape transcriptomic data (e.g., promoters of strongly drought‑induced genes).
Design pattern:
pDrought_inducible::VvDREB_homolog
pDrought_inducible::VvNAC_homolog
This ensures drought‑tolerance pathways are activated under stress, not constitutively.
4. Promoter logic for safety and cultural governance
To align with Yuman governance and biosafety:
Prefer native Vitis promoters (STS, PAL, berry‑specific, stress‑responsive) over viral promoters (e.g., 35S), to keep the design cisgenic/intragenic as much as possible. SpringerUniProt
Use marker‑free systems:
Cre/lox or recombinase‑based excision of selection markers after transformation.
For example, a promoter responsive to a specific agro‑ecological practice (e.g., a foliar spray allowed only within community‑controlled fields) that can down‑regulate key TFs if needed.
5. Microbial biocuir module – key elements
Even though the search was grape‑focused, we can specify a reasonable synthetic design for Komagataeibacter:
Genes:
Cellulose synthase operon (bcsABCD) – native, but we can upregulate via stronger promoters.
Secreted AMPs:
Short, plant‑inspired peptides (e.g., defensin‑like) with broad‑spectrum but low‑toxicity profiles.
Promoters:
Constitutive medium‑strength promoter for cellulose (to avoid metabolic overload).
Inducible promoter for AMPs:
Sugar‑inducible (e.g., arabinose‑like logic) or pH‑responsive, activated only during biocuir growth phase.
Design pattern:
pConstitutive_medium::bcs_operon (if needed for boost)
pInducible_pomace_sugar::AMP1‑secreted
If you’d like, next step I can:
Turn this into a schematic genetic circuit map (module by module), or
Draft a methods‑style section (for a paper or grant) describing how these constructs would be built and tested in phases.
Here’s a text‑based schematic genetic circuit map for the main modules. Think of each as a layer you can redraw later in vector form.
Output: cellulose‑based biocuir with embedded antimicrobial phenolics + secreted AMPs.
5. Safety / governance layer (conceptual)
[Community-controlled trigger] ──► pOffSwitch
│
└──► Repressor of key TF (e.g. VviMYB14)
↓
Downregulation of STS module
This layer is where Yuman governance can be encoded: a controllable “dial” on how intense the antimicrobial/secondary metabolism is, tied to practices only allowed within community‑managed systems.
Week 4: Protein design I
PART A of HW: Answer any NINE of the following questions from Shuguang Zhang
1. How many molecules of amino acids do you take with a piece of 500 grams of meat?
Using Anthropic Clude I got the following answere and did some editions: There are more molecules of amino acids than the number of stars in the observable universe (~10²³). The calculation made by this IA modelis as follow:
Protein content of meat: ~25% by weight → 125 g of protein
Average amino acid molecular weight: ~100 Da (100 g/mol)
Moles of amino acids: $$\frac{125 \text{ g}}{100 \text{ g/mol}} = 1.25 \text{ mol}$$
Number of molecules (using Avogadro’s number, 6.022 × 10²³): $$1.25 \times 6.022 \times 10^{23} \approx \textbf{7.5 × 10}^{23} \textbf{ molecules}$$
Result: You consume roughly ~7.5 × 10²³ amino acid residues — about **1.25 times Avogadro’s number
2. Why are there only 20 natural amino acids?
Using Anthropic Clude I got the following answere and did some editions: We do not really know. However,there are some theories. Five of those are:
I. 🧬 The Genetic Code Constrains It: The standard genetic code uses 64 codons (4³) to encode only 20 amino acids + stop signals. The code is redundant but not expansive — evolution “froze” it early. Changing the code once life is established is catastrophically costly (one codon reassignment would corrupt thousands of proteins simultaneously). This is called the “frozen accident” hypothesis (Francis Crick, 1968).
II. ⚗️ Chemical Sufficiency: 20 amino acids already cover an enormous chemical diversity:
Charged (+ and −)
Hydrophobic / hydrophilic
Tiny (Gly) to bulky (Trp)
Flexible vs. rigid (Pro)
Nucleophilic (Cys, Ser, His)
This diversity is apparently sufficient to fold proteins into almost any 3D shape and catalyze almost any reaction. More variety may simply not be needed.
III. 🌍 Prebiotic Availability: The amino acids found in the genetic code largely overlap with those found in meteorites and Miller-Urey-type experiments — suggesting the 20 were simply the ones available on early Earth when life was getting started. Notably, the “later” amino acids (Trp, His, Cys) are rarer in prebiotic chemistry — hinting at a gradual expansion from perhaps 10 → 20 over early evolution.
IV. 📐 Diminishing Returns: Theoretical and computational studies suggest that adding a 21st amino acid yields rapidly diminishing gains in protein foldability and function. The 20 already allow ~10¹³⁰ possible protein sequences — more than enough diversity.
V. 🔢 It’s Not Strictly 20 — Biology Cheats a Little (Nature has actually expanded the code in special cases):
Selenocysteine (#21) — encoded by a repurposed UGA stop codon, used in some enzymes
Pyrrolysine (#22) — found in some archaea
Synthetic biology labs have engineered organisms with expanded genetic codes (up to 23+ amino acids)
This shows the limit isn’t absolute — but expanding it has a high evolutionary cost.
3. Can you make other non-natural amino acids? Design some new amino acids.
Using Anthropic Clude I got the following answere and did some editions: Yes - Synthetic biology labs have engineered organisms with expanded genetic codes (up to 23+ amino acids).
4. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Using Anthropic Clude I got the following answere and did some editions: Left. The Logic: Normal L-amino acids form right-handed α-helices. This isn’t arbitrary — it’s a direct consequence of the stereochemistry at the Cα atom. D-amino acids are the mirror image of L-amino acids. Therefore, a helix built from D-amino acids is the mirror image of a normal α-helix → a left-handed helix.
5. Can you discover additional helices in proteins?
Using Anthropic Clude I got the following answere and did some editions: Yes — Several Avenues Exist. Some of those are:
I. 🤖 AI-Driven Structure Prediction
AlphaFold2/3 and RoseTTAFold have predicted millions of structures, and researchers are data-mining them for unusual backbone geometries never seen in experimental structures. This is arguably the most promising current approach.
II. 🧪 Exotic Amino Acid Compositions
β-peptides (backbone extended by one carbon) form helices with completely different geometries — 14-helix, 12-helix, etc. — that don’t exist in natural proteins
D-amino acids form mirror-image helices
Peptoids (N-substituted glycines) form their own helix families
These aren’t natural, but they reveal the chemical space of possible helices is much larger than biology uses.
III. 🔬 Cryo-EM Resolution Revolution
Cryo-EM now routinely reaches 1.5–2 Å resolution, allowing detection of subtle backbone conformations that were invisible before. The π-helix was long thought to be a curiosity until cryo-EM showed it’s functionally important in ion channels and enzymes.
IV. 🧬 Extremophile & Novel Organism Proteomes
Proteins from hyperthermophiles, deep-sea organisms, or newly sequenced microbial dark matter sometimes adopt unusual folds. Systematic structural surveys of these proteomes could reveal new helix types.
V. 📐 Computational Helix Hunting
You can define a helix purely mathematically (repeating φ/ψ dihedral angles on the Ramachandran plot) and ask: which repeating geometries are sterically allowed but biologically unobserved? Some groups are doing exactly this.
6. Why are most molecular helices right-handed?
Using Anthropic Clude I got the following answere and did some editions: Most biological helices are right-handed because life uses L-amino acids, and the stereochemistry of L-amino acids energetically favors right-handed helical geometry.
7. Why do β-sheets tend to aggregate?
Using Anthropic Clude I got the following answere and did some editions: β-sheet aggregation it’s at the heart of some of the most devastating diseases known.
The core structure is a *ross-β spine:
Strands run perpendicular to the fibril axis
H-bonds run parallel to the fibril axis
Two tightly mated β-sheets form a “steric zipper” — interdigitated side chains with almost no water inside
β-sheet aggregation is the molecular basis of a whole class of diseases:
Disease
Protein
Alzheimer’s
Aβ peptide + Tau
Parkinson’s
α-synuclein
Type II Diabetes
IAPP (islet amyloid)
Prion diseases (CJD, BSE)
PrP
Huntington’s
Huntingtin (polyQ)
Systemic amyloidosis
Transthyretin, immunoglobulin light chain
Spider silk: β-sheet aggregation may not be purely pathological — some organisms exploit it. Also, the same property that makes amyloid a disease culprit — extreme stability and cooperative assembly — makes it a remarkable material when controlled. Spider silk is a controlled amyloid-like β-sheet material with extraordinary mechanical properties
8. What is the driving force for β-sheet aggregation?
Using Anthropic Clude I got the following answere and did some editions:
The tendency to aggregate is essentially baked into β-sheet geometry:
Life has spent billions of years evolving ways to suppress this tendency — and when those mechanisms fail, disease follows.
9. Why do many amyloid diseases form β-sheets?
Using Anthropic Clude I got the following answere and did some editions: This is actually a subtle question — it’s almost the inverse of the previous one. The question isn’t just why do β-sheets aggregate?" but"why do so many different, unrelated proteins converge on the same amyloid structure? Yet they all form the same cross-β amyloid architecture. Why?
Dobson’s view suggests that life is in a constant battle against amyloid. The entire proteostasis network — chaperones, the proteasome, autophagy, the unfolded protein response — exists largely to keep proteins away from their thermodynamic ground state. Amyloid disease is not a failure of a specific protein. It is a failure of the system that keeps all proteins from doing what they thermodynamically want to do. Aging is, in part, the slow losing of that battle.
10. Can you use amyloid β-sheets as materials?
Using Anthropic Clude I got the following answere and did some editions: Yes. The very properties that make amyloid dangerous in disease make it extraordinarily useful as a material. Some emarkable intrinsic properties are:
Property
Value
Comparison
Young’s modulus
~10–20 GPa
Similar to bone (~20 GPa)
Tensile strength
~0.1–1 GPa
Comparable to steel
Width
5–10 nm
Truly nanoscale
Persistence length
~1–10 μm
Very stiff at nanoscale
Thermal stability
>100°C
Resists boiling
Chemical stability
Resists proteases, detergents
Extraordinary durability
All of this self-assembles from cheap, abundant protein under mild aqueous conditions. No factory needed.
Nature already uses amyloid as a material, proving biocompatibility:
Organism
Functional Amyloid
Purpose
Bacteria
Curli fibers (E. coli)
Biofilm scaffolding, surface adhesion
Fungi
Chaplins, hydrophobins
Waterproof spore coatings
Humans
Pmel17
Melanin synthesis scaffold
Spiders
Silk β-sheet crystals
Structural fiber
Mussels
Adhesive plaques
Underwater adhesion
Sea cucumbers
Catch collagen
Variable-stiffness tissue
Mussel adhesion is particularly interesting — they stick to rocks underwater using amyloid-like structures, inspiring wet adhesives for surgery and marine engineering.
🌱 Sustainability Angle
Amyloid materials are compelling from an environmental perspective:
Made from protein waste streams (whey, soy, egg white)
Self-assemble in water at mild temperatures — low energy process
Fully biodegradable under the right conditions
Could replace petroleum-based plastics in some applications
11. Design a β-sheet motif that forms a well-ordered structure
Using Copilot I got the following idea and did some editions to develop a biocuir with antimicrobial functiones:
Turn the β‑sheet motif into an AMP-like sequence
Key AMP traits to build in: cationic, amphipathic, and sufficiently hydrophobic to interact with membranes. FrontiersNature
A concrete β‑sheet–forming, antimicrobial‑oriented motif:
Ac–KFKFKFKF–NH2 (8‑mer, net charge +4 at neutral pH)
Alternating pattern: Phe (F, hydrophobic/aromatic) and Lys (K, cationic) favor β‑sheet assembly and amphipathic surfaces.
Cationic surface: Multiple Lys residues promote electrostatic attraction to negatively charged bacterial membranes and biofilms.
End-capping: Ac-/–NH2 keeps the assembly more ordered and reduces terminal fraying.
You can use this as a “hard” antimicrobial layer interwoven with cellulose or with the more neutral structural peptide.
Biocuir-specific hybrid motif
To keep β‑sheet order, add a short cellulose-contact and crosslinking segment:
Segment 2 (cellulose-contact):YGYG Aromatic + H‑bonding for interaction with glucan surfaces.
Segment 3 (handle):K Extra positive charge and a site for chemical/enzymatic crosslinking to oxidized cellulose or other polymers.
This should give you nanofibrillar β‑sheet assemblies that can both integrate into the cellulose network and present a cationic, antimicrobial surface.
Design rules you can tune
Net charge: Aim for overall +3 to +7 per peptide to keep strong antimicrobial character without extreme toxicity. Frontiers
Hydrophobic fraction: Roughly 30–50% hydrophobic/aromatic residues (F, W, L, I, V) to ensure membrane insertion but avoid uncontrolled aggregation. MDPI
β‑sheet bias: Use alternating hydrophobic/cationic pattern (e.g., K–F, K–L, R–W) to favor β‑sheets over α‑helices.
How to test it in a biocuir context
In solution:
CD for β‑sheet signature (negative band ~218 nm).
MIC/kill curves against E. coli and S. aureus to confirm antimicrobial activity.
In the biocuir:
Incorporate peptide during cellulose growth or post‑treat pellicles.
Test zone of inhibition around biocuir disks and biofilm formation on the surface.
PART B of HW: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
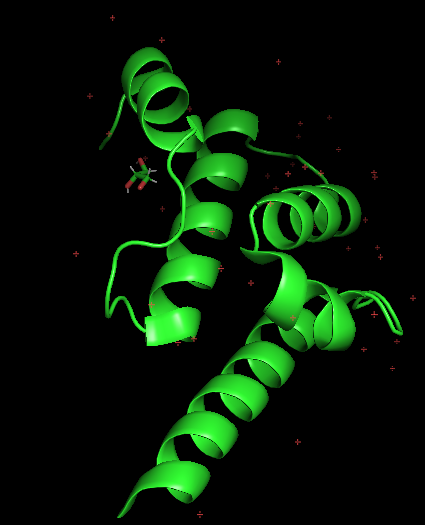
Briefly describe the protein you selected and why you selected it: 🧠 Protein Arc — The Brain’s “Viral Memory”
Arc (Activity-Regulated Cytoskeleton-associated protein) Master regulator of synaptic plasticity that self-assembles into virion-like capsids that encapsulate RNAs and mediate intercellular RNA transfer in the nervous system. ARC protein is released from neurons in extracellular vesicles that mediate the transfer of ARC mRNA into new target cells, where ARC mRNA can undergo activity-dependent translation. ARC capsids are endocytosed and are able to transfer ARC mRNA into the cytoplasm of neurons. Acts as a key regulator of synaptic plasticity: required for protein synthesis-dependent forms of long-term potentiation (LTP) and depression (LTD) and for the formation of long-term memory. Regulates synaptic plasticity by promoting endocytosis of AMPA receptors (AMPARs) in response to synaptic activity: this endocytic pathway maintains levels of surface AMPARs in response to chronic changes in neuronal activity through synaptic scaling, thereby contributing to neuronal homeostasis. Acts as a postsynaptic mediator of activity-dependent synapse elimination in the developing cerebellum by mediating elimination of surplus climbing fiber synapses. Accumulates at weaker synapses, probably to prevent their undesired enhancement. This suggests that ARC-containing virion-like capsids may be required to eliminate synaptic material. Required to transduce experience into long-lasting changes in visual cortex plasticity and for long-term memory (By similarity). (https://www.uniprot.org/uniprotkb/Q7LC44/entry#sequences)
What is the most frequent amino acid? Glutamic Acid (E)
Rank
Amino Acid
Count
Percentage
🥇 1st
Glutamic Acid (E)
46
11.6%
2nd
Leucine (L)
36
9.0%
3rd
Glycine (G)
30
7.5%
4th
Proline (P)
29
7.3%
5th
Glutamine (Q)
28
7.0%
Why is Glutamic Acid (E) so abundant?
Negatively charged at physiological pH — contributes to ARC’s electrostatic interactions
Important for protein-protein interactions, which makes sense given ARC’s role as a synaptic scaffolding protein
Involved in binding and signaling within the postsynaptic density
The high E content (~11.6%) is notably above its average abundance in typical human proteins (~6.4%), suggesting it plays a structural or functional role specific to ARC’s activity in synaptic plasticity and memory formation. (Anthropic Claude using the promt: What is the most frecuent Aminoacit in the ARC protein? MELDHRTSGGLHAYPGPRGGQVAKPNVILQIGKCRAEMLEHVRRTHRHLLAEVSKQVERE LKGLHRSVGKLESNLDGYVPTSDSQRWKKSIKACLCRCQETIANLERWVKREMHVWREVF
YRLERWADRLESTGGKYPVGSESARHTVSVGVGGPESYCHEADGYDYTVSPYAITPPPAA)
Does your protein belong to any protein family? ARC belongs to the retroviral Gag protein superfamily (Anthroppc Calude promt: Does ARC protein belongs to any protein family?)
Structural Homology: ARC has a fold that resembles the proteins retroviruses use to form their protective protein shells (capsids).
Just like retroviral Gag proteins, ARC can: Self-assemble, Encapsulate RNA (including its own mRNA, Form extracellular vesicles, that transfer RNA between neurons — remarkably similar to how retroviruses package and deliver genetic material
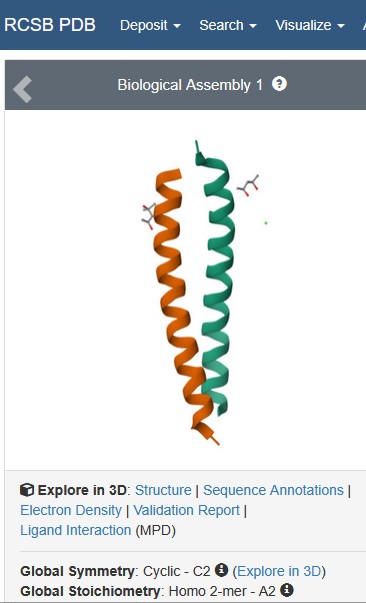
Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å): YES Resolution: 0.95 Å
Are there any other molecules in the solved structure apart from protein? YES, 1) Activity-regulated cytoskeleton-associated protein, 2) SMALL: (4S)-2-METHYL-2,4-PENTANEDIOL / C6 H14 O2 & SVTBMSDMJJWYQN-YFKPBYRVSA-N 3) SMALL (4S)-2-METHYL-2,4-PENTANEDIOL / C6 H14 O2 / SVTBMSDMJJWYQN-YFKPBYRVSA-N
Does your protein belong to any structure classification family?: Gag protein superfamily
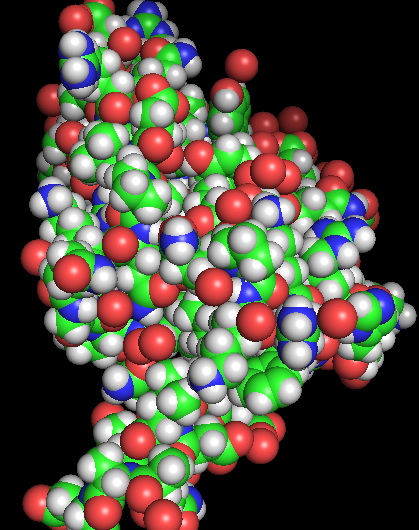
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
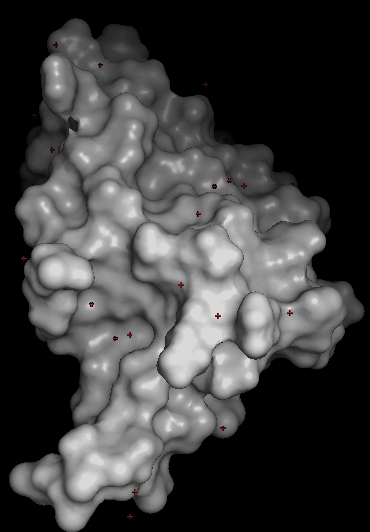
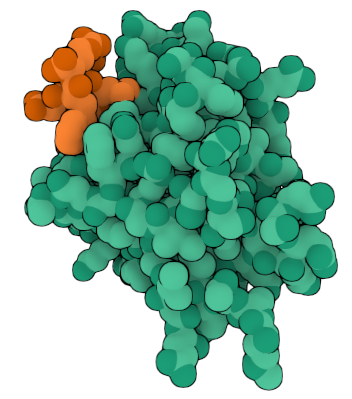
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)? YES (Copilot). The details about ARC’s binding cavity come directly from the structural description on the RCSB entry for 6TN7 rcsb.org.
Guide to visualizing the ARC surface in PyMOL. The surface view is the best way to inspect whether the protein has cavities or depressions (Copilot).
🧬 Does ARC (6TN7) have holes or binding pockets? NO HOLES. The ARC’s C‑lobe is a compact capsid‑homology domain. It does not contain deep tunnels or internal cavities. YES BIDING CAVITY that recognizes short linear motifs (SLiMs) from proteins such as stargazin and GKAP. This means ARC has a functional pocket, but it is shallow and surface‑exposed, not a deep “hole.”
What the cavity is used for
The cavity binds:
Stargazin peptides
GKAP repeat peptides
Other postsynaptic density motifs
This is central to ARC’s role in synaptic plasticity.
🧭 Practical PyMOL workflow to inspect ARC’s binding cavity
When you rotate the structure, the main cavity PyMOL highlights will correspond to the peptide‑binding cavity described in the RCSB entry.
PART C HOMEWORK. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling using ARC as example
Deep Mutational Scans
-Use ESM2 to generate an unsupervised deep mutational scan of your protein (ARC) based on language model likelihoods.
-Can you explain any particular pattern? (choose a residue and a mutation that stands out)
-(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
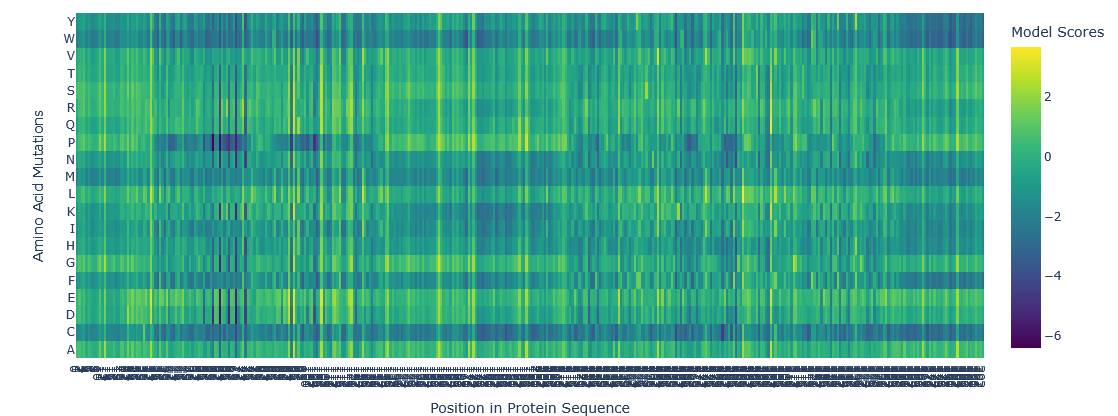
ESM2 result for ARC:
Asfar as I understand the A row in Y is more likely for the mutation to happen because is more yellow, meaning a more positive value. Whereas in the B row is less likely to happen because is blue, meaning a more negative value. This is a 2D representation of a 3D molecule, then is important to analize each position value to be sure about what is the best position for the mutation to occur."
Using the gemmini IA emmbeded in the notebook I got the following analysis about best and worst palces for mutations:
Most favorable mutation (highest LLR):
LLR value: 3.6797
Position in protein sequence: 290 (0-indexed, corresponding to 291 in 1-indexed view)
Original amino acid: E
Mutated to: L
Most unfavorable mutation (lowest LLR):
LLR value: -6.4074
Position in protein sequence: 60 (0-indexed, corresponding to 61 in 1-indexed view)
Original amino acid:
Mutated to: P
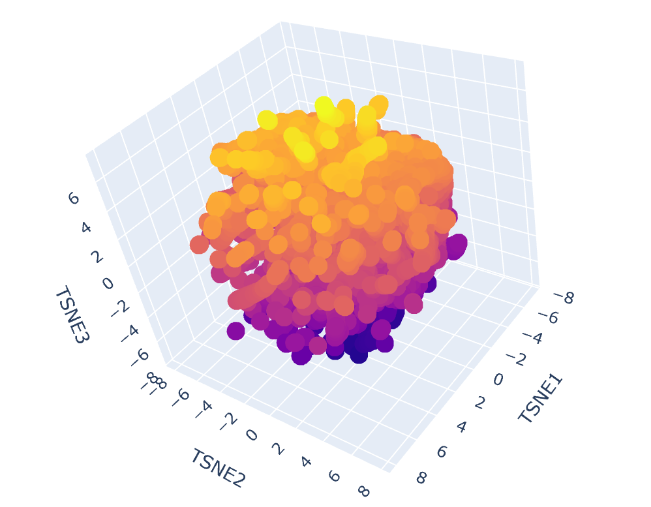
Latent Space Analysis
-Use the provided sequence dataset to embed proteins in reduced dimensionality.
-Analyze the different formed neighborhoods: do they approximate similar proteins?
-Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
 and a most unfavorable mutation with an LLR of -6.4074 (at position 60, changing an unknown original amino acid to ‘P’).
The protein exhibits a mixed resilience to mutations. While some positions show a degree of flexibility and can accommodate or even benefit from certain changes, other positions appear to be highly constrained, where specific mutations are strongly disfavored. The overall resilience would depend on the prevalence of such constrained versus flexible sites across the entire sequence. If many positions show low LLRs for most mutations, the protein would be considered less resilient.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
For the Inverse-Folding I got the following sequences:
I asked Gemini for hel to compare the predicted structure with the original and got the following answere:
Original Protein (Blue): ID=predicted_sequence_fold_552c4, PTM=0.321, pLDDT=73.364
Predicted Protein (Red): ID=predicted_sequence_fold_552c4, PTM=0.888, pLDDT=94.589
It appears that the predicted sequence, despite being significantly different from the original, yielded a structure with much higher confidence metrics (PTM and pLDDT).
pTM (predicted TM-score): The predicted protein has a significantly higher pTM (0.888 vs 0.321). A higher pTM indicates that the predicted structure of the new sequence is likely much closer to a true, stable structure than the original prediction of the sequence provided to ESMFold, or that the original sequence was a poor prediction. A pTM closer to 1 suggests a very good structural prediction, implying that the ProteinMPNN-designed sequence adopted a stable and well-defined fold. This is a very positive result, suggesting that the designed sequence is likely to fold well.
pLDDT (predicted Local Distance Difference Test): Similarly, the predicted protein has a much higher average pLDDT (94.589 vs 73.364). A pLDDT value close to 100 indicates very high confidence in the local structure of the protein. The significant increase in pLDDT suggests that the designed sequence leads to a more confidently predicted and likely more stable structure across its residues compared to the original. High pLDDT values generally correlate with well-folded and experimentally verifiable structures.
From a visual perspective, you can compare the blue (original) and red (predicted) structures. Given the large differences in pTM and pLDDT, you might observe that the red structure appears more compact or well-defined, while the blue structure might have regions of lower confidence that appear more flexible or disordered.
In summary, the ProteinMPNN-designed sequence, when folded by ESMFold, appears to result in a highly confident and presumably more stable protein structure, as indicated by the substantially improved pTM and pLDDT scores compared to the original sequence’s prediction.
Part D. Group Brainstorm on Bacteriophage Engineering
Find a group of ~3–4 students
Read through the Phage Reading material listed under “Reading & Resources” below.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why do you think those tools might help solve your chosen sub-problem?
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Include a schematic of your pipeline.
This resource may be useful: HTGAA Protein Engineering Tools
Each individually put your plan on your HTGAA website:
Proposal: Computational Stabilization of the L Protein by Preventing Protease Cleavage (With the advice of PhD Armando Garcia head of the Nanobiolab at UNAN and Copilot)
Project Goal
This project focuses on increasing the stability of the L protein by preventing its degradation by host proteases during recombinant expression. The working hypothesis, aligned with Armando García’s recommendations, is that a specific bacterial protease recognizes and cleaves a short motif in the L protein, leading to loss of function. By identifying this cleavage site and introducing targeted mutations that avoid protease recognition—while preserving the protein’s fold—we aim to computationally design a stabilized variant.
Computational Strategy
Identify the protease and its cleavage motif
The first step is to determine which bacterial protease is likely responsible for degrading the L protein. Sequence based protease site predictors (e.g., PROSPER, DeepProtease) will be used to scan the L protein for motifs recognized by common E. coli proteases such as Lon, ClpXP, OmpT, or DegP. This defines the minimal region where mutations should be introduced.
Generate mutation candidates using Protein Language Models
Protein Language Models (PLMs) such as ESM 2 or ProtT5 will be used to perform in silico mutagenesis at the predicted cleavage site. PLMs score each mutation by sequence likelihood, which correlates with foldability and evolutionary plausibility. Mutations that reduce protease recognition while maintaining high PLM likelihood will be prioritized. This step provides a computationally efficient filter before structural modeling.
Predict structural effects of mutations with AlphaFold2
Following Armando García’s guidance, AlphaFold2 will be used to predict the structure of both the wild type L protein and the top PLM ranked mutants. Structural comparisons will evaluate:
• RMSD between WT and mutant models
• pLDDT and PAE confidence metrics
• Local structural perturbations around the mutated region
Only mutations that preserve the global fold and maintain high structural confidence will advance.
Evaluate thermodynamic stability using ΔΔG calculations
To quantify the stabilizing or destabilizing effect of each mutation, FoldX or Rosetta ΔΔG calculations will be performed. Mutations predicted to lower the folding free energy (ΔΔG < 0) while preserving structure will be considered strong candidates for increasing stability.
Functional and biophysical sanity checks
Final candidates will be evaluated for:
• Solvent exposure and aggregation propensity
• Disorder predictions
• Potential disruption of functional or membrane associated regions
This ensures that stabilization does not compromise biological activity.
Why These Tools Are Appropriate
PLMs provide a rapid, sequence level exploration of mutational space; AlphaFold2 offers a structural safeguard to ensure mutations do not distort the fold; and ΔΔG tools quantify stability changes. Together, these methods directly address the mechanism described by García: mutate the protease sensitive region and verify that the structure remains intact.
Potential Pitfalls
• AlphaFold2 limitations: If the L protein is highly novel or disordered, structural predictions may be unreliable.
• Mutation induced misfolding: Some mutations may block protease recognition but still disrupt the fold, reducing stability rather than improving it.
• Context dependence: Protease cleavage may depend on local flexibility or exposure, not just sequence, complicating prediction.
Pipeline Schematic
WT L protein sequence
|
v
[Protease-site Prediction]
Identify likely bacterial protease cleavage motifs
|
v
[Protein Language Models]
Mutate cleavage site
Rank mutations by sequence likelihood
|
v
[AlphaFold2 Modeling]
Predict WT and mutant structures
Compare RMSD, pLDDT, PAE
|
v
[ΔΔG Stability Scoring]
FoldX/Rosetta evaluation
Select stabilizing, structure-preserving variants
|
v
[Biophysical Filters]
Aggregation, disorder, solvent exposure
|
v
Shortlist of stabilized L protein variants
Include your group’s short plan for engineering a bacteriophage (I was not able to work with a Nodo´s group due to timeshortage and probles for coordinating with my team)
Week 5: Protein design 2
HOMEWORK
Part A: SOD1 Binder Peptide Design
Context
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme -protein- that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
1. Design short peptides -aminoacit chain going form 2 to 50 aprox- that bind mutant SOD1.
2. Then decide which ones are worth advancing toward therapy.
What is a peptide binder? short proteins that bind to larger proteins. Peptide binders are high specific and cheap to synthesize. Therefor they are a prime candidate for drug design. In this case to bind -or fix- mutant SOD1.
You will use three models:
-PepMLM: target sequence-conditioned peptide generation via masked language modeling. Here is an opena ccess article for reference: https://www.nature.com/articles/s41587-025-02761-2
-PeptiVerse: therapeutic property prediction. Here is an opena ccess article for reference: https://www.biorxiv.org/content/10.64898/2025.12.31.697180v1.full.pdf
-moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM). Here is an opena ccess article for reference: https://www.biorxiv.org/content/10.1101/2024.07.31.606098v2
Instructions
Part 1: Generate Binders with PepMLM
-Looking for the PepMLM model I found a webpage called TamarindoBio, wich seems to be very helpfull since integrates different models in one singe platform for computational biology: https://www.tamarind.bio/
Begin by retrieving the human SOD1 sequence from UniProt (P00441)
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Binder Perplexity
TRGEDLELLLEGLWG 16.156637 (ATKAVCVLISG a common mutant SOD1 sequence)
index,Binder,Pseudo Perplexity
0,WTRGETEEENWL,20.351590911595604
1,KPRGETTVVNWR,10.28386335939656
2,RTTDDEEVEEPL,13.21296682161187
3,WPTEEEEEEEPL,7.778752281638014 (This is the lowest. Why?)
FLYRWLPSRRGG SOD1 comparion peptide conditioned on the mutant SOD1 sequence provided by the instructor:
index,Binder,Pseudo Perplexity
0,KPEGEEEVEPGE,14.948628502503167
1,RRRDEEPVEEPE,11.433765492062454
2,RREGETPEEPWR,11.150938004006173
3,WTTDEEELEEWR,22.782388379405774 (This is the highest. Why?)
Part 2: Evaluate Binders with AlphaFold3 This model led to the nobel prize winners in chemestry for the 2024. Here is a paper explaining how it works: https://www.nature.com/articles/s41586-024-07487-w
Instructions Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind.
What is the pTM and the ipTM? In the frequently asked questions of the AlfaFold server I found the following answer: the predicted template modeling (pTM) score and the interface predicted template modeling (ipTM) score are both derived from a measure called the template modeling (TM) score. This measures the accuracy of the entire structure (Zhang and Skolnick, 2004; Xu and Zhang, 2010). A pTM score above 0.5 means the overall predicted fold for the complex might be similar to the true structure. ipTM measures the accuracy of the predicted relative positions of the subunits within the complex. Values higher than 0.8 represent confident high-quality predictions, while values below 0.6 suggest likely a failed prediction. ipTM values between 0.6 and 0.8 are a gray zone where predictions could be correct or incorrect. TM score is very strict for small structures or short chains, so pTM assigns values less than 0.05 when fewer than 20 tokens are involved; for these cases PAE or pLDDT may be more indicative of prediction quality.
Does it localize near the N-terminus where A4V sits?
Does it engage the β-barrel region or approach the dimer interface?
Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
I used Anthropic Claude trying to understand the results I got running the binnder ranked with the lower Perplexity index (WPTEEEEEEEPL,7.778752281638014) and the higher (WTTDEEELEEWR,22.782388379405774). Not sure why in both cases the ipTM was the same: 0.41. However, the PTM changed a bit, for the former was 0.86 and the later 0.87. I will present Anthropic interpretation only for the higher:
Results Interpretation: WTTDEEELEEWR + SOD1 A4V
Scores Received
MetricScoreInterpretationipTM0.41⚠️ Low confidence interfacepTM0.87✅ High confidence overall fold
What These Scores Mean
pTM = 0.87 ✅
The overall structure prediction is high quality
SOD1 A4V is folding correctly and confidently
This is expected — SOD1 is a well-characterized protein
ipTM = 0.41 ⚠️
This is the critical score for protein-peptide interaction
Falls in the low confidence zone (threshold for reliable interaction is typically >0.60)
Suggests the peptide does not form a stable, well-defined complex with SOD1 A4V
The binding pose may be ambiguous or non-specific
ipTM Score Scale Reference
0.0 0.2 0.4 0.6 0.8 1.0
↑
0.41
WTTDEEELEEWR
(Low/Uncertain)
↑
Strong hit
needed here
Biological Interpretation
ObservationImplicationLow ipTM (0.41)Peptide likely does not bind specifically to SOD1 A4VHigh pTM (0.87)SOD1 structure is well-predicted — not a modeling artifactScore near 0.40 cutoffInteraction is borderline — could be weak/transientPeptide character (acidic) May not complement SOD1 surface well at this site.
Regarding the N-terminus / A4V Site Question
Given the low ipTM of 0.41, you should still check the structure visually, but: The peptide likely does not localize confidently near Val4. Any placement near the N-terminus would be unreliable at this confidence level.
The acidic stretch (DEEELEEE) of the peptide may be electrostatically incompatible with the local SOD1 surface near A4V
Summary Statement for Your Report
“The WTTDEEELEEWR peptide yielded an ipTM score of 0.41 against SOD1 A4V, indicating low confidence in the predicted protein-peptide interface. While the overall SOD1 structure was well-modeled (pTM = 0.87), the interaction does not appear to be specific or stable. Visual inspection of the model is needed to determine whether the peptide localizes near the A4V mutation site at the N-terminus, though the low ipTM score suggests this interaction may not be physiologically meaningful.”
What is the difference between the N and the C terminuous? I found a video where is well explained: https://youtu.be/y3G_W-3J_8k. Here is an screean shoot of it:
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Intructions:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Results:
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Comparison: AlphaFold3 and PeptiVerse show low affinity for WTTDEEELEEWR + SOD1 A4V. The choosen peptide is soluble, however, is non-hemolytic, maybe the latter characteristic is the reason for its weak binding affinity.
Choose one peptide you would advance and justify your decision briefly.
Non of my designed pepdides showed affinity. Therefore I will procede with WTTDEEELEEWR in order to verify if is posible to optimize it wiht moPPIt.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card. Make a copy and switch to a GPU runtime. In the notebook: Paste your A4V mutant SOD1 sequence. Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch). Set peptide length to 12 amino acids. Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
Results moPPIt-v3 aims to design peptide binders for the A4V mutant SOD1 sequence
/content/moPPIt/flow_matching/path/path_sample.py:45: SyntaxWarning: invalid escape sequence ‘\s’
x_t (Tensor): the sample along the path :math:X_t \sim p_t.
/content/moPPIt/flow_matching/path/scheduler/scheduler.py:72: SyntaxWarning: invalid escape sequence ‘\s’
SchedulerOutput: :math:\alpha_t,\sigma_t,\frac{\partial}{\partial t}\alpha_t,\frac{\partial}{\partial t}\sigma_t
/content/moPPIt/flow_matching/path/scheduler/scheduler.py:79: SyntaxWarning: invalid escape sequence ‘\k’
Computes :math:t from :math:\kappa_t.
Target Motifs: [1, 2, 3, 4, 5, 6, 7, 8, 9, 10]
tensor([ 1, 2, 3, 4, 5, 6, 7, 8, 9, 10], device=‘cuda:0’)
Some weights of EsmModel were not initialized from the model checkpoint at facebook/esm2_t33_650M_UR50D and are newly initialized: [’esm.pooler.dense.bias’, ’esm.pooler.dense.weight’]
You should probably TRAIN this model on a down-stream task to be able to use it for predictions and inference.
Some weights of EsmModel were not initialized from the model checkpoint at facebook/esm2_t33_650M_UR50D and are newly initialized: [’esm.pooler.dense.bias’, ’esm.pooler.dense.weight’]
You should probably TRAIN this model on a down-stream task to be able to use it for predictions and inference.
Some weights of EsmModel were not initialized from the model checkpoint at facebook/esm2_t33_650M_UR50D and are newly initialized: [’esm.pooler.dense.bias’, ’esm.pooler.dense.weight’]
You should probably TRAIN this model on a down-stream task to be able to use it for predictions and inference.
NFE: 99: 100%|██████████| 0.9990000128746033/0.9990000128746033 [12:46<00:00, 767.09s/it]
NFE: 100: 100%|██████████| 0.9990000128746033/0.9990000128746033 [12:46<00:00, 767.09s/it]
NFE: 100: 100%|██████████| 0.9990000128746033/0.9990000128746033 [12:46<00:00, 767.09s/it]
2026-04-12 12:15:43.585473: E external/local_xla/xla/stream_executor/cuda/cuda_fft.cc:467] Unable to register cuFFT factory: Attempting to register factory for plugin cuFFT when one has already been registered
WARNING: All log messages before absl::InitializeLog() is called are written to STDERR
E0000 00:00:1775996143.819521 8847 cuda_dnn.cc:8579] Unable to register cuDNN factory: Attempting to register factory for plugin cuDNN when one has already been registered
E0000 00:00:1775996143.879429 8847 cuda_blas.cc:1407] Unable to register cuBLAS factory: Attempting to register factory for plugin cuBLAS when one has already been registered
W0000 00:00:1775996144.365984 8847 computation_placer.cc:177] computation placer already registered. Please check linkage and avoid linking the same target more than once.
W0000 00:00:1775996144.366062 8847 computation_placer.cc:177] computation placer already registered. Please check linkage and avoid linking the same target more than once.
W0000 00:00:1775996144.366066 8847 computation_placer.cc:177] computation placer already registered. Please check linkage and avoid linking the same target more than once.
W0000 00:00:1775996144.366069 8847 computation_placer.cc:177] computation placer already registered. Please check linkage and avoid linking the same target more than once.
2026-04-12 12:15:44.415070: I tensorflow/core/platform/cpu_feature_guard.cc:210] This TensorFlow binary is optimized to use available CPU instructions in performance-critical operations.
To enable the following instructions: AVX2 AVX512F FMA, in other operations, rebuild TensorFlow with the appropriate compiler flags.
[‘KCWKQKSFGKEI’]
[0.931668795645237, 0.75, 6.4554948806762695, 0.3713146150112152]
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
I asked Gemini IA linked to the Colab notebook to answere the question got the folloging answere:
How would you evaluate these peptides before advancing them to clinical studies?
Evaluating peptides before advancing them to clinical studies involves a rigorous multi-step process to ensure safety, efficacy, and suitability for human use. Based on the properties optimized by moPPIt-v3, here’s a general approach:
Confirmation of moPPIt-v3 Optimized Properties (In Vitro & Early In Vivo):
Binding Affinity and Specificity: Thoroughly validate the binding affinity to the target and specificity against off-targets using techniques like SPR (Surface Plasmon Resonance), ITC (Isothermal Titration Calorimetry), ELISA, or competitive binding assays.
Motif Binding: If a specific motif is targeted, confirm its interaction and functional consequence.
Solubility and Stability: Assess solubility in various physiological buffers and evaluate stability (e.g., against proteolysis, aggregation) over time and under different environmental conditions.
Hemolysis and Cytotoxicity: Conduct in vitro assays (e.g., red blood cell lysis, cell viability assays) to confirm low or no hemolytic and cytotoxic effects, which moPPIt-v3 aims to minimize.
Non-Fouling: Evaluate the peptide's resistance to non-specific binding to surfaces or other biomolecules, particularly if it's designed for use in complex biological environments or materials.
Half-Life (Early PK): Begin preliminary in vivo pharmacokinetic (PK) studies in animal models to understand the peptide's absorption, distribution, metabolism, and excretion (ADME) profile, especially its circulating half-life, which moPPIt-v3 aims to optimize.
Comprehensive Safety Assessment:
General Toxicology: Conduct acute, sub-acute, and chronic toxicity studies in relevant animal models to identify any adverse effects.
Immunogenicity: Assess the potential for the peptide to elicit an immune response (antibody formation) in preclinical models, which could lead to reduced efficacy or adverse reactions.
Genotoxicity and Carcinogenicity: Evaluate if the peptide can cause DNA damage or has carcinogenic potential, usually through in vitro and in vivo tests.
Efficacy and Pharmacodynamics (PD):
In Vitro Efficacy: Confirm biological activity in cell-based assays or tissue models that mimic the disease state.
In Vivo Efficacy: Demonstrate the therapeutic effect of the peptide in relevant animal models of the target disease. This includes dose-response studies and evaluation of appropriate biomarkers.
Mechanism of Action (MoA): Fully elucidate how the peptide exerts its therapeutic effect.
Formulation and Delivery:
Develop a Stable Formulation: Design a pharmaceutical formulation that ensures the peptide's stability, bioavailability, and appropriate route of administration (e.g., injection, oral, topical).
Manufacturing Feasibility: Ensure the peptide can be produced at scale with consistent quality and purity.
Regulatory Considerations:
GLP/GMP Compliance: All preclinical studies (safety and efficacy) must be conducted under Good Laboratory Practice (GLP) regulations, and manufacturing processes must adhere to Good Manufacturing Practice (GMP) standards.
IND-Enabling Studies: Compile all the necessary data and documentation to support an Investigational New Drug (IND) application to regulatory authorities (e.g., FDA in the US, EMA in Europe), which is required before human clinical trials can commence.
By systematically addressing these points, researchers can build a strong case for a peptide’s potential and mitigate risks before it enters the expensive and complex clinical trial phases.
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Using Copilot I got the following answere and did some editions: Phusion High‑Fidelity PCR Master Mix is a 2X ready‑to‑use mix designed to simplify PCR setup while providing extremely high fidelity. There are five componets for specific porpuses:
Component
Purpose
Phusion DNA Polymerase
High‑fidelity, proofreading DNA synthesis
HF or GC Buffer
Optimizes reaction conditions; HF for fidelity, GC for difficult templates
MgCl₂
Required cofactor for polymerase activity
dNTPs
Nucleotide building blocks for DNA amplification
DMSO (optional)
Enhances amplification of GC‑rich or structured templates
What are some factors that determine primer annealing temperature during PCR?
Primer Annealing Temperature: The primer melting temperature is the estimate of the DNA-DNA hybrid stability and critical in determining the annealing temperature. Too high Ta will produce insufficient primer-template hybridization resulting in low PCR product yield. Too low Ta may possibly lead to non-specific products caused by a high number of base pair mismatches,. Mismatch tolerance is found to have the strongest influence on PCR specificity. *This information was taken from:*https://www.premierbiosoft.com/tech_notes/PCR_Primer_Design.html
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Promp Copilot: In simple terms compare PCR and and restriction enzyme digests to créate linear fragments of DNA and explain when one may be preferable to use over the other and what are the protocol diferences.
Copilot answere: PCR creates DNA fragments by copying a specific region using primers and a polymerase.
Restriction digests cut DNA at specific short sequences using restriction enzymes.
PCR is best when you want to amplify or create a fragment that doesn’t already exist as a clean piece.
Restriction digestion is best when you want to cut an existing plasmid or DNA sample at precise known sites.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Following the lab protocol here are some considerations to be taken when PCR is uded to amplify regions of DNA:
Length: 18–22 bp core binding region, plus overhang if needed.
Tm Range: ~52–58 °C for optimal annealing; pairs should be within 5 °C.
GC-clamp: Add G or C bases at the 3’ end of primers (GC clamp) to promote specific binding. More than 3 G’s or C’s should be avoided in the last 5 bases at the 3’ end of the primer.
Secondary Structures: Avoid strong hairpins or dimers (check with software).
How does the plasmid DNA enter the E. coli cells during transformation?
Following the lab protocol there are to main methods to be considered:
1.Heat shock: Generate pores in bacterial cell wall with an abrupt temperature change
2.Electroporation: Generate pores in bacterial cell wall with high electrical voltage
In either of these methods, we shock the cells (either by applying an electrical shock or heat shock) which causes the cell membrane to “open up”. The plasmid now enters the cells by diffusion. After the shock, the cells are fed with SOC growth media and incubated for 1 hour in 37C. That one hour incubation filled with nutrients allows the cells to recover and to start multiplying, some with the plasmid inside. Last, we plate the transformed cells onto a selective media plate, which contains antibiotics.
Only cells that had successfully received the plasmid will survive the antibiotics and grow. After 1-2 days, we should see the colors expressed by our inserted gene.
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences..
Promp Copilot: Explain how the Golden gate assembly Works in an short way
Golden Gate assembly is a one‑pot DNA cloning method that uses Type IIS restriction enzymes (like BsaI or BsmBI) to cut outside their recognition site, creating custom overhangs that fit together like puzzle pieces.
🧬 Short, simple explanation
Type IIS enzymes cut outside their recognition sequence, generating overhangs you design.
Each DNA fragment is built with matching overhangs, so they assemble in a specific order.
The reaction cycles between:
37°C → enzyme cuts and creates overhangs
16°C → ligase joins the fragments
Because the recognition sites are removed during assembly, the enzyme cannot cut the final product again, making the reaction self‑correcting.
The result is a seamless, scarless, correctly ordered DNA construct assembled from many parts in a single tube.
In short: Golden Gate lets you assemble multiple DNA fragments in a precise order in one reaction, without extra scars or leftover restriction sites.
…plus diagrams (either handmade or online).
Copilot:
[Fragment A] [Fragment B] [Fragment C]
┌──────────────┐ ┌──────────────┐ ┌──────────────┐
| BsaI site | | BsaI site | | BsaI site |
| + designed | | + designed | | + designed |
| overhangs | | overhangs | | overhangs |
└──────┬────────┘ └──────┬────────┘ └──────┬────────┘
| | |
v v v
BsaI cuts BsaI cuts BsaI cuts
┌──────────────┐ ┌──────────────┐ ┌──────────────┐
| A overhang | | B overhang | | C overhang |
└──────┬────────┘ └──────┬────────┘ └──────┬────────┘
| | |
| Matching | Matching |
| overhangs | overhangs |
v v v
DNA Ligase joins matching pieces
(like puzzle pieces snapping together)
┌──────────────────────────────┐
| Final assembled product |
| A — B — C (correct order) |
└──────────────────────────────┘
Important:
The BsaI recognition sites are removed during assembly.
Final product cannot be cut again → reaction becomes self‑correcting.
Assignment: Asimov Kernel
Kernel is a service provided by Asimov, wich is An integrated suite of cells, genes, and software to power advanced genetic design: https://www.asimov.com/
Asimov Kernel Integrated software for data-driven genetic design
Kernel connects Asimov’s models, bio-tools, and expertise into one interface for designing and optimizing genetic constructs—enabling scientists to independently design, optimize, and deploy high-performing expression systems: https://www.asimov.com/kernel
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
Week 7: Genetic circuits part II
Assignment Part 1: Intracellular Artificial Neural Networks
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Assignment Part 3: First DNA Twist Order
Review the Individual Final Project documentation guidelines.
Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs.
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.