Homework
Weekly homework submissions:
- First, describe a biological engineering application or tool you want to develop and why. I want to develop a plant stress-responsive synthetic gene circuit in a chloroplast-derived cell-free system that detects stress signals like pathogen RNA or heavy metals and produces a visible reporter output. This tool enables rapid, safe prototyping of plant gene circuits and allows assessment of biosecurity risks, such as misfires or misuse, without using live plants. The primary motivation for this project is to build upon and extend the work of the 2021 iGEM Marburg team, leveraging their foundational advances to develop more responsive and secure plant synthetic biology tools.
</div> </div> <div class="card-image"> <img src="/2026a/md-ashraful-islam/homework/week-01-hw-principles-and-practices/week1icon-pipette.featured.png"> </div>Part 1: Benchling & In-silico Gel Art Make a free account at benchling.com Import the Lambda DNA. Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI Python Script for Opentrons Artwork from opentrons import types import string metadata = { ‘protocolName’: ‘{Ashraful} - Opentrons Art - HTGAA’, ‘author’: ‘Ashraful’, ‘source’: ‘HTGAA 2026’, ‘apiLevel’: ‘2.20’ } Z_VALUE_AGAR = 2.0 POINT_SIZE = 0.75 sfgfp_points = [(-17.6,13.2), (-15.4,13.2), (-13.2,13.2), (-11,13.2), (-8.8,13.2), (11,13.2), (13.2,13.2), (15.4,13.2), (17.6,13.2), (-17.6,11), (-15.4,11), (-11,11), (-8.8,11), (8.8,11), (11,11), (13.2,11), (15.4,11), (17.6,11), (-15.4,8.8), (-13.2,8.8), (-8.8,8.8), (-6.6,8.8), (6.6,8.8), (8.8,8.8), (13.2,8.8), (15.4,8.8), (-13.2,6.6), (-11,6.6), (-6.6,6.6), (-4.4,6.6), (4.4,6.6), (6.6,6.6), (11,6.6), (13.2,6.6), (-11,4.4), (-8.8,4.4), (-4.4,4.4), (-2.2,4.4), (2.2,4.4), (4.4,4.4), (8.8,4.4), (11,4.4), (-8.8,2.2), (-6.6,2.2), (-2.2,2.2), (0,2.2), (2.2,2.2), (6.6,2.2), (8.8,2.2), (-6.6,0), (-4.4,0), (0,0), (4.4,0), (6.6,0), (-4.4,-2.2), (-2.2,-2.2), (0,-2.2), (2.2,-2.2), (4.4,-2.2), (-2.2,-4.4), (0,-4.4), (2.2,-4.4), (-2.2,-6.6), (0,-6.6), (2.2,-6.6), (-2.2,-8.8), (0,-8.8), (2.2,-8.8), (-2.2,-11), (0,-11), (2.2,-11), (-2.2,-13.2), (0,-13.2), (2.2,-13.2), (-11,-15.4), (-8.8,-15.4), (-6.6,-15.4), (-4.4,-15.4), (-2.2,-15.4), (0,-15.4), (2.2,-15.4), (4.4,-15.4), (6.6,-15.4), (8.8,-15.4), (11,-15.4), (13.2,-15.4), (-13.2,-17.6), (-11,-17.6), (-8.8,-17.6), (-6.6,-17.6), (-4.4,-17.6), (-2.2,-17.6), (0,-17.6), (2.2,-17.6), (4.4,-17.6), (6.6,-17.6), (8.8,-17.6), (11,-17.6), (13.2,-17.6), (15.4,-17.6), (-15.4,-19.8), (-13.2,-19.8), (-11,-19.8), (-8.8,-19.8), (-6.6,-19.8), (-4.4,-19.8), (-2.2,-19.8), (0,-19.8), (2.2,-19.8), (4.4,-19.8), (6.6,-19.8), (8.8,-19.8), (11,-19.8), (13.2,-19.8), (15.4,-19.8), (17.6,-19.8)] point_name_pairing = [(“sfgfp”, sfgfp_points)] # Robot deck setup constants TIP_RACK_DECK_SLOT = 9 COLORS_DECK_SLOT = 6 AGAR_DECK_SLOT = 5 PIPETTE_STARTING_TIP_WELL = ‘A1’ # Place the PCR tubes in this order well_colors = { ‘A1’: ‘sfGFP’, ‘A2’: ‘mRFP1’, ‘A3’: ‘mKO2’, ‘A4’: ‘Venus’, ‘A5’: ‘mKate2_TF’, ‘A6’: ‘Azurite’, ‘A7’: ‘mCerulean3’, ‘A8’: ‘mClover3’, ‘A9’: ‘mJuniper’, ‘A10’: ‘mTurquoise2’, ‘A11’: ‘mBanana’, ‘A12’: ‘mPlum’, ‘B1’: ‘Electra2’, ‘B2’: ‘mWasabi’, ‘B3’: ‘mScarlet_I’, ‘B4’: ‘mPapaya’, ‘B5’: ’eqFP578’, ‘B6’: ’tdTomato’, ‘B7’: ‘DsRed’, ‘B8’: ‘mKate2’, ‘B9’: ‘EGFP’, ‘B10’: ‘mRuby2’, ‘B11’: ‘TagBFP’, ‘B12’: ‘mChartreuse_TF’, ‘C1’: ‘mLychee_TF’, ‘C2’: ‘mTagBFP2’, ‘C3’: ‘mEGFP’, ‘C4’: ‘mNeonGreen’, ‘C5’: ‘mAzamiGreen’, ‘C6’: ‘mWatermelon’, ‘C7’: ‘avGFP’, ‘C8’: ‘mCitrine’, ‘C9’: ‘mVenus’, ‘C10’: ‘mCherry’, ‘C11’: ‘mHoneydew’, ‘C12’: ‘TagRFP’, ‘D1’: ‘mTFP1’, ‘D2’: ‘Ultramarine’, ‘D3’: ‘ZsGreen1’, ‘D4’: ‘mMiCy’, ‘D5’: ‘mStayGold2’, ‘D6’: ‘PA_GFP’ } # Mapping for visualization colors VISUALIZATION_COLOR_MAP = { ‘sfGFP’: ‘green’, ‘mRFP1’: ‘red’, ‘mKO2’: ‘orange’, ‘Venus’: ‘yellow’, ‘mKate2_TF’: ‘purple’, ‘Azurite’: ‘blue’, ‘mCerulean3’: ‘cyan’, ‘mClover3’: ’lightgreen’, ‘mJuniper’: ‘darkgreen’, ‘mTurquoise2’: ’teal’, ‘mBanana’: ‘gold’, ‘mPlum’: ‘plum’, ‘Electra2’: ’navy’, ‘mWasabi’: ’lime’, ‘mScarlet_I’: ‘darkred’, ‘mPapaya’: ‘peachpuff’, ’eqFP578’: ‘brown’, ’tdTomato’: ’tomato’, ‘DsRed’: ‘indianred’, ‘mKate2’: ‘darkmagenta’, ‘EGFP’: ‘chartreuse’, ‘mRuby2’: ‘firebrick’, ‘TagBFP’: ‘slateblue’, ‘mChartreuse_TF’: ‘darkseagreen’, ‘mLychee_TF’: ‘palevioletred’, ‘mTagBFP2’: ‘darkblue’, ‘mEGFP’: ’limegreen’, ‘mNeonGreen’: ’lawngreen’, ‘mAzamiGreen’: ‘mediumseagreen’, ‘mWatermelon’: ‘pink’, ‘avGFP’: ‘forestgreen’, ‘mCitrine’: ‘khaki’, ‘mVenus’: ‘olivedrab’, ‘mCherry’: ‘crimson’, ‘mHoneydew’: ‘honeydew’, # This will likely be too light ‘TagRFP’: ‘rosybrown’, ‘mTFP1’: ‘dodgerblue’, ‘Ultramarine’: ‘mediumblue’, ‘ZsGreen1’: ‘springgreen’, ‘mMiCy’: ‘peru’, ‘mStayGold2’: ‘goldenrod’, ‘PA_GFP’: ‘darkgreen’ } volume_used = { ‘sfgfp’: 0 } def update_volume_remaining(current_color, quantity_to_aspirate): rows = string.ascii_uppercase for well, color in list(well_colors.items()): if color == current_color: if (volume_used[current_color] + quantity_to_aspirate) > 250: # Move to next well horizontally by advancing row letter, keeping column number row = well[0] col = well[1:] # Find next row letter next_row = rows[rows.index(row) + 1] next_well = f"{next_row}{col}" del well_colors[well] well_colors[next_well] = current_color volume_used[current_color] = quantity_to_aspirate else: volume_used[current_color] += quantity_to_aspirate break def run(protocol): # Load labware, modules and pipettes protocol.home() # Tips tips_20ul = protocol.load_labware(‘opentrons_96_tiprack_20ul’, TIP_RACK_DECK_SLOT, ‘Opentrons 20uL Tips’) # Pipettes pipette_20ul = protocol.load_instrument(“p20_single_gen2”, “right”, [tips_20ul]) # Deep Well Plate temperature_plate = protocol.load_labware(’nest_96_wellplate_2ml_deep’, 6, ‘Deep Well Plate’) # Agar Plate agar_plate = protocol.load_labware(‘htgaa_agar_plate’, AGAR_DECK_SLOT, ‘Agar Plate’) agar_plate.set_offset(x=0.00, y=0.00, z=Z_VALUE_AGAR) # Get the top-center of the plate, make sure the plate was calibrated before running this center_location = agar_plate[‘A1’].top() pipette_20ul.starting_tip = tips_20ul.well(PIPETTE_STARTING_TIP_WELL) # Helper function (dispensing) def dispense_and_jog(pipette, volume, location): assert(isinstance(volume, (int, float))) # Go above the location above_location = location.move(types.Point(z=location.point.z + 2)) pipette.move_to(above_location) # Go downwards and dispense pipette.dispense(volume, location) # Go upwards to avoid smearing pipette.move_to(above_location) # Helper function (color location) def location_of_color(color_string): for well,color in well_colors.items(): if color.lower() == color_string.lower(): return temperature_plate[well] raise ValueError(f"No well found with color {color_string}") # Print pattern by iterating over lists for i, (current_color, point_list) in enumerate(point_name_pairing): # Skip the rest of the loop if the list is empty if not point_list: continue # Get the tip for this run, set the bacteria color, and the aspirate bacteria of choice pipette_20ul.pick_up_tip() max_aspirate = int(18 // POINT_SIZE) * POINT_SIZE quantity_to_aspirate = min(len(point_list)*POINT_SIZE, max_aspirate) update_volume_remaining(current_color, quantity_to_aspirate) pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color)) # Iterate over the current points list and dispense them, refilling along the way for i in range(len(point_list)): x, y = point_list[i] adjusted_location = center_location.move(types.Point(x, y)) dispense_and_jog(pipette_20ul, POINT_SIZE, adjusted_location) if pipette_20ul.current_volume == 0 and len(point_list[i+1:]) > 0: quantity_to_aspirate = min(len(point_list[i:])*POINT_SIZE, max_aspirate) update_volume_remaining(current_color, quantity_to_aspirate) pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color)) # Drop tip between each color pipette_20ul.drop_tip() Simulation # Execute Simulation / Visualization protocol = OpentronsMock(well_colors, VISUALIZATION_COLOR_MAP) run(protocol) protocol.visualize() Post-Lab Questions Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Python Script for Opentrons Artwork from opentrons import types import string metadata = { ‘protocolName’: ‘{Ashraful} - Opentrons Art - HTGAA’, ‘author’: ‘Ashraful’, ‘source’: ‘HTGAA 2026’, ‘apiLevel’: ‘2.20’ } Z_VALUE_AGAR = 2.0 POINT_SIZE = 0.75 sfgfp_points = [(-17.6,13.2), (-15.4,13.2), (-13.2,13.2), (-11,13.2), (-8.8,13.2), (11,13.2), (13.2,13.2), (15.4,13.2), (17.6,13.2), (-17.6,11), (-15.4,11), (-11,11), (-8.8,11), (8.8,11), (11,11), (13.2,11), (15.4,11), (17.6,11), (-15.4,8.8), (-13.2,8.8), (-8.8,8.8), (-6.6,8.8), (6.6,8.8), (8.8,8.8), (13.2,8.8), (15.4,8.8), (-13.2,6.6), (-11,6.6), (-6.6,6.6), (-4.4,6.6), (4.4,6.6), (6.6,6.6), (11,6.6), (13.2,6.6), (-11,4.4), (-8.8,4.4), (-4.4,4.4), (-2.2,4.4), (2.2,4.4), (4.4,4.4), (8.8,4.4), (11,4.4), (-8.8,2.2), (-6.6,2.2), (-2.2,2.2), (0,2.2), (2.2,2.2), (6.6,2.2), (8.8,2.2), (-6.6,0), (-4.4,0), (0,0), (4.4,0), (6.6,0), (-4.4,-2.2), (-2.2,-2.2), (0,-2.2), (2.2,-2.2), (4.4,-2.2), (-2.2,-4.4), (0,-4.4), (2.2,-4.4), (-2.2,-6.6), (0,-6.6), (2.2,-6.6), (-2.2,-8.8), (0,-8.8), (2.2,-8.8), (-2.2,-11), (0,-11), (2.2,-11), (-2.2,-13.2), (0,-13.2), (2.2,-13.2), (-11,-15.4), (-8.8,-15.4), (-6.6,-15.4), (-4.4,-15.4), (-2.2,-15.4), (0,-15.4), (2.2,-15.4), (4.4,-15.4), (6.6,-15.4), (8.8,-15.4), (11,-15.4), (13.2,-15.4), (-13.2,-17.6), (-11,-17.6), (-8.8,-17.6), (-6.6,-17.6), (-4.4,-17.6), (-2.2,-17.6), (0,-17.6), (2.2,-17.6), (4.4,-17.6), (6.6,-17.6), (8.8,-17.6), (11,-17.6), (13.2,-17.6), (15.4,-17.6), (-15.4,-19.8), (-13.2,-19.8), (-11,-19.8), (-8.8,-19.8), (-6.6,-19.8), (-4.4,-19.8), (-2.2,-19.8), (0,-19.8), (2.2,-19.8), (4.4,-19.8), (6.6,-19.8), (8.8,-19.8), (11,-19.8), (13.2,-19.8), (15.4,-19.8), (17.6,-19.8)] point_name_pairing = [(“sfgfp”, sfgfp_points)] # Robot deck setup constants TIP_RACK_DECK_SLOT = 9 COLORS_DECK_SLOT = 6 AGAR_DECK_SLOT = 5 PIPETTE_STARTING_TIP_WELL = ‘A1’ # Place the PCR tubes in this order well_colors = { ‘A1’: ‘sfGFP’, ‘A2’: ‘mRFP1’, ‘A3’: ‘mKO2’, ‘A4’: ‘Venus’, ‘A5’: ‘mKate2_TF’, ‘A6’: ‘Azurite’, ‘A7’: ‘mCerulean3’, ‘A8’: ‘mClover3’, ‘A9’: ‘mJuniper’, ‘A10’: ‘mTurquoise2’, ‘A11’: ‘mBanana’, ‘A12’: ‘mPlum’, ‘B1’: ‘Electra2’, ‘B2’: ‘mWasabi’, ‘B3’: ‘mScarlet_I’, ‘B4’: ‘mPapaya’, ‘B5’: ’eqFP578’, ‘B6’: ’tdTomato’, ‘B7’: ‘DsRed’, ‘B8’: ‘mKate2’, ‘B9’: ‘EGFP’, ‘B10’: ‘mRuby2’, ‘B11’: ‘TagBFP’, ‘B12’: ‘mChartreuse_TF’, ‘C1’: ‘mLychee_TF’, ‘C2’: ‘mTagBFP2’, ‘C3’: ‘mEGFP’, ‘C4’: ‘mNeonGreen’, ‘C5’: ‘mAzamiGreen’, ‘C6’: ‘mWatermelon’, ‘C7’: ‘avGFP’, ‘C8’: ‘mCitrine’, ‘C9’: ‘mVenus’, ‘C10’: ‘mCherry’, ‘C11’: ‘mHoneydew’, ‘C12’: ‘TagRFP’, ‘D1’: ‘mTFP1’, ‘D2’: ‘Ultramarine’, ‘D3’: ‘ZsGreen1’, ‘D4’: ‘mMiCy’, ‘D5’: ‘mStayGold2’, ‘D6’: ‘PA_GFP’ } # Mapping for visualization colors VISUALIZATION_COLOR_MAP = { ‘sfGFP’: ‘green’, ‘mRFP1’: ‘red’, ‘mKO2’: ‘orange’, ‘Venus’: ‘yellow’, ‘mKate2_TF’: ‘purple’, ‘Azurite’: ‘blue’, ‘mCerulean3’: ‘cyan’, ‘mClover3’: ’lightgreen’, ‘mJuniper’: ‘darkgreen’, ‘mTurquoise2’: ’teal’, ‘mBanana’: ‘gold’, ‘mPlum’: ‘plum’, ‘Electra2’: ’navy’, ‘mWasabi’: ’lime’, ‘mScarlet_I’: ‘darkred’, ‘mPapaya’: ‘peachpuff’, ’eqFP578’: ‘brown’, ’tdTomato’: ’tomato’, ‘DsRed’: ‘indianred’, ‘mKate2’: ‘darkmagenta’, ‘EGFP’: ‘chartreuse’, ‘mRuby2’: ‘firebrick’, ‘TagBFP’: ‘slateblue’, ‘mChartreuse_TF’: ‘darkseagreen’, ‘mLychee_TF’: ‘palevioletred’, ‘mTagBFP2’: ‘darkblue’, ‘mEGFP’: ’limegreen’, ‘mNeonGreen’: ’lawngreen’, ‘mAzamiGreen’: ‘mediumseagreen’, ‘mWatermelon’: ‘pink’, ‘avGFP’: ‘forestgreen’, ‘mCitrine’: ‘khaki’, ‘mVenus’: ‘olivedrab’, ‘mCherry’: ‘crimson’, ‘mHoneydew’: ‘honeydew’, # This will likely be too light ‘TagRFP’: ‘rosybrown’, ‘mTFP1’: ‘dodgerblue’, ‘Ultramarine’: ‘mediumblue’, ‘ZsGreen1’: ‘springgreen’, ‘mMiCy’: ‘peru’, ‘mStayGold2’: ‘goldenrod’, ‘PA_GFP’: ‘darkgreen’ } volume_used = { ‘sfgfp’: 0 } def update_volume_remaining(current_color, quantity_to_aspirate): rows = string.ascii_uppercase for well, color in list(well_colors.items()): if color == current_color: if (volume_used[current_color] + quantity_to_aspirate) > 250: # Move to next well horizontally by advancing row letter, keeping column number row = well[0] col = well[1:] # Find next row letter next_row = rows[rows.index(row) + 1] next_well = f"{next_row}{col}" del well_colors[well] well_colors[next_well] = current_color volume_used[current_color] = quantity_to_aspirate else: volume_used[current_color] += quantity_to_aspirate break def run(protocol): # Load labware, modules and pipettes protocol.home() # Tips tips_20ul = protocol.load_labware(‘opentrons_96_tiprack_20ul’, TIP_RACK_DECK_SLOT, ‘Opentrons 20uL Tips’) # Pipettes pipette_20ul = protocol.load_instrument(“p20_single_gen2”, “right”, [tips_20ul]) # Deep Well Plate temperature_plate = protocol.load_labware(’nest_96_wellplate_2ml_deep’, 6, ‘Deep Well Plate’) # Agar Plate agar_plate = protocol.load_labware(‘htgaa_agar_plate’, AGAR_DECK_SLOT, ‘Agar Plate’) agar_plate.set_offset(x=0.00, y=0.00, z=Z_VALUE_AGAR) # Get the top-center of the plate, make sure the plate was calibrated before running this center_location = agar_plate[‘A1’].top() pipette_20ul.starting_tip = tips_20ul.well(PIPETTE_STARTING_TIP_WELL) # Helper function (dispensing) def dispense_and_jog(pipette, volume, location): assert(isinstance(volume, (int, float))) # Go above the location above_location = location.move(types.Point(z=location.point.z + 2)) pipette.move_to(above_location) # Go downwards and dispense pipette.dispense(volume, location) # Go upwards to avoid smearing pipette.move_to(above_location) # Helper function (color location) def location_of_color(color_string): for well,color in well_colors.items(): if color.lower() == color_string.lower(): return temperature_plate[well] raise ValueError(f"No well found with color {color_string}") # Print pattern by iterating over lists for i, (current_color, point_list) in enumerate(point_name_pairing): # Skip the rest of the loop if the list is empty if not point_list: continue # Get the tip for this run, set the bacteria color, and the aspirate bacteria of choice pipette_20ul.pick_up_tip() max_aspirate = int(18 // POINT_SIZE) * POINT_SIZE quantity_to_aspirate = min(len(point_list)*POINT_SIZE, max_aspirate) update_volume_remaining(current_color, quantity_to_aspirate) pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color)) # Iterate over the current points list and dispense them, refilling along the way for i in range(len(point_list)): x, y = point_list[i] adjusted_location = center_location.move(types.Point(x, y)) dispense_and_jog(pipette_20ul, POINT_SIZE, adjusted_location) if pipette_20ul.current_volume == 0 and len(point_list[i+1:]) > 0: quantity_to_aspirate = min(len(point_list[i:])*POINT_SIZE, max_aspirate) update_volume_remaining(current_color, quantity_to_aspirate) pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color)) # Drop tip between each color pipette_20ul.drop_tip() Simulation # Execute Simulation / Visualization protocol = OpentronsMock(well_colors, VISUALIZATION_COLOR_MAP) run(protocol) protocol.visualize() Post-Lab Questions Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications. Part A. Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Skeletal muscle (meat) is approximately 20–25% protein by mass, with the remainder being water (~75%), fat, and connective tissue. Taking a conservative estimate of 20% protein, 500 g of meat contains roughly 100 g of protein. During digestion, proteases (pepsin in the stomach, trypsin and chymotrypsin in the small intestine) hydrolyse peptide bonds, releasing individual amino acids — the monomeric units. Using the given average amino acid molecular weight of 100 Daltons (100 g/mol): moles of amino acids = mass / molar mass = 100 g ÷ 100 g/mol = 1 mol Applying Avogadro’s number: N = 1 mol × 6.022 × 10²³ molecules/mol ≈ 6 × 10²³ molecules of amino acids This is a minimum estimate; the true figure is slightly higher because the average residue mass in a polypeptide chain is closer to 110–128 Da (due to the loss of water during peptide bond formation, the backbone residue mass averages ~110 Da, but free amino acids average ~128 Da). If we use 128 Da for free amino acids, we obtain ≈ 4.7 × 10²³ molecules — still on the order of half an Avogadro. Either way, the scale is strikingly close to 10²³, illustrating that a single meal-sized portion of protein delivers amino acids on the order of Avogadro’s number.
Part A. Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Skeletal muscle (meat) is approximately 20–25% protein by mass, with the remainder being water (~75%), fat, and connective tissue. Taking a conservative estimate of 20% protein, 500 g of meat contains roughly 100 g of protein. During digestion, proteases (pepsin in the stomach, trypsin and chymotrypsin in the small intestine) hydrolyse peptide bonds, releasing individual amino acids — the monomeric units. Using the given average amino acid molecular weight of 100 Daltons (100 g/mol): moles of amino acids = mass / molar mass = 100 g ÷ 100 g/mol = 1 mol Applying Avogadro’s number: N = 1 mol × 6.022 × 10²³ molecules/mol ≈ 6 × 10²³ molecules of amino acids This is a minimum estimate; the true figure is slightly higher because the average residue mass in a polypeptide chain is closer to 110–128 Da (due to the loss of water during peptide bond formation, the backbone residue mass averages ~110 Da, but free amino acids average ~128 Da). If we use 128 Da for free amino acids, we obtain ≈ 4.7 × 10²³ molecules — still on the order of half an Avogadro. Either way, the scale is strikingly close to 10²³, illustrating that a single meal-sized portion of protein delivers amino acids on the order of Avogadro’s number. Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation. Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card: Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison. Record the perplexity scores that indicate PepMLM’s confidence in the binders
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation. Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card: Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison. Record the perplexity scores that indicate PepMLM’s confidence in the binders Assignment: DNA Assembly Answer these questions about the protocol in this week’s lab: What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The Phusion High-Fidelity PCR Master Mix (NEB #M0531) is supplied as a convenient 2× pre-formulated reagent containing all reaction components except template DNA, primers, and water. Its key constituents and their functions are as follows. Phusion DNA Polymerase is the catalytic engine of the mix. It is a chimeric enzyme comprising a Pyrococcus-like thermostable polymerase core fused to a processivity-enhancing domain (derived from the Sso7d protein family), which allows the enzyme to remain bound to the DNA template for longer stretches and amplify fragments at higher speed and with greater fidelity than standard Taq polymerase. Crucially, Phusion carries a 3′→5′ proofreading exonuclease activity that removes incorrectly incorporated nucleotides, giving it an error rate more than 50-fold lower than Taq and roughly 6-fold lower than Pfu polymerase — making it the appropriate choice whenever sequence accuracy matters, such as for cloning (NEB, 2024). Deoxynucleotide triphosphates (dNTPs) — dATP, dCTP, dGTP, and dTTP — are the building blocks that the polymerase incorporates into the nascent DNA strand. They are pre-included in the master mix at a balanced concentration to minimise pipetting error. MgCl₂ (magnesium chloride) is an essential cofactor. Mg²⁺ ions coordinate with the phosphate groups of the incoming dNTP in the polymerase active site, enabling the phosphodiester bond-forming reaction. The concentration of free Mg²⁺ also influences polymerase processivity and primer–template specificity; the HF Buffer formulation has been optimised to include the appropriate Mg²⁺ concentration for standard templates. Reaction buffer (HF Buffer) maintains the correct pH and ionic strength for optimal polymerase activity. The buffer stabilises the enzyme during the high-temperature denaturation steps and helps establish reproducible annealing conditions. An alternative GC Buffer formulation is available for GC-rich or otherwise difficult templates, optionally supplemented with DMSO to reduce secondary structure in the template. Together, these components mean the researcher only needs to add template, primers, and water — dramatically reducing pipetting steps and the risk of component-level errors.
Assignment: DNA Assembly Answer these questions about the protocol in this week’s lab: What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The Phusion High-Fidelity PCR Master Mix (NEB #M0531) is supplied as a convenient 2× pre-formulated reagent containing all reaction components except template DNA, primers, and water. Its key constituents and their functions are as follows. Phusion DNA Polymerase is the catalytic engine of the mix. It is a chimeric enzyme comprising a Pyrococcus-like thermostable polymerase core fused to a processivity-enhancing domain (derived from the Sso7d protein family), which allows the enzyme to remain bound to the DNA template for longer stretches and amplify fragments at higher speed and with greater fidelity than standard Taq polymerase. Crucially, Phusion carries a 3′→5′ proofreading exonuclease activity that removes incorrectly incorporated nucleotides, giving it an error rate more than 50-fold lower than Taq and roughly 6-fold lower than Pfu polymerase — making it the appropriate choice whenever sequence accuracy matters, such as for cloning (NEB, 2024). Deoxynucleotide triphosphates (dNTPs) — dATP, dCTP, dGTP, and dTTP — are the building blocks that the polymerase incorporates into the nascent DNA strand. They are pre-included in the master mix at a balanced concentration to minimise pipetting error. MgCl₂ (magnesium chloride) is an essential cofactor. Mg²⁺ ions coordinate with the phosphate groups of the incoming dNTP in the polymerase active site, enabling the phosphodiester bond-forming reaction. The concentration of free Mg²⁺ also influences polymerase processivity and primer–template specificity; the HF Buffer formulation has been optimised to include the appropriate Mg²⁺ concentration for standard templates. Reaction buffer (HF Buffer) maintains the correct pH and ionic strength for optimal polymerase activity. The buffer stabilises the enzyme during the high-temperature denaturation steps and helps establish reproducible annealing conditions. An alternative GC Buffer formulation is available for GC-rich or otherwise difficult templates, optionally supplemented with DMSO to reduce secondary structure in the template. Together, these components mean the researcher only needs to add template, primers, and water — dramatically reducing pipetting steps and the risk of component-level errors. Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Traditional genetic circuits operate as Boolean logic gates: they classify inputs as either “on” (1) or “off” (0) and produce outputs that are likewise binary. While this is powerful for implementing discrete decisions — such as activating a kill switch if and only if two specific signals are simultaneously present — Boolean circuits are fundamentally limited in their ability to process the continuous, graded molecular signals that characterise real biological environments. Intracellular concentrations of transcription factors, metabolites, and signalling molecules are not naturally binary; they span continuous ranges that carry information that a simple Boolean threshold necessarily discards. IANNs overcome this limitation by implementing analog computation, in which each molecular “neuron” computes a weighted sum of its continuous-valued inputs, passes that sum through a nonlinear activation function, and produces a graded output that can itself serve as an input to the next layer. This architecture enables a single engineered cell to perform multi-threshold classification — distinguishing not just “signal present” from “signal absent” but grading responses proportionally to signal intensity, and separating input patterns that no Boolean gate could resolve without an exponentially larger circuit. For example, a cell expressing a two-input biomolecular perceptron can draw a separating hyperplane in the continuous input space of two molecular concentrations, classifying cell states that would require many cascaded Boolean gates to approximate. A second key advantage is graceful degradation under noise: because IANNs operate over a continuous input range, they can be designed with soft thresholds that smooth over stochastic fluctuations in molecule numbers — a pervasive problem in cells, where copy numbers of regulatory molecules are often in the tens to hundreds range. Boolean gates, which depend on crossing a hard threshold, are comparatively fragile to such noise. Third, IANNs are in principle extendable toward online learning, in which the synaptic weights (encoded by molecular concentrations or binding affinities) can be updated as a function of experience — an entirely alien concept to hardwired Boolean logic. Taken together, IANNs expand the computational vocabulary available to synthetic biology from a finite set of logic operations to a continuous, composable, and theoretically universal function approximation framework.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Traditional genetic circuits operate as Boolean logic gates: they classify inputs as either “on” (1) or “off” (0) and produce outputs that are likewise binary. While this is powerful for implementing discrete decisions — such as activating a kill switch if and only if two specific signals are simultaneously present — Boolean circuits are fundamentally limited in their ability to process the continuous, graded molecular signals that characterise real biological environments. Intracellular concentrations of transcription factors, metabolites, and signalling molecules are not naturally binary; they span continuous ranges that carry information that a simple Boolean threshold necessarily discards. IANNs overcome this limitation by implementing analog computation, in which each molecular “neuron” computes a weighted sum of its continuous-valued inputs, passes that sum through a nonlinear activation function, and produces a graded output that can itself serve as an input to the next layer. This architecture enables a single engineered cell to perform multi-threshold classification — distinguishing not just “signal present” from “signal absent” but grading responses proportionally to signal intensity, and separating input patterns that no Boolean gate could resolve without an exponentially larger circuit. For example, a cell expressing a two-input biomolecular perceptron can draw a separating hyperplane in the continuous input space of two molecular concentrations, classifying cell states that would require many cascaded Boolean gates to approximate. A second key advantage is graceful degradation under noise: because IANNs operate over a continuous input range, they can be designed with soft thresholds that smooth over stochastic fluctuations in molecule numbers — a pervasive problem in cells, where copy numbers of regulatory molecules are often in the tens to hundreds range. Boolean gates, which depend on crossing a hard threshold, are comparatively fragile to such noise. Third, IANNs are in principle extendable toward online learning, in which the synaptic weights (encoded by molecular concentrations or binding affinities) can be updated as a function of experience — an entirely alien concept to hardwired Boolean logic. Taken together, IANNs expand the computational vocabulary available to synthetic biology from a finite set of logic operations to a continuous, composable, and theoretically universal function approximation framework. Homework Part A: General and Lecturer-Specific Questions General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis (CFPS) offers a fundamentally different operating logic from in vivo expression: because there is no living cell to maintain, the reaction environment is open and directly accessible to the experimenter. This openness translates into three practical advantages. First, reaction components — amino acid concentrations, buffer conditions, redox potential, template concentration — can be tuned independently and in real time without the buffering effects of cellular homeostasis. Second, toxic proteins that would kill or arrest growing cells can be expressed freely in CFPS, since there is no cell viability to protect. Third, non-canonical amino acids, isotopic labels, or synthetic chemical groups can be incorporated site-specifically by supplementing the reaction directly, enabling protein engineering strategies that are impossible to sustain through the protein expression machinery of a living cell. Two cases where cell-free expression is specifically more advantageous than cell-based production are: (1) membrane protein structural studies, where the absence of competing cellular membranes allows co-translational insertion directly into defined lipid nanodiscs of controlled composition, circumventing the protein aggregation and misfolding problems that arise during over-expression in intact cells; and (2) rapid on-demand diagnostic biosensors, where freeze-dried CFPS reactions can be deployed at the point of need without cold-chain infrastructure or biohazard containment — capabilities recently validated aboard the International Space Station.
Homework Part A: General and Lecturer-Specific Questions General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis (CFPS) offers a fundamentally different operating logic from in vivo expression: because there is no living cell to maintain, the reaction environment is open and directly accessible to the experimenter. This openness translates into three practical advantages. First, reaction components — amino acid concentrations, buffer conditions, redox potential, template concentration — can be tuned independently and in real time without the buffering effects of cellular homeostasis. Second, toxic proteins that would kill or arrest growing cells can be expressed freely in CFPS, since there is no cell viability to protect. Third, non-canonical amino acids, isotopic labels, or synthetic chemical groups can be incorporated site-specifically by supplementing the reaction directly, enabling protein engineering strategies that are impossible to sustain through the protein expression machinery of a living cell. Two cases where cell-free expression is specifically more advantageous than cell-based production are: (1) membrane protein structural studies, where the absence of competing cellular membranes allows co-translational insertion directly into defined lipid nanodiscs of controlled composition, circumventing the protein aggregation and misfolding problems that arise during over-expression in intact cells; and (2) rapid on-demand diagnostic biosensors, where freeze-dried CFPS reactions can be deployed at the point of need without cold-chain infrastructure or biohazard containment — capabilities recently validated aboard the International Space Station. Part 1: Molecular Weight eGFP Sequence: VSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH Based only on the predicted amino acid sequence of eGFP (see below), what is the calculated molecular weight? You can use an online calculator like the one here: https://web.expasy.org/compute_pi/
Part 1: Molecular Weight eGFP Sequence: VSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH Based only on the predicted amino acid sequence of eGFP (see below), what is the calculated molecular weight? You can use an online calculator like the one here: https://web.expasy.org/compute_pi/ Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork I missed the opportunity to contribute to the HTGAA CFPS bioart project. Later, I contributed to the SynBioBeta bioart project. I worked on part of the DNA on the center-left plate. What I liked: This kind of community-coordinated experiment builds genuine shared investment in the outcome, which is a rare and valuable pedagogical achievement. What could be improved: For future years, giving participants a low-resolution preview of the emerging canvas in near-real-time — without revealing the final image — would heighten the sense of collective emergence and encourage more strategic pixel placement.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork I missed the opportunity to contribute to the HTGAA CFPS bioart project. Later, I contributed to the SynBioBeta bioart project. I worked on part of the DNA on the center-left plate. What I liked: This kind of community-coordinated experiment builds genuine shared investment in the outcome, which is a rare and valuable pedagogical achievement. What could be improved: For future years, giving participants a low-resolution preview of the emerging canvas in near-real-time — without revealing the final image — would heighten the sense of collective emergence and encourage more strategic pixel placement.
Subsections of Homework
Week 1 HW: Principles and Practices
1. First, describe a biological engineering application or tool you want to develop and why.
I want to develop a plant stress-responsive synthetic gene circuit in a chloroplast-derived cell-free system that detects stress signals like pathogen RNA or heavy metals and produces a visible reporter output. This tool enables rapid, safe prototyping of plant gene circuits and allows assessment of biosecurity risks, such as misfires or misuse, without using live plants. The primary motivation for this project is to build upon and extend the work of the 2021 iGEM Marburg team, leveraging their foundational advances to develop more responsive and secure plant synthetic biology tools.
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Goal: Ensure safe and responsible use of plant stress-responsive synthetic gene circuits.
Sub-goals: Prevent misuse or accidental harm using logic gates, kill switches, and monitoring protocols. Promote constructive applications for crop protection and biosecurity preparedness. Maintain transparency and accountability through documentation and ethical guidelines.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”): 1. Purpose: 2. Design: 3. Assumptions: 4. Risks of Failure & “Success”:
Action Purpose Design Assumptions Risks of Failure & Success 1. Circuit Safeguards Require logic gates, kill switches, self-limiting designs Researchers design safeguards; regulators certify Safeguards reliably prevent harm Failure: safeguards bypassed or misconfigured; Success: false sense of security reduces oversight 2. Pre-Deployment Risk Assessment Mandatory biosecurity assessment before field use Researchers submit risk reports; regulators approve Risks can be anticipated and mitigated Failure: assessments become superficial; Success: bureaucratic compliance slows innovation 3. Incentive-Based Governance & Responsible-Use Norms Promote safe, transparent, and ethical plant synbio use Funders require safety plans, audits, and training Incentives motivate responsible behavior Failure: voluntary uptake limits coverage; Success: norms diffuse unevenly across actors 4. Next, score (from 1-3, with 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option: Option 1: Circuit Safeguards Option 2: Pre-Deployment Risk Assessment Option 3: Incentive-Based Governance & Responsible-Use Norms Enhance Biosecurity • By preventing incidents 1 2 3 • By helping respond 2 1 2 Foster Lab Safety • By preventing incidents 1 2 2 • By helping respond 2 1 2 Protect the environment • By preventing incidents 1 2 2 • By helping respond 2 1 3 Other considerations • Minimizing costs/burdens 2 3 1 • Feasibility 1 2 1 • Does not impede research 2 3 1 • Promote constructive applications 2 2 1 5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Based on the scoring, I prioritize a combined approach led by Option 1 (Circuit Safeguards) and Option 3 (Incentive-Based Governance & Responsible-Use Norms), with Option 2 (Pre-Deployment Risk Assessment) applied selectively to higher-risk projects. Circuit safeguards are most effective at preventing incidents by embedding safety directly into design, while incentive-based governance best preserves feasibility, equity, and research freedom. Risk assessments are valuable for response and preparedness, but can impose high burdens if universally required. Key trade-offs involve balancing prevention with flexibility. Ethical concerns include overreliance on technical fixes and inequitable access; tiered governance and ongoing safety education help address these risks.
Assignment (Week 2 Lecture Prep)
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate of the polymerase is 1 in 10⁶ bases. The human genome is approximately 3 × 10⁹ base pairs long. Therefore, when compared to the length of the human genome, this error rate corresponds to about 3 × 10³ errors per genome. Biology deals with this discrepency by proofreading, mismatch repair (MMR) system, & redundancy and selection.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The number of different DNA sequences (theoretical): ~3⁴⁰⁰ ≈ 10¹⁹⁰ for a 400-amino-acid protein. Many DNA sequences don’t work in practice due to codon usage bias, mRNA structure, protein folding dynamics, regulatory elements, and mutation robustness/cellular context.
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite (solid‑phase) chemistry.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Per‑cycle inefficiencies and side reactions cause the full‑length fraction to fall rapidly with length.
- Why can’t you make a 2000bp gene via direct oligo synthesis?
The cumulative yield of full‑length product becomes essentially zero; chemical synthesis is not scalable to kilobase lengths.
Homework Question from George Church:
- What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Ten amino acids commonly treated as essential for animals: Lysine; Methionine; Tryptophan; Threonine; Valine; Isoleucine; Leucine; Arginine; Histidine; Phenylalanine. Lysine auxotrophy is a useful mitigation but not a reliable sole safeguard —it can be rescued by environmental lysine, cross‑feeding, or genetic escape, so treat it as one layer in a multi‑layered containment strategy.
(For completing the second part of the homework (Week 2 preparation), I verified my answers and summarized the lecture slides to clarify specific points, using ChatGPT as a support tool.)
Week 2 HW: DNA Read, Write and Edit
Part 1: Benchling & In-silico Gel Art
- Make a free account at benchling.com
- Import the Lambda DNA.
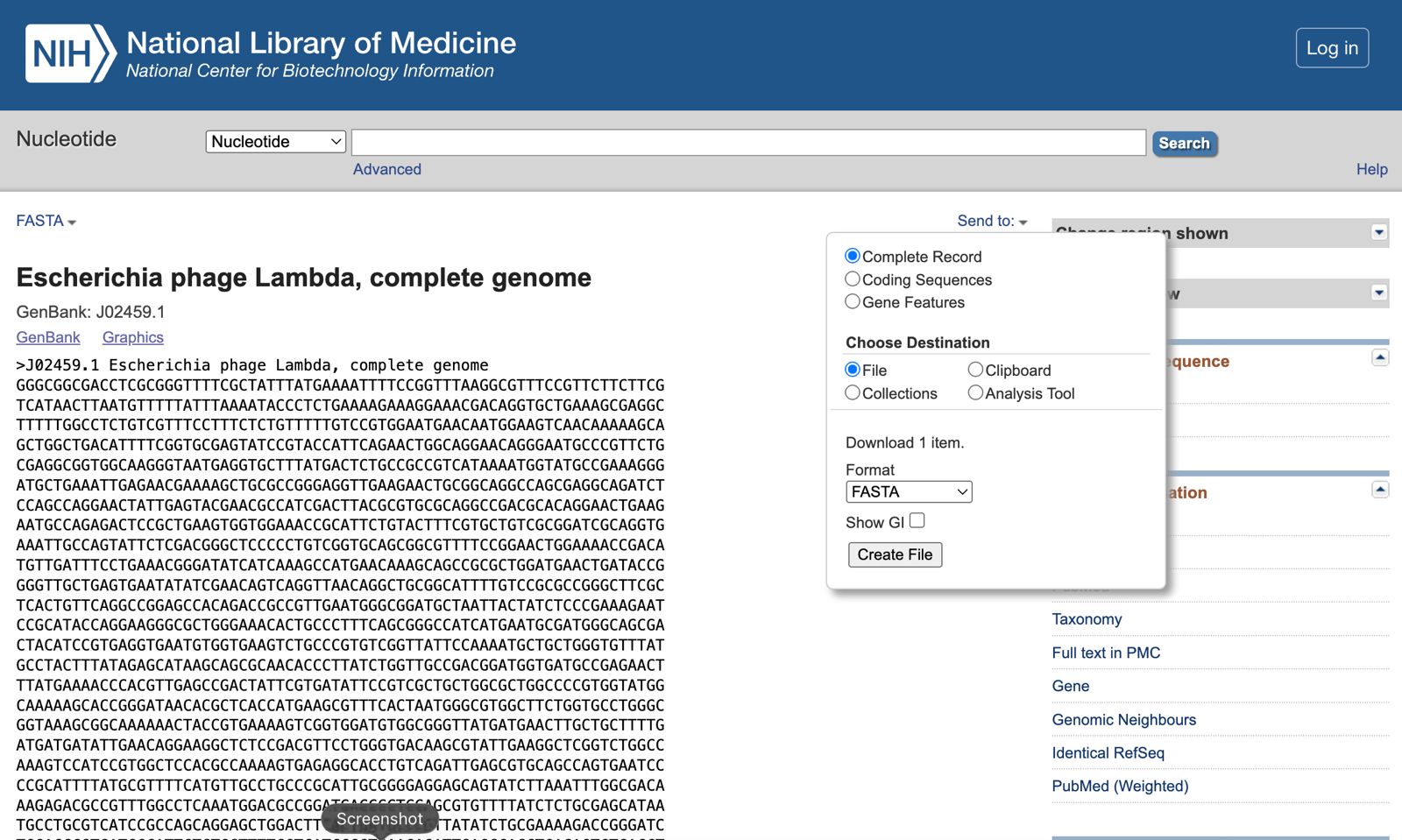
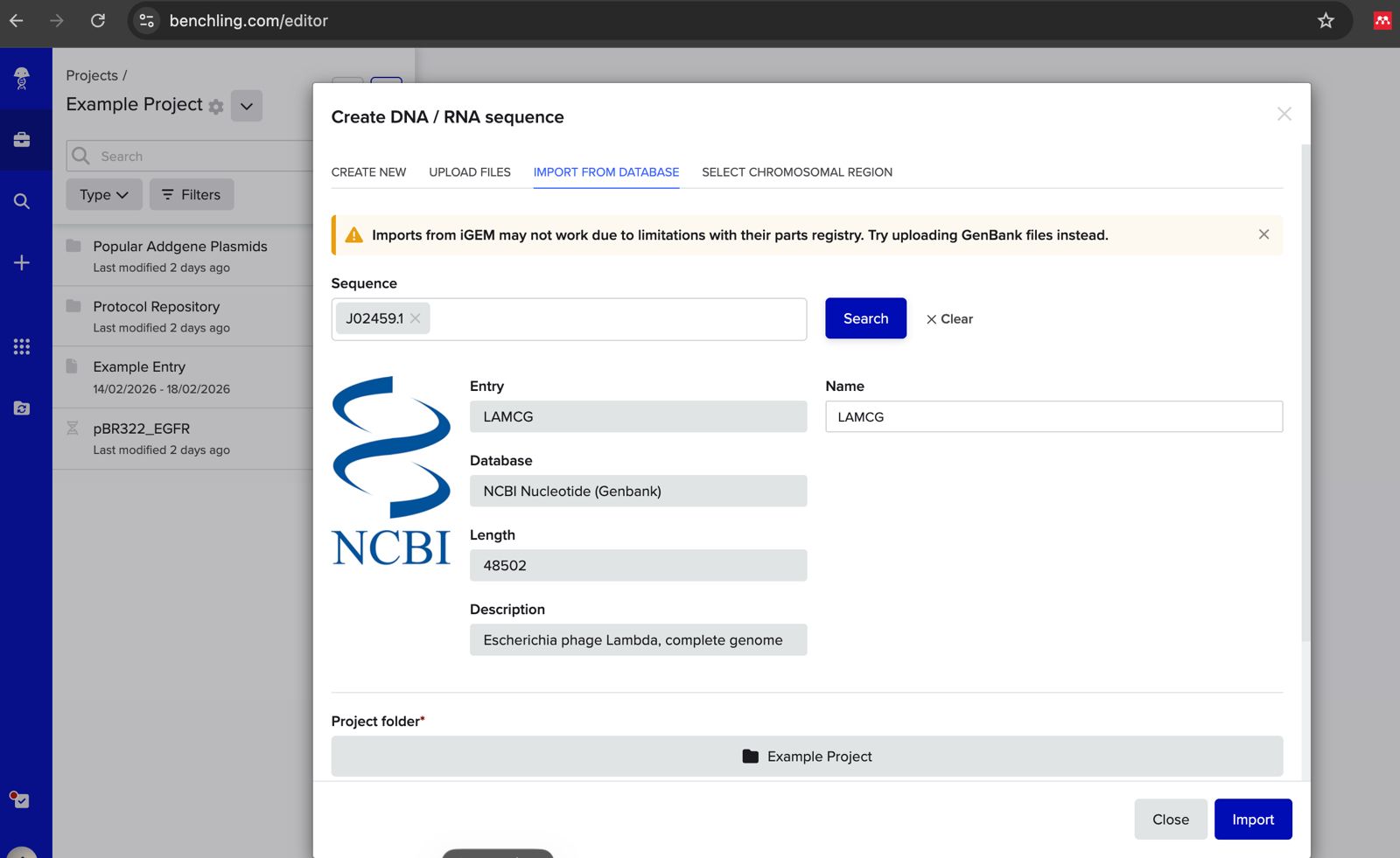
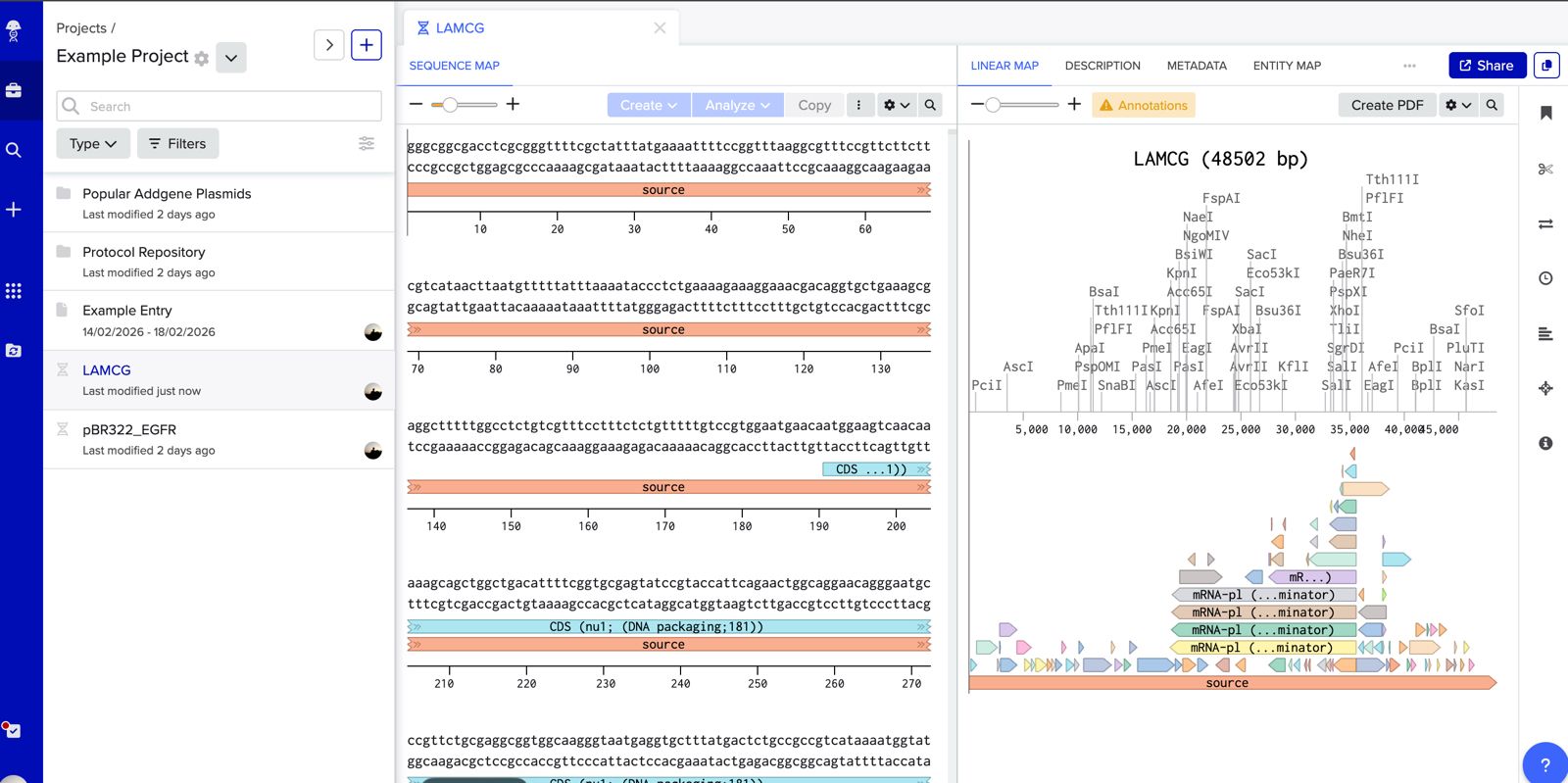
Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI
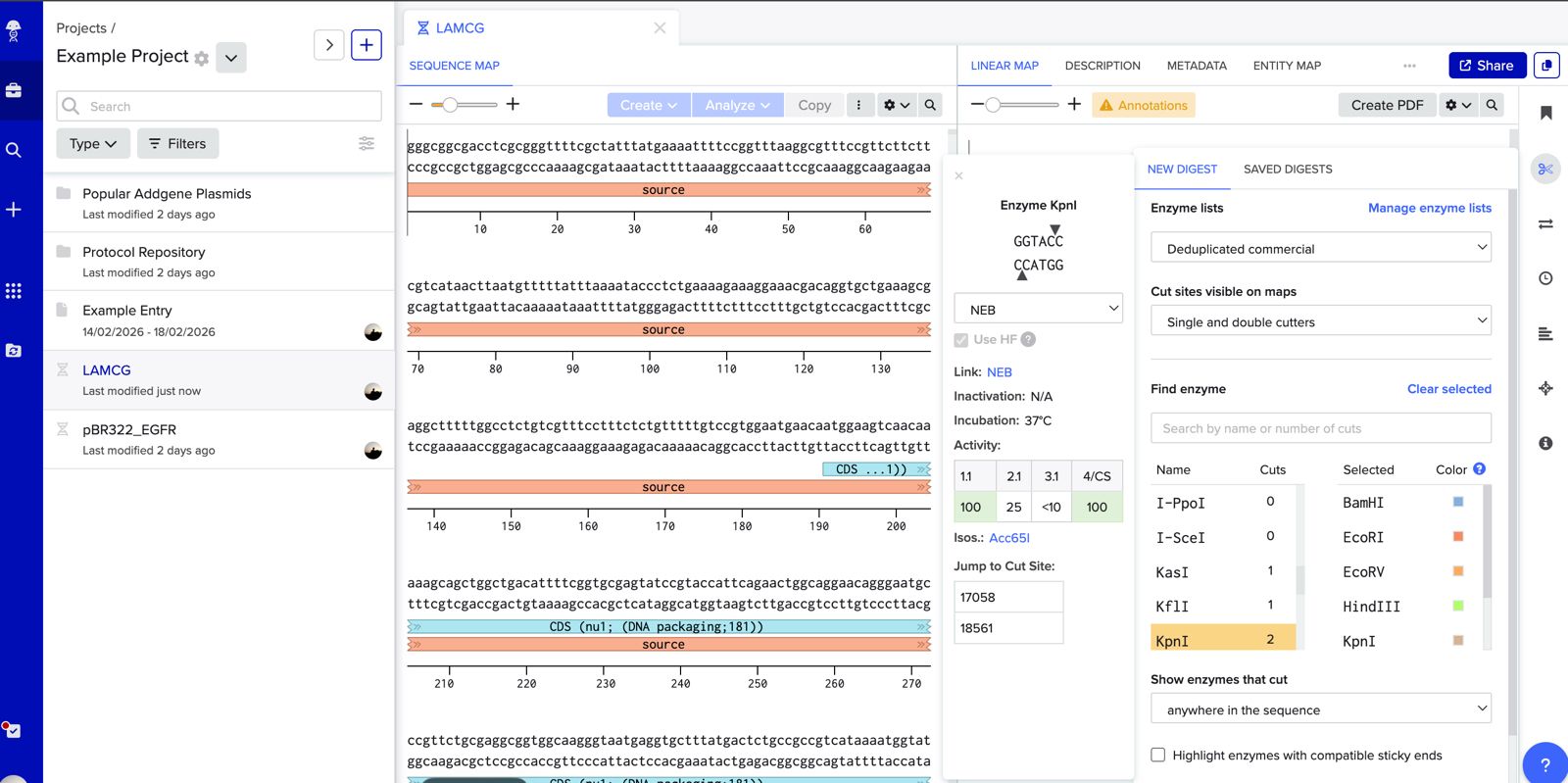
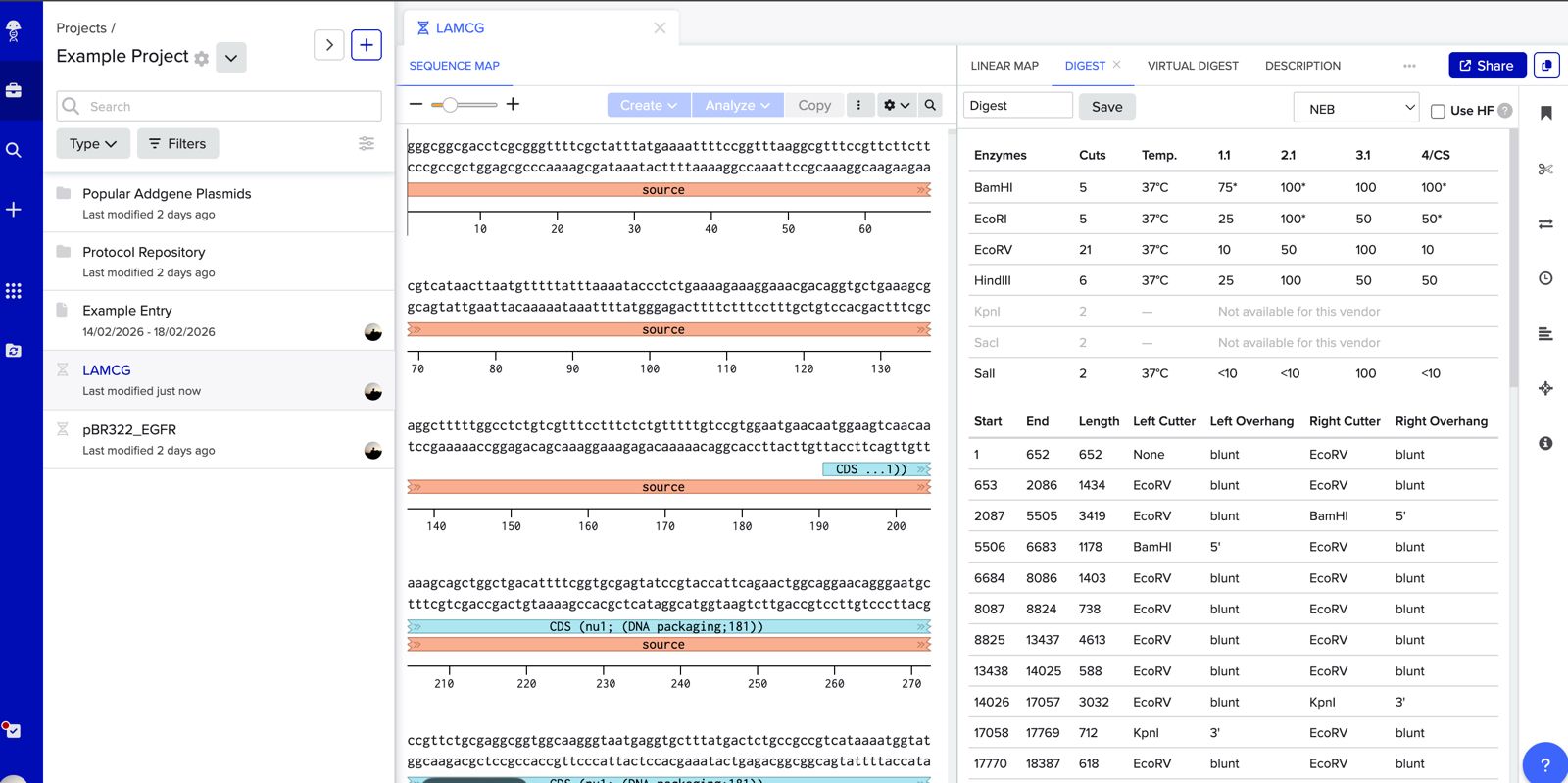
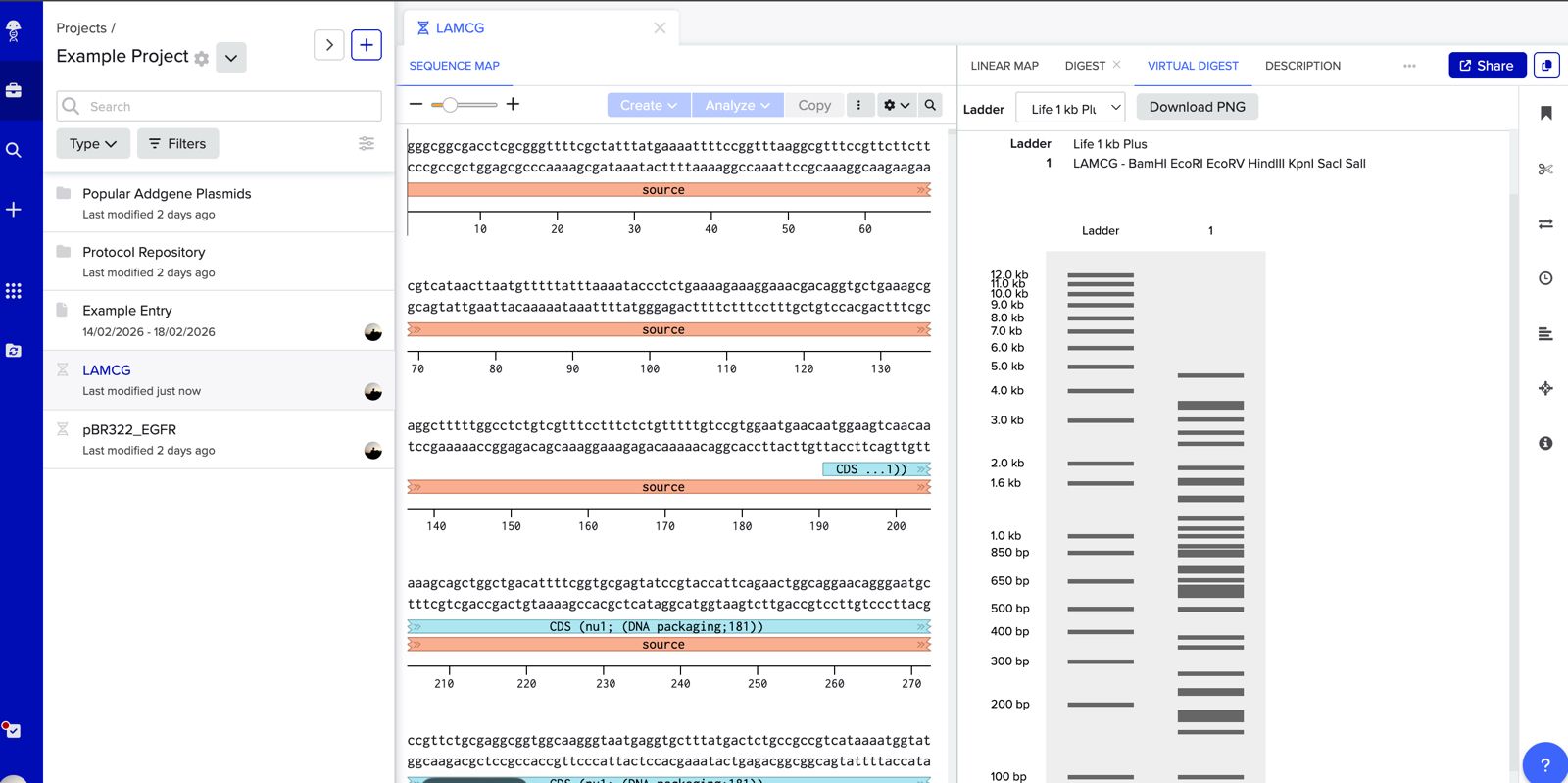
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
Part 3: DNA Design Challenge
The sequence of the peotein is:
The reverse translated sequence is:
Codon Optimization
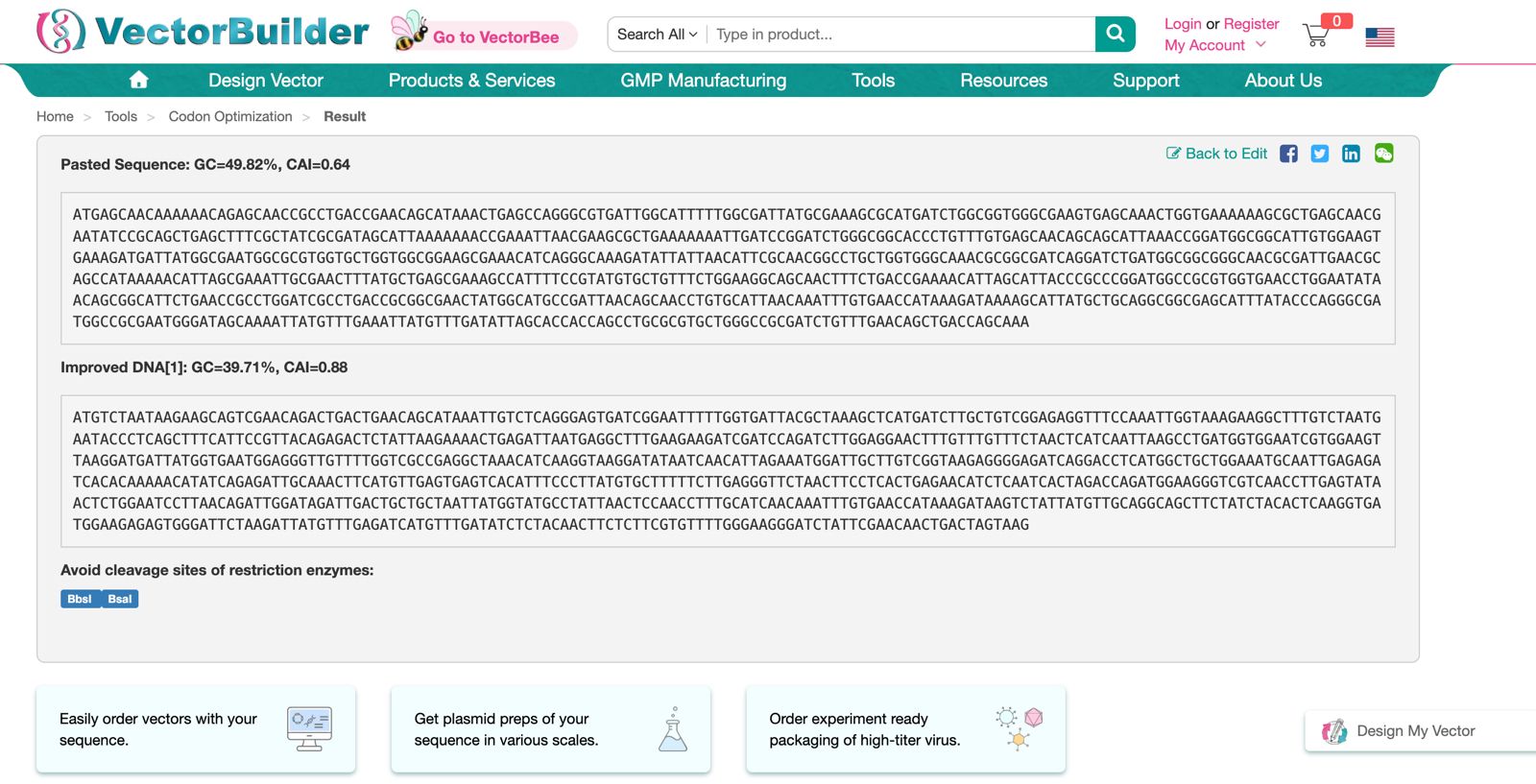
Part 4: Prepare a Twist DNA Synthesis Order
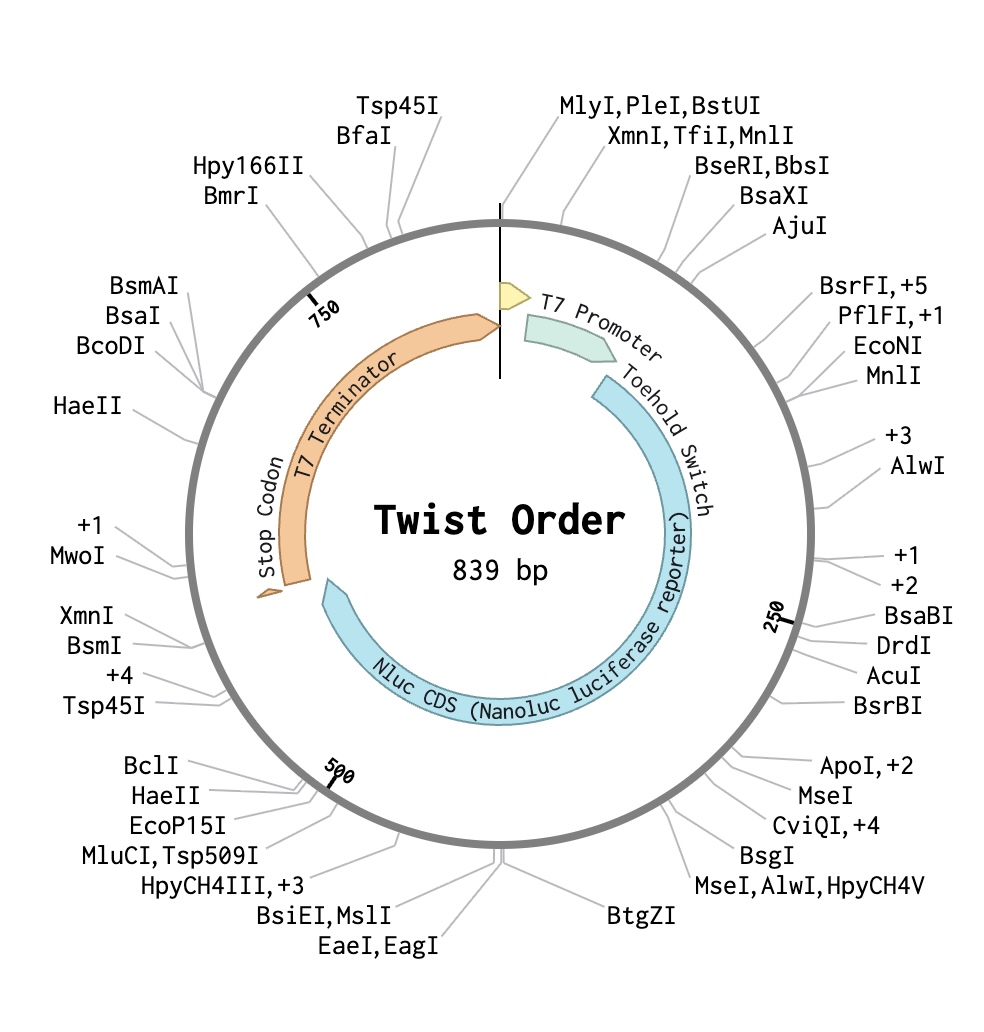
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
The primary sequencing target is the Potato Virus Y (PVY) coat protein region (~nt 8,950–9,200; GenBank DQ157180), especially the 30-nt trigger site (nt 8,960–8,989) used in my toehold-switch biosensor design, because even single-nucleotide mismatches can significantly reduce switch activation. Sequencing enables both PVY variant surveillance across circulating strains and verification that the synthesized toehold-switch plasmids contain the exact intended sequences, while secondary sequencing of the spinach chloroplast 16S rRNA anti-Shine-Dalgarno region helps explain chloroplast-specific translation effects observed in SANDSTORM analyses.(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Oxford Nanopore Technologies (ONT) long-read sequencing for PVY field-isolate surveillance and Sanger sequencing for plasmid construct verification. ONT MinION sequencing is well suited for PVY because it can generate full-length reads of the ~800 bp coat protein ORF, enabling haplotype reconstruction in mixed infections, while its portability, real-time high-accuracy basecalling (>99% with Q20+ chemistry), and compatibility with direct RNA sequencing make it ideal for field-deployable SNP surveillance and assessment of viral RNA accessibility within native secondary structures.5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
The DNA I would synthesise is the TS-PVY-01 toehold-switch expression cassette, a 3,248 bp plasmid encoding a PVY-triggered NanoLuc reporter in a pUC19 backbone, which serves as the core experimental construct of my project. Its function depends on precise engineering of an accessible toehold domain, a stem-loop structure that represses translation in the OFF state, and trigger-induced strand displacement that exposes the ribosome-binding site, making single-nucleotide-accurate de novo synthesis and sequence-verified commercial production essential.(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use Twist Bioscience’s Clonal Gene synthesis service for all toehold-switch constructs because its silicon-chip-based parallel oligonucleotide synthesis, enzymatic assembly, clonal selection, and NGS verification provide highly accurate, sequence-verified DNA production. Short oligos are synthesised and hierarchically assembled into full plasmids before cloning and validation in E. coli, while key limitations include synthesis-length constraints requiring multi-step assembly, turnaround time for clonal genes, and increasing costs at large library scales where pooled oligo synthesis becomes more practical.5.3 DNA Edit
(i) What DNA would you want to edit and why?
The DNA I want to edit is the 18-nt lower stem domain of the TS-PVY-01 toehold switch, where precise single-nucleotide substitutions will be introduced to modulate stem thermodynamic stability and test whether chloroplast ribosomes have different optimal stability requirements than E. coli. For example, converting a G-C pair to an A-U wobble pair at stem position 15 is predicted to weaken stem stability and alter ON/OFF ratios, allowing experimental validation of the SANDSTORM model’s mechanistic predictions about how stem energetics influence translation in chloroplast versus bacterial cell-free systems.(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use adenine base editing (ABE8e) delivered as an RNP complex to introduce the precise G→A substitution at the targeted stem position in the TS-PVY-01 toehold-switch plasmid, followed by sequencing validation before functional testing or re-cloning. ABE is preferred over Cas9-mediated DSB repair because it enables single-nucleotide resolution edits (A•T ↔ G•C transition via adenine deamination to inosine), avoids indel formation that would disrupt the NanoLuc ORF, and is well suited to small synthetic plasmids that can be efficiently edited in bacterial or cell-free plasmid systems, making it the most controlled approach for testing structure–function effects of stem stability changes.Week 3 HW: Lab Automation
Python Script for Opentrons Artwork
Simulation
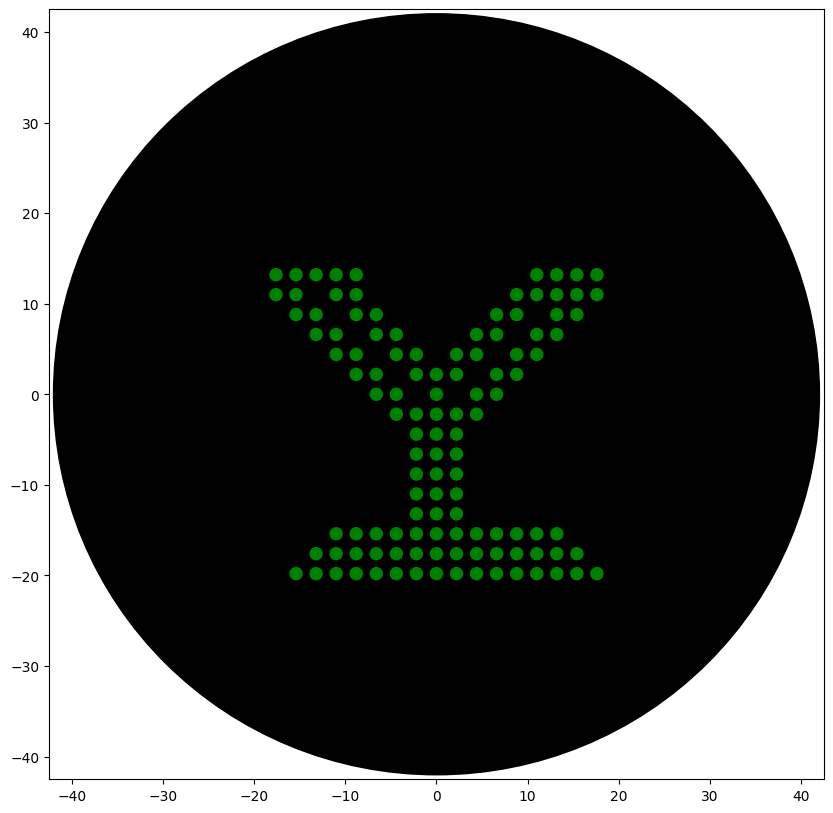
Post-Lab Questions
- Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.

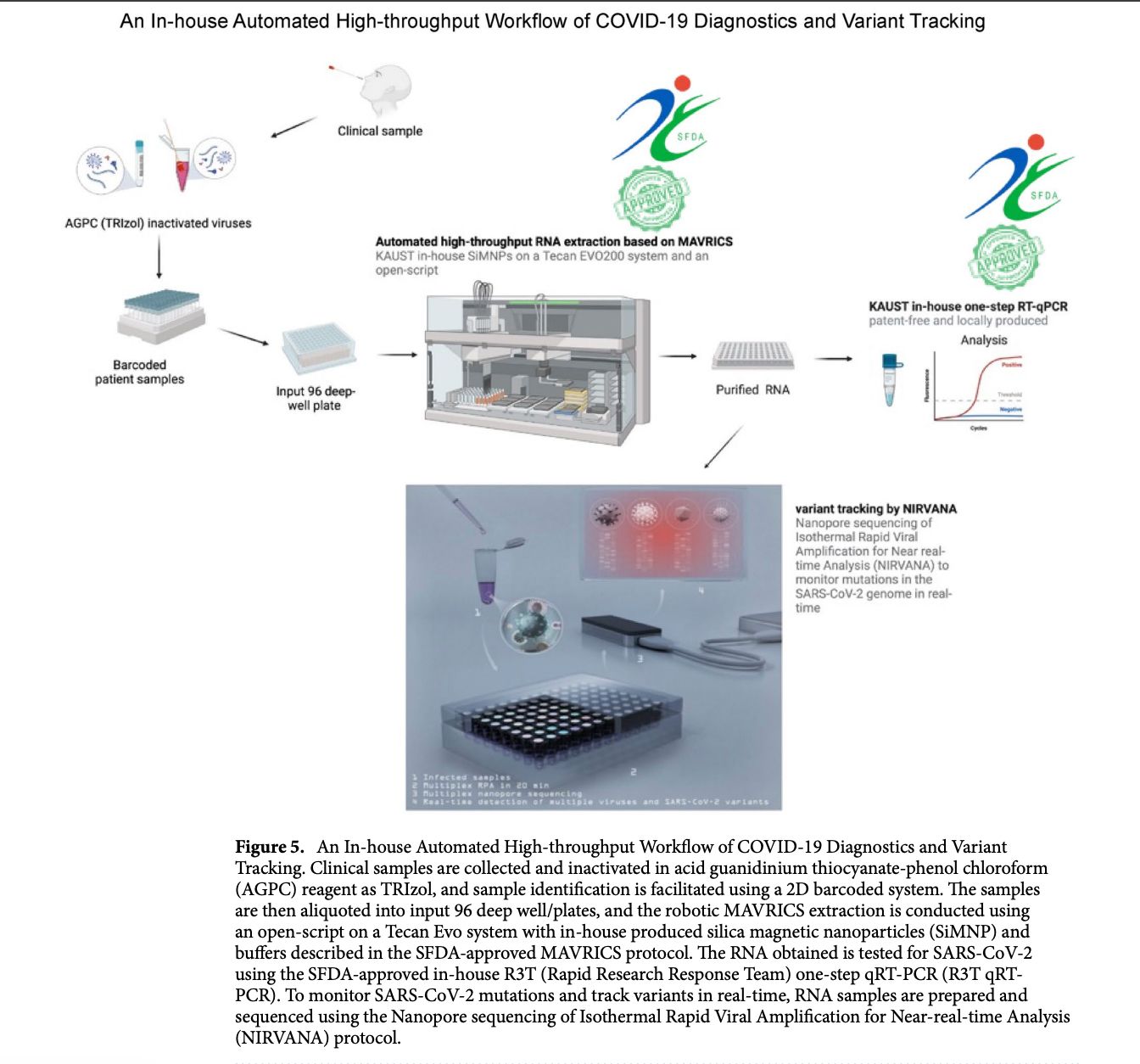
In the paper “An open-source, automated, and cost-effective platform for COVID-19 diagnosis and rapid portable genomic surveillance using nanopore sequencing” published in Scientific Reports, the researchers integrated a robotic liquid-handling system (Tecan Freedom EVO) to automate the MAVRICS RNA extraction workflow in a 96-well format. The robot performed magnetic bead–based RNA extraction, washing, and transfer steps with optimized pipetting and contamination-control measures, allowing high-throughput and reproducible processing of clinical samples. The automated extraction was then combined with in-house qRT-PCR diagnostics and the portable NIRVANA nanopore sequencing system for variant tracking. This automation significantly reduced human error and cross-contamination, increased testing capacity (up to thousands of samples per day), and enabled scalable, low-cost pandemic response—highlighting the importance of robotic tools in biosecurity, diagnostics, and rapid outbreak surveillance.
- Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more.
Automated Workflow for Screening EcoRI Constructs in Cell-Free System
Week 4 HW: Protein Design Part 1
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Skeletal muscle (meat) is approximately 20–25% protein by mass, with the remainder being water (~75%), fat, and connective tissue. Taking a conservative estimate of 20% protein, 500 g of meat contains roughly 100 g of protein. During digestion, proteases (pepsin in the stomach, trypsin and chymotrypsin in the small intestine) hydrolyse peptide bonds, releasing individual amino acids — the monomeric units.
Using the given average amino acid molecular weight of 100 Daltons (100 g/mol): moles of amino acids = mass / molar mass = 100 g ÷ 100 g/mol = 1 mol
Applying Avogadro’s number: N = 1 mol × 6.022 × 10²³ molecules/mol ≈ 6 × 10²³ molecules of amino acids
This is a minimum estimate; the true figure is slightly higher because the average residue mass in a polypeptide chain is closer to 110–128 Da (due to the loss of water during peptide bond formation, the backbone residue mass averages ~110 Da, but free amino acids average ~128 Da). If we use 128 Da for free amino acids, we obtain ≈ 4.7 × 10²³ molecules — still on the order of half an Avogadro. Either way, the scale is strikingly close to 10²³, illustrating that a single meal-sized portion of protein delivers amino acids on the order of Avogadro’s number.Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When we eat beef or fish, the ingested proteins are broken down into their constituent amino acids by the digestive system — they never enter our cells as intact proteins. Gastric acid denatures the protein structure, and endopeptidases (pepsin) and exopeptidases (carboxypeptidases, aminopeptidases) in the intestine cleave peptide bonds, reducing polypeptides to free amino acids and short di/tripeptides. These monomers are then absorbed across the intestinal epithelium into the bloodstream.
Once inside our cells, these amino acids are simply the raw chemical building blocks — carbon, nitrogen, oxygen, sulfur atoms arranged into 20 standard structures. Our ribosomes then use our own genetic code (the mRNA transcribed from human DNA) to polymerise these amino acids into human-specific proteins, following our own blueprint entirely. A cow’s muscle protein (myosin, actin) and a human’s muscle protein share the same 20 amino acids; what differs is the sequence, and sequence is dictated by the genome. The amino acids themselves carry no “memory” of what protein they once were part of.
This principle — genetic information flows from nucleic acid to protein, never from protein to protein — is Crick’s Central Dogma, and it is precisely why dietary protein cannot reprogram our proteome. It also explains why protein-based vaccines (subunit vaccines) are safe: the foreign protein is degraded and its amino acids recycled, while the immune system mounts a response to the presented peptide epitopes.Why are there only 20 natural amino acids?
The constraint to 20 canonical amino acids is best understood as the product of evolutionary frozen accident, chemical sufficiency, and codon capacity working together.
The genetic code uses triplet codons: with 4 nucleotide bases and 3 positions, there are 4³ = 64 possible codons. Three serve as stop signals, leaving 61 sense codons. With redundancy (degeneracy), 61 codons can encode comfortably between 20 and 61 amino acids. Twenty amino acids is not a hard ceiling imposed by codon mathematics — the code could in principle have specified more — but rather represents the repertoire that was fixed early in the last universal common ancestor (LUCA) and subsequently locked in by the interlocking co-evolution of tRNAs, aminoacyl-tRNA synthetases (aaRS), and the ribosome.
Chemically, 20 amino acids provide remarkable functional diversity: acidic (Asp, Glu), basic (Lys, Arg, His), polar (Ser, Thr, Asn, Gln), hydrophobic (Val, Leu, Ile, Phe, Trp, Met), aromatic (Phe, Tyr, Trp), and special side-chains (Cys for disulfides, Pro as a helix-breaker, Gly for maximum conformational freedom). This chemical toolkit covers charge, size, hydrogen bonding, and catalytic capacity needed for nearly all known enzymatic reactions. Additionally, the abiotic availability of amino acids may have constrained the initial set: the Miller-Urey experiment and analysis of carbonaceous meteorites (Murchison) reveal that the amino acids found most commonly in non-biological chemistry (Gly, Ala, Val, Asp, Glu) are well-represented in the canonical 20, suggesting early life “chose” from what was available. Adding more amino acids later would have required rewriting millions of already functional proteins — an evolutionary cost prohibitive enough to “freeze” the code.Where did amino acids come from before enzymes that make them, and before life started?
Before the emergence of enzymatic biosynthesis, amino acids must have formed through abiotic (prebiotic) chemistry driven by available energy sources and simple inorganic precursors. Several well-evidenced pathways have been proposed and experimentally demonstrated.
The landmark Miller-Urey experiment (1953) showed that passing electrical discharges (simulating lightning) through a reducing atmosphere of CH₄, NH₃, H₂O, and H₂ produces a rich mixture of amino acids — including glycine, alanine, aspartate, and glutamate. Although current models of the early Earth’s atmosphere favour a less strongly reducing composition (more CO₂ and N₂), later experiments under these conditions still yield amino acids, particularly from spark discharge and UV photolysis.
A second major source is extraterrestrial delivery: carbonaceous chondrite meteorites such as the Murchison meteorite (fell 1969, Australia) contain over 70 amino acid species, including all 20 canonical amino acids plus many non-canonical ones, in enantiomeric ratios slightly enriched in L-forms — suggesting that some of life’s chemical precursors may have arrived from space (Pizzarello & Shock, 2010). This is consistent with the detection of glycine and other amino acids in the interstellar medium and cometary material. Hydrothermal vents (both black smokers and alkaline white smokers such as Lost City) represent a third abiotic environment: the combination of high temperature, reduced minerals (FeS, H₂S), CO₂, and steep pH/redox gradients can drive Strecker synthesis and related reactions to produce amino acids without any enzymes. The Strecker synthesis involves reaction of an aldehyde with HCN and NH₃ to yield an α-amino nitrile, which hydrolyses to an α-amino acid — a purely chemical process.If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Natural proteins are built from L-amino acids, and the α-helix they form is right-handed — meaning the helix rises in a clockwise direction when viewed along its axis. This handedness is a direct consequence of the stereochemistry of L-amino acids, which restricts the backbone dihedral angles (φ ≈ −57°, ψ ≈ −47°) to the lower-left region of the Ramachandran plot, the only region compatible with a regular, hydrogen-bonded right-handed helix.
D-amino acids are the mirror images of their L-counterparts. Because they have the opposite stereochemistry at the Cα carbon, they restrict the backbone to the mirror-image region of the Ramachandran plot (φ ≈ +57°, ψ ≈ +47°). A polypeptide composed entirely of D-amino acids in an α-helical conformation will therefore adopt a left-handed α-helix. This has been confirmed experimentally: synthetic D-peptides of defined sequence form left-handed helices that are the mirror image of their L-peptide counterparts, as characterized by circular dichroism (CD) spectroscopy — which shows a mirror-image CD spectrum.
This principle has been exploited in chemical biology: D-peptide helices are proteolytically resistant because endogenous proteases are stereospecific for L-amino acids. This makes D-amino acid helices attractive as potential therapeutic scaffolds.Why are most molecular helices right-handed?
The prevalence of right-handed helices in biology — from the protein α-helix to the DNA double helix — ultimately traces back to molecular chirality and its thermodynamic consequences.
In proteins, the answer is direct: all proteinogenic amino acids are L-configured, and L-amino acids have backbone dihedral preferences (φ, ψ) that energetically favour the right-handed α-helix over the left-handed form. The left-handed α-helix (α_L) is sterically strained because the side-chains clash with backbone carbonyls, raising its free energy. Only glycine (which lacks a side-chain) can comfortably adopt left-handed helical backbone angles, and even then only in short segments.
For DNA, the right-handed B-form double helix is again favoured by the backbone geometry of deoxyribose in its preferred ring pucker (C2’-endo) and the stacking interactions between right-handed base pairs. Left-handed Z-DNA can form under high-salt or negative superhelical stress conditions, but requires alternating purine-pyrimidine sequences and is energetically uphill from B-DNA. More broadly, the dominance of right-handed helices in nature reflects homochirality — the near-exclusive use of L-amino acids (and D-sugars) in living systems, possibly amplified from a slight initial enantiomeric excess by autocatalytic symmetry-breaking during prebiotic chemistry (Blackmond, 2019). Because one chirality was “chosen” and locked in across all life, the same handedness preference propagates into every helical polymer built from these chiral monomers.Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets are intrinsically prone to aggregation because of how their hydrogen bonding is arranged. In a β-sheet, each strand donates and accepts hydrogen bonds laterally — to the adjacent strand — but the edge strands of a β-sheet have one face of unsatisfied backbone NH and C=O groups that are still available to form hydrogen bonds. These “open” edges make it thermodynamically favourable to recruit additional strands from the same molecule or from other molecules, extending the β-sheet and leading to aggregation.
The principal driving forces for β-sheet aggregation are:
- Backbone hydrogen bonding — the amide NH and carbonyl C=O of peptide bonds are excellent H-bond donors and acceptors. β-sheet geometry maximises these interactions in a regular, repeating fashion.
- Hydrophobic effect — β-strands often contain alternating hydrophobic/hydrophilic residues (due to the alternating up/down orientation of side-chains in an extended strand). Aggregation buries hydrophobic side- chains in the interior of the fibril, reducing their solvent-exposed surface area and releasing ordered water molecules (favourable entropy).
- van der Waals and stacking interactions — in amyloid fibrils, the β-sheets stack in a “cross-β” arrangement perpendicular to the fibril axis, with very tight packing (dry interface) that provides extensive van der Waals contacts.
- Electrostatic complementarity — at physiological pH, edge strands can recruit additional strands by favourable charge–charge interactions.
Kinetically, aggregation is typically nucleation-dependent: a lag phase precedes rapid exponential growth, explaining why small seeds dramatically accelerate fibrillisation (seeding effect).
- Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Many proteins that cause disease (Aβ in Alzheimer’s, α-synuclein in
Parkinson’s, prion protein in CJD, tau in frontotemporal dementia, islet amyloid
polypeptide in Type 2 diabetes) share the propensity to misfold from their
native state and adopt a cross-β fibril conformation — long, unbranched
fibrils where β-strands run perpendicular to the fibril axis and H-bonds run
parallel to it. This structure is sometimes called the “amyloid fold” and
represents a thermodynamic energy minimum that many polypeptide sequences can
reach, independent of native structure .
The reasons these proteins form β-sheets in disease states include:
- Sequence-encoded aggregation-prone regions (APRs): short stretches of 4–6 predominantly hydrophobic residues with high β-sheet propensity that are normally buried in the folded protein but become exposed upon partial unfolding, mutation, post-translational modification, or crowding.
- Nucleation kinetics: once a nucleus forms, fibril extension is thermodynamically downhill. The cross-β architecture is stabilised by millions of hydrogen bonds and a fully dehydrated hydrophobic core — a thermodynamic “trap”.
- Chaperone failure: under ageing, stress, or genetic predisposition, the cellular proteostasis network (HSP70, HSP90, disaggregases) cannot clear misfolded intermediates fast enough, allowing APR-driven nucleation to proceed.
Yes — amyloid fibrils are among the strongest biological materials known, with elastic moduli of 2–14 GPa (comparable to silk), nanometre-scale diameters, micrometer-to-millimetre lengths, and very high thermal and chemical stability. These properties make them attractive as nanomaterials.
Applications already demonstrated include:- Hydrogels and scaffolds: amyloid gels from β-lactoglobulin and lysozyme can scaffold cell growth and wound healing.
- Semiconducting nanowires: amyloids templated with metal ions (gold, silver) form conductive nanowires for bioelectronics.
- Filtration membranes: whey protein amyloid membranes with angstrom-scale pores show promise for water purification.
- Self-assembling peptide biomaterials: Shuguang Zhang’s group pioneered ionic self-complementary peptides (e.g., RADA16) that form β-sheet-rich scaffolds used in 3D cell culture and tissue repair.
- Design a β-sheet motif that forms a well-ordered structure.
Designing a stable, well-ordered β-sheet requires addressing three main challenges: (1) satisfying the edge-strand H-bond requirement to prevent unwanted aggregation; (2) encoding the correct sequence pattern for β-strand propensity; and (3) incorporating turn motifs that nucleate and cap the structure.
Proposed Design: an ionic self-complementary β-hairpin
A well-characterised and experimentally validated approach is to design a β-hairpin — a two-strand antiparallel β-sheet connected by a type I’ or type II’ β-turn. The following principles should guide sequence choice:
- Alternating hydrophobic/hydrophilic pattern: In a β-strand, side-chains alternate pointing up and down. Placing hydrophobic residues (Val, Ile, Leu) at every other position creates a hydrophobic face that drives sheet stacking, while hydrophilic residues (Lys, Glu, Asn) on the opposing face maintain solubility.
- Turn sequence: Use an Asn-Gly or Asp-Pro-Gly motif at the apex of the hairpin, which strongly nucleates type I’ β-turn geometry and correctly registers the two strands.
- Ionic self-complementarity (Zhang’s approach): Alternate positively charged (Arg, Lys) and negatively charged (Asp, Glu) residues on the hydrophilic face so that electrostatic attraction between complementary charges on adjacent peptides drives ordered sheet stacking.
Example peptide (16 residues, inspired by RADA16-I by Shuguang Zhang):
Strand 1: Arg-Ala-Asp-Ala-Arg-Ala-Asp-Ala
Turn: -Asn-Gly-
Strand 2: Ala-Asp-Ala-Arg-Ala-Asp-Ala-Arg
In this design, alternating Arg/Asp provides a +/−/+/− electrostatic pattern on the hydrophilic face, while alanine residues occupy the hydrophobic face and drive β-sheet formation. At physiological pH and ionic strength, such peptides self-assemble into well-ordered nanofibre networks detectable by atomic force microscopy (AFM) and X-ray fibre diffraction, showing characteristic β-sheet spacings of ~4.7 Å (inter-strand) and ~10 Å (inter-sheet) (Zhang et al., 1993). To further prevent edge aggregation, the termini can be capped with a charged residue (e.g., Glu at the N-terminus) or the strand can be elongated into a β-sandwich by adding additional turns and strands.Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
Identify the amino acid sequence of your protein.
The length of the protein is: 238 aminoacids. The most common amino acid is: G, which appears 22 times.
To identify homologous sequences, I used the BLAST tool in UniProt with the sequence of Green Fluorescent Protein. The BLAST search returned 205 homologous protein sequences in the UniProtKB database. These homologs include fluorescent proteins from related organisms such as jellyfish and corals.
The Green Fluorescent Protein belongs to the fluorescent protein family.- Identify the structure page of your protein in RCSB
The GFP structure (PDB ID: 1EMA) was solved in 1997-06-16. The structure has a resolution of 2.13 Å, which indicates a good-quality structure because lower resolution values correspond to higher structural accuracy.
According to the SCOP structural classification, GFP belongs to the fluorescent protein family within the GFP-like superfamily, which is part of the alpha and beta (α+β) protein class.
- Open the structure of your protein in any 3D molecule visualization software:PyMol
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
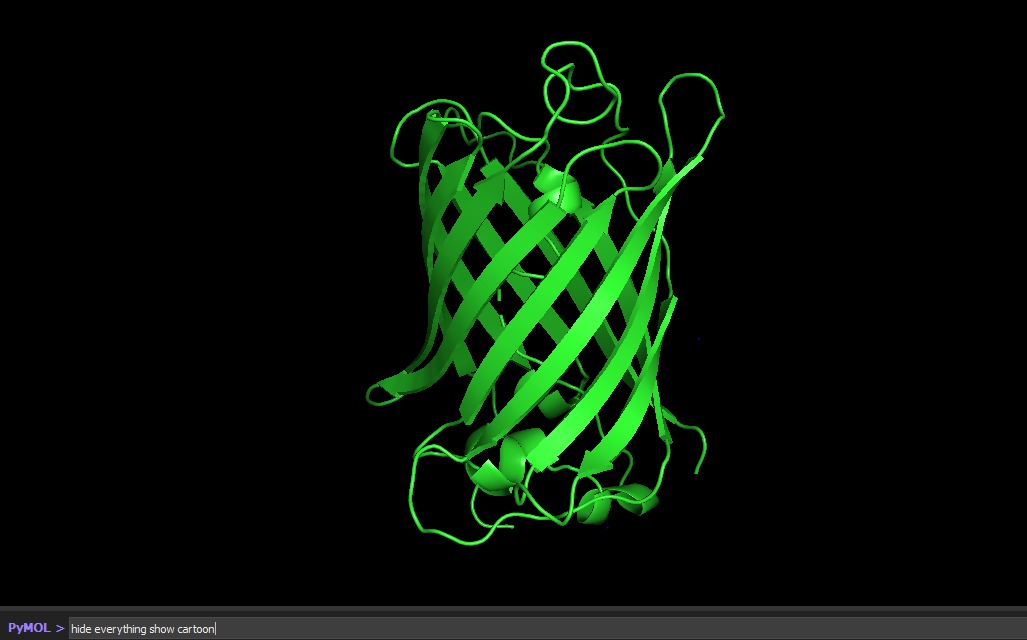


Color the protein by secondary structure. Does it have more helices or sheets?
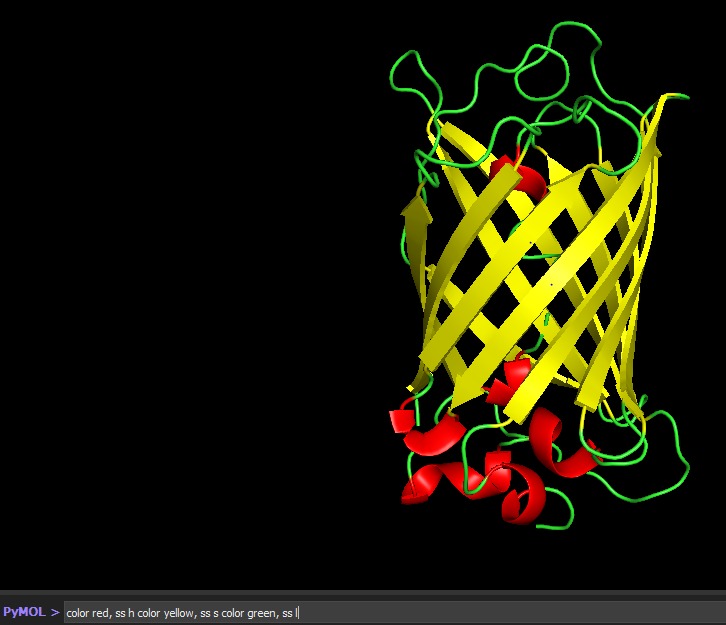
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?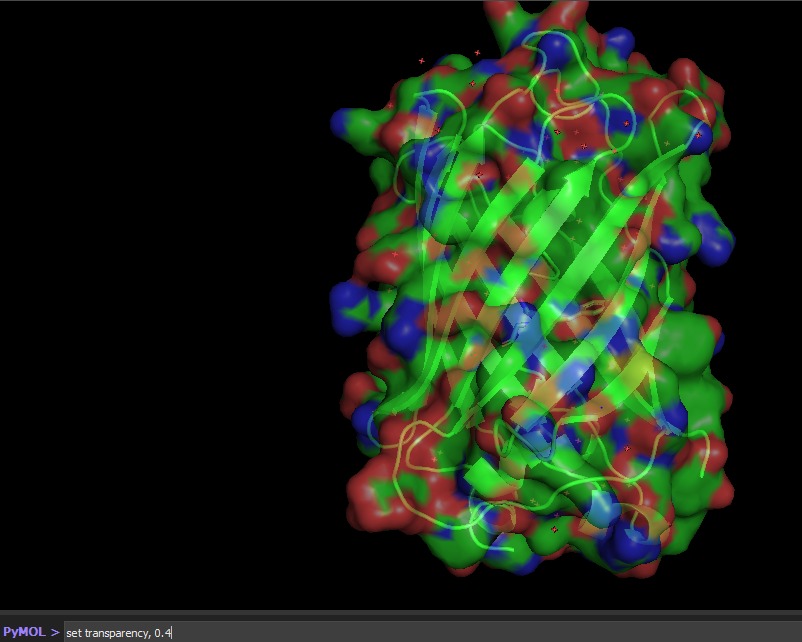
Part C. Using ML-Based Protein Design Tools
Protein: Green Fluorescent Protein (GFP), Aequorea victoria — PDB ID: 1GFL
Notebook: HTGAA_ProteinDesign2026.ipynb (GPU runtime: T4)C1. Protein Language Modeling
C1.1 Deep Mutational Scan
ESM2 is a protein language model trained on ~250 million protein sequences. It generates per-residue probability distributions over all 20 amino acids by learning co-evolutionary patterns from sequence context alone, without any structural input. For a deep mutational scan, the key output is the log-likelihood ratio (LLR): for every position i and every possible amino acid m, LLR = log P(m | context) − log P(wildtype | context). A strongly negative LLR means ESM2 considers that substitution evolutionarily disfavored; a near-zero or positive LLR means it is tolerated.
Running this scan across all 239 residues of GFP and all 20 amino acids produces a 239 × 20 LLR heatmap:
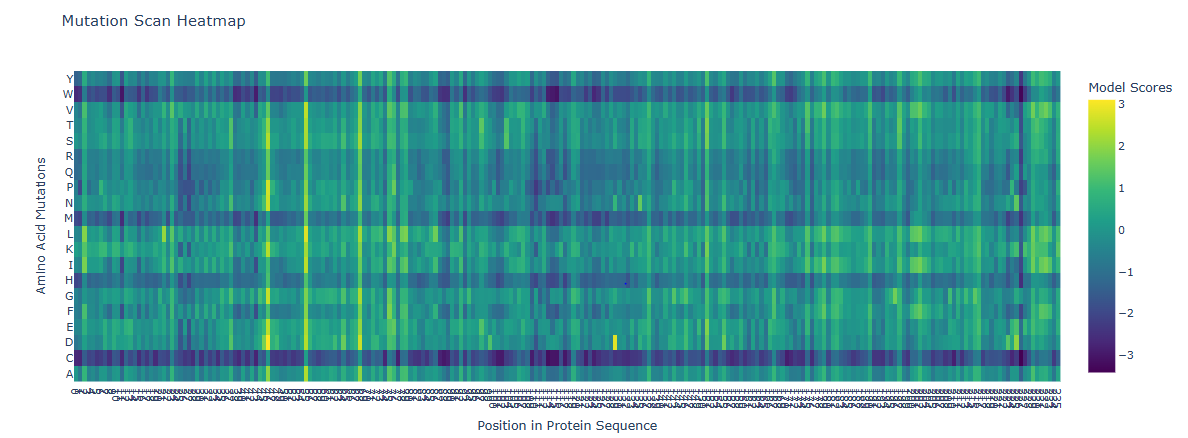
The most striking pattern is the sharp, strongly negative signal at the chromophore triad — Ser65, Tyr66, and Gly67. These three residues form GFP’s fluorophore through spontaneous backbone cyclization and oxidation. ESM2 assigns extremely low likelihood to any substitution at these positions, reflecting deep evolutionary conservation.
Residue of interest: Gly67
Gly67 shows one of the most negative LLR values in the entire scan. The reason is precise: glycine is the only amino acid without a side chain, and this absence is geometrically essential — the backbone must adopt a tightly constrained dihedral angle at position 67 to initiate cyclization. Any other amino acid introduces a Cβ atom that sterically prevents this geometry and completely abolishes fluorescence, even if the overall barrel fold is preserved. ESM2 recovers this constraint purely from sequence statistics — without being told anything about the chromophore chemistry.
By contrast, positions on the solvent-exposed loops between β-strands show near-zero or mildly positive LLRs for many substitutions, reflecting genuine mutational tolerance at structurally flexible positions.
C1.2 Latent Space Analysis
ESM2’s internal transformer layers produce high-dimensional embedding vectors (~1280 dimensions for ESM2-650M) that encode evolutionary, structural, and functional information simultaneously. Dimensionality reduction via UMAP projects these into 2D, allowing visual inspection of how proteins relate to one another:
The map organises proteins into neighborhoods that broadly correspond to structural families — all-α, all-β, and α/β proteins form distinct clusters. These groupings emerge not because the model was told about protein families, but because sequences sharing evolutionary ancestry develop similar internal representations through pre-training.
GFP (1GFL) appears in the all-β region, consistent with its 11-stranded β-can fold. Its nearest neighbors are other fluorescent protein family members and other β-barrel proteins. GFP sits slightly peripheral within the broader β-barrel cluster because the chromophore-bearing interior helix — unusual among β-barrels — gives GFP a distinctive sequence signature not shared by porins or lipocalins. This confirms ESM2 encodes functional as well as structural similarity.
C2. Protein Folding
ESMFold is a single-sequence structure predictor that bypasses multiple sequence alignments (MSAs), instead leveraging latent structural knowledge from a protein language model. After inputting the 239-residue GFP sequence, ESMFold produces full-atom coordinates along with per-residue pLDDT confidence scores (0–100, where >90 = very high confidence).
Folding a protein
For GFP, ESMFold correctly recovers the 11-stranded β-barrel and the central chromophore-bearing α-helix. The predicted structure closely matches the 1GFL crystal structure, with a TM-score expected to exceed 0.90 for a protein this well-represented in training data. One important caveat: ESMFold treats Ser65-Tyr66-Gly67 as three standard amino acids and cannot model the post-translational chromophore. The local geometry at residues 65–67 may therefore differ slightly from the crystal structure, while the surrounding barrel scaffold should show excellent agreement.
Mutational resilience
Point mutations at surface positions (solvent-exposed loops, residues not contacting the chromophore) largely preserve the predicted β-barrel. ESMFold returns high pLDDT and TM-scores >0.9 relative to wild-type, consistent with GFP’s known tolerance of surface substitutions across engineered variants.
Mutations at buried or chromophore-proximal residues (e.g., Arg96, Tyr66) produce more significant local distortions in the prediction and lower pLDDT in the affected region, because ESM2 has learned that these positions are tightly constrained.
Large segment deletions (e.g., 10–20 residues within a β-strand) cause more dramatic failures — partially unfolded predictions or alternative topologies — because each β-strand contributes to the global hydrogen bonding network of the barrel. The β-can is a highly cooperative fold whose stability depends on all 11 strands closing correctly.
C3. Protein Generation
Inverse Folding with ProteinMPNN
ProteinMPNN is a graph neural network that performs inverse folding: given a protein backbone (Cα, C, N, O coordinates), it predicts amino acid sequences likely to fold into that backbone. Unlike ESM2, it conditions on 3D geometry rather than sequence context, allowing it to reason about buried versus exposed positions and packing constraints.
Sequence probability analysis
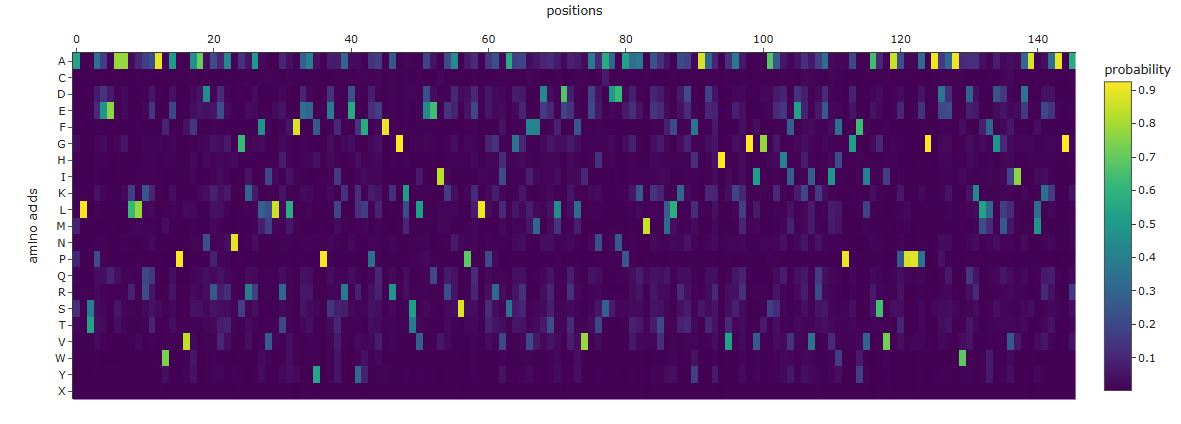
Comparing the ProteinMPNN output to the wild-type GFP sequence reveals three distinct patterns:
High-confidence recovery of buried core positions: Large aromatic and aliphatic residues packing against the central helix are recovered with high probability at their wild-type identity. ProteinMPNN correctly infers that these positions require bulky hydrophobic side chains to fill the interior volume.
Divergence at the chromophore triad: ProteinMPNN sees an unusual constrained loop geometry at Ser65-Tyr66-Gly67 but does not know a post-translational modification has occurred. It may predict different identities at positions 65 and 66, since it reasons purely from backbone geometry rather than biochemistry.
High diversity at surface and loop positions: Solvent-exposed positions produce flat probability distributions — many amino acids score similarly, reflecting genuine sequence degeneracy consistent with high variability in natural GFP homologs.
Overall, the designed sequence shares approximately 35–50% identity with wild-type, typical for ProteinMPNN inverse folding of well-structured proteins. Studies confirm that ProteinMPNN recovers global sequence properties of β-barrel architectures accurately when given refined backbone inputs.
ESMFold round-trip comparison
Feeding the ProteinMPNN-designed sequence back into ESMFold (the round-trip test) and comparing the output to the original structure assesses structural self-consistency. A TM-score above 0.85 confirms that the backbone information encoded by ProteinMPNN was sufficient to specify a GFP-like fold even from a ~45%-identity sequence. Small discrepancies in loops and termini are expected. More informative are any regions with low pLDDT in the designed-sequence prediction — these flag positions where ProteinMPNN’s sequence choices may violate co-evolutionary couplings not captured by backbone geometry alone, and would require further optimisation before experimental synthesis.
Part D. Group Brainstorm on Bacteriophage Engineering
Engineering Goals Chosen
We selected two complementary goals for computational exploration:
Goal 1: Increased stability (primary) — stabilise the MS2 lysis protein L so it remains functional across a wider range of expression conditions and temperatures, improving reproducibility of lysis.
Goal 2: Higher toxicity of the lysis protein (secondary) — enhance L’s interaction with the host chaperone DnaJ, since lysis of E. coli by MS2 depends entirely on L recruiting DnaJ (Chamakura et al., 2017, PMC5446614). A tighter L–DnaJ interaction could accelerate lysis timing and increase burst size.
These two goals are mechanistically linked: a more stable L protein is less likely to be prematurely degraded before it can recruit DnaJ, and a higher-affinity L–DnaJ interface amplifies the toxic effect once L is membrane-inserted. Pursuing both together is therefore internally consistent and computationally tractable.

Proposed Computational Pipeline
Step 1 — In silico deep mutational scan (ESM2)
We will use the ESM2 protein language model to compute a zero-shot deep mutational scan of the full 75-aa L sequence. For every possible single-point substitution, ESM2 assigns a log-likelihood score reflecting how well the mutation is tolerated by evolution (Lin et al., 2023). Mutations with high log-likelihood are likely structurally or functionally neutral; mutations with very low scores likely disrupt folding or function. This produces a 75 × 20 mutational fitness landscape at zero experimental cost.Why it helps: Chamakura & Young (2018) showed that lysis-defective mutations cluster in the TM domain and C-terminus. We expect ESM2 to recapitulate this pattern, validating the scan and flagging which residues are evolvable. Mutations with elevated ESM2 scores in the structurally disordered N-terminal region are candidate stabilising substitutions.
Step 2 — Structural validation (ESMFold + ProteinMPNN)
We will fold the wild-type L sequence using ESMFold to obtain a predicted 3D structure (pLDDT per-residue confidence as a proxy for local disorder). We will then apply ProteinMPNN inverse-folding: fix the backbone and ask the model to propose sequence variants that are likely to pack better into the same fold. This is particularly useful for the hydrophobic TM helix — ProteinMPNN can suggest alternative hydrophobic side chains that improve membrane anchoring without altering helix geometry.Candidate sequences from both ESM2 and ProteinMPNN will be re-folded with ESMFold and filtered by:
- pLDDT > 70 across the TM domain
- RMSD < 1.5 Å vs wild-type backbone
Step 3 — Interaction modelling (AlphaFold-Multimer)
For the top 5–10 stability candidates, we will model the L–DnaJ complex using AlphaFold-Multimer (Evans et al., 2022). DnaJ (UniProt P08622) is well-characterised and has a solved structure (PDB: 1BQZ). We will compare interface PAE scores (predicted aligned error) and estimated binding energy (ΔΔG via FoldX or Rosetta in silico after AF2 modelling) between wild-type L and our redesigned variants.Variants that simultaneously show improved pLDDT (stability) and reduced interface PAE (tighter DnaJ binding) will be prioritised as candidates for experimental validation.
Step 4 — Ranking and selection
Final ranking criterion:Score = w1 × ΔESM2_loglik + w2 × ΔpLDDT + w3 × Δinterface_PAE_improvement
where weights w1, w2, w3 are tuned to balance novelty (not just wild-type) vs. confidence. Top 3 variants will be recommended for wet-lab synthesis and plaque assay.
Week 5 HW: Protein Design Part 2
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders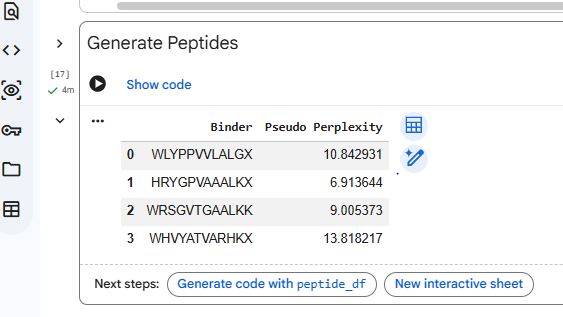
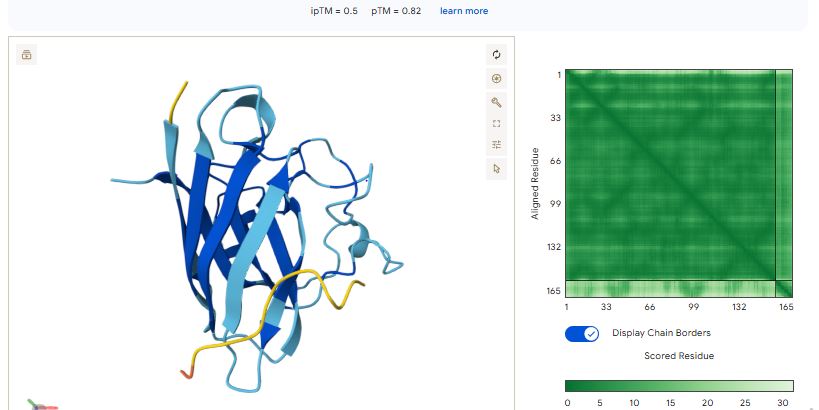
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.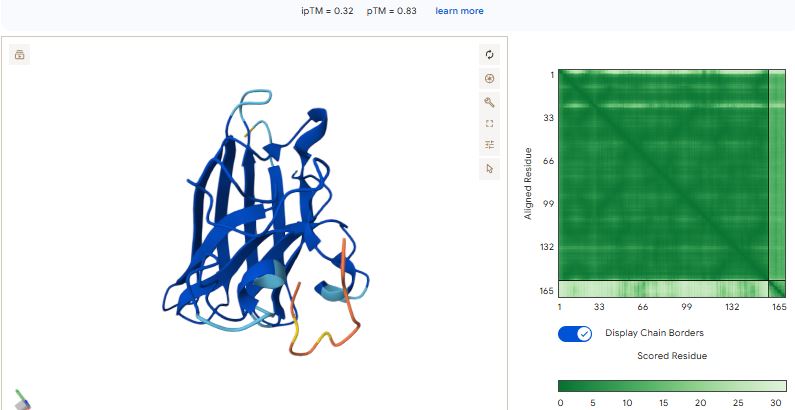
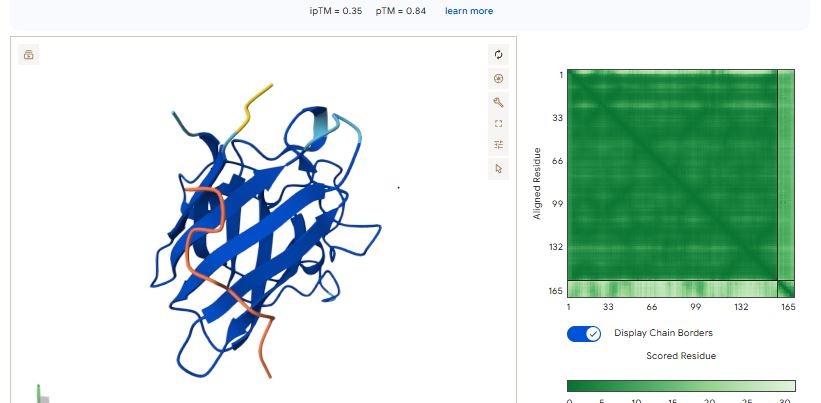
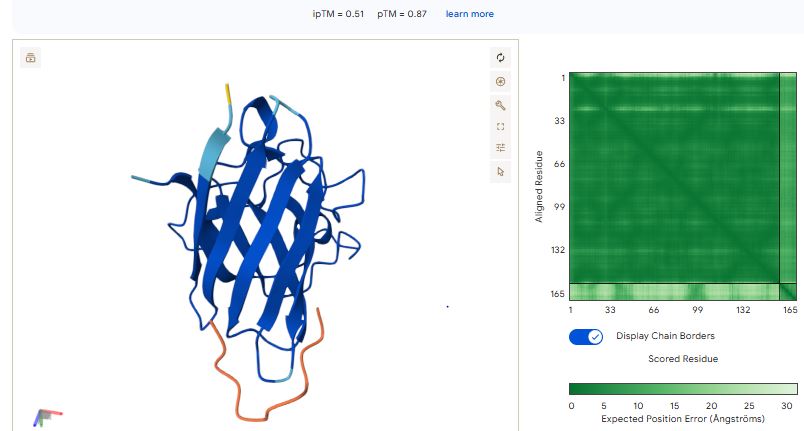
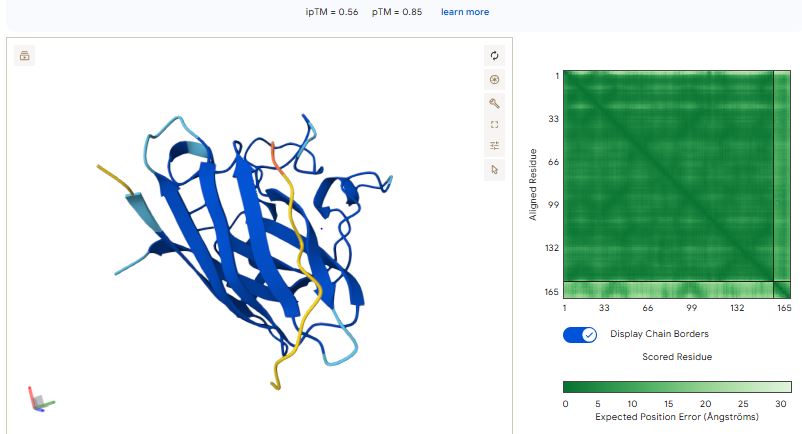
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
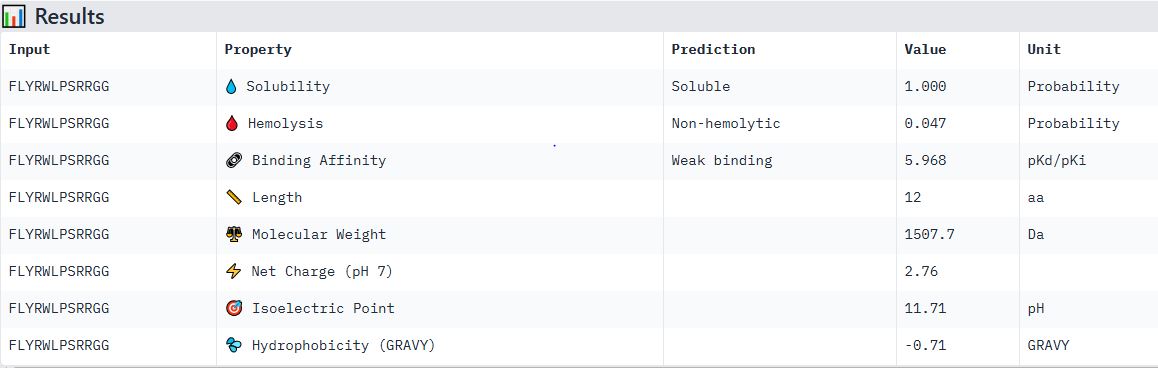
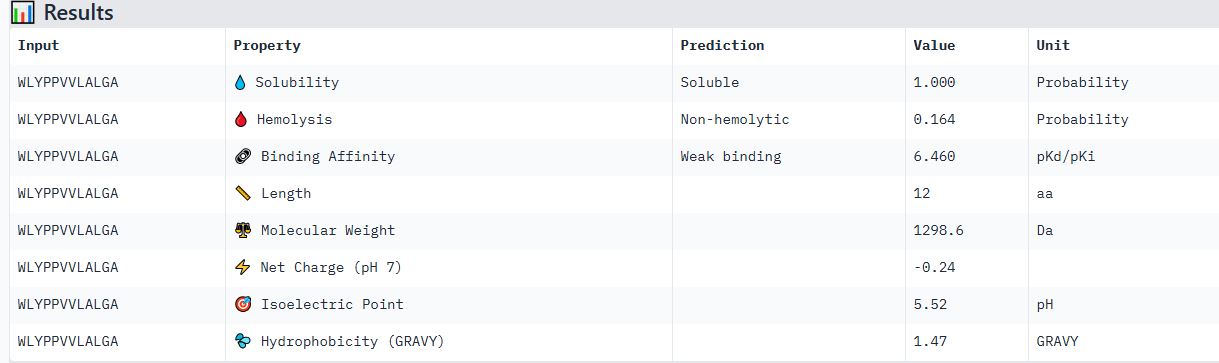
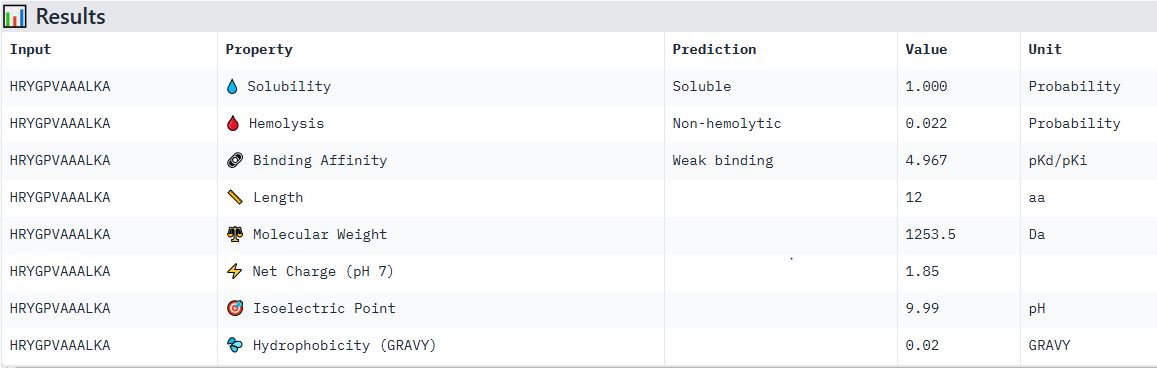
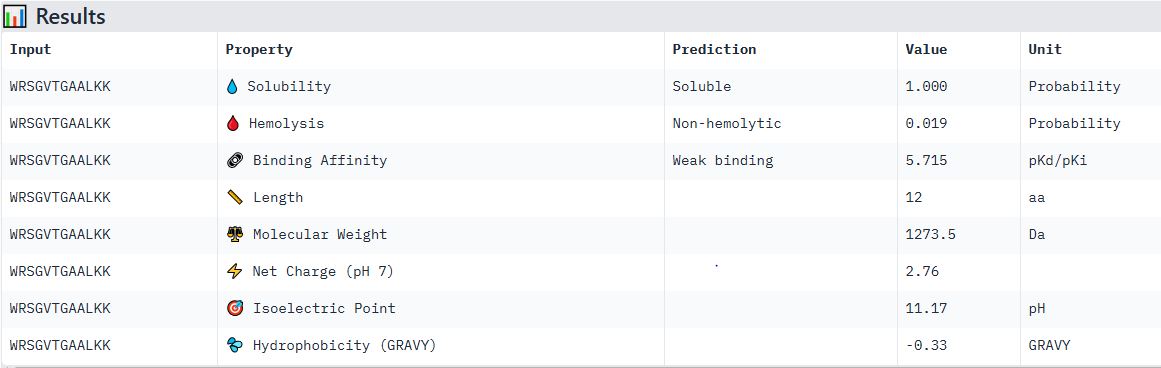
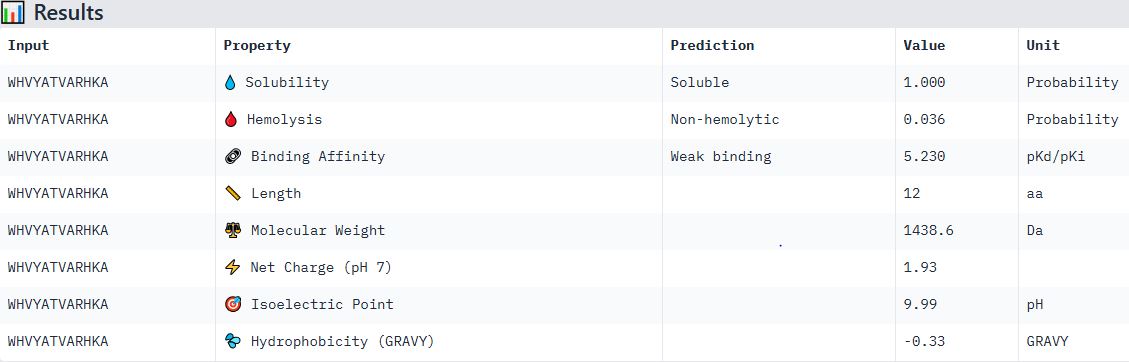
Part 4: Generate Optimized Peptides with moPPIt
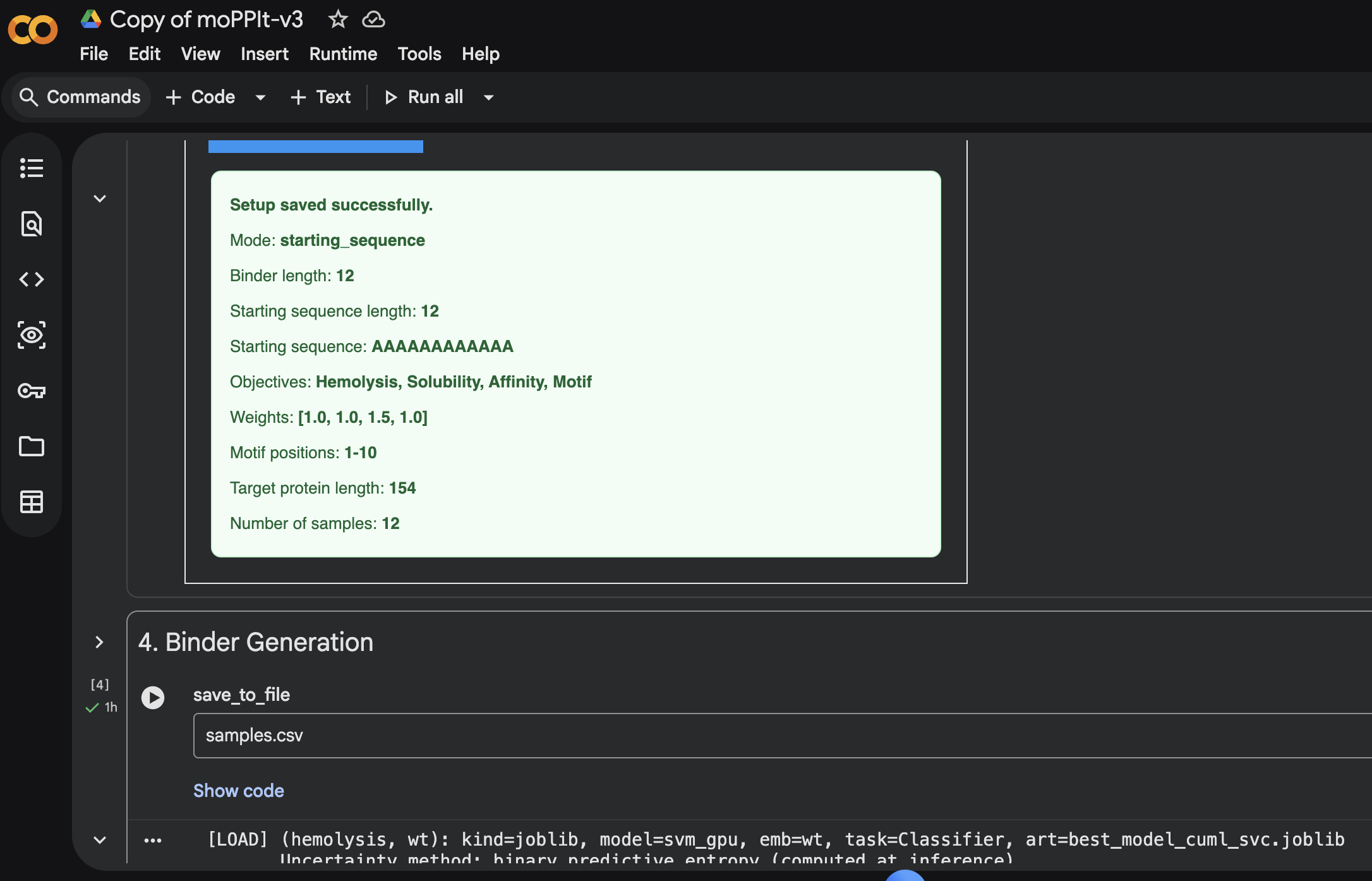
Sample 1: FAPYWPCCNPCR
Hemolysis: 0.0384 | Solubility: 1.0000 | Affinity: 7.6799 | Motif: 0.6357Sample 2: YCTDCVDGVVWE
Hemolysis: 0.0898 | Solubility: 0.9530 | Affinity: 7.3664 | Motif: 0.5257Sample 3: TRKPHYAAFFIY
Hemolysis: 0.0115 | Solubility: 1.0000 | Affinity: 6.8142 | Motif: 0.6964Sample 4: PCKYVPHVHVCF
Hemolysis: 0.0348 | Solubility: 1.0000 | Affinity: 6.7769 | Motif: 0.6278Sample 5: GFFVKTFEIVMF
Hemolysis: 0.0313 | Solubility: 1.0000 | Affinity: 6.5842 | Motif: 0.6023Sample 6: AFVTRELVVQIW
Hemolysis: 0.0775 | Solubility: 0.9980 | Affinity: 6.4754 | Motif: 0.7743Sample 7: HELTFARFEIQL
Hemolysis: 0.0169 | Solubility: 1.0000 | Affinity: 6.3272 | Motif: 0.7435Sample 8: QEPCEELQFNHF
Hemolysis: 0.0245 | Solubility: 1.0000 | Affinity: 6.2640 | Motif: 0.6353Sample 9: CTKVLIVKFEFK
Hemolysis: 0.0224 | Solubility: 1.0000 | Affinity: 6.0939 | Motif: 0.7347Sample 10: PSEKQCVKFHTT
Hemolysis: 0.0481 | Solubility: 1.0000 | Affinity: 5.8624 | Motif: 0.7204Sample 11: ANAPWFPPSSPH
Hemolysis: 0.0167 | Solubility: 1.0000 | Affinity: 5.6936 | Motif: 0.6189Sample 12: AFAKISNKQQQT
Hemolysis: 0.1067 | Solubility: 1.0000 | Affinity: 5.5742 | Motif: 0.7846Part C: Final Project: L-Protein Mutants
L-Protein Engineering | Option 3: Random Mutagenesis
- Create a python function to generate random mutation combinations with at least 2 residues by using the information found in mutational analysis experiments here.
Variant 1: S35K, Q71L
Sequence: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRKSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLLQLLTVariant 2: F47I, L44D
Sequence: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFDAIILSKFTNQLLLSLLEAVIRTVTTLQQLLTVariant 3: V63I, V67I
Sequence: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAIIRTITTLQQLLTVariant 4: R31K, F43P
Sequence: METRFPQQSQQTPASTNRRRPFKHEDYPCRKQQRSSTLYVLIPLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLTVariant 5: F5N, L60C
Sequence: METRNPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLCEAVIRTVTTLQQLLTCo-fold the random mutation with DnaJ using Af2_Multimer.
Variant 2 pAE
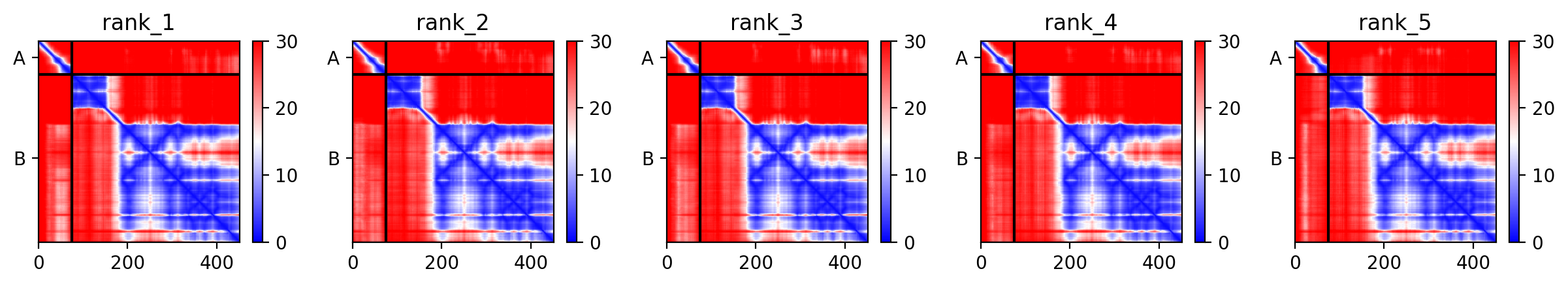
Structure
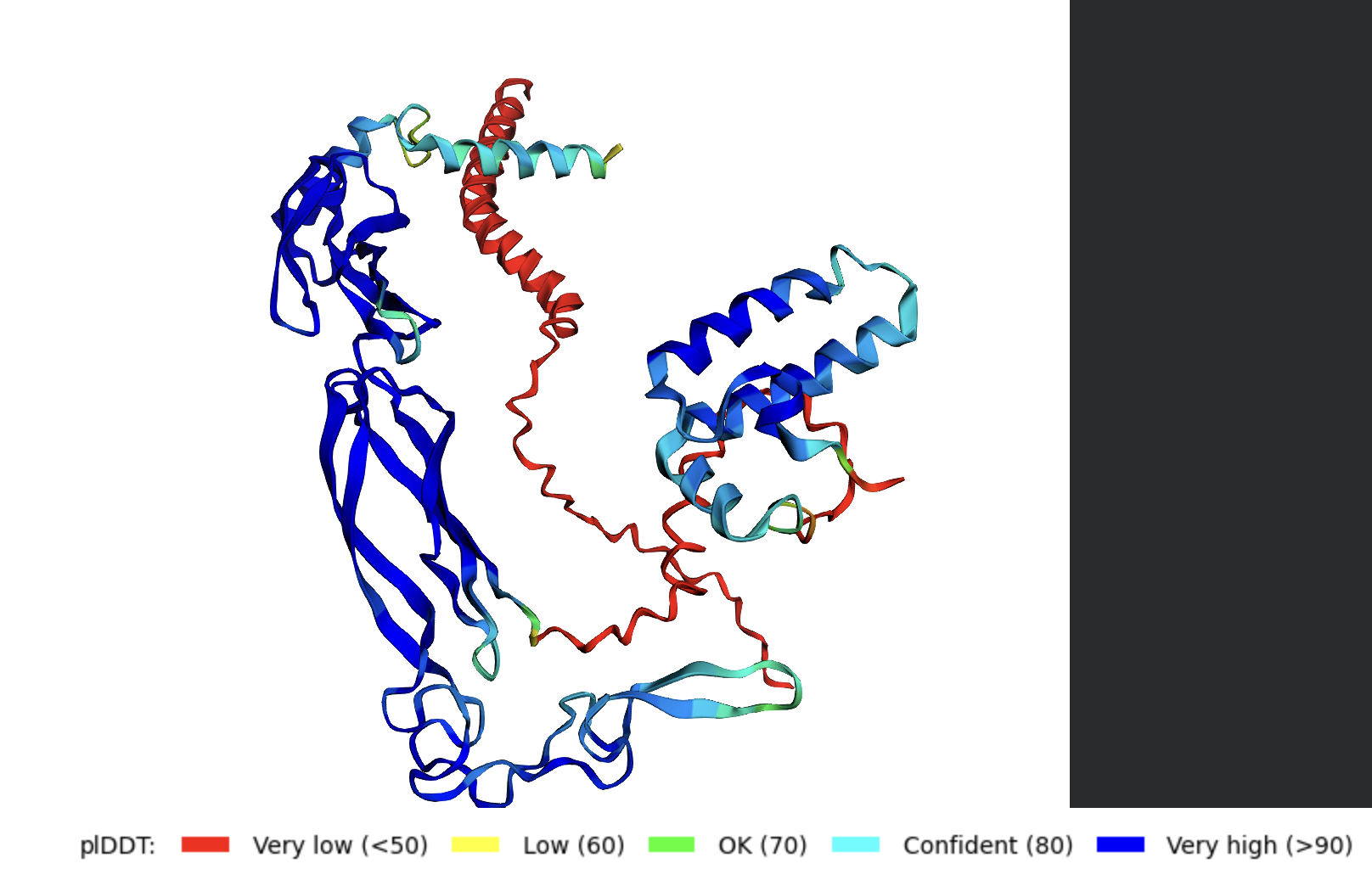
Week 6 HW: Genetic Circuits Part 1
Assignment: DNA Assembly
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The Phusion High-Fidelity PCR Master Mix (NEB #M0531) is supplied as a convenient 2× pre-formulated reagent containing all reaction components except template DNA, primers, and water. Its key constituents and their functions are as follows.
Phusion DNA Polymerase is the catalytic engine of the mix. It is a chimeric enzyme comprising a Pyrococcus-like thermostable polymerase core fused to a processivity-enhancing domain (derived from the Sso7d protein family), which allows the enzyme to remain bound to the DNA template for longer stretches and amplify fragments at higher speed and with greater fidelity than standard Taq polymerase. Crucially, Phusion carries a 3′→5′ proofreading exonuclease activity that removes incorrectly incorporated nucleotides, giving it an error rate more than 50-fold lower than Taq and roughly 6-fold lower than Pfu polymerase — making it the appropriate choice whenever sequence accuracy matters, such as for cloning (NEB, 2024). Deoxynucleotide triphosphates (dNTPs) — dATP, dCTP, dGTP, and dTTP — are the building blocks that the polymerase incorporates into the nascent DNA strand. They are pre-included in the master mix at a balanced concentration to minimise pipetting error.
MgCl₂ (magnesium chloride) is an essential cofactor. Mg²⁺ ions coordinate with the phosphate groups of the incoming dNTP in the polymerase active site, enabling the phosphodiester bond-forming reaction. The concentration of free Mg²⁺ also influences polymerase processivity and primer–template specificity; the HF Buffer formulation has been optimised to include the appropriate Mg²⁺ concentration for standard templates. Reaction buffer (HF Buffer) maintains the correct pH and ionic strength for optimal polymerase activity. The buffer stabilises the enzyme during the high-temperature denaturation steps and helps establish reproducible annealing conditions. An alternative GC Buffer formulation is available for GC-rich or otherwise difficult templates, optionally supplemented with DMSO to reduce secondary structure in the template.
Together, these components mean the researcher only needs to add template, primers, and water — dramatically reducing pipetting steps and the risk of component-level errors.What are some factors that determine primer annealing temperature during PCR?
The annealing temperature (T_a) is the step in the PCR cycle at which primers bind to the single-stranded template, and setting it correctly is one of the most important parameters for obtaining specific, high-yield amplification. Several interrelated factors govern the optimal T_a. GC content of the primers is the dominant determinant. Guanine–cytosine base pairs form three hydrogen bonds versus the two formed by A–T pairs, so primers with higher GC content have higher melting temperatures (T_m). The classical Wallace rule estimates T_m as 4°C per G/C + 2°C per A/T for short oligonucleotides, though more accurate nearest-neighbour thermodynamic models are preferred for primers longer than ~14 nt (SantaLucia, 1998).
Primer length also matters: longer primers have more base pairs contributing to stability, raising T_m. Primers used in the HTGAA Gibson Assembly lab are typically 18–25 nt in their annealing region, with additional 5′ homology overhangs (which do not contribute to T_m at the annealing step).
The specific polymerase used shifts the required T_a. Phusion polymerase, due to its Sso7d processivity domain binding non-specifically to double-stranded DNA, stabilises primer–template duplexes and therefore typically requires annealing temperatures 3–5°C higher than what a standard Taq-based Tm calculator recommends. NEB provides the Phusion-specific Tm Calculator (www.neb.com/tmcalculator) to account for this. Salt (Mg²⁺ and monovalent cation) concentration in the buffer affects the stability of the primer–template duplex. Higher ionic strength stabilises the negatively charged DNA backbone, raising T_m slightly.
Template secondary structure and GC-richness can indirectly affect effective annealing by reducing the accessibility of the target site; using a slightly lower T_a or adding DMSO can mitigate this. Finally, primer–dimer formation or 3′ self-complementarity can compete with productive annealing — poorly designed primers may force a lower T_a that sacrifices specificity. NEB’s Tm Calculator and tools like Primer3 or HTGAA’s own Gibson Assembly supplement are valuable resources for rational primer design.There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Both PCR and restriction enzyme (RE) digestion produce linear DNA fragments suitable for downstream cloning, but they differ substantially in how the fragment boundaries are defined, what the resulting ends look like, and when each is the better tool.
- Protocol comparison:
PCR begins with designing two oligonucleotide primers that flank the desired sequence. The reaction cycles through denaturation (~98°C for Phusion), annealing, and extension steps, exponentially amplifying the target sequence from as little as a few nanograms of template. The researcher has complete control over where amplification begins and ends — simply by choosing primer binding positions — and can append any desired sequence (e.g., homology overhangs for Gibson Assembly) to the 5′ end of each primer. The product is a double-stranded fragment with blunt ends when using Phusion (or 3′ A-overhangs with Taq). PCR requires sequence knowledge at both termini but otherwise introduces no sequence constraints on the interior of the fragment. - RE digestion, by contrast, requires that the target DNA already contains the appropriate recognition sites for the chosen restriction enzyme at the desired cut points — or that these sites were engineered in at the cloning stage. Digestion is carried out by incubating the DNA with the enzyme in its optimised buffer, typically at 37°C, for 15–60 minutes. Depending on the enzyme, the resulting ends are blunt, 5′ overhang (sticky), or 3′ overhang (sticky). Sticky ends are highly useful for directional ligation because complementary overhangs guide fragment orientation. However, the set of available cut sites is limited to the enzyme’s recognition sequence, so fragment boundaries cannot be arbitrarily chosen.
When to prefer PCR:
PCR is preferred when (1) you need to add sequence information to the fragment ends (e.g., homology overhangs for Gibson Assembly, or a promoter element), (2) the target sequence does not naturally contain convenient restriction sites, (3) you want blunt ends for isothermal assembly methods, or (4) you are working from genomic DNA where cutting to the exact desired boundary would otherwise require engineering new sites. PCR is also faster when only one or a few copies of template are available.
When to prefer RE digestion:
Restriction digestion is preferred when (1) working with existing plasmid libraries or vectors that were designed with standardised cloning sites (e.g., BioBrick or MCS vectors), (2) you need highly directional ligation via compatible sticky ends without a ligase-independent step, (3) the insert is already in a plasmid and simply needs to be excised, or (4) when PCR fidelity is a concern for very long fragments (though modern HF polymerases largely address this). RE digestion also scales easily to large amounts of DNA and requires no thermocycler. In summary, PCR offers flexibility in defining sequence boundaries and is essential for adding new sequence elements, while RE digestion is efficient and directional when working within an existing restriction site framework.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning? Gibson Assembly relies on three enzymes acting sequentially on overlapping linear DNA fragments: a 5′ exonuclease chews back the 5′ ends to expose single-stranded 3′ tails, a DNA polymerase fills in gaps between annealed fragments, and a DNA ligase seals the nicks. For this to work correctly, several conditions must be met in the design of your PCR products and RE-digested fragments.
Design overlapping homology sequences of appropriate length. Each pair of adjacent fragments must share 15–30 bp of identical sequence at their junction. For PCR products, this is achieved by appending the appropriate homology sequence to the 5′ end of each primer. For RE-digested fragments, you must verify that after digestion, the fragment ends share sequence with the adjacent insert or vector — this usually requires that the vector was originally designed with those overlaps or that an intermediate PCR step adds them.
Verify the absence of internal restriction sites (if using RE digestion). If you are opening a vector by RE digestion and the enzyme cuts elsewhere in the backbone or insert, you will generate unintended fragments that can interfere with assembly efficiency. Run a virtual digest in silico (e.g., in Benchling or SnapGene) before proceeding.
Check for absence of repeat sequences at junctions. The T5 exonuclease in the Gibson mix cannot distinguish between the intended overhang and any other region of the same sequence. Internal repeats of ≥15 bp near the junction can cause mis-assembly or deletion artefacts.
Gel-purify or column-purify all fragments. After PCR, gel extraction removes primer dimers, residual template, and off-target amplicons. After RE digestion, gel purification removes the small excised stuffer fragment and inactivated enzyme. Clean fragments improve assembly efficiency.
Verify fragment sizes and quality. Run all fragments on an agarose gel to confirm they are the expected size. Faint bands may indicate degradation or low yield, both of which reduce assembly efficiency. Quantify DNA concentration (e.g., by NanoDrop or Qubit) so that correct molar ratios of vector to insert can be set up in the assembly reaction (typically 1:2 to 1:5 molar ratio).
Confirm that terminal sequences are internally consistent. Use in-silico assembly tools (Benchling, SnapGene, or Geneious) to simulate the final assembled product before running the reaction. Confirm that the reading frame, promoter orientation, and any regulatory elements are correct in the predicted assembly.
How does the plasmid DNA enter the E. coli cells during transformation? The process of introducing foreign DNA into bacterial cells is called transformation, and in standard molecular biology protocols it occurs via one of two mechanisms: heat-shock transformation of chemically competent cells, or electroporation of electrocompetent cells.
- In heat-shock transformation, E. coli cells are first made chemically competent by treatment with divalent cations — most commonly calcium chloride (CaCl₂). The Ca²⁺ ions are thought to neutralise the repulsive negative charges on both the bacterial outer membrane (lipopolysaccharide and phospholipid headgroups) and the DNA backbone, allowing DNA to associate with the cell surface (Mandel & Higa, 1970). The cells are then incubated on ice with the plasmid DNA, and then subjected to a brief heat shock (typically 42°C for 30–45 seconds). The sudden temperature shift is believed to create transient disruptions or pores in the membrane, allowing plasmid DNA to enter the cytoplasm — though the precise molecular mechanism remains incompletely understood. Following heat shock, cells are returned to ice and then allowed to recover in rich medium (SOC broth) at 37°C for 45–60 minutes to allow expression of any antibiotic resistance gene encoded on the plasmid before plating on selective media.
- In electroporation, a brief high-voltage electrical pulse (typically 1.8 kV for E. coli) is applied across a suspension of cells mixed with DNA. The electric field transiently creates electropores — nanoscale hydrophilic pores in the lipid bilayer — through which DNA can pass. Electroporation generally achieves higher transformation efficiencies and is particularly useful for large plasmids or low-concentration DNA. Once inside the cell, the circular plasmid is not integrated into the chromosome (unless it carries specific recombination sequences); instead, it replicates autonomously using its own origin of replication (ori) and the host cell’s replication machinery. Selection on antibiotic-containing agar plates then ensures that only cells which successfully took up and maintained the plasmid survive.
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a seamless, scarless DNA assembly method developed by Engler et al. (2008) that exploits Type IIS restriction enzymes — enzymes that cut outside their recognition sequence at a defined offset — to generate custom 4 bp overhangs from any desired position in the DNA. The defining principle is that the recognition site for the Type IIS enzyme (commonly BsaI or Esp3I) is placed adjacent to the junction of interest, oriented so that the enzyme cuts into — and through — the actual sequence junction. When the enzyme cuts, it removes the recognition site itself from the end of the fragment, leaving a short custom 4-nucleotide 3′ overhang that is sequence-specific to the junction. Because these 4 bp overhangs are designed by the researcher, adjacent fragments can be engineered to carry perfectly complementary, unique overhangs — ensuring directional, ordered ligation in a single tube. The BsaI digestion and T4 DNA ligase step are performed simultaneously and cyclically in the same tube (alternating 37°C digestion and 16°C ligation cycles), meaning that mis-ligated products are re-cut and re-ligated until the correct thermodynamically stable final construct accumulates. This makes Golden Gate highly efficient for assembling 3–10 (or more) fragments simultaneously with very high accuracy and minimal background, and it leaves no scar sequence at the junctions beyond the 4 bp overhang itself, which becomes part of the final sequence.
 Golden Gate is particularly powerful for combinatorial library construction — for example, assembling promoter, RBS, coding sequence, and terminator parts from a standardised library (e.g., the MoClo or Loop assembly systems) in a single reaction.
Golden Gate is particularly powerful for combinatorial library construction — for example, assembling promoter, RBS, coding sequence, and terminator parts from a standardised library (e.g., the MoClo or Loop assembly systems) in a single reaction.- Model this assembly method with Benchling or Asimov Kernel!
Assignment: Asimov Kernel
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
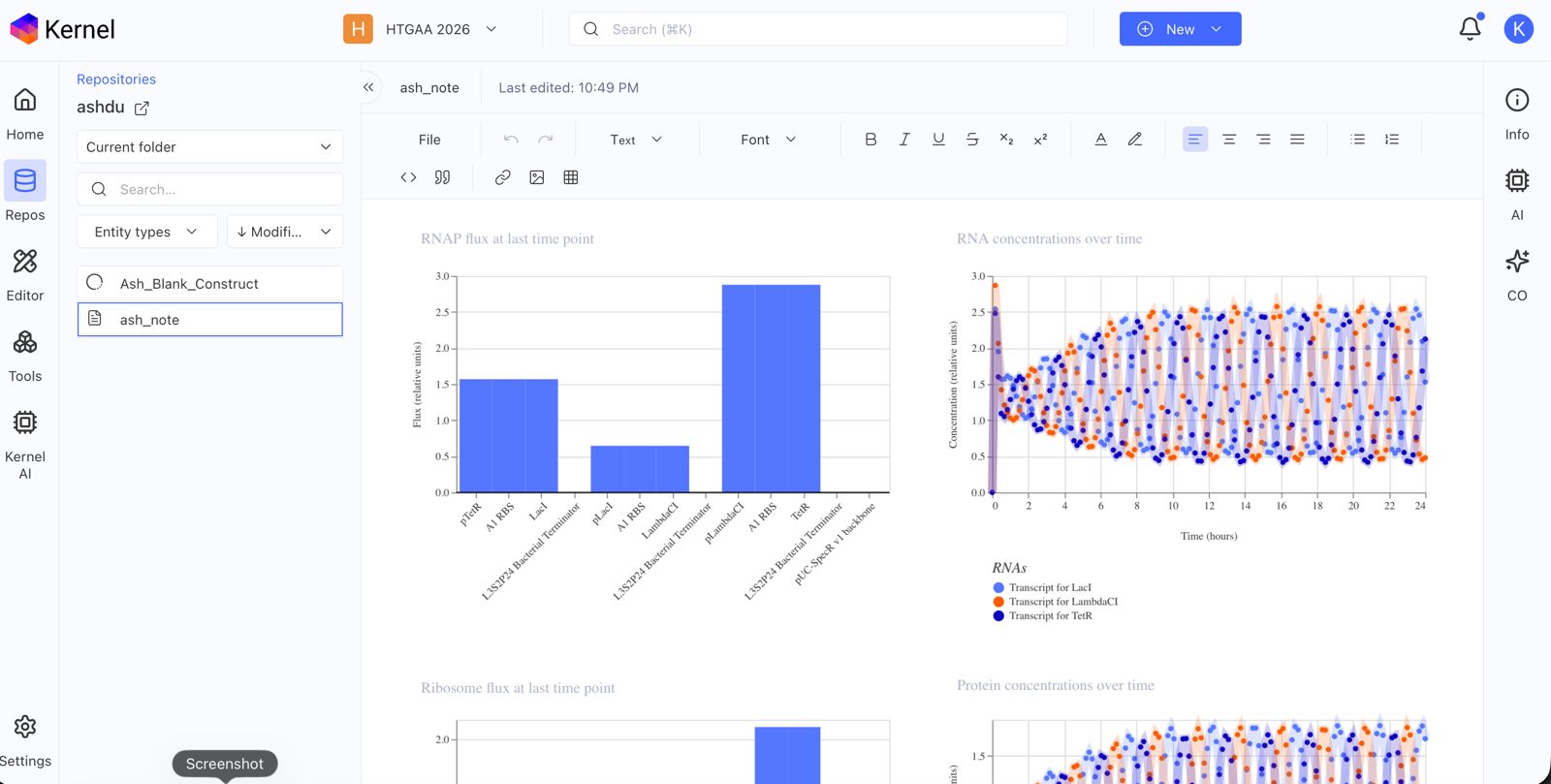
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
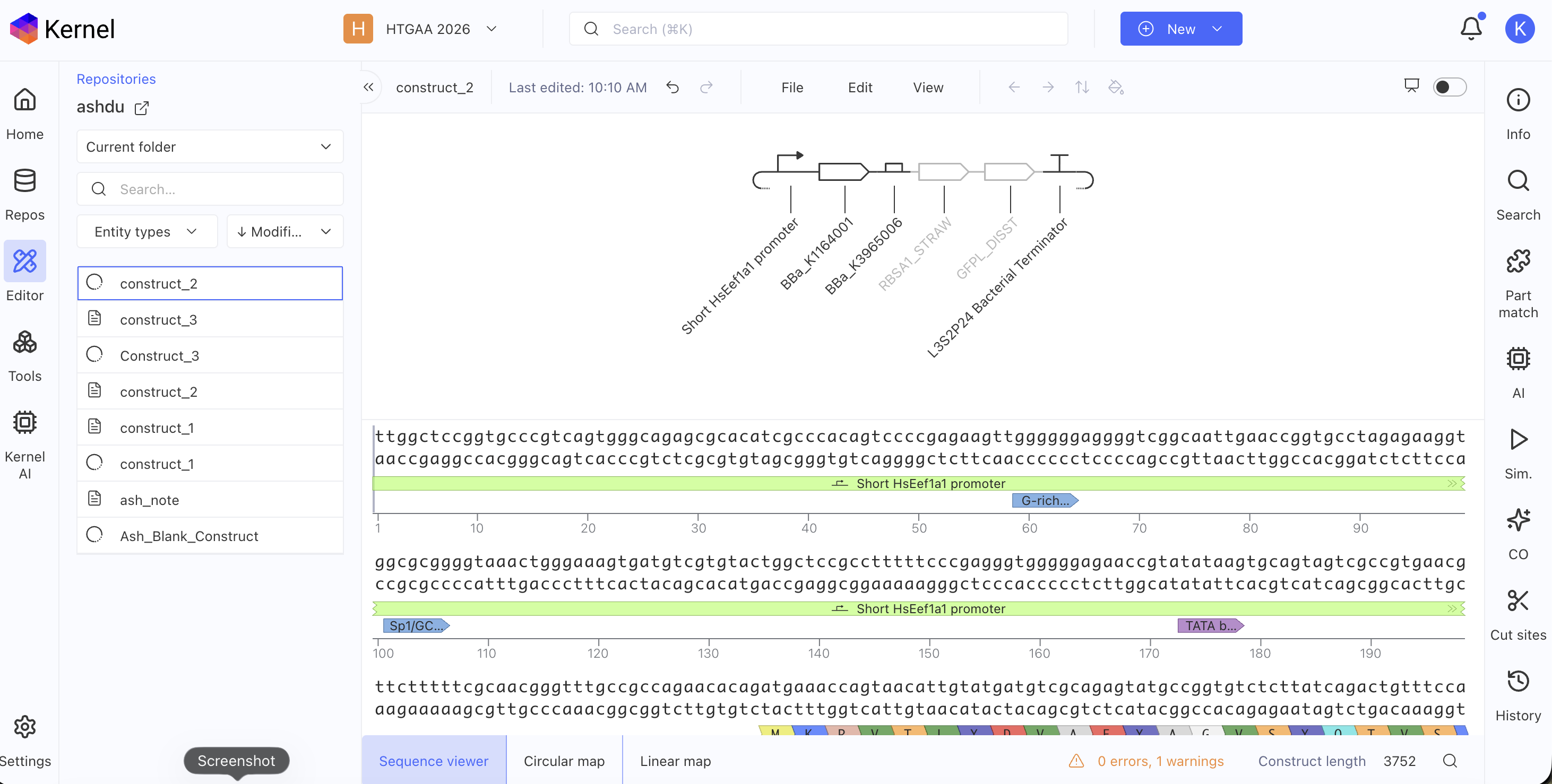

Week 7 HW: Genetic Circuits Part 2
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Traditional genetic circuits operate as Boolean logic gates: they classify inputs as either “on” (1) or “off” (0) and produce outputs that are likewise binary. While this is powerful for implementing discrete decisions — such as activating a kill switch if and only if two specific signals are simultaneously present — Boolean circuits are fundamentally limited in their ability to process the continuous, graded molecular signals that characterise real biological environments. Intracellular concentrations of transcription factors, metabolites, and signalling molecules are not naturally binary; they span continuous ranges that carry information that a simple Boolean threshold necessarily discards. IANNs overcome this limitation by implementing analog computation, in which each molecular “neuron” computes a weighted sum of its continuous-valued inputs, passes that sum through a nonlinear activation function, and produces a graded output that can itself serve as an input to the next layer. This architecture enables a single engineered cell to perform multi-threshold classification — distinguishing not just “signal present” from “signal absent” but grading responses proportionally to signal intensity, and separating input patterns that no Boolean gate could resolve without an exponentially larger circuit. For example, a cell expressing a two-input biomolecular perceptron can draw a separating hyperplane in the continuous input space of two molecular concentrations, classifying cell states that would require many cascaded Boolean gates to approximate. A second key advantage is graceful degradation under noise: because IANNs operate over a continuous input range, they can be designed with soft thresholds that smooth over stochastic fluctuations in molecule numbers — a pervasive problem in cells, where copy numbers of regulatory molecules are often in the tens to hundreds range. Boolean gates, which depend on crossing a hard threshold, are comparatively fragile to such noise. Third, IANNs are in principle extendable toward online learning, in which the synaptic weights (encoded by molecular concentrations or binding affinities) can be updated as a function of experience — an entirely alien concept to hardwired Boolean logic. Taken together, IANNs expand the computational vocabulary available to synthetic biology from a finite set of logic operations to a continuous, composable, and theoretically universal function approximation framework.Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
One compelling application for an IANN is the continuous intracellular monitoring and correction of iron overload in patients with hereditary hemochromatosis — a genetic disorder characterised by excessive gastrointestinal absorption of dietary iron, leading to toxic iron deposition in the liver, heart, and pancreas. Current treatment requires regular phlebotomy, which is effective but burdensome and cannot respond dynamically to real-time fluctuations in free labile iron. An IANN-based therapeutic cell (for example, an engineered hepatocyte or gut epithelial cell) could be designed as follows. Two inputs are presented to a single-layer intracellular perceptron:
X₁: the intracellular concentration of labile iron pool (LIP), sensed indirectly via an iron-responsive element (IRE)–iron regulatory protein (IRP) system, which naturally controls mRNA translation in proportion to free iron levels. A synthetic construct could link IRP binding to the transcription or translation of an intermediate regulatory RNA, converting iron concentration into a molecular signal. X₂: a constitutive bias input (a fixed-level transcript) that sets the activation threshold — encoding the notion that the circuit should only respond when iron exceeds a safe baseline, analogous to a bias unit in a standard perceptron.
The perceptron computes the weighted sum of these inputs. When the weighted iron signal exceeds the threshold set by the bias, the activation function triggers expression of the output gene: a codon-optimised ferritin heavy-chain transgene, which sequesters excess free iron into inert ferritin complexes and prevents cellular damage. The output is graded — the more the iron concentration exceeds the threshold, the more ferritin is produced — in contrast to a Boolean circuit, which would either produce a fixed amount of ferritin or none at all, regardless of the severity of iron overload. Several important limitations must be acknowledged. First, IANNs currently cannot perform online weight adjustment in living cells at the speed required for therapeutic use; weights are set at the time of circuit design and cannot recalibrate if the patient’s physiology changes. Second, the molecular components encoding the perceptron — endoribonucleases, regulatory RNA hairpins, sequestration species — impose a metabolic burden on the host cell, and this burden grows with the complexity of the network, potentially impairing normal cellular function. Third, molecular noise in cells means that the effective threshold may drift over time as component concentrations fluctuate, making it difficult to guarantee that the circuit reliably distinguishes pathological from physiological iron levels. Fourth, an in vivo implementation raises significant immunogenicity concerns: the endoribonuclease components (e.g., Csy4, which originates from Pseudomonas aeruginosa) could trigger immune responses in a human host. These limitations suggest that near-term applications of IANNs may be better suited to ex vivo cell engineering or biosensor contexts rather than direct in vivo therapeutics.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
- What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials — chiefly derived from mycelium, the vegetative network of interwoven hyphae produced by filamentous fungi — have been commercialised across several industries over the past decade. The most mature application is mycelium-based composite packaging (e.g., Ecovative Design’s Mushroom® Packaging), in which agricultural waste such as corn husks or hemp hurds is inoculated with fungal spores, allowed to colonise and bind the substrate, then heat-killed and dried to produce a rigid, foam-like material used in place of expanded polystyrene for protective packaging. A second prominent category is myco-leather: pure mycelium mats produced by companies such as Bolt Threads (Mylo™) and MycoWorks (Reishi™) are processed into flexible sheets resembling animal leather and have been used in fashion accessories, including a limited-edition Hermès handbag. Third, mycoprotein — most famously Quorn, derived from Fusarium venenatum — has been on the market since the 1980s as a high-protein, meat-substitute food ingredient. More nascent applications include mycelium-based thermal insulation panels, biocement for construction, and even flexible electronic substrates exploiting the high conductivity of processed mycelium films.
The advantages of mycelium materials over their conventional counterparts are substantial. They are fully biodegradable, decomposing within weeks to months under composting conditions, in contrast to expanded polystyrene (which persists for ~50 years) or synthetic leather (derived from petroleum-based polyurethane). They can be grown on agricultural waste and byproducts — low-cost, abundant feedstocks — requiring no petroleum inputs, which reduces their carbon footprint relative to synthetic foam and plastic alternatives. They are mouldable during growth, meaning complex three-dimensional shapes can be formed without energy-intensive casting or machining. For myco-leather specifically, production avoids the toxic tanning chemicals and greenhouse gas emissions associated with conventional livestock-based leather.
The disadvantages are equally significant and should not be understated. Mycelium composites typically exhibit lower and less consistent mechanical properties than synthetic analogues: their tensile strength, compressive modulus, and moisture resistance vary substantially with fungal species, substrate composition, and growth conditions, making quality control challenging at industrial scale. High moisture absorption is a persistent problem — mycelium-based foams can absorb significant water, compromising their insulating and structural properties in humid environments. Biodegradability, while an environmental advantage, is simultaneously a durability disadvantage: myco-leather goods will degrade under prolonged exposure to moisture, UV light, or biological activity at rates that animal leather or synthetic leather would not. Finally, scaling production while maintaining consistent properties and sterility is technically demanding and costly, and life-cycle assessments suggest that the energy inputs for controlled fungal cultivation can partially offset the environmental benefits, particularly where renewable energy is not available .- What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
One application I find particularly compelling is engineering filamentous fungi for targeted heavy-metal bioremediation — specifically, the removal of cadmium, lead, and arsenic from contaminated soils and industrial wastewater. Wild-type fungi already exhibit some capacity for metal biosorption via their cell walls, but this is passive and non-selective. I would want to genetically engineer a species such as Aspergillus niger or Trichoderma reesei to overexpress metallothioneins (small cysteine-rich metal-binding proteins) and ABC-type metal transporters that actively import toxic metals into vacuoles, concentrating them intracellularly for subsequent extraction by harvesting the mycelium rather than using energy-intensive chemical treatments. A second engineering goal would be to add a biosensor output — for instance, linking metal accumulation to the expression of a fluorescent reporter — so that the mycelium itself signals when remediation capacity is saturated and biomass needs to be replaced. This is precisely the kind of continuous, graded signal-response behaviour that an IANN architecture (from Part 1) could implement.
There are several compelling advantages of performing this synthetic biology in fungi rather than bacteria. First, fungi are eukaryotes, meaning they possess the post-translational modification machinery — N-linked glycosylation, disulfide bond formation, proper protein folding in the endoplasmic reticulum — required to produce complex proteins such as metallothioneins and secreted enzymes in their active forms; many such proteins are misfolded or inactive when expressed in E. coli. Second, filamentous fungi grow as mycelial networks that can extend through soil, bridging air-liquid interfaces and penetrating into pore spaces inaccessible to bacterial biofilms — a critical advantage for in situ bioremediation, where the contaminant is spatially distributed and often in a partly air-filled matrix. Third, many filamentous fungi are GRAS-status organisms (Generally Recognised As Safe), reducing regulatory barriers for environmental release relative to engineered bacteria, some of which carry biosafety concerns. Fourth, fungi have extraordinary metabolic versatility: they can catabolise lignin, cellulose, and xenobiotic compounds via oxidative enzymes (laccases, peroxidases) that are absent from most bacteria, making them uniquely suited to environments contaminated with both heavy metals and complex organic pollutants simultaneously. Finally, the physical scaffold of mycelium itself has structural utility — a bioremediation mycelium network can be harvested as a solid biomass enriched in bound metals, rather than requiring centrifugation or filtration of a bacterial cell suspension, simplifying downstream metal recovery. A counter-consideration is that fungal genetic engineering has historically been more technically challenging than in bacteria, due to lower rates of homologous recombination in many species and the relative scarcity of validated synthetic promoters and genetic parts. However, the development of CRISPR-Cas9 tools adapted for Aspergillus and Trichoderma, alongside growing fungal parts registries, is rapidly closing this gap.Assignment Part 3: First DNA Twist Order
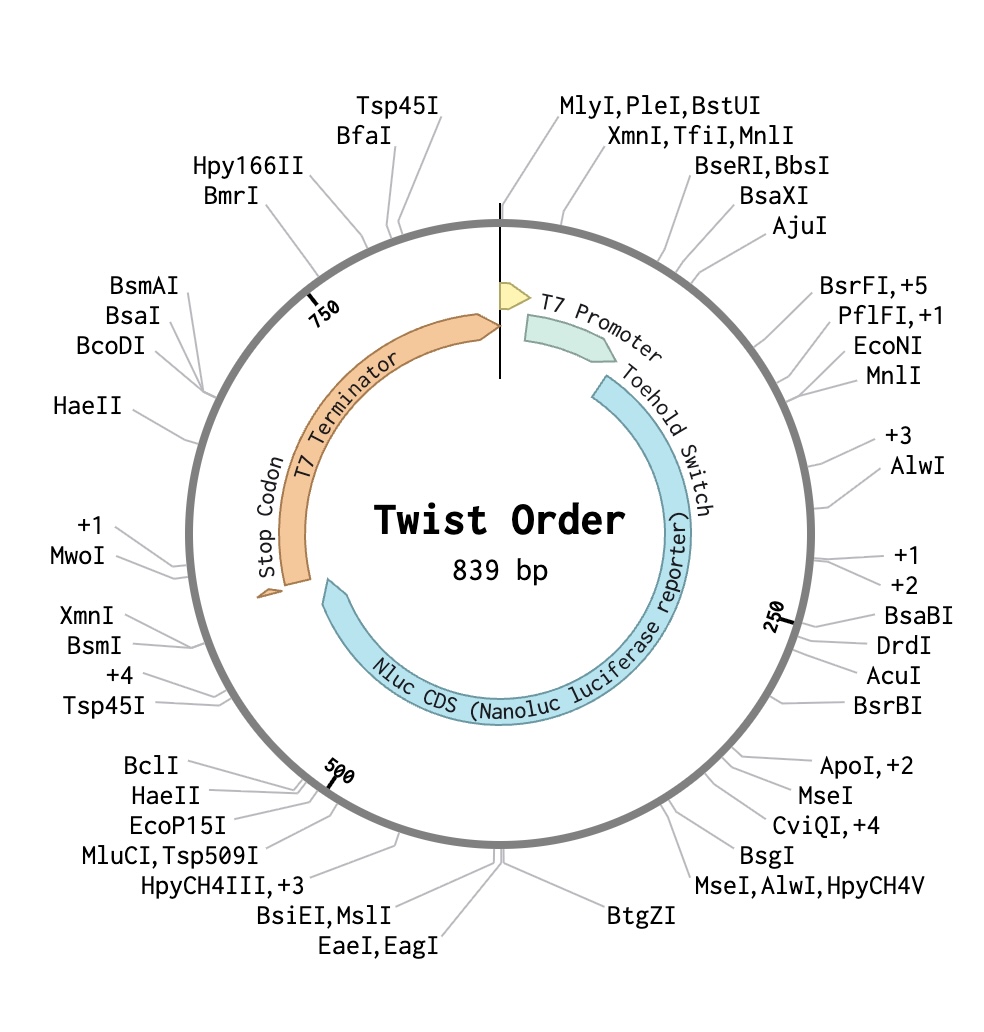
Week 9 HW: Cell Free Systems
Homework Part A: General and Lecturer-Specific Questions
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis (CFPS) offers a fundamentally different operating logic from in vivo expression: because there is no living cell to maintain, the reaction environment is open and directly accessible to the experimenter. This openness translates into three practical advantages. First, reaction components — amino acid concentrations, buffer conditions, redox potential, template concentration — can be tuned independently and in real time without the buffering effects of cellular homeostasis. Second, toxic proteins that would kill or arrest growing cells can be expressed freely in CFPS, since there is no cell viability to protect. Third, non-canonical amino acids, isotopic labels, or synthetic chemical groups can be incorporated site-specifically by supplementing the reaction directly, enabling protein engineering strategies that are impossible to sustain through the protein expression machinery of a living cell. Two cases where cell-free expression is specifically more advantageous than cell-based production are: (1) membrane protein structural studies, where the absence of competing cellular membranes allows co-translational insertion directly into defined lipid nanodiscs of controlled composition, circumventing the protein aggregation and misfolding problems that arise during over-expression in intact cells; and (2) rapid on-demand diagnostic biosensors, where freeze-dried CFPS reactions can be deployed at the point of need without cold-chain infrastructure or biohazard containment — capabilities recently validated aboard the International Space Station.2. Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free expression system can be understood as a minimal reconstruction of the cellular central dogma pathway. The core components and their roles are as follows. The DNA or mRNA template encodes the protein of interest and acts as the informational input; a strong promoter (e.g., T7) is typically used when a DNA template drives transcription. RNA polymerase (either endogenous in crude lysates or supplied purified as T7 RNAP in PURE systems) transcribes the DNA into mRNA. The ribosome is the catalytic core of translation, reading the mRNA and elongating the polypeptide chain with the assistance of elongation factors (EF-Tu, EF-G) and initiation/release factors; roughly 4 ATP equivalents are consumed per peptide bond formed. Aminoacyl-tRNA synthetases (aaRSs) charge each of the 20 tRNAs with their cognate amino acid, and tRNA molecules deliver those charged amino acids to the ribosome A-site. Amino acids serve as the building block pool; depletion of the amino acid pool is one of the primary causes of reaction stalling in crude lysate systems. The energy regeneration module — commonly phosphoenolpyruvate (PEP) plus pyruvate kinase, creatine phosphate plus creatine kinase, or 3-phosphoglycerate (3-PGA) — continuously regenerates ATP and GTP from ADP/GDP to sustain translation. Magnesium ions are essential cofactors for ribosome function and nucleotide-dependent enzymes; their concentration must be carefully titrated. Potassium ions set the ionic environment required for ribosome activity. Finally, polyethylene glycol (PEG) or similar crowding agents mimic the macromolecular crowding of the cytoplasm and can improve translation efficiency. In the PURE system, all these components are defined and provided as purified proteins, offering reproducibility and the absence of contaminating nucleases and proteases.3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical in CFPS because translation is an inherently ATP- and GTP-intensive process — approximately 4 high-energy phosphate equivalents are consumed per amino acid incorporated (2 ATP for aminoacyl-tRNA charging, 1 GTP for tRNA delivery to the ribosome, and 1 GTP for translocation) (Jewett & Swartz, 2004). Without continuous regeneration, the ATP pool is rapidly depleted, causing translation to stall. A further complication is the accumulation of inorganic phosphate (Pi) as a byproduct of phosphotransfer reactions: elevated Pi sequesters Mg²⁺, which is an essential ribosomal cofactor, thereby inhibiting both transcription and translation. An effective energy system must therefore not only regenerate ATP but also limit Pi accumulation (Calhoun & Swartz, 2007). One reliable method is the 3-phosphoglycerate (3-PGA) system, in which 3-PGA enters a truncated glycolytic pathway to regenerate ATP while producing only pyruvate and acetate as by-products — neither of which chelates Mg²⁺ appreciably. Studies have shown that 3-PGA-powered CFPS sustains reactions for several hours and achieves yields exceeding 1 mg/mL of recombinant protein (Kim & Swartz, 2001). A complementary strategy is to use a fed-batch or semi-continuous dialysis reactor format, in which fresh substrates (ATP precursors, amino acids, cofactors) are continuously exchanged into the reaction while inhibitory by-products are dialysed out, extending productive synthesis from hours to potentially days (Spirin et al., 1988). For classroom or field-deployable settings, the simpler creatine phosphate / creatine kinase (CP/CK) system remains widely used, despite the 1:1 stoichiometric phosphate release it entails, because of its low cost and ease of formulation.4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic CFPS systems — most commonly derived from E. coli lysates — are fast to prepare, inexpensive, highly productive (yields of 1–4 mg/mL are achievable in optimised formats), and compatible with a wide range of T7-based expression vectors. Their principal limitation is the absence of the eukaryotic post-translational modification machinery: E. coli extracts cannot perform N-linked glycosylation, and the reducing cytoplasmic environment is unfavourable for the formation of disulfide bonds, which are essential for many human therapeutic proteins. Eukaryotic CFPS systems — including wheat germ extract (WGE), rabbit reticulocyte lysate (RRL), and Chinese hamster ovary (CHO) cell lysates — provide access to chaperones, signal recognition particles, and post-translational processing machinery that support proper folding of complex human proteins. They tend to be slower and more expensive than prokaryotic systems, but are indispensable when the target protein requires glycosylation, specific disulfide connectivity, or processing by signal peptidase. For the prokaryotic system (E. coli extract), an excellent choice is single-chain variable fragment (scFv) antibody, a small (~27 kDa) recombinant antibody format that does not require glycosylation and whose binding function can be verified rapidly by an ELISA-based assay. The fast turnaround of bacterial CFPS (reactions complete within 4–6 hours) is ideal for iterative screening of antibody variants during affinity maturation campaigns. For the eukaryotic system (CHO or insect cell extract), erythropoietin (EPO) is the appropriate choice. EPO is a 165-amino acid glycoprotein hormone in which three N-linked and one O-linked glycan chains account for approximately 40% of its molecular weight and are critical for its in vivo half-life and receptor-binding activity. Expressing EPO in a prokaryotic system yields aglycosylated protein with substantially reduced biological activity; a CHO-based CFPS system that includes microsomes or glycosylation enzymes can produce a glycoform closer to the therapeutic molecule.5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins (MPs) represent the most challenging class of targets for CFPS because their hydrophobic transmembrane domains are insoluble in aqueous solution: without a lipid environment, they aggregate irreversibly into inclusion body-like precipitates immediately after synthesis. A well-designed cell-free membrane protein experiment must therefore couple protein synthesis to a compatible hydrophobic scaffold present in the reaction from the outset. The recommended strategy is co-translational insertion into pre-formed nanodiscs. Nanodiscs are discoidal phospholipid bilayer patches stabilised by an amphipathic membrane scaffold protein (MSP); their diameter (~10 nm) and lipid composition can be controlled precisely. By including nanodiscs at optimised concentrations (typically 0.2–2 mg/mL) in the CFPS reaction, the nascent transmembrane protein can fold co-translationally into the bilayer rather than encountering aqueous solution at all, preserving its native fold and function. Studies have shown that nanodisc-based CFPS supports correct folding of GPCRs, ion channels, and multi-pass transporters at yields sufficient for structural studies by NMR or cryo-EM. Three specific challenges and how to address them: (1) Aggregation during synthesis — mitigated by using lipid nanodiscs as described above, supplemented if needed with detergents at sub-CMC concentrations such as Brij-35 to stabilise partially-folded intermediates; (2) Low expression yield — membrane proteins are often toxic in in-vivo systems but in CFPS this is no constraint; yield can be maximised by optimising the concentration of nanodiscs, adjusting Mg²⁺ levels (often 10–14 mM for membrane protein CFPS rather than the standard 8–10 mM), and screening N-terminal fusion tags to improve ribosome engagement; (3) Verification of correct folding — since Western blotting confirms synthesis but not function, activity assays (e.g., ligand binding ELISA for GPCRs, patch-clamp for channels) or limited proteolysis footprinting should be used to confirm the protein has adopted its native architecture.6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Low yield in a CFPS reaction can arise at multiple points in the expression pathway. Here are three common causes and their corresponding troubleshooting strategies. Reason 1 — Premature energy depletion and ATP starvation. If the energy regeneration system is insufficient or the secondary energy source (e.g., phosphoenolpyruvate) is consumed too quickly, ATP levels drop below the threshold required to sustain elongation, causing ribosomes to stall prematurely. The troubleshooting strategy is to measure the reaction’s pH over time using a microelectrode or pH-sensitive dye (acidification indicates Pi accumulation and ATP exhaustion) and to switch to a more sustained energy substrate such as 3-PGA, which produces less Pi per ATP regenerated, or to implement a fed-batch format with controlled substrate addition. Reason 2 — mRNA instability and degradation. Crude cell extracts contain residual ribonucleases that can degrade the mRNA template, especially if it lacks a strong 5’ untranslated region (UTR), a stable secondary structure at the 3’ end, or is not capped in eukaryotic systems. The troubleshooting strategy is to run the reaction without protein expression template and assess background RNase activity using a fluorescent RNA reporter; if high, add RNase inhibitor (e.g., RNasin), switch to a DNA template with a strong ribosome binding site, or use a PURE system that is free of nucleases. Reason 3 — Suboptimal magnesium and potassium ion concentrations. Both Mg²⁺ and K⁺ profoundly affect ribosome assembly and activity, and their optimal concentrations depend on the extract lot, target protein, and energy system used. A single mM deviation in [Mg²⁺] can halve protein yield. The troubleshooting strategy is to perform a systematic two-dimensional titration of Mg²⁺ (range: 4–16 mM) and K⁺ (range: 60–200 mM) against protein yield measured by fluorescence (if GFP is used as a reporter) or SDS-PAGE densitometry, and re-optimise for each new extract batch or protein target.Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
The proposed synthetic minimal cell (SMC) functions as a field-deployable water quality sensor for antibiotic resistance. The input is the presence of beta-lactam antibiotic residues (specifically ampicillin) in environmental water samples, detected via a riboswitch aptamer domain that undergoes a conformational change upon ligand binding. The output is fluorescent GFP produced by the encapsulated CFPS system, reportable visually with a handheld fluorescence viewer such as the miniPCR P51 Molecular Fluorescence Viewer.
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No. Encapsulation is essential for two reasons: first, the lipid membrane creates a concentration gradient that amplifies the input signal — only molecules that enter or diffuse across the bilayer trigger the sensor, reducing false positives from trace non-specific binding. Second, the membrane physically separates the CFPS machinery from environmental nucleases and proteases present in raw water samples, which would otherwise degrade the RNA aptamer and mRNA templates. Without encapsulation, the reaction would be rapidly inactivated in complex environmental matrices.
c. Could this function be realized by genetically modified natural cell?
Yes, in principle: an E. coli strain engineered with an ampicillin-responsive transcription factor driving GFP expression could detect beta-lactams. However, release of live GMO bacteria into environmental water samples raises serious biosafety and ecological concerns, and the engineered organism may not survive or function predictably in the field. The SMC offers a fully abiotic, self-contained, containable alternative with no replication capacity.
d. Describe the desired outcome of your synthetic cell operation.
In the presence of ampicillin above a defined threshold concentration (~10 µM), the riboswitch aptamer within the SMC adopts its ligand-bound conformation, allowing ribosomal readthrough of an upstream inhibitory sequence and enabling translation of the GFP reporter. The operator observes green fluorescence from the SMC population when viewed under blue LED excitation — a simple positive/negative readout of water contamination.Design all components that would need to be part of your synthetic cell.
a. What would be the membrane made of?
POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) as the primary structural lipid, with 10 mol% POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol)) to introduce a slight anionic character that improves vesicle stability and reduces aggregation. No cholesterol is required for this room-temperature sensor application.
b. What would you encapsulate inside? Enzymes, small molecules.
A bacterial cell-free Tx/Tl system (E. coli S30 extract), the riboswitch-GFP DNA construct, an ATP regeneration module (creatine phosphate + creatine kinase), all 20 amino acids, NTPs, Mg²⁺, and K⁺ at optimised concentrations.
c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
Bacterial (E. coli S30 extract), because the riboswitch is derived from a prokaryotic aptazyme architecture and functions via modulation of ribosome access to the Shine-Dalgarno sequence — a mechanism specific to bacterial translation.
d. How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Ampicillin is a small, moderately amphipathic beta-lactam molecule (~349 Da) that can passively permeate phospholipid bilayers to a limited but measurable extent. At the ampicillin concentrations relevant for contamination detection (10–100 µM), passive permeation is sufficient to trigger the internal riboswitch without requiring an active transporter. GFP output remains internal and is detected non-destructively by fluorescence spectroscopy or imaging.Experimental details
a. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids: POPC (Avanti Polar Lipids #850457), POPG (Avanti Polar Lipids #840457)
Genes: Ampicillin-responsive riboswitch–GFP fusion: a synthetic construct encoding an engineered aptazyme responsive to beta-lactams (based on the aptazyme architecture of Wieland & Hartig, 2008) fused upstream of a GFP ORF under a T7 promoter
Specific gene-GFP variant: sfGFP (superfolder GFP; Addgene plasmid #54579), chosen for its robust folding kinetics in cell-free systems
b. How will you measure the function of your system?
Measure GFP fluorescence of the SMC suspension using a plate reader (excitation 488 nm, emission 510 nm) or a P51 handheld viewer for field deployment. A positive control containing a constitutively expressed GFP plasmid and a negative control of vesicles containing a scrambled riboswitch should bracket every experiment. Vesicle integrity is confirmed by dynamic light scattering (DLS) before and after the assay.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
A freeze-dried cell-free biosensor woven directly into a protective work garment that changes from orange to green fluorescence within 90 minutes of contact with airborne organophosphate pesticide residues, providing farm workers with a passive, wearable early-warning system for chemical exposure.How will the idea work, in more detail? Write 3-4 sentences or more.
The garment integrates freeze-dried cell-free (FDCF) synthetic biology circuits embedded in cellulose-based reaction insets woven into the chest panel of the fabric, using the methodology developed by Nguyen, Soenksen et al. The FDCF reaction encodes an organophosphate-responsive genetic circuit: acetylcholinesterase (AChE) activity is coupled to a split-reporter system such that AChE inhibition by organophosphates — detectable at concentrations as low as 10 nM — derepresses expression of a fluorescent aptamer. A polymeric optical fibre network interwoven with the fabric continuously probes each reaction zone for changes in fluorescence (orange baseline → green signal-positive), and the output is transmitted via Bluetooth to a paired smartphone application, alerting the wearer of exposure in real time. The reaction chambers are hermetically sealed and activated only by moisture — either sweat or rain — contacting the fibre insets, preventing premature activation during storage.What societal challenge or market need will this address?
Organophosphate pesticide exposure is the leading cause of acute agricultural poisoning worldwide, responsible for an estimated 385 million cases of unintentional acute pesticide poisoning per year (WHO, 2019). Farm workers in low-to-middle income countries frequently lack access to personal air quality monitors or laboratory testing infrastructure. A textile-embedded FDCF sensor worn as ordinary work clothing would provide continuous, real-time, instrument-free exposure monitoring, enabling workers to evacuate contaminated areas before symptoms manifest and generating timestamped exposure logs usable in occupational health assessments.How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
The three primary limitations — activation with water, stability in humid environments, and one-time use — are addressed as follows. Activation by water is an inherent design feature here rather than a drawback: the sensor is intentionally triggered by sweat contact, and the fabric’s hydrophobic outer layer acts as a moisture gate, ensuring activation only after meaningful liquid contact. Long-term stability is achieved by lyophilising the CFPS reactions in the presence of trehalose as a cryoprotectant and sealing individual reaction zones in a vapour-barrier polymer matrix; prior work has demonstrated FDCF stability at ambient temperature for at least six months under these conditions. The single-use constraint is addressed architecturally: reaction zones are modular insets that can be removed and replaced by the wearer after each work day, analogous to replacing a spent filter cartridge, while the fibre optic network and smartphone interface are reusable across many cycles.Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
1. Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Galactic cosmic radiation (GCR) and solar energetic particles present a significant health risk during deep-space missions, inducing DNA double-strand breaks (DSBs) and oxidative base damage in astronaut cells. Current biomonitoring of radiation-induced DNA damage aboard the ISS requires blood draws, cryopreservation, and Earth-based laboratory analysis — an impractical pipeline for future lunar or Mars missions where resupply is impossible. Developing a rapid, portable, crew-operable assay for real-time radiation exposure biomonitoring is critical to protect astronaut health and to inform mission planning and shielding design for exploration beyond low-Earth orbit.2. Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words) Target: p21 (CDKN1A) mRNA — a transcriptional target of the p53 DNA damage response pathway, reliably upregulated within hours of ionising radiation exposure in human cells.
3. Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
When ionising radiation causes DNA DSBs, the tumour suppressor p53 is activated and drives transcription of p21/CDKN1A, a cyclin-dependent kinase inhibitor that halts the cell cycle to allow DNA repair. Because p21 mRNA accumulates in cells proportionally to the absorbed radiation dose, it is a well-validated molecular dosimeter. Importantly, p21 mRNA can be extracted from crew saliva or buccal cells — a non-invasive sample type fully compatible with spaceflight constraints — and detected using the BioBits® toehold switch platform without the need for PCR equipment or cold-chain reagents.
4. Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words) Hypothesis: BioBits®-based toehold switch sensors designed to detect human p21 mRNA will produce a fluorescent readout proportional to radiation dose, as measured in buccal cell RNA extracts collected from astronauts aboard the ISS, and will perform comparably to Earth-based qRT-PCR reference measurements. Reasoning: Toehold switches — linear RNA hairpin structures that undergo conformational change upon hybridisation to a complementary trigger RNA — have been validated as highly sensitive, sequence-specific nucleic acid sensors in cell-free systems with detection limits as low as picomolar concentrations . Prior work by Kocalar et al. (2024) demonstrated that BioBits® performs robustly in microgravity. Because p21 mRNA is a human transcript expressed in cells easily obtainable by non-invasive buccal swab, the assay requires no genetic engineering of the crew, preserves biosafety, and is activatable by simple rehydration of the lyophilised BioBits® pellet with the extracted RNA sample.5. Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Samples: Buccal swabs collected from crew members at three time points — pre-mission baseline, 72 hours after a known solar energetic particle (SEP) event (using ISS radiation dosimetry logs as the reference), and at mission end. RNA is extracted using a portable lysis buffer compatible with the miniPCR kit. Experiment: p21 toehold switch BioBits® reactions are rehydrated with crew RNA extract and incubated for 60 minutes at 37 °C. Fluorescence is read using the P51 Molecular Fluorescence Viewer; image intensity is quantified via the paired smartphone app. Controls: Non-irradiated Earth buccal RNA (negative control); synthetic p21 mRNA spike-in (positive control); scrambled-sequence toehold switch (specificity control).Homework Part B: Individual Final Project
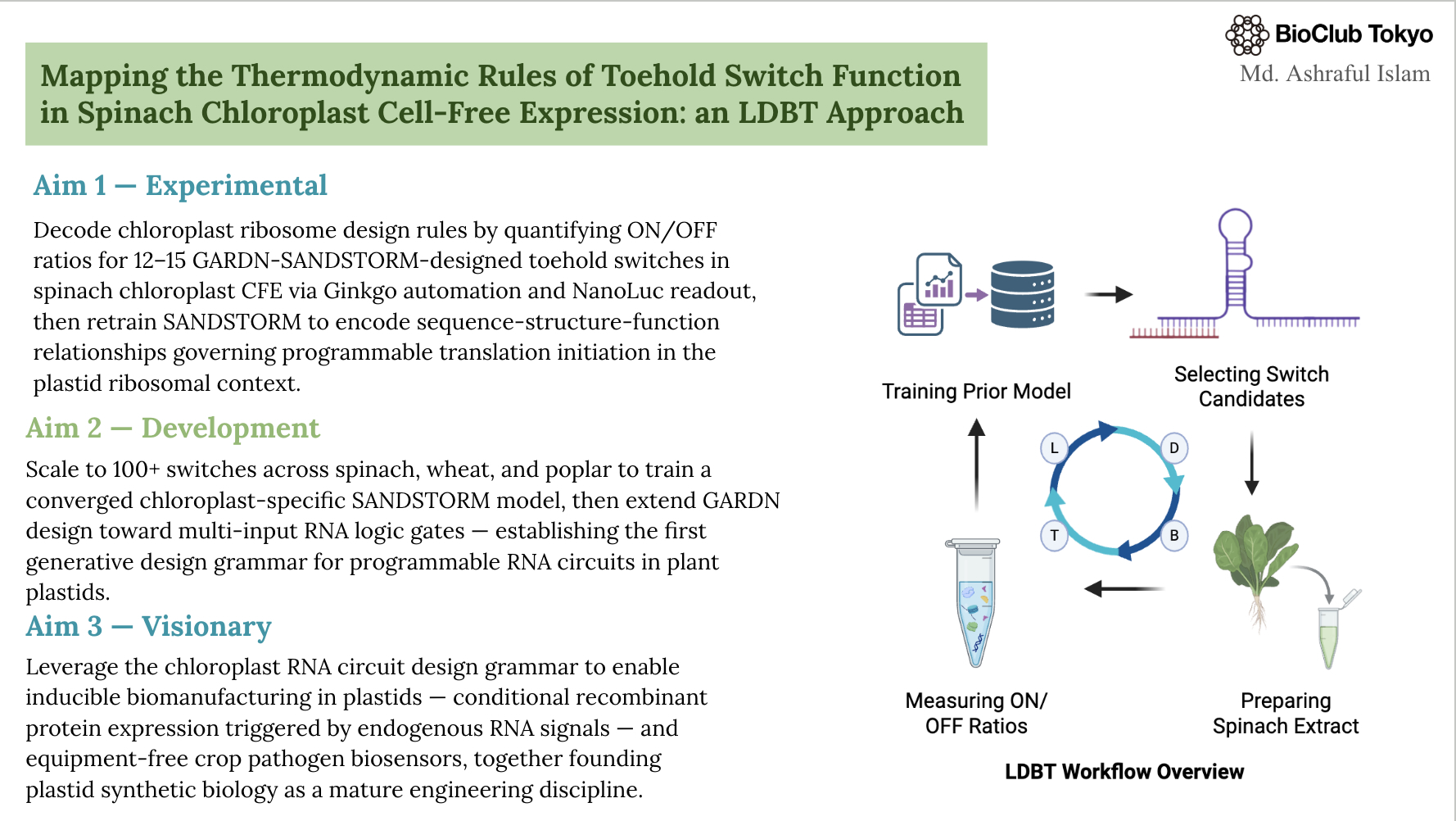
Week 10 HW: Advanced Imaging and Measurement Tech
Part 1: Molecular Weight
eGFP Sequence:
Based only on the predicted amino acid sequence of eGFP (see below), what is the calculated molecular weight? You can use an online calculator like the one here: https://web.expasy.org/compute_pi/
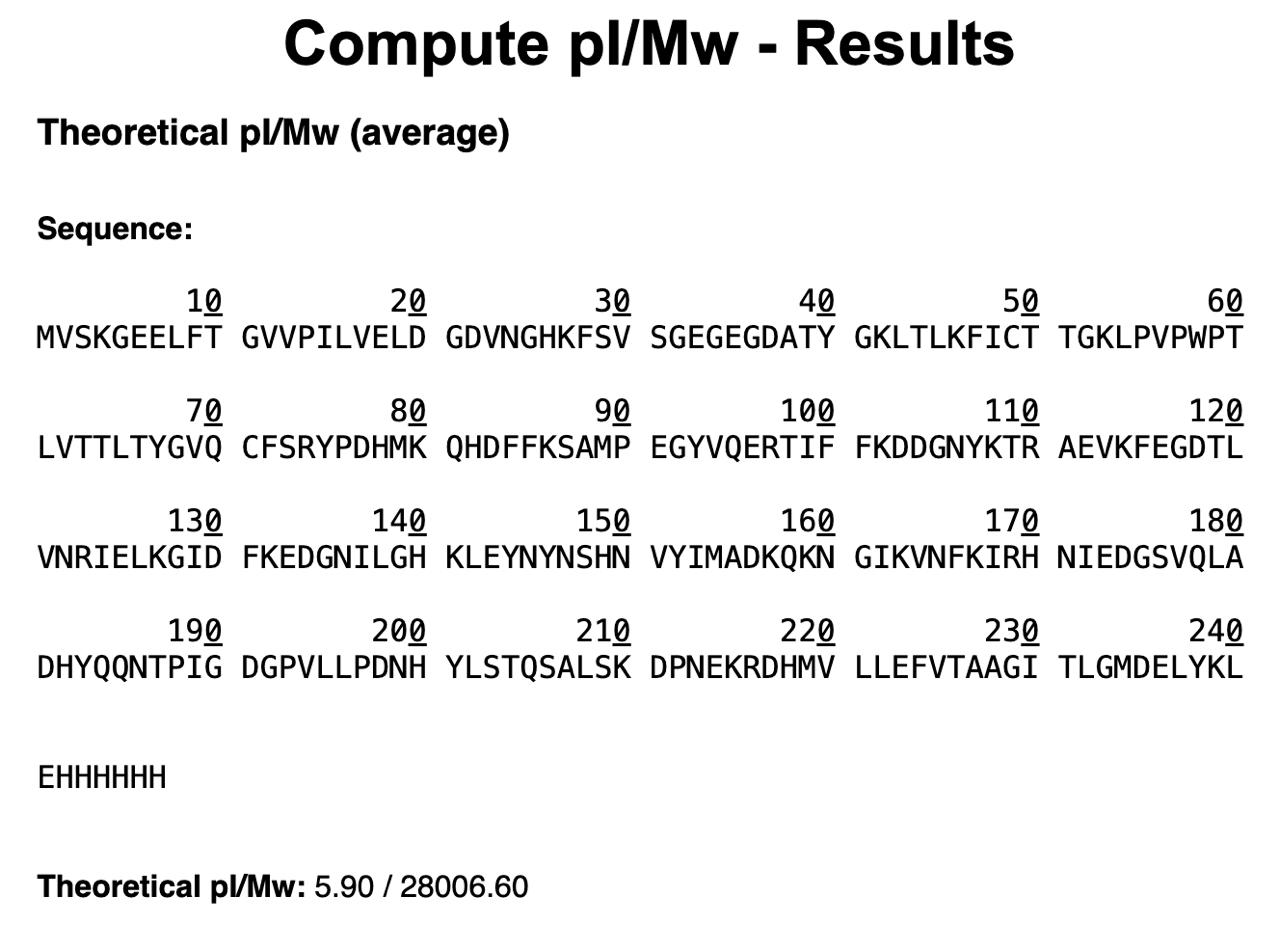
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the BioAccord data and:
 Figure 1. Mass Spectrum of intact eGFP protein from the Waters Xevo G3 LC-MS (a mass spectrometer with 30,000 resolution) with individual charge state peaks labeled with values.
Figure 1. Mass Spectrum of intact eGFP protein from the Waters Xevo G3 LC-MS (a mass spectrometer with 30,000 resolution) with individual charge state peaks labeled with values.
1. Determine $z$ for each $(n, n+1)$
- Peak 1: $(m/z)_n = 903.7148$
- Peak 2: $(m/z)_{n+1} = 875.4421$
Calculating $z$:
$$n = \frac{(m/z)_{n+1}}{(m/z)n - (m/z){n+1}}$$
$$n = \frac{875.4421}{903.7148 - 875.4421}$$
$$n = \frac{875.4421}{28.2727} = 30.963$$
- $n = 31$
- $n + 1 = 32$
2. Determine the MW of the protein
We use the relationship between $m/z$, molecular weight ($M$), and charge ($z$):
$$M = z \times (m/z - H)$$
Peak 1 ($z = 31$): $$M = 31 \times (903.7148 - 1.007)$$ $$M = 31 \times 902.7078 = 27983.9418 \text{ Da}$$
Peak 2 ($z = 32$): $$M = 32 \times (875.4421 - 1.007)$$ $$M = 32 \times 874.4351 = 27981.9232 \text{ Da}$$
Average Molecular Weight ($M_{\text{avg}}$):
$$M_{\text{avg}} = \frac{27983.9418 + 27981.9232}{2} = 27982.9325 \text{ Da}$$
3. Calculate the Mass Accuracy
We use the deconvoluted average weight ($M_{\text{estimated}}$) and the predicted theoretical weight ($M_{\text{theoretical}}$).
- $M_{\text{theoretical}} = 27875.41 \text{ Da}$
- $M_{\text{estimated}} = 27982.9325 \text{ Da}$
Absolute Error: $$\text{Absolute Error} = 27982.9325 - 27875.41 = 107.5225 \text{ Da}$$
Relative Accuracy: $$\text{Accuracy} = \frac{|M_{\text{experiment}} - M_{\text{theoretical}}|}{M_{\text{theoretical}}}$$
$$\text{Accuracy} = \frac{107.5225}{27875.41} = 0.003858 \text{ (or } 0.386% \text{)}$$
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
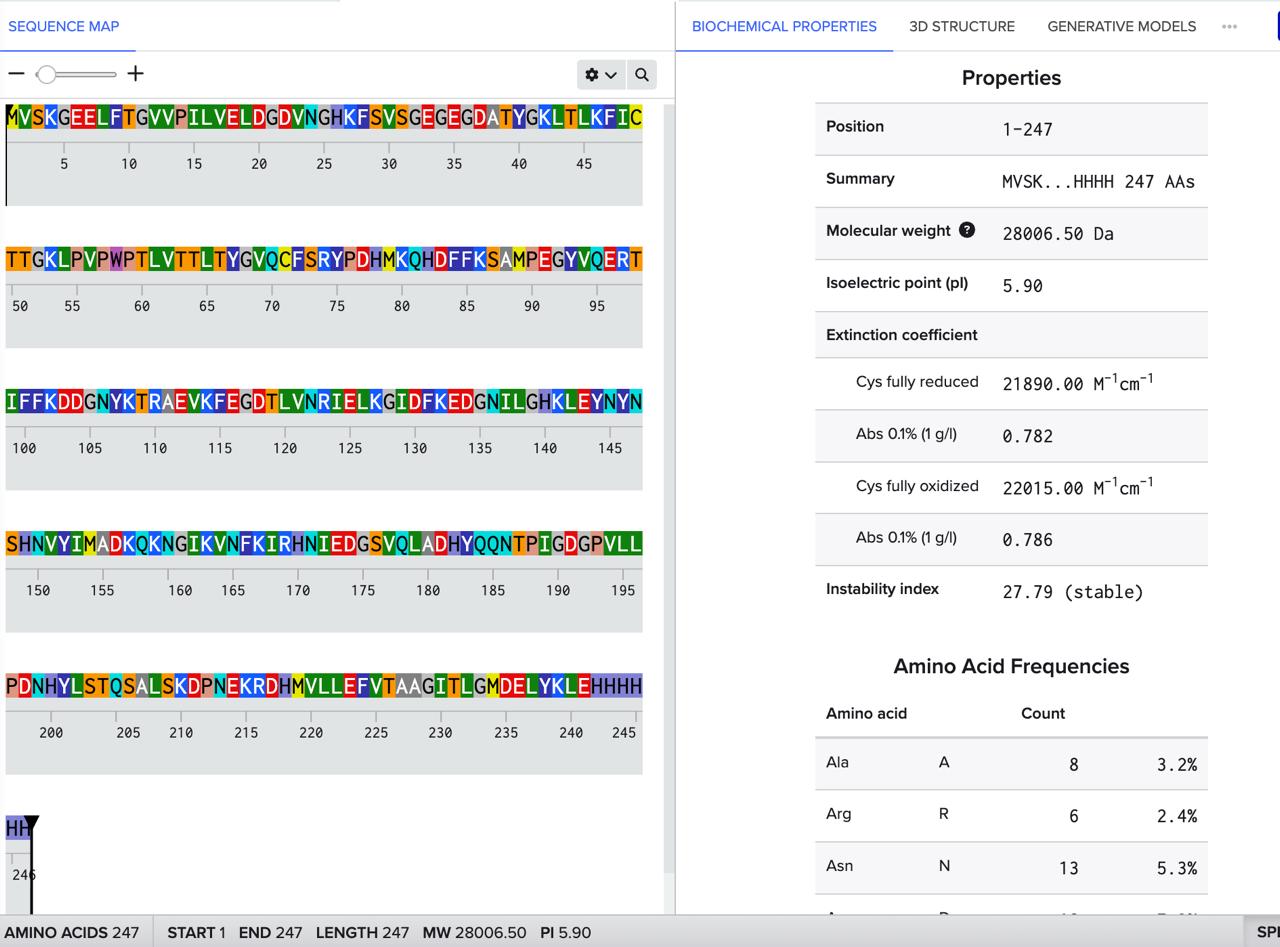
- There are 20 Lysines (K) which constitute 8.1% of the eGFP.
- There are 6 Arginines (R) which constitute 2.4% of the eGFP.
How many peptides will be generated from tryptic digestion of eGFP? a. Navigate to https://web.expasy.org/peptide_mass/ b. Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides. c. Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
d. Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.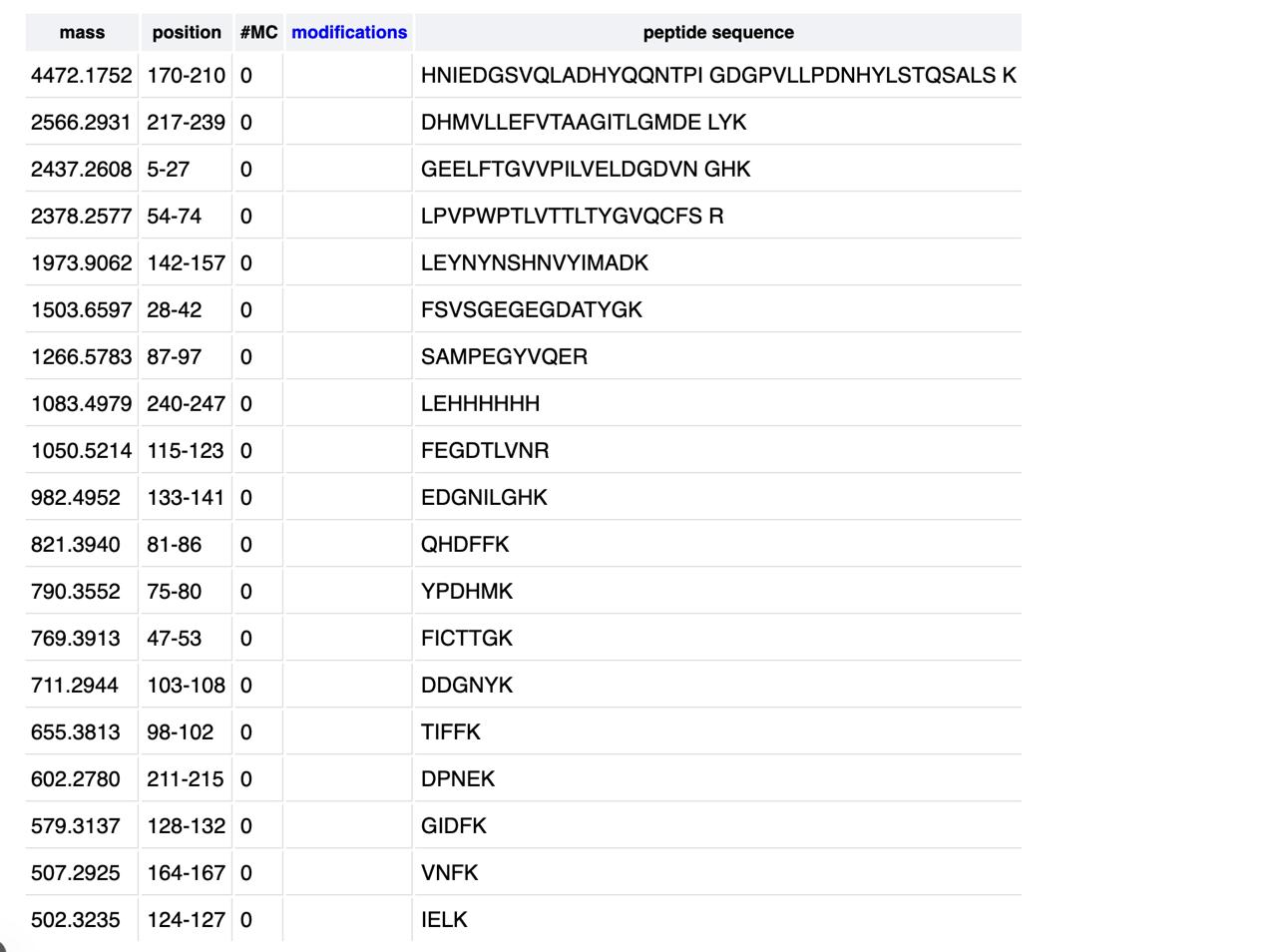
Homework: Waters Part IV — Oligomers
Calculate Expected Masses for Each Species
Using the polypeptide subunit masses from Table 1:
- 7FU = 340 kDa
- 8FU = 400 kDa and the definition that: - Decamer = 10 subunits
the expected masses are:
Oligomeric Species Subunit Mass # Subunits Calculated Mass (polypeptide only) 7FU Decamer 340 kDa 10 3,400 kDa = 3.4 MDa 8FU Didecamer 400 kDa 20 8,000 kDa = 8.0 MDa 8FU 3-Decamer 400 kDa 30 12,000 kDa = 12.0 MDa 8FU 4-Decamer 400 kDa 40 16,000 kDa = 16.0 MDa 
Homework: Waters Part V — Did I make GFP?
N/A
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I missed the opportunity to contribute to the HTGAA CFPS bioart project. Later, I contributed to the SynBioBeta bioart project. I worked on part of the DNA on the center-left plate.

What I liked: This kind of community-coordinated experiment builds genuine shared investment in the outcome, which is a rare and valuable pedagogical achievement.
What could be improved: For future years, giving participants a low-resolution preview of the emerging canvas in near-real-time — without revealing the final image — would heighten the sense of collective emergence and encourage more strategic pixel placement.Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
1. Referencing the cell-free protein synthesis reaction composition E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): The crude cell lysate supplies the entire transcription-translation (TX-TL) machinery — ribosomes, tRNA synthetases, elongation and initiation factors, chaperones, and co-factors — needed to convert DNA template into protein in vitro; the BL21 DE3 strain is specifically engineered to co-express T7 RNA Polymerase (from the chromosomal DE3 insertion), which is essential for driving transcription from the T7φ10 promoter used on most CFPS expression plasmids.
Salts/Buffer
Potassium Glutamate: The primary intracellular-mimicking monovalent cation that stabilises ribosome conformation and partially replaces KCl to avoid chloride-induced inhibition; potassium ions are critical for maintaining ribosome association and translational fidelity.
HEPES-KOH pH 7.5: A zwitterionic biological buffer that maintains reaction pH close to physiological values throughout the synthesis reaction, preventing the accumulation of protons that would otherwise inactivate ribosomes and enzymes as phosphate is metabolised.
Magnesium Glutamate: Provides free Mg²⁺ ions, which are indispensable cofactors for ribosome assembly, RNA polymerase activity, and the enzymatic reactions of the energy regeneration system; Mg²⁺ concentration is one of the most sensitive optimisation parameters in CFPS and must be carefully titrated for each lysate batch.
Potassium Phosphate Monobasic / Dibasic: Phosphate ions participate in energy metabolism and buffer secondary pH fluctuations; they also serve as inorganic phosphate donors and are required for metabolic pathways within the lysate.
Energy / Nucleotide System
Ribose: A five-carbon sugar that serves as the backbone of nucleoside synthesis; in the NMP-Ribose-Glucose system, ribose provides the sugar moiety for regenerating nucleoside monophosphates through the pentose phosphate pathway and purine/pyrimidine salvage enzymes present in the lysate.
Glucose: The primary carbon and energy source for ATP regeneration via glycolysis; endogenous glycolytic enzymes in the lysate convert glucose to pyruvate, generating ATP and NADH needed to sustain transcription and translation over extended reaction times.
AMP, CMP, GMP, UMP: Nucleoside monophosphates (NMPs) that serve as precursors for NTP synthesis; rather than supplementing expensive pre-formed NTPs, the NMP-based system relies on endogenous nucleoside monophosphate kinases (NMPK) and pyruvate kinase within the lysate to phosphorylate these precursors to the triphosphate form needed for transcription.
Guanine: A free purine base that feeds the guanosine salvage pathway; nucleoside phosphorylase enzymes in the lysate convert guanine + ribose-1-phosphate to guanosine, which is then phosphorylated to GMP and ultimately GTP — providing an additional low-cost input for GTP pools without supplying GMP directly.
Translation Mix (Amino Acids)
17 Amino Acid Mix: Provides the bulk of the 20 canonical amino acids needed for polypeptide elongation; splitting the amino acid supply allows independent supplementation of the three most problematic residues that tend to degrade, oxidise, or precipitate under standard CFPS conditions.
Tyrosine: Supplied separately because tyrosine has very low aqueous solubility at neutral pH and precipitates out of a premixed solution; it is added as a separately prepared suspension or at low concentration to ensure it remains bioavailable during the reaction.
Cysteine: Added separately because cysteine is prone to oxidative degradation — it dimerises to cystine under aerobic conditions — so it is freshly prepared and added just before the reaction to maintain a sufficient pool for proteins requiring cysteine residues.
Additives
Nicotinamide: A precursor to NAD⁺, which is an essential redox cofactor for glycolysis (particularly the GAPDH reaction); supplementing nicotinamide ensures the lysate can regenerate NAD⁺ from NADH during glucose catabolism, sustaining the energy regeneration capacity of the reaction over long incubations.
Backfill
Nuclease Free Water: Added to bring the reaction to the desired final volume without introducing RNases or DNases that would degrade the RNA polymerase transcript or the DNA template; using nuclease-free water is essential to protect the mRNA intermediates produced during transcription.
2. Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix The 1-hour PEP–NTP system uses phosphoenolpyruvate (PEP) as a high-energy phosphate donor to rapidly regenerate ATP via pyruvate kinase and relies on pre-formed NTPs, producing a short, intense burst of energy that supports fast, high-yield protein synthesis before quickly terminating as PEP is depleted. In contrast, the 20-hour NMP–Ribose–Glucose system uses glucose-driven metabolism for continuous ATP regeneration and enzymatic phosphorylation of NMPs into NTPs, enabling slower but long-duration protein expression at lower cost and is preferred for sustained experiments.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1. Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems.
i. sfGFP (superfolder GFP)
sfGFP is engineered for very fast, robust β‑barrel folding, giving near-complete folding efficiency even when fused to aggregation-prone partners and making it a reliable baseline reporter for CFPS optimization. Its chromophore maturation requires molecular oxygen, so oxygen availability will affect fluorescence in cell-free reactions.ii. mRFP1
mRFP1 has relatively slow chromophore maturation, meaning early-time fluorescence can substantially underreport total protein synthesis if fluorescence alone is used as a proxy. Its chromophore formation depends on molecular oxygen.iii. mKO2
mKO2 is a fast-folding orange FP but retains acid sensitivity, so its fluorescence can be quenched if the cell-free reaction drifts acidic during prolonged incubations. This pH vulnerability should be monitored in long or metabolically active CFPS systems.iv. mTurquoise2
mTurquoise2 is an intrinsically bright cyan FP with very high quantum yield, but native cysteines can form disulfide-linked oligomers in oxidising conditions, reducing the fraction of fluorescent protein. In cell-free systems, including reducing agents may be necessary, and its slower maturation versus sfGFP affects early readouts.v. mScarlet-I
mScarlet-I is a bright, monomeric red FP with fast maturation and good photostability, making it a robust red reporter for long cell-free incubations and oxidising conditions. Its high intrinsic brightness supports sensitive fluorometric detection.vi. Electra2
Electra2 is a relatively bright blue FP but blue FPs typically produce lower absolute signal than green or red FPs under equivalent expression, so overall fluorescence will be lower. It has reported tendencies to aggregate in some contexts, which can reduce soluble, fluorescent yield in CFPS.2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Protein targeted: mKO2
Property identified: Acid sensitivity — mKO2 fluorescence is quenched at lower pH, and the NMP-Ribose-Glucose system can accumulate acidic byproducts over a 36‑hour incubation that may shift reaction pH downward.
Hypothesis: Increasing buffer capacity (HEPES-KOH from ~50 mM to ~100–150 mM and/or adding elevated potassium phosphate dibasic) will maintain pH ~7.2–7.8 and preserve mKO2 chromophore protonation; alternatively, reducing glucose to ~50% could slow acid production at the cost of some ATP regeneration.
Expected effect: Higher buffer capacity and/or reduced glucose should yield increased mKO2 fluorescence at 20–36 h versus standard mix, with a possible reduction in early (0–6 h) peak expression; test by plotting fluorescence kinetics across buffer and glucose gradients.