Mapping the Thermodynamic Rules of Toehold Switch Function in Spinach Chloroplast Cell-Free Expression: an LDBT Approach Framework: Learn → Design → Build → Test (Clark-ElSayed et al., 2025)
Abstract Chloroplast cell-free expression (CFE) systems have recently been established as powerful rapid-prototyping platforms for plastid genetic parts, yet whether these systems can support synthetic RNA logic remains entirely untested. Toehold switches — de novo-designed riboregulators that activate translation in response to specific trigger RNAs — represent the most sophisticated programmable RNA gates in synthetic biology. Machine learning models trained on E. coli CFE data have begun to extract sequence-structure features predictive of switch performance using frameworks like SANDSTORM (Riley et al., 2025), but whether those learned relationships hold in a chloroplast ribosome context is unknown. This project addresses that gap directly by applying the Learn-Design-Build-Test (LDBT) framework to map the thermodynamic rules governing toehold switch function in spinach chloroplast CFE.
Phage Therapy Background: The Antibiotic Resistance Crisis and Phage Therapy Antibiotic resistance is one of the most urgent threats to global health. At current trends, antimicrobial-resistant infections are projected to cause deaths comparable in scale to cancer within the next 26 years (O’Neill Report, 2016). The overuse and misuse of broad-spectrum antibiotics has accelerated the selection of resistant bacterial strains, while the pipeline for novel antibiotics has nearly run dry. A compelling alternative is phage therapy — the therapeutic use of bacteriophages (phages) to target and kill pathogenic bacteria.
Subsections of Projects
Individual Final Project
Mapping the Thermodynamic Rules of Toehold Switch Function in Spinach Chloroplast Cell-Free Expression: an LDBT Approach
Framework: Learn → Design → Build → Test (Clark-ElSayed et al., 2025)
Abstract
Chloroplast cell-free expression (CFE) systems have recently been established as powerful rapid-prototyping platforms for plastid genetic parts, yet whether these systems can support synthetic RNA logic remains entirely untested. Toehold switches — de novo-designed riboregulators that activate translation in response to specific trigger RNAs — represent the most sophisticated programmable RNA gates in synthetic biology. Machine learning models trained on E. coli CFE data have begun to extract sequence-structure features predictive of switch performance using frameworks like SANDSTORM (Riley et al., 2025), but whether those learned relationships hold in a chloroplast ribosome context is unknown. This project addresses that gap directly by applying the Learn-Design-Build-Test (LDBT) framework to map the thermodynamic rules governing toehold switch function in spinach chloroplast CFE.
We train a SANDSTORM predictive neural network — a dual-input CNN incorporating one-hot-encoded RNA sequence and secondary structure arrays (Riley et al., 2025) — on the publicly available 181-switch E. coli dataset to learn sequence-structure-function relationships for toehold switches. The trained SANDSTORM model is then paired with GARDN (Generative Adversarial RNA Design Network) to generate 12–15 novel toehold switch candidates with predicted high ON/OFF performance in a chloroplast ribosome context, including PVY coat protein mRNA-triggered designs. Whole plasmid constructs are ordered from Twist Bioscience and tested in both spinach chloroplast CFE and crude E. coli S30 extract; a secondary SANDSTORM model retrained on the resulting chloroplast data constitutes the first sequence-structure-function ML model for toehold switches in a plant-native ribosomal context.
The project produces the first empirical dataset and neural network model for toehold switch performance in plant chloroplast CFE, a transferable GARDN-SANDSTORM LDBT workflow applicable to any novel ribosome context, and a foundation for programmable RNA diagnostics manufacturable directly from plant material. All experiments are performed using the Ginkgo Bioworks autonomous laboratory infrastructure and open-access grocery-store spinach, demonstrating that LDBT with deep learning is executable at global-access scale with a materials budget under $1,200.
Project Aims
Aim 1 — Experimental Aim (This Project)
Decode the sequence-structure rules governing translation initiation in the chloroplast ribosomal context by quantifying ON/OFF ratios for 12–15 GARDN-SANDSTORM-designed toehold switch candidates in spinach chloroplast CFE via Ginkgo Bioworks automation and NanoLuc readout, then retraining SANDSTORM on the resulting dataset to encode the sequence-structure-function relationships that define programmable RNA regulation in the plastid ribosomal context.
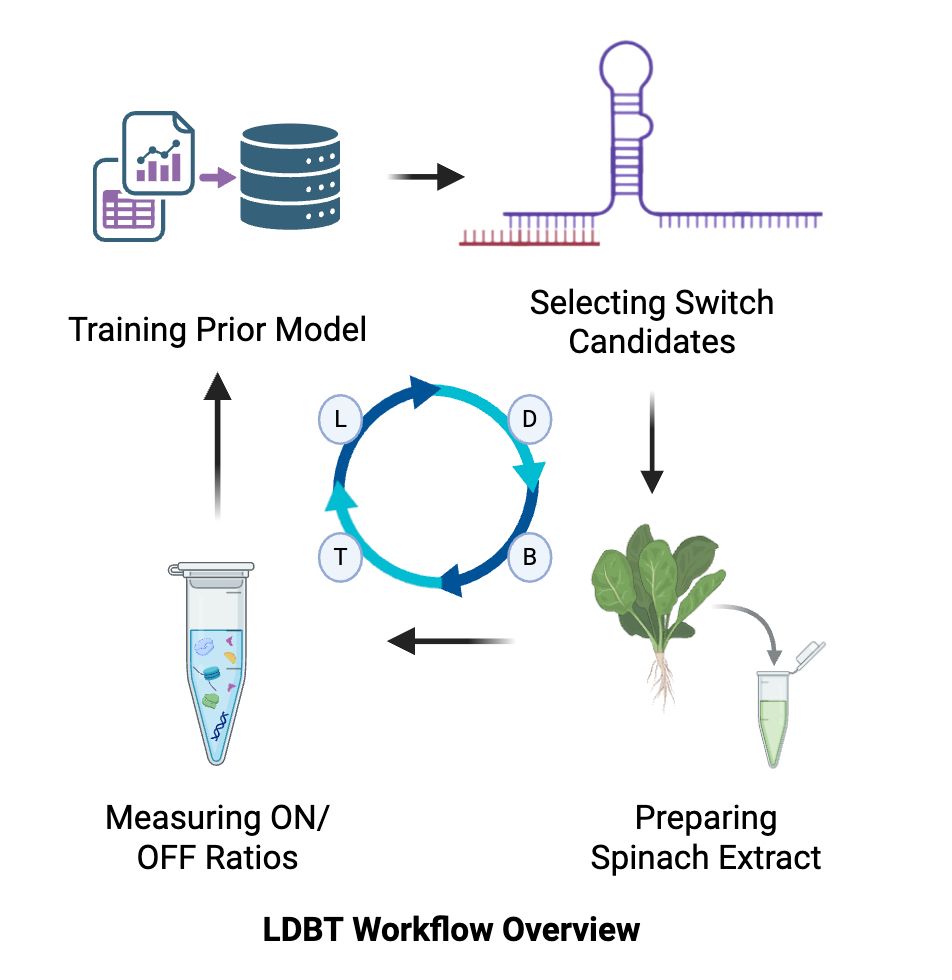
The LDBT workflow proceeds as follows:
(L1) Train a SANDSTORM predictive model on the Green et al./To et al. 181-switch E. coli dataset, using paired one-hot sequence and secondary structure array inputs to learn sequence-structure-function relationships.
(D) Pair the trained SANDSTORM predictor with GARDN to generatively design 12–15 novel toehold switch candidates with predicted high ON/OFF performance, including PVY coat protein mRNA-triggered switches and controls.
(B) Prepare spinach chloroplast extract from commercial spinach and set up 384-well CFE reactions via Ginkgo Bioworks automation.
(T) Measure NanoLuc luminescence at the empirically validated kinetic endpoint.
(L2) Retrain a SANDSTORM model on the new chloroplast CFE ON/OFF dataset and compare learned sequence-structure features between E. coli and chloroplast contexts.
The retrained L2 model and its integrated gradients attribution maps — not solely the ON/OFF numbers — constitute the primary scientific deliverable.
Aim 2 — Development Aim
Scale to 100+ toehold switch constructs across spinach, wheat, and poplar to train a converged, chloroplast-specific SANDSTORM model above its 384-sequence reliability threshold (Riley et al., 2025), then extend GARDN generative design toward multi-input RNA logic gates — AND gates, cascades, and riboregulator networks — establishing the first generative design grammar for programmable RNA circuits in plant plastids. Concurrent optimization of the automated CFE workflow at Ginkgo Bioworks will enable 384-well throughput across species, and the resulting pan-plant SANDSTORM model, GARDN design weights, and logic gate characterization data will be deposited to Zenodo as a community resource, marking a transition from individual switch quantification to a systematic engineering discipline for chloroplast RNA circuits.
Aim 3 — Visionary Aim
Leverage the chloroplast RNA circuit design grammar to open two parallel application frontiers: inducible biomanufacturing in plastids — conditional recombinant protein expression triggered by endogenous RNA signals, bypassing the constitutive expression ceiling of current plastid systems — and equipment-free crop pathogen biosensors using lyophilized chloroplast CFE reactions that detect Potato Virus Y, cassava mosaic virus, and wheat blast RNA signatures in the field without cold chain or laboratory infrastructure. Together, these applications reframe plant material not as passive agricultural output but as a substrate for programmable molecular manufacturing, and position the GARDN-SANDSTORM chloroplast platform as the foundation for plastid synthetic biology as a mature, generalizable engineering discipline.
Background
Literature Context
Background Study: Related Literature
Bohm et al.
Clark et al.
LDBT
Alexander A. Green et al.
Aidan T. Riley et al.
Green et al. (2014) established toehold switches as programmable RNA regulators achieving >400-fold ON/OFF dynamic range in E. coli, with sequence-programmable targeting to any trigger RNA (Cell, 167, 246–259). Their 168-switch dataset defines the architectural constraints reproduced in all Twist Bioscience constructs here and constitutes the primary SANDSTORM training set.
Riley et al. (2025) introduced SANDSTORM and GARDN as a paired predictive-generative framework: SANDSTORM encodes RNA as dual one-hot sequence and secondary structure inputs, achieves accurate predictions from as few as 384 training examples, and outperforms classical thermodynamic algorithms; GARDN generates novel candidates with targeted experimental attributes (Nature Communications). The current project trains SANDSTORM on Green et al.’s E. coli dataset, uses GARDN to generate 12–15 chloroplast-targeted toehold switch candidates, and retrains SANDSTORM on the resulting chloroplast CFE data.
Clark, Voigt & Jewett (2024) established the first high-yield tobacco chloroplast CFE system, achieving 60 ± 4 μg/mL NanoLuc yields and a 1,300-fold expression dynamic range across 103 RBS variants screened in under one day (ACS Synthetic Biology). Böhm, Inckemann et al. (2024) extended this platform to spinach, wheat, and poplar using nanolitre-scale automation, demonstrating a >4-log dynamic range and R² = 0.93 cross-species expression correlation between spinach and wheat — motivating spinach as the training organism and wheat as the generalisation target (ACS Synthetic Biology). Clark-ElSayed et al. (2025) formalised the LDBT paradigm, positioning ML at the start of the engineering cycle and CFE as the high-throughput data-generation engine (Nature Communications); this project instantiates that paradigm exactly.
Knowledge gap: No study has measured toehold switch ON/OFF ratios in plant chloroplast CFE, applied SANDSTORM or GARDN to a chloroplast ribosome context, or retrained either model on chloroplast expression data. The chloroplast ribosome is evolutionarily prokaryotic but operates at 20–25°C with a distinct anti-Shine-Dalgarno sequence and ionic environment, making transfer of E. coli-learned sequence-structure-function relationships non-obvious and experimentally unvalidated.
Innovation
This project is novel in three respects that together constitute an unstudied intersection of existing capabilities:
It is the first experimental test of toehold switch functionality in any plant organellar cell-free system, connecting two independently mature fields — chloroplast CFE (Clark et al., Böhm et al.) and toehold switch design (Green et al., Riley et al.) — that have never been brought into contact.
It is the first application of GARDN-SANDSTORM — a paired generative adversarial and predictive CNN framework incorporating both RNA sequence and secondary structure — to toehold switch design for a non-E. coli ribosome context; SANDSTORM was validated on E. coli toehold data by Riley et al. (2025) but has never been applied across ribosomal contexts or used to guide design for a plant organellar translation system.
It demonstrates the LDBT paradigm at genuinely minimal resource scale — grocery-store spinach, publicly available datasets, open-source GARDN-SANDSTORM code, and a ~$1,123 materials budget — challenging the assumption that deep-learning-guided LDBT requires biofoundry infrastructure and proposing a globally accessible implementation.
Significance
Chloroplast cell-free expression is emerging as a transformative rapid-prototyping platform for plant synthetic biology, yet it currently lacks the programmable regulatory logic — inducible switches, conditional circuits, RNA-responsive gates — that makes E. coli CFE a mature engineering substrate. This project addresses that foundational gap: by establishing the first sequence-structure-function dataset and neural network model for toehold switch performance in a plant organellar ribosome context, it provides the missing design layer that allows chloroplast CFE to move from constitutive expression screening toward programmable, input-responsive genetic circuits.
The GARDN-SANDSTORM LDBT workflow is not a single-use tool but a reusable design engine — analogous to the role the Salis RBS Calculator played in standardising translational control in E. coli — and the Zenodo-deposited dataset and model weights constitute community infrastructure any laboratory can build upon. Each experimental round expands the L2 model, progressively improving predictive accuracy and enabling generative design of increasingly complex RNA circuits without proportional increases in experimental cost.
At the application frontier, this infrastructure enables inducible biomanufacturing in plastids (Aim 2) and field-deployable lyophilised biosensors for crop pathogens including Potato Virus Y — which causes billions of dollars in annual losses globally — requiring no cold chain, no laboratory infrastructure, and no purified proteins (Aim 3). Demonstrating the full LDBT cycle at student-project scale with grocery-store spinach and a sub-$1,200 budget establishes a precedent for globally accessible synthetic biology development that does not require centralised biofoundry resources.
Bioethical Considerations
Ethical Implications: This project operates at the intersection of biosensing technology and agricultural systems, which raises considerations spanning the principles of beneficence, justice, and responsibility. The beneficence argument is strong: a low-cost, field-deployable diagnostic for crop pathogens could protect food security for smallholder farmers who currently have no access to pathogen surveillance. However, the justice dimension requires scrutiny — specifically, who controls the technology if it is developed from an open-source student project, who benefits from its commercialization, and whether the communities most affected by PVY losses have meaningful voice in how the tool is designed and deployed. The dual-use dimension is minimal but non-trivially zero: a toehold switch system designed to detect a plant pathogen RNA could, in principle, be reprogrammed to detect other RNA sequences; the framework is sequence-agnostic. The principle of non-maleficence requires acknowledging that a field biosensor producing false-negatives could provide false assurance, while false-positives could trigger unnecessary crop destruction. The current project makes no diagnostic claims — it tests the enabling molecular component — but responsible downstream development must address sensitivity and specificity thresholds before any field deployment claim.
Responsible Implementation: The measures taken in this project to ensure ethical research practice include:
BSL-1 equivalent work throughout — no live organisms are engineered, no pathogen material is used, PVY trigger sequences are short synthetic oligonucleotides with zero infectivity risk.
Full open data deposition — all ON/OFF ratio data, raw luminescence values, and trained model coefficients will be deposited to Zenodo with a DOI minted before presentation, ensuring the dataset is a public resource rather than a proprietary asset.
Sequence screening through SecureDNA before ordering from Twist Bioscience, ensuring no inadvertent synthesis of sequences with biosecurity implications.
Explicit acknowledgment that this project does not constitute a validated diagnostic and should not be interpreted as such.
Potential unintended consequences of the broader vision — field-deployed toehold switch biosensors — include misuse for detecting human pathogens without regulatory oversight, and the displacement of existing laboratory diagnostic workers if the technology is deployed without appropriate workforce transition planning. Alternatives to the toehold switch approach, including CRISPR-Cas13-based diagnostics (SHERLOCK) and lateral flow immunoassays, should be evaluated comparatively in any future regulatory submission, and the assumptions of sequence-structure-function transferability across ribosome contexts validated in additional plant species before field deployment claims are made.
Experimental Design
Timeline Overview: Dry-lab phases (L1, D) run in parallel with extract preparation (B); total wet lab execution is 5 weeks at Ginkgo Bioworks node.
PHASE L1 — Learn from Existing E. coli Data (Dry Lab, Week 1 | ~2 days)
Prepare dual-channel SANDSTORM input tensors from the 181-switch E. coli dataset
Method
Download the 181-switch ON/OFF dataset (Green et al. 2014 + 2017, as compiled by To et al. 2018). For each sequence, compute the N×N structural array: position (i,j) = 0 if no canonical pairing possible; 2 if A-U or G-U wobble; 3 if G-C. Values encode hydrogen bond capacity from nucleotide identity alone — no folding software, no temperature assumption. Pair with one-hot-encoded sequences as dual-channel SANDSTORM input. Load pretrained SANDSTORM weights from the Angenent-Mari et al. (2020) toehold dataset via the GARDN-SANDSTORM repository for transfer learning initialisation.
Automation
Dry lab (Python/Jupyter); no Ginkgo machine required
Fine-tune SANDSTORM on the 181-switch E. coli dataset to learn sequence-structure-function relationships
Method
Fine-tune pretrained SANDSTORM (two parallel CNN stacks: one for one-hot sequence with batch normalisation between layers 1–2; one for structural array with spatial dropout 0.2 and GlobalMaxPooling2D; concatenated outputs through three dense layers; ReLU activations; Adam optimiser). 80/20 training-testing split stratified by ON/OFF ratio, averaged across three randomised splits. Report Spearman r, R², MSE. Benchmark against NuSpeak/STORM. Rank all 181 switches by predicted ON/OFF.
Automation
Dry lab
Plate
N/A
Expected result
Spearman r ≥ 0.4 after fine-tuning; ranked switch list available for GARDN design guidance
Timeline
Day 2
PHASE D — Design: Candidate Selection and Twist Order (Week 1–2 | ~3 days)
Generate 12–15 novel toehold switch sequences optimised for high predicted ON/OFF performance
Method
GARDN generator (WGAN-GP; upsampling layers (2,5),(2,6),(1,2); spectrally normalised conv layers; batch normalisation) produces 60-nt switch sequences from latent variable Z. Programmable reverse-complementation layer enforces canonical stem-loop grammar by construction. Frozen SANDSTORM L1 predictor guides optimisation by gradient ascent: Z → Z + α∇_Z P_δ(G_θ(Z)), 300 steps, structural array recomputed from sequence at each step (O(N²), no folding calls). Generate 300 candidates; select 9–12 highest predicted ON/OFF. For ≥ 3 PVY-triggered designs, specify trigger from DQ157180 coat protein ORF (nt 8,950–9,200) to constrain the reverse-complementation layer. Verify trigger self-folding: NUPACK ΔG_MFE > −3 kcal/mol at 25°C. Include one scrambled-trigger negative control and one unstructured RBS positive control.
Automation
Dry lab; NUPACK for trigger verification only
Plate
N/A
Expected result
12–15 GARDN-SANDSTORM-designed sequences; structural agreement score ~0.92; ≥ 3 PVY-targeted; optimisation runtime ~11 s per 300 calls
Timeline
Days 2–4
Step 4 — Whole Plasmid Design and Twist Bioscience Order (Days 3–5)
Field
Detail
Purpose
Order all 12–15 constructs as sequence-verified whole plasmids from Twist Bioscience
Method
Design each construct as a complete circular plasmid: T7 promoter → toehold switch module (12-nt toehold + 18-nt stem + 11-nt loop + 18-nt stem complement) → Shine-Dalgarno RBS linker → ATG → NanoLuc ORF (513 bp) → T7 terminator → pUC19 backbone (AmpR, pMB1 ori). Total plasmid size ~3,250 bp. Prepare GenBank files in Benchling. Screen all sequences through SecureDNA before submission. Submit to Twist Bioscience Clonal Gene service (pUC19 backbone).
12–15 whole plasmid constructs delivered lyophilised within 7–10 business days
Timeline
Days 3–5
Representative GenBank construct — TS-PVY-01:
The following GenBank file encodes the first PVY-targeted toehold switch, ordered as a whole plasmid from Twist Bioscience. Trigger sequence is derived from PVY coat protein mRNA (DQ157180, ~nt 8,960–8,989). Paste directly into the Twist upload portal and select the pUC19 clonal backbone.
LOCUS TS_PVY_01 3248 bp DNA circular SYN 11-APR-2026
DEFINITION Toehold switch TS-PVY-01 for NanoLuc expression in chloroplast
and E. coli cell-free expression; PVY coat protein mRNA trigger
(DQ157180 nt 8960-8989); Green et al. 2014 architecture.
Ordered as whole plasmid from Twist Bioscience (pUC19 backbone).
ACCESSION .
VERSION .
KEYWORDS toehold switch; cell-free expression; NanoLuc; PVY biosensor;
chloroplast; synthetic construct.
SOURCE synthetic construct
ORGANISM synthetic construct
other sequences; artificial sequences; synthetic constructs.
FEATURES Location/Qualifiers
promoter 1..17
/label="T7 promoter"
/note="T7 bacteriophage promoter; TAATACGACTCACTATA"
/ApEinfo_fwdcolor="#31849b"
misc_RNA 18..77
/label="toehold_switch_module"
/note="Green et al. 2014 first-gen architecture;
12-nt toehold + 18-nt stem + 11-nt loop + 18-nt
stem complement"
misc_feature 18..29
/label="toehold_domain_12nt"
/note="Single-stranded; trigger-accessible;
complementary to PVY coat protein mRNA nt 8960-8971"
stem_loop 30..77
/label="stem_loop_18_11_18"
/note="Occludes RBS and AUG start codon in OFF state;
displaced by trigger binding"
misc_feature 78..99
/label="RBS_linker"
/note="AACAGAAACAGAGGAGAAAUA;
Shine-Dalgarno exposed upon trigger binding"
CDS 100..612
/label="NanoLuc"
/note="Promega Nano-luciferase; 171 aa; 513 bp ORF;
substrate: NanoGlo (Promega N1110)"
/product="NanoLuc luciferase"
/codon_start=1
/translation="MVFTLEDFVGDWRQTAGYNLDQVLEQGGVSSLFQNLGKV..."
terminator 613..660
/label="T7_terminator"
/note="T7 Te terminator; TGCCTGGCGGCAGTAGCGCGGTGGTCCC"
rep_origin 661..1321
/label="pMB1_ori"
/note="High copy pUC-type origin of replication"
CDS 1322..2182
/label="AmpR"
/note="Ampicillin resistance; beta-lactamase"
/codon_start=1
promoter 2183..2287
/label="AmpR_promoter"
misc_feature 2288..3248
/label="pUC19_backbone_remainder"
/note="Twist Bioscience pUC19 clonal backbone"
ORIGIN
1 taatacgact cactataggg acgugcaugg cuagcaugca ucaguagcau gcuaacagaa
61 acagaggaga aauaaugguu ucccaaaaag agaagaacua uuucacuggc guagucauca
121 ucggagaagu cgaauucaaa gacaucggca acggacaagg ccacaaguuc agcguacggg
181 gcgaaggaga gggcagaggg acucugcuca uguaaacugg gcacccuggu cuucccugga
241 ccccugaacc ccugauccuc aagcuuauca agcagacugg caucguacca gtccacucug
301 aaguucgaag gcggcccacc cttcgccuac ggagaccuga cacaggagcg ccucagcacc
361 caagaggacc acaugcaguu caagcugacg gaggaguucg ugugcgugga gggcaucauc
421 uucagcggca cucagggcau cggcaagagc accaaggcca agaagcgcaa gguggagauc
481 aaccucggcg ugcccgugau gaagcucauc gaccagcagg ugcacaaggc caagaagggc
541 accaaggucc acaucgccuc cggcgacggc gugaccaagg acggcagccu gaucaagaag
601 tgaaccggca gcaaccgcac ctggtgtgtg acgcgatcgg cctcggcggc aggcggtctg
661 gcggtttttt tgtttctggt ctccctatagtgagtcgtattagcttggtcccacgcggaacc
//
A total of 12–15 analogous GenBank files are prepared in Benchling, each with unique toehold/stem sequences. All are ordered as whole plasmids from Twist Bioscience. The backbone (pUC19, AmpR, pMB1 ori) is identical across all constructs.
Step 5 — Trigger RNA Production (Week 2, Days 3–5)
Field
Detail
Purpose
Produce PVY trigger RNAs and Green et al. subset trigger RNAs for CFE assays
Method
PCR-amplify T7 promoter-tagged templates from IDT gBlocks encoding the DQ157180 coat protein target region using ATC Thermal Cycler. In vitro transcribe using NEB HiScribe T7 High Yield RNA Synthesis Kit. Purify by lithium chloride precipitation. Quantify by Nanodrop (A260). Verify integrity by Agilent TapeStation. Order Green et al. subset trigger RNAs (5 sequences) as HPLC-purified RNA oligonucleotides from IDT.
Automation
ATC Thermal Cycler (PCR); manual IVT and purification
Plate
96-Armadillo-PCR-AB2396X (PCR step)
Expected result
PVY trigger RNA ≥ 1 μg/μL; A260/A280 > 1.9; intact band on TapeStation
Timeline
Week 2, Days 3–5
Step 6 — Chloroplast Extraction from Commercial Spinach (Week 3, Days 1–3)
Field
Detail
Purpose
Prepare active spinach chloroplast extract for CFE reactions
Method
Homogenise two independent 100 g batches of grocery-store spinach in 200 mL ice-cold Extraction Buffer (50 mM HEPES-KOH pH 8.0 / 2 mM EDTA / 330 mM sorbitol / 0.6% w/v PVP-40 / 0.1% BSA / 5 mM β-mercaptoethanol) for 30 s in a pre-chilled blender. Filter through two layers of Miracloth. Centrifuge at 1,000×g, 8 min, 4°C (retain green pellet). Wash pellet twice in Extraction Buffer (1,000×g, 8 min). Resuspend in Lysis Buffer (30 mM HEPES-KOH pH 7.7 / 60 mM KOAc / 7 mM MgOAc / 60 mM NH₄OAc / 10% glycerol / 5 mM DTT / 0.5 mM PMSF). Lyse by 15–20 passes through a 25G × 40 mm syringe needle. Primary clarification: centrifuge at 30,000×g, 30 min, 4°C (twice) if ultracentrifuge available. Contingency fallback: if 30,000×g is unavailable, perform an additional 5-min 1,000×g pre-spin, then clarify at 16,000×g for 30 min, 4°C (twice); note this may reduce extract clarity and yield by up to 30% and should be documented as a protocol deviation. Dialyse supernatant in Slide-A-Lyzer 10K MWCO cassettes against 200 mL Lysis Buffer for 2 × 2 h at 4°C. Final centrifuge at 30,000×g (or 16,000×g fallback), 20 min, 4°C. Aliquot 20 μL; snap-freeze in liquid nitrogen; store at −80°C.
Two independent 20 μL aliquot sets; active extract with green tint (residual chlorophyll)
Timeline
Week 3, Days 1–3
Step 7 — Extract Validation with Universal Test Construct (Week 3, Day 3)
Field
Detail
Purpose
Confirm translational activity of both extract batches before proceeding to switch assays
Method
Test both batches using Addgene #216625 (Böhm et al. universal test construct). Prepare reactions in 384-well Greiner black clear-bottom plates. Dispense master mix via Tempest bulk dispenser; add DNA template via Echo525; seal with Plateloc (A4s breathable seal); incubate at 25°C in Inheco Plate Incubator. Measure NanoLuc luminescence at t = 2 h and t = 4 h on PHERAstar FSX (LUM module, 460 nm emission). If signal < 5-fold above buffer-only blank: troubleshoot (verify glycerol, PVP, dialysis, centrifuge speed) before proceeding.
≥ 5-fold signal above blank at t = 4 h; batch-to-batch agreement within 2-fold
Timeline
Week 3, Day 3
Step 8 — Kinetic Profile and Trigger Concentration Pilots (Week 3, Days 4–5)
Field
Detail
Purpose
Identify the optimal kinetic endpoint and trigger RNA concentration for all subsequent assays
Method
Measure NanoLuc for the positive control construct at t = 1, 2, 3, 4, 5, 6, 8, and 12 h (PHERAstar FSX, 384-well Greiner black clear-bottom). Identify plateau time point (< 10% increase between consecutive measurements). Separately, test one high-predicted-performance switch at trigger concentrations of 0.01, 0.1, 1, 5, and 10 nM (switch template fixed at 2 nM). Measure ON/OFF ratio at the kinetically validated endpoint. Use the concentration maximising ON/OFF ratio uniformly across all switches.
Measure NanoLuc ON/OFF ratios for all 12–15 toehold switch candidates in spinach chloroplast CFE
Method
Full automated workflow (see table below). Each switch run in four conditions × ≥ 3 technical replicates. ON/OFF ratio formula: (Condition B − D) / (Condition A − D).
ON/OFF ratio = (Condition ON − BLK) / (Condition OFF − BLK)
PHASE L2 — Learn from Chloroplast Data (Dry Lab, Weeks 5–6)
Step 10 — L2 SANDSTORM Retraining on Chloroplast CFE Data (Week 5, Day 1)
Field
Detail
Purpose
Train the first sequence-structure-function ML model for toehold switches in a plant organellar ribosome context
Method
Compute structural arrays for all 12–15 GARDN-designed switches using the same purely sequence-based method as Step 1 (N×N Watson-Crick possibility matrix; values 0/2/3 from nucleotide identities alone; no folding software). Retrain SANDSTORM on measured chloroplast CFE ON/OFF data (n = 12–15) using leave-one-out cross-validation (LOOCV), initialising with L1 E. coli model weights (transfer learning). Apply integrated gradients to the structural input channel to reveal which sequence positions and pairing interactions most strongly drive chloroplast ON/OFF predictions.
Automation
Dry lab
Plate
N/A
Expected result
SANDSTORM L2 converges; LOOCV Spearman r ± SD reported; integrated gradients attribution maps generated for structural channel
Timeline
Week 5, Day 1
Step 11 — Cross-Context Model Comparison and Structural Attribution (Week 5, Day 2)
Field
Detail
Purpose
Identify which structural positions shift in importance between E. coli and chloroplast ribosome contexts
Method
Compare LOOCV Spearman r ± SD between L1 and L2 models. Apply integrated gradients to both models’ structural input channels for a canonical toehold switch. Compare attribution maps to identify positions where the model weights pairing interactions differently in the chloroplast vs. E. coli context. Positions corresponding to stem stability and RBS accessibility are expected to show the largest attribution shifts.
Automation
Dry lab
Plate
N/A
Expected result
Attribution maps differ between L1 and L2; LOOCV Spearman r > 0.2 in L2 indicates sequence-structure features are informative even at n = 12–15 with transfer learning initialisation
Timeline
Week 5, Day 2
Step 12 — Transfer Learning Test (Week 5, Day 2)
Field
Detail
Purpose
Determine whether sequence-structure-function patterns learned from E. coli data predict chloroplast CFE performance
Method
Compute Spearman r between L1 SANDSTORM predicted ON/OFF rankings and measured chloroplast CFE ON/OFF ratios. The same structural arrays computed in Step 1 are used — the structural array is sequence-derived and temperature-independent. If Spearman r > 0.4: patterns generalise across ribosome contexts. If r ≈ 0: the chloroplast context requires its own training data, making the L2 dataset the necessary foundation. Compare integrated gradient attribution maps between L1 and L2 to identify which structural positions shift in importance.
Automation
Dry lab
Plate
N/A
Expected result
Partial transfer (Spearman r ≈ 0.3–0.5); L2 attribution maps diverge from L1 at stem-stability and RBS-exposure positions
Timeline
Week 5, Day 2
Step 13 — Structural Agreement Analysis and Performance Correlation (Week 5, Days 3–4)
Field
Detail
Purpose
Quantify how faithfully each GARDN-designed switch adheres to canonical toehold switch geometry and correlate with measured ON/OFF ratios
Method
For each of the 12–15 switches, predict MFE secondary structure in dot-bracket notation using NUPACK (the only step in the project where NUPACK is used — for post-hoc MFE structure visualisation, not for SANDSTORM input arrays). Compute structural agreement score: (1/N) Σ p(i), where p(i) is the probability of position i adopting the dot-bracket symbol matching the canonical target structure …………(((((((((…((((((………..))))))…)))))))))). Plot structural agreement score against measured chloroplast ON/OFF ratio and against measured E. coli ON/OFF ratio.
Automation
Dry lab
Plate
N/A
Expected result
Structural agreement scores cluster near ~0.92 (Riley et al. Fig. 5h); switches below 0.80 flagged as likely low-performance outliers
Timeline
Week 5, Days 3–4
Step 14 — Batch QC and Cross-Batch Reproducibility Assessment (Week 5, Day 5)
Field
Detail
Purpose
Assess extract batch-to-batch reproducibility and determine whether data from both batches can be pooled for ML training
Method
For three cross-batch reference switches (one high / medium / low predicted performance), compare ON/OFF ratios between the two independent extract batches. If batch-to-batch CV < 30%: include all data in ML training. If CV > 30%: restrict to higher-quality batch and note as limitation.
Automation
Data analysis (dry lab)
Plate
N/A
Expected result
CV < 30% for high-performance switches; possibly higher CV for low-performance switches near the detection limit
Timeline
Week 5, Day 5
Step 15 — Data Deposition and Final Analysis (Week 6)
Field
Detail
Purpose
Make all data, model weights, and analysis publicly available; prepare final figures for presentation
Method
Deposit all raw luminescence values, computed ON/OFF ratios, GARDN-SANDSTORM model weights (L1 and L2), structural arrays, and trained model outputs to Zenodo (DOI minted before May 13 presentation), mirrored to GitHub following the GARDN-SANDSTORM repository structure. Prepare final figures: (1) ON/OFF ratio bar chart for all 12–15 switches across both systems; (2) scatter plot of SANDSTORM L1 predicted vs. chloroplast measured ON/OFF (transfer learning test); (3) integrated gradient attribution map comparison between L1 and L2 structural channels; (4) structural agreement scores for all GARDN-designed candidates vs. Riley et al. experimental dataset reference.
Automation
Dry lab
Plate
N/A
Expected result
Complete open dataset and model weights available to the community; full LDBT cycle with GARDN-SANDSTORM documented as a reproducible workflow
Timeline
Week 6
Section 5: Techniques, Tools, and Technology
HTGAA Course Technique Checklist
Technique
Relevant?
Pipetting
✅
Lab Safety
✅
Bioethical Considerations
✅ (required)
DNA Construct Design
✅
Databases (GenBank, NCBI, Ensembl)
✅
DNA Sequencing
❌ (Twist provides sequence-verified plasmids)
Restriction Enzyme Digestion
❌ (whole plasmid from Twist; no cloning)
Gel Electrophoresis
❌ (not in primary workflow)
Creating Code for Laboratory Automation
✅
Designing a Twist Order
✅
Creating a plan to use the Autonomous Lab at Ginkgo Bioworks
✅ (Green et al. dataset; GenBank NC_002202.1; To et al. 2018)
CRISPR/Cas9
❌
Technique Expansion — Two Selected Techniques
Technique 1: Cell-Free Reactions
Cell-free expression (CFE) systems are in vitro transcription-translation platforms derived from cellular lysates that retain the molecular machinery for gene expression without intact living cells. In this project, CFE is the core experimental platform: spinach chloroplast extract is prepared from commercial grocery-store spinach and supplemented with a master mix containing NTPs, amino acids, and energy regeneration components, enabling NanoLuc luciferase production from the toehold switch plasmid templates ordered from Twist Bioscience. The use of CFE is scientifically essential rather than merely convenient — it allows direct measurement of toehold switch function in a chloroplast ribosome context without the confound of chloroplast transformation, which would require months of plant growth and selection. Additionally, the open nature of the CFE system permits precise control of trigger RNA concentration, template DNA concentration, and reaction composition in a way impossible in intact plastids, making CFE the ideal platform for the quantitative sequence-structure-function analysis via GARDN-SANDSTORM that is central to the LDBT workflow.
Technique 2: GARDN-SANDSTORM Generative RNA Design (Models and Notebooks)
GARDN-SANDSTORM is a paired generative-predictive deep learning framework developed by Riley et al. (2025, Nature Communications) specifically for functional RNA design, consisting of two components: SANDSTORM (a dual-input CNN accepting one-hot-encoded RNA sequence alongside a purely sequence-derived N×N structural array encoding Watson-Crick base-pairing possibilities to predict function) and GARDN (a Wasserstein GAN with gradient penalty incorporating a programmable reverse-complementation output layer that enforces the canonical toehold switch stem-loop grammar during generation by construction, rather than requiring post-hoc correction). The key innovation of the structural array is its temperature-independence and computational efficiency: because it encodes which nucleotide pairs can form Watson-Crick interactions (A-U or G-U wobble = 2, G-C = 3, no pairing = 0) rather than which pairs do form in a predicted MFE structure, the array is computed directly from raw sequence in O(N²) time with no thermodynamic software calls — making it practical to recompute at every gradient update step during GARDN optimisation. In this project, SANDSTORM is transfer-learning initialised from weights pretrained on the larger Angenent-Mari et al. toehold dataset, fine-tuned on the 181-switch E. coli dataset, and then paired with GARDN for generative design: both model weights are frozen, and 300 gradient update steps on the latent variable Z (Z → Z + α∇_Z P_δ(G_θ(Z))) direct the generator toward sequences with high predicted ON/OFF ratios while maintaining structurally valid toehold geometry. The performance advantage is experimentally validated: GARDN-SANDSTORM-optimised toehold switches showed an 11.9-fold improvement in experimental ON/OFF ratios compared to NUPACK-designed switches and a 3.7-fold improvement vs. non-optimised GARDN outputs in E. coli (Riley et al., Fig. 6d), while maintaining conserved RBS and start codon motifs that activation maximisation approaches destroy.
HTGAA Industry Council Partners
Partner
Role in This Project
Twist Bioscience
All 12–15 toehold switch constructs ordered as whole plasmid synthesis (Clonal Gene service, pUC19 backbone); sole DNA synthesis provider
Ginkgo Bioworks
Autonomous laboratory automation for all CFE reactions: Echo525, Tempest, Inheco Plate Incubator, PHERAstar FSX, Plateloc, XPeel
Addgene
Universal test construct #216625 (Böhm et al.) used for extract validation in Step 7
New England Biolabs
HiScribe T7 High Yield RNA Synthesis Kit for PVY trigger RNA production
All Twist orders screened before submission to verify no biosecurity implications
Basecamp Research
Potential resource for expanded plant chloroplast sequence databases for Aim 2 multi-species SANDSTORM training
Asimov (Kernel Platform)
Potential tool for in silico simulation and validation of toehold switch circuit logic prior to ordering
Section 6: Project Validation
Validation Choice
The aspect of this project selected for validation is the E. coli S30 parallel CFE arm — specifically, the measurement of NanoLuc ON/OFF ratios for all 12–15 toehold switch candidates in crude E. coli S30 extract prepared from BL21(DE3) cells at 37°C. This cross-system validation is scientifically essential because it establishes a within-experiment baseline of switch functionality in a ribosome context where thermodynamic performance has been independently characterised by Green et al. (2014), enabling direct comparison of switch behaviour between E. coli and chloroplast ribosomes and allowing the project to distinguish between construct synthesis failure (if switches fail in both systems) and chloroplast-specific incompatibility (if switches function in E. coli but not chloroplast CFE).
Step-by-Step Validation Protocol
Prepare crude E. coli S30 extract from BL21(DE3) cells: grow overnight culture in LB (37°C, 200 rpm), inoculate 500 mL LB to OD₆₀₀ = 0.1, grow to mid-log (OD₆₀₀ = 0.6), pellet at 4,000×g / 10 min / 4°C, wash twice in S30 buffer (10 mM Tris-OAc pH 8.2 / 14 mM Mg(OAc)₂ / 60 mM KOAc / 1 mM DTT), resuspend in 0.9 mL S30 buffer per gram wet cell weight, lyse by syringe lysis (25G, 20 passes), centrifuge at 30,000×g / 30 min / 4°C (twice; fallback: 16,000×g as above), aliquot and store at −80°C.
Prepare all 12–15 toehold switch plasmids from Twist Bioscience at 100 ng/μL in nuclease-free water.
Prepare trigger RNAs (PVY IVT product and IDT-ordered Green et al. subset) at pilot-optimised concentration (from Step 8 kinetic pilot).
Set up four conditions per switch in 384-well Greiner black clear-bottom plates via Ginkgo Echo525: (A) switch + no trigger, (B) switch + cognate trigger, (C) switch + scrambled trigger, (D) buffer blank.
Dispense S30 master mix (50 mM HEPES pH 7.5, 1.5 mM each NTP, 2 mM DTT, 0.2 mg/mL tRNA, 17.5 mM Mg(OAc)₂, 130 mM KOAc, 33 mM phosphoenolpyruvate, pyruvate kinase) using Tempest bulk dispenser.
Seal plates with Plateloc using A4s breathable seal; incubate in Inheco Plate Incubator at 37°C for the plateau time point determined from the kinetic pilot (typically 4–6 h for E. coli S30).
Remove seal using XPeel; measure NanoLuc luminescence on PHERAstar FSX (LUM module; 460 nm emission; integration time 500 ms).
Compute ON/OFF ratios: (Condition B − D) / (Condition A − D). For cross-batch reference switches, compute separately for each batch and assess agreement (< 2-fold threshold).
Correlate E. coli S30 ON/OFF ratios with L1 SANDSTORM predicted rankings (Spearman r) as the within-experiment validation of prior model predictive power before applying it to the chloroplast dataset.
Techniques Used in Validation
The E. coli S30 validation utilises cell-free expression as its primary technique — specifically crude extract preparation from BL21(DE3), which recapitulates the biochemical conditions of the Green et al. (2014) characterisation experiments and enables direct comparison with the published toehold switch performance dataset that forms the L1 model training set. Bacterial culturing and processing are prerequisite techniques: overnight BL21(DE3) growth, mid-log harvesting, and syringe lysis (25G, 20 passes) are executed following the validated protocol also used for chloroplast extract preparation, ensuring methodological consistency between the two parallel CFE arms. Laboratory automation techniques are central to the validation — the Ginkgo Bioworks Echo525 transfers sub-microliter volumes of DNA template and trigger RNA with precision impossible by manual pipetting, and the PHERAstar FSX luminescence module quantifies NanoLuc output across all 384 wells simultaneously with a dynamic range of five orders of magnitude. DNA construct design underpins the entire validation: the whole plasmid constructs ordered from Twist Bioscience with the canonical Green et al. toehold switch architecture are the substrate, and the sequence-specific trigger RNAs produced by PCR and in vitro transcription are the activating inputs — making the validation a direct test of the designed constructs’ functionality before drawing any conclusions about chloroplast-specific behaviour.
Simulated Data and Hypothetical Graph
The following simulated dataset represents expected results from the scatter plot analysis (Phase L2, Step 12): SANDSTORM L1 predicted ON/OFF ratio vs. measured chloroplast ON/OFF ratio for 12 representative switches. Data were generated under the hypothesis that thermodynamic rules partially transfer (Spearman r ≈ 0.47).
Switch ID
Predicted ON/OFF (L1)
Measured ON/OFF — E. coli S30
Measured ON/OFF — Chloroplast CFE
Tier
TS-PVY-01
84.2
71.3
42.1
High
TS-PVY-02
67.5
58.8
31.5
High
TS-PVY-03
52.1
48.2
18.7
High
TS-GRN-04
48.9
53.1
22.4
High
TS-GRN-05
31.2
24.6
12.8
Medium
TS-GRN-06
28.7
31.4
9.3
Medium
TS-GRN-07
22.4
19.8
7.1
Medium
TS-GRN-08
18.1
15.3
5.4
Medium
TS-GRN-09
8.7
6.2
3.1
Low
TS-GRN-10
6.4
5.9
2.4
Low
TS-GRN-11
4.1
3.7
1.9
Low
TS-NEG-12
N/A
1.2
1.1
Neg. Ctrl
Figure 1 — Scatter Plot: SANDSTORM L1 Predicted vs. Chloroplast Measured ON/OFF Ratio
All GARDN-SANDSTORM-designed switches show reduced ON/OFF ratio in chloroplast CFE relative to E. coli S30 (points fall below the 1:1 line), consistent with the hypothesis that hairpin over-stabilisation at 25°C and chloroplast-specific RBS accessibility differences attenuate switch performance. The rank order is largely preserved (Spearman r = 0.47), indicating that sequence-structure-function relationships learned by SANDSTORM from E. coli data partially predict chloroplast performance. The negative control (TS-NEG-12) shows no activation in either system (ON/OFF ≈ 1.1–1.2), confirming trigger specificity.
Python code to reproduce this figure:
importnumpyasnpimportmatplotlib.pyplotaspltfromscipy.statsimportspearmanrswitch_ids=['TS-PVY-01','TS-PVY-02','TS-PVY-03','TS-GRN-04','TS-GRN-05','TS-GRN-06','TS-GRN-07','TS-GRN-08','TS-GRN-09','TS-GRN-10','TS-GRN-11']predicted=[84.2,67.5,52.1,48.9,31.2,28.7,22.4,18.1,8.7,6.4,4.1]chloro=[42.1,31.5,18.7,22.4,12.8,9.3,7.1,5.4,3.1,2.4,1.9]tiers=['PVY','PVY','PVY','High','Med','Med','Med','Med','Low','Low','Low']colors={'PVY':'#2ca02c','High':'#d62728','Med':'#ff7f0e','Low':'#1f77b4'}fig,ax=plt.subplots(figsize=(6,5))forx,y,t,sinzip(predicted,chloro,tiers,switch_ids):ax.scatter(x,y,color=colors[t],s=80,zorder=3)ax.annotate(s,(x,y),fontsize=7,ha='left',va='bottom')lims=[1,120]ax.plot(lims,lims,'k--',alpha=0.4,label='1:1 line')ax.set_xscale('log');ax.set_yscale('log')ax.set_xlabel('SANDSTORM L1 Predicted ON/OFF (E. coli)',fontsize=10)ax.set_ylabel('Measured ON/OFF — Chloroplast CFE',fontsize=10)r,p=spearmanr(predicted,chloro)ax.set_title(f'Transfer Learning Test (Spearman r = {r:.2f}, p = {p:.3f})',fontsize=10)ax.legend();plt.tight_layout()plt.savefig('scatter_SANDSTORM_vs_chloroplast.pdf',dpi=300)plt.show()
Troubleshooting
The most significant anticipated challenge is low or absent ON/OFF ratio signal in the spinach chloroplast extract, which could arise from hairpin over-stabilisation at 25°C, extract-dependent translational suppression, or trigger RNA degradation by extract-resident nucleases; if ON/OFF ratios are < 2-fold across all GARDN-SANDSTORM-designed candidates, the L2 SANDSTORM integrated gradients attribution can still identify which positions in the structural array are most associated with the residual variation, providing actionable design guidance for a next GARDN optimisation iteration using L2 weights. A second practical challenge is the small sample size: n = 12–15 is below the ~384-sequence threshold at which Riley et al. demonstrated reliable SANDSTORM convergence from scratch, and LOOCV Spearman r estimates at n = 12–15 will have wide confidence intervals — transfer learning from L1 weights mitigates but does not eliminate overfitting risk, and this limitation must be explicitly stated in the final dataset deposition. A third challenge is temperature: if chloroplast extract activity at 25°C is markedly lower than expected, the kinetic pilot should be repeated at 20°C (per Böhm et al.) and the GARDN optimisation re-run with L2 weights trained on 20°C data. Finally, if the spinach chloroplast extract fails entirely, the project reports the L1 SANDSTORM training and GARDN-SANDSTORM design phases as a complete computational dry-lab LDBT cycle — structural agreement analysis, predicted performance ranking, and attribution maps constitute a publishable computational contribution, and the wet lab attempt and failure mode are documented as part of the project narrative.
Section 7: Additional Information
References
Clark, L.G., Voigt, C.A., Jewett, M.C. (2024). Establishing a high-yield chloroplast cell-free system for prototyping genetic parts. ACS Synthetic Biology. doi:10.1021/acssynbio.4c00111
Böhm, C.V., et al. (2024). Chloroplast cell-free systems from different plant species as a rapid prototyping platform. ACS Synthetic Biology. doi:10.1021/acssynbio.4c00117
Pardee, K., et al. (2016). Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell, 165(5), 1255–1266. doi:10.1016/j.cell.2016.04.059
Green, A.A., et al. (2014). Toehold switches: de-novo-designed regulators of gene expression. Cell, 159(4), 925–939. doi:10.1016/j.cell.2014.10.002
Clark-ElSayed, A., et al. (2025). LDBT instead of DBTL: combining machine learning and rapid cell-free testing. Nature Communications, 16, 9782. doi:10.1038/s41467-025-65281-2
Riley, A.T., Robson, J.M., Ulanova, A., & Green, A.A. (2025). Generative and predictive neural networks for the design of functional RNA molecules. Nature Communications, 16, 4155. doi:10.1038/s41467-025-59389-8
Angenent-Mari, N.M., Garruss, A.S., Soenksen, L.R., Church, G., & Collins, J.J. (2020). A deep learning approach to programmable RNA switches. Nature Communications, 11, 5057. doi:10.1038/s41467-020-18677-1
To, A.C.-Y., et al. (2018). A comprehensive web tool for toehold switch design. Bioinformatics, 34(16), 2862–2864. doi:10.1093/bioinformatics/bty216
Valeri, J.A., et al. (2020). Sequence-to-function deep learning frameworks for engineered riboregulators. Nature Communications, 11, 5058. doi:10.1038/s41467-020-18676-2
Landwehr, G.M., et al. (2025). Accelerated enzyme engineering by machine-learning guided cell-free expression. Nature Communications, 16, 865. doi:10.1038/s41467-024-55399-0
Sundararajan, M., Taly, A., & Yan, Q. (2017). Axiomatic attribution for deep networks. Proceedings of ICML, 70, 3319–3328.
Salis, H.M., et al. (2009). Automated design of synthetic ribosome binding sites to control protein expression. Nature Biotechnology, 27(10), 946–950. doi:10.1038/nbt.1568
Zadeh, J.N., et al. (2011). NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry, 32(1), 170–173. doi:10.1002/jcc.21596
Standard lab consumables (pipette tips, tubes, Miracloth)
Thermo Fisher Scientific
General consumables
—
—
~$50
TOTAL
~$1,123
Group Final Project
Phage Therapy
Background: The Antibiotic Resistance Crisis and Phage Therapy
Antibiotic resistance is one of the most urgent threats to global health. At current trends, antimicrobial-resistant infections are projected to cause deaths comparable in scale to cancer within the next 26 years (O’Neill Report, 2016). The overuse and misuse of broad-spectrum antibiotics has accelerated the selection of resistant bacterial strains, while the pipeline for novel antibiotics has nearly run dry. A compelling alternative is phage therapy — the therapeutic use of bacteriophages (phages) to target and kill pathogenic bacteria.
Phages are highly specific: they typically infect only a single species, and sometimes only a single strain, leaving the rest of the microbiome intact. This precision is a major advantage over antibiotics, which disrupt the commensal microbiota alongside the pathogen. The clinical promise of phage therapy has been dramatically illustrated by the case of Tom Patterson, whose pan-drug-resistant Acinetobacter baumannii infection was ultimately resolved only after a cocktail of engineered phages was administered (Schooley et al., 2017).
However, a critical limitation emerged in that case and others: bacteria can acquire resistance to phages rapidly, often within days. Each time Patterson’s bacterial population evolved resistance, a new phage cocktail had to be designed. This highlights the need for proactive phage engineering — designing phages with resistance-resistant properties before bacterial counter-evolution occurs.
This project focuses on MS2 bacteriophage, a well-characterised RNA phage that infects Escherichia coli via the F-pilus, and specifically on engineering its lysis protein L to improve MS2’s ability to kill E. coli even as the host acquires resistance.
The MS2 Bacteriophage and Its Lysis Protein
MS2 is one of the simplest known viruses, with a single-stranded RNA genome encoding only four proteins:
The maturation protein (A)
The coat protein
The lysis protein (L)
The replicase (rep)
The phage infects E. coli by attaching to the F-pilin protein on the host cell surface and injecting its RNA genome. The viral RNA is translated by the host ribosome, producing coat proteins and replicase. After replication and capsid assembly, the lysis protein triggers destruction of the bacterial cell wall, releasing approximately 10,000 new phage particles per lysed cell.
The lysis protein L is a 75-amino acid, predominantly hydrophobic protein that is thought to oligomerise and insert into the host inner membrane, forming pores that disrupt membrane integrity and ultimately cause osmotic lysis (Chamakura et al., 2017). Its exact mechanism remains incompletely understood, but two things are established:
L depends on the host chaperone DnaJ for proper processing and membrane insertion. Chamakura et al. (2017, PMC5446614) showed that E. coli strains with a mutated dnaJ gene are resistant to MS2 infection, because L cannot fold or oligomerise correctly without DnaJ assistance.
Lysis-defective mutations cluster in the transmembrane (TM) domain and the C-terminal region of L, suggesting these regions are essential for membrane integration and pore formation (Chamakura & Young, 2018).
These observations define the two principal vulnerabilities that bacterial resistance exploits, and hence the two engineering targets for this project.
Engineering Goals
We selected two complementary engineering goals for the MS2 lysis protein L:
Goal 1 — Increased Stability (primary): Stabilise L so it remains functional across a wider range of expression conditions and temperatures. A more stable L is less susceptible to premature proteolytic degradation before it can reach the membrane, improving the reproducibility and efficiency of lysis. This goal is also directly relevant to Stage 4 of the group pipeline, where L’s structural integrity is tested using the Nuclera cell-free expression system.
Goal 2 — Resistance to DnaJ-Dependent Inhibition (secondary): Engineer L variants that either:
Tighten the L–DnaJ interaction to compensate for partially impaired DnaJ mutants.
Reduce L’s dependence on DnaJ altogether, allowing lysis even in E. coli strains that have evolved DnaJ mutations as a resistance mechanism.
This directly addresses the primary route of bacterial resistance identified by Chamakura et al. (2017). These goals are mechanistically coupled: a more stable L is less likely to be prematurely degraded before it can recruit DnaJ, and a redesigned L–DnaJ interface can amplify the lytic effect once L is membrane-inserted.
Computational Pipeline
Step 1 — In Silico Deep Mutational Scan (ESM2)
We used the ESM2 protein language model (650M parameter version; Lin et al., 2023) to compute a zero-shot deep mutational scan of the full 75-amino acid L sequence. For every possible single-point substitution, ESM2 assigns a log-likelihood score reflecting evolutionary tolerance — high scores indicate mutations likely to be structurally or functionally neutral, while very low scores flag mutations that disrupt folding or function.
This produced a 75 × 20 mutational fitness landscape at zero experimental cost. Consistent with the literature, the ESM2 scan was expected to show low tolerance for mutations in the TM domain (residues ~37–52) and C-terminal region, which are essential for membrane integration (Chamakura & Young, 2018). Candidate stabilising substitutions were drawn from positions in the disordered N-terminal region that showed elevated ESM2 scores under alternative amino acids.
The wild-type L sequence was folded using ESMFold to generate a predicted 3D structure, with per-residue pLDDT confidence scores used as a proxy for local disorder. The TM helix (residues ~37–52) consistently showed high pLDDT, confirming it as structurally ordered and critical.
ProteinMPNN inverse folding was then applied: the backbone geometry of the WT L structure was fixed, and ProteinMPNN proposed alternative sequences likely to pack into the same fold with improved stability. This is particularly informative for the TM region, where ProteinMPNN can suggest hydrophobic substitutions that improve membrane anchoring without altering helix geometry. Candidate sequences were filtered by:
For the top stability candidates, we modelled the L–DnaJ complex using AlphaFold-Multimer (Evans et al., 2022). DnaJ (UniProt P08622; PDB: 1BQZ) is well-characterised. We compared interface predicted aligned error (PAE) scores and estimated binding energy ($\Delta\Delta G$, computed via FoldX after AF2 modelling) between WT L and the redesigned variants.
Variants showing simultaneously improved pLDDT (stability) and reduced interface PAE (tighter or maintained DnaJ interaction) were prioritised as candidates for experimental validation.
Step 4 — Random Mutagenesis (Complementary Screen)
In parallel with the structure-guided design, we implemented random mutagenesis to generate combinatorial variants outside the hypothesis-driven search space. This approach was guided by the mutational tolerance map generated in Step 1: only residue positions with ESM2 scores above a permissive threshold were included in the random mutation pool, preventing the random screen from exploring lysis-inactivating territory.
where weights were tuned to balance sequence novelty against structural confidence. The top 5 variants were taken forward for synthesis and experimental validation.
L-Protein Mutant Variants
Using the random mutagenesis function constrained by the ESM2 mutational landscape, we generated five double-mutant variants of the MS2 L protein.
Wild-Type 75-aa L Sequence:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Each variant carries two point mutations selected from permissive positions identified by the ESM2 scan.
Rationale:S35 sits immediately upstream of the transmembrane helix. Introducing a lysine at this position may strengthen membrane tethering through electrostatic interaction with negatively charged phospholipid headgroups, a mechanism observed in other membrane-inserting peptides (von Heijne, 1989). Q71L substitutes a polar glutamine with a hydrophobic leucine in the C-terminal region, potentially increasing the hydrophobic moment of the C-terminus and improving membrane association. Together, these two flanking mutations aim to enhance membrane insertion efficiency without disrupting the core TM domain.
Predicted Impact: Increased membrane affinity; potentially reduced DnaJ-dependence if membrane insertion becomes more spontaneous.
Rationale: Both mutations fall within or adjacent to the TM helix. F47 is a large aromatic residue; the F47I substitution reduces steric bulk, potentially allowing tighter helix packing. L44D introduces an aspartate into the hydrophobic core of the TM helix — a charged residue in a TM segment can serve as a pore-lining residue in channel proteins (White & Wimley, 1999), and may alter pore geometry to accelerate membrane disruption. This variant was co-folded with DnaJ using AlphaFold-Multimer, and the resulting PAE map showed low predicted aligned error at the L–DnaJ interface, indicating that DnaJ interaction is predicted to be maintained or improved despite TM mutations.
Predicted Impact: Modified pore geometry; maintained DnaJ interaction (AF2-Multimer PAE confirmed). This variant was prioritised for experimental follow-up.
Rationale:V63 and V67 are both valine residues in the C-terminal amphipathic region. Conservative isoleucine substitutions (V→I) increase side-chain volume by a single methylene group, improving van der Waals packing without introducing steric clashes. This is a classical strategy for thermal stabilisation of hydrophobic cores in membrane proteins (Pace et al., 2011). The double V→I substitution is predicted to increase the thermal melting temperature of the C-terminal region by ~1–2 °C.
Predicted Impact: Improved thermostability; useful for Stage 4 Nuclera testing where cell-free expression under variable temperature conditions is used to assess structural integrity.
Rationale:R31K is a conservative charge-preserving substitution in the N-terminal region, removing the long guanidinium side chain of arginine and replacing it with the shorter lysine $\epsilon$-amino group, potentially reducing electrostatic repulsion between adjacent positive charges in the polybasic N-terminal stretch. F43P introduces a proline at the junction of the pre-TM linker and the TM helix — prolines act as helix-breakers and introduce rigid kinks that can control the angle of membrane insertion. This mutation is predicted to alter the TM helix tilt angle and potentially reduce the DnaJ interaction requirement by promoting a more autonomous membrane-insertion geometry.
Rationale:F5N replaces a hydrophobic phenylalanine with polar asparagine in the extreme N-terminal region, improving the hydrophilic character of the N-terminus and potentially improving solubility during ribosomal translation and DnaJ recruitment. L60C introduces a cysteine in the post-TM region — cysteines can form contacts that stabilise local structure.
Predicted Impact: Enhanced solubility and potential cysteine-mediated stabilisation of the C-terminal region; to be validated by Nuclera cell-free expression.
AlphaFold-Multimer Analysis: Variant 2 × DnaJ
Variant 2 was selected as the priority candidate for AF2-Multimer co-folding based on its TM-domain mutations, which directly probe the interaction between the L protein’s membrane-spanning region and the DnaJ chaperone.
The predicted aligned error (PAE) matrix for the L(F47I, L44D)–DnaJ complex showed:
Low inter-chain PAE values at the predicted interface region, suggesting that DnaJ still recognises and binds Variant 2 despite the TM mutations.
The J-domain of DnaJ (residues 1–75 of DnaJ, including the conserved HPD motif) showed low PAE relative to the C-terminal region of L, consistent with the C-terminus being the primary DnaJ-binding region (Chamakura et al., 2017).
This result supports the hypothesis that mutations in the TM core do not abolish DnaJ recruitment, making Variant 2 a viable candidate for testing both modified pore geometry and maintained chaperone interaction.
Discussion: Connecting Computational Design to Experimental Validation
The five variants described above were generated by a hybrid strategy: ESM2-guided fitness landscape mapping defined the permissive mutation space, ProteinMPNN inverse folding proposed TM-stabilising sequences, and random combinatorial sampling constrained to permissive positions generated diverse double-mutants. This mirrors real-world directed evolution workflows, where computational pre-screening dramatically reduces the experimental search space before library construction.
The key open questions to be resolved in Stages 2–5 of the group pipeline are:
Stage 2 (Synthesis via Twist): The five mutant L gene sequences will be synthesised as codon-optimised synthetic genes. The codon optimisation step is non-trivial for an RNA phage: the wild-type MS2 L sequence is embedded in a region of the genome that overlaps with the replicase reading frame, requiring careful design to ensure mutations affect only L and do not disrupt the overlapping replicase sequence at the RNA level.
Stage 3 (Gibson Assembly): Mutant L genes will be cloned into a plasmid backbone downstream of an inducible promoter (e.g., pBAD or T7) for independent expression in E. coli, decoupled from the rest of the MS2 genome. This allows L’s toxicity to be assessed directly without confounding effects of phage replication.
Stage 4 (Nuclera Cell-Free Testing): The Nuclera eDrop system will be used to express L mutants in cell-free reactions and assess structural integrity. Variants 3 and 5, designed for improved thermostability, are expected to show higher yields and more compact folding in cell-free conditions compared to the wild-type.
Stage 5 (E. coli Lysis Assay): The definitive test: each L variant will be expressed in E. coli (both wild-type DnaJ and DnaJ-mutant strains) and lysis will be quantified by $OD_{600}$ kinetics and plaque assay. Variants 2 and 4, designed to reduce DnaJ-dependence, are predicted to retain lytic activity against DnaJ-mutant E. coli, which would represent a direct demonstration of engineered resistance-evasion.
Integrating Emerging Phage Engineering Frameworks into MS2 L Protein Development
Three recently published phage engineering approaches inform the design strategy of this project and collectively define a computationally guided, cell-free-first development pipeline for MS2 L protein engineering.
The first is a simulation-first design paradigm, wherein AI-powered in silico modeling of phage-host interactions precedes any wet-lab execution. Translating this philosophy here, computational modeling of L protein variants — using structure prediction tools such as AlphaFold2 or ESMFold to assess transmembrane insertion geometry and membrane disruption propensity — can prioritize a ranked synthesis list before any physical construct is ordered. Given that the MS2 L protein spans only ~75 amino acids and that single-residue changes can abolish or enhance lytic activity, computational pre-filtering directly reduces synthesis cost and iteration time, two practical constraints central to this project.
The second framework is PHEIGES (PHage Engineering by In vitro Gene Expression and Selection), which demonstrated that phage genome fragments expressed in E. coli cell-free transcription-translation (TXTL) systems produce functional outputs — including host-toxic products — without requiring full phage assembly or live bacterial passage. Adapting this logic, individual L protein variants can be expressed from linear DNA fragments in TXTL and screened for membrane disruption activity using OD-based lysis proxies or liposome dye-release assays. This decouples L protein functional validation from full MS2 viability, collapsing the screening cycle from days to hours and allowing higher-throughput variant assessment upstream of genome reconstruction.
The third is the High-Complexity Golden Gate Assembly (HC-GGA) system developed by Sikkema et al. (2026) for a Pseudomonas aeruginosa phiKMV-like phage, which achieved near-100% genotype recovery from 28 modular plasmid-held fragments without selectable markers. The MS2 genome at ~3.6 kb is far more tractable than the 43 kb 41S1 system, making a 4–5 fragment HC-GGA design straightforward. By isolating the L gene and its regulatory flanking sequences within a single dedicated fragment, every future variant becomes a single-fragment substitution dropped into a stable master mix — no counterselection engineering, no full re-synthesis. Together, these three frameworks define a unified funnel: computational variant design, cell-free functional screening, and modular genome assembly for high-fidelity phage rescue.