Class assignment 1. First, describe a biological engineering application or tool you want to develop and why. I’m heavily inspired by Professor Jacobson’s call for a “bio-FPGA” tool, as well as his lecture about cellular automata. I’d like to develop a bio-FPGA that can be programmed to grow into arbitrary 2D patterns on a petri dish, using the machine learning technique mentioned in the lecture to reverse learn the cellular automata rules for growing a specific pattern. The learned CA rules can be encoded by genetically programming the bio-FPGA then using bacteria with the genes to grow an actual cell culture into the pattern, like the butterfly wing letter patterns in the lecture. If this is feasible, 3D patterns would be the next step, and one might even imagine a wild future of programmable plants that grow into the shapes of houses and furniture.
Part 0: Basics of Gel Electrophoresis I watched the lecture, recitation, and read the lab. Essentially, we use the negative charge of DNA to pull DNA fragments towards a positive anode in a porous agarose gel. Larger DNA fragments move slower in the agarose gel.
Part 1: Benchling & In-silico Gel Art I spent some time playing around with Ronan’s gel art site to make a pattern (below on the left). I noticed that some of the restriction enzymes in the gel art tool weren’t on the HTGAA enzyme list, so I didn’t use them.
Opentrons Artwork My artwork is here: https://rcdonovan.com/?id=vmns94wqt45wpqc
I used Ronan’s tool to make this. I uploaded an image of tomatoes but it didn’t render well, so I modified it significantly by hand with the editor.
Then, I attended the Saturday session on Zoom with Ronan, Michelle, and Ice at Ginkgo Bioworks. Here’s the end result:
Part A: Conceptual questions Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) A dalton is 1.66053906892(52)*10−23 g, so 500g = 500 / 1.66053906892(52)*10−23 = 3.0110704e+25 daltons.
Part A: SOD1 Binder Peptide Design (From Pranam) Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The mix includes: Phusion DNA Polymerase, deoxynucleotides and reaction buffer that has been optimized and includes MgCl2.
DNA Polymerase builds the DNA sequence from deoxynucleotides starting with the template strands and primer. It’s high-fidelity, so it won’t make as many errors as Taq.
Reaction buffer allows the reaction to take place.
What are some factors that determine primer annealing temperature during PCR? For primer annealing, we want the template to be high enough for the template DNA to be denatured and but low enough that some of the primer is annealed. So the lab protocol suggests to use temperatures near the Tm of the primer (temp where the DNA is 50/50 double/single stranded).
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? IANNs are analog, instead of digital (e.g. boolean/binary logic), which means they work on numerical values. This means that their behavior can be finely adjusted: they can output specific quantities, and they can take into account the specific/relative magnitudes of inputs. This is well suited for biological applications because biological systems exhibit homeostatic behaviors.
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell free systems are much simpler to control because the reaction does not have to happen within a cell-wall, so scientists have direct control over concentrations, monitoring, and timing. Cell based production is much harder to control, since you need to control the expression and inputs of the cell. For example, if you need to do experiments to optimize production, or you need to produce proteins that would kill living cells.
Subsections of Homework
Week 1 HW: Principles and Practices
Class assignment
1. First, describe a biological engineering application or tool you want to develop and why.
I’m heavily inspired by Professor Jacobson’s call for a “bio-FPGA” tool, as well as his lecture about cellular automata. I’d like to develop a bio-FPGA that can be programmed to grow into arbitrary 2D patterns on a petri dish, using the machine learning technique mentioned in the lecture to reverse learn the cellular automata rules for growing a specific pattern. The learned CA rules can be encoded by genetically programming the bio-FPGA then using bacteria with the genes to grow an actual cell culture into the pattern, like the butterfly wing letter patterns in the lecture. If this is feasible, 3D patterns would be the next step, and one might even imagine a wild future of programmable plants that grow into the shapes of houses and furniture.
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
My primary goal is to protect health and safety. The sub-goals are:
Prevent the development of biological weapons.
Prevent outbreak of harmful bacteria.
Maximize productive use-cases.
A bio-FPGA has the possibility to be used for great benefit with many applications, but could also be abused.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Action 1: Engineer a genetic “off switch” into the bio-FPGA to stop proliferation any time.
Purpose: We would build a genetic off switch to immediately turn off all genes of the bio-FPGA.
Design: This would involve researchers and industry as actors to build this prior to releasing the bio-FPGA. The government could also regulate by requiring all bio-FPGA and adjacent tools have such a fail-safe.
Assumptions: This assumes that such a technological solution could be reliably engineered and triggered.
Risks: The risks are that the technical fail-safe does not work, or could even cause problems if it does work because it could be abused to disable legitimate use cases.
Action 2: Regulate against use for biological warfare.
Purpose: Although there are already regulations in place, we could craft regulation to specifically account for bio-FPGA technology.
Design: This would involve the government to understand the technology, the dangers, and pass appropriate laws preventing malicious use of bio-FPGAs.
Assumptions: This assumes that lawmakers would be motivated to pass regulation and that the public would be accepting of such regulation. It also assumes that lawmakers are able to craft good laws or adapt accordingly.
Risks: The risk is that excessive regulation could stifle adoption and research for beneficial use cases. Another risk is that lawmakers don’t understand the science and pass inappropriate laws.
Action 3: Host a conference for researchers and industry to share new developments.
Purpose: To share beneficial use cases, foster collaboration, and disseminate research learnings.
Design: This requires coordinating and organizing the research and industry community, as well as raising funds to host a venue.
Assumptions: I assume that researchers would be interested in attending and discussing.
Risks: The conference could be used to develop malicious use-cases, or ethics could be overlooked in favor of scientific progress at all costs.
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
Does the action:
Action 1
Action 2
Action 3
Enhance Biosecurity
• By preventing incidents
3
1
2
• By helping respond
1
3
3
Foster Lab Safety
• By preventing incident
3
1
2
• By helping respond
1
3
3
Protect the environment
• By preventing incidents
3
1
2
• By helping respond
1
3
3
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
1
• Feasibility?
2
1
1
• Not impede research
1
3
1
• Promote constructive applications
3
1
1
Week 2 lecture prep
Homework Questions from Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
According to the slides, the error rate of polymerase is 1:106 (online it suggest that it can be even worse depending on the polymerase), or 1 in 1 million. The length of the human genome is 3.2 Gbp (3.2 * 109), so at that rate there would be ~3.2 * 103 (3,200) errors in the human genome per copy. That would be a lot of errors, but there are additional pathways that perform error correction, such as MutS.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The slides mention the average human protein is 1036 base pairs. That is 345 codons, so 345 amino acids.
Amino acids can have multiple corresponding codons. There are 20 amino acids and 4 possible nucleotides, so there are about 3 possible codons per amino acid.
That is an estimate of 3345 ways to code for a given protein, a huge number.
However, despite a synonymous codon coding for the same amino acid, the base pairs choice can affect the chemical bonds of the mRNA structure, affecting RNA cleavage rules.
Another view is that there are 4^1035 possible nucleotides, which are very unlikely to code for the specific protein even with synonymous codons due to sheer possibility space.
Homework Questions from Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
The phosphoramidite method.
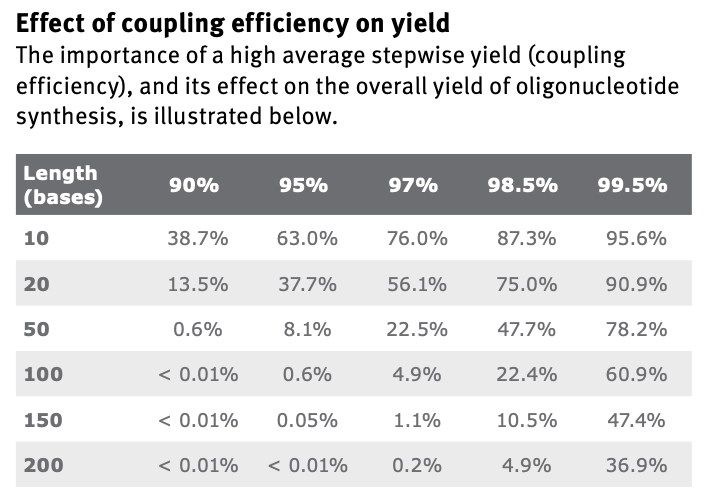
Why is it difficult to make oligos longer than 200nt via direct synthesis?
The coupling step is not possible to have perfect efficiency. That step is repeated per cycle, and each additional base requires the cycle to repeat. This means that longer oligos become dramatically harder to make, even with extremely high efficiencies:
Why can’t you make a 2000bp gene via direct oligo synthesis?
The above answer explains why we can’t synthesize longer oligos. At 2000bp, the probabilities become near impossible even at the highest efficiencies.
Homework Question from George Church
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
I’m answering question 1.
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Arginine (R), sometimes considered conditionally essential.
The “Lysine Contingency” is apparently from Jurassic Park, which was a plot element in the movie that was a genetic modification to make the dinosaurs not to be able to produce Lysine so they would die off without human provided Lysine supplements.
However, Lysine is one of the 10 essential amino acids so animals cannot
produce it, making this is a scientifically dubious plot point (the genetic modification would have done nothing).
[Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions?
[(Advanced students)] Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or devise one of your own:
I watched the lecture, recitation, and read the lab. Essentially, we use the negative charge of DNA to pull DNA fragments towards a positive anode in a porous agarose gel. Larger DNA fragments move slower in the agarose gel.
Part 1: Benchling & In-silico Gel Art
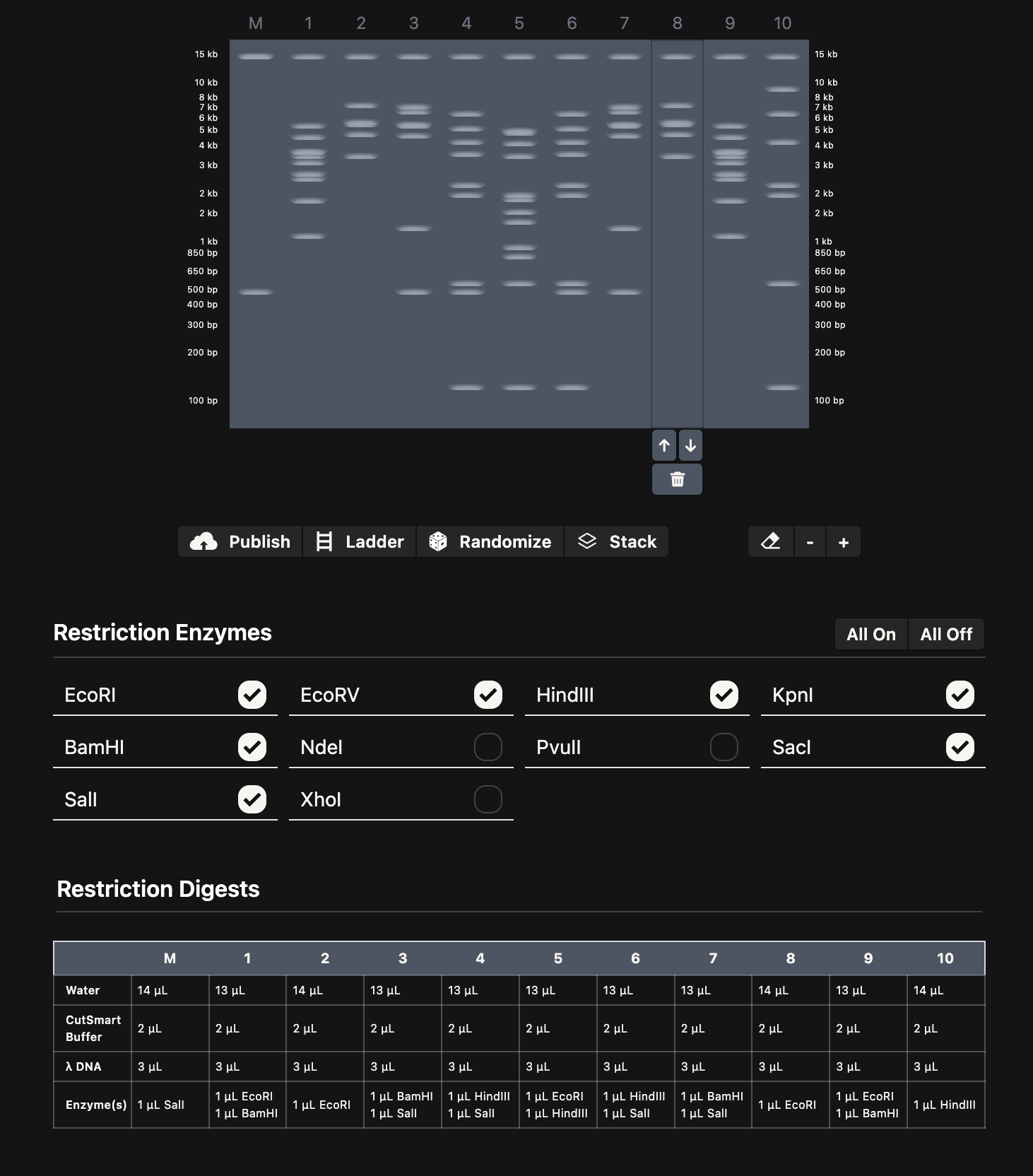
I spent some time playing around with Ronan’s gel art site to make a pattern (below on the left). I noticed that some of the restriction enzymes in the gel art tool weren’t on the HTGAA enzyme list, so I didn’t use them.
I think it looks kind of like Darth Vader.
Then, I added the Lambda DNA to Benchling. I made a custom enzyme list with the EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI. Then, I added the restriction enzymes from the gel art tool to make a virtual digest (below on the right). I had some difficulty ordering the digests properly, so I saved them with the names and ordered them by dragging the tabs after.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis (Wet Lab)
N/A: This is optional for committed listeners and I didn’t have access to a wet lab this week.
Part 3: DNA Design Challenge
3.1. Choose your protein
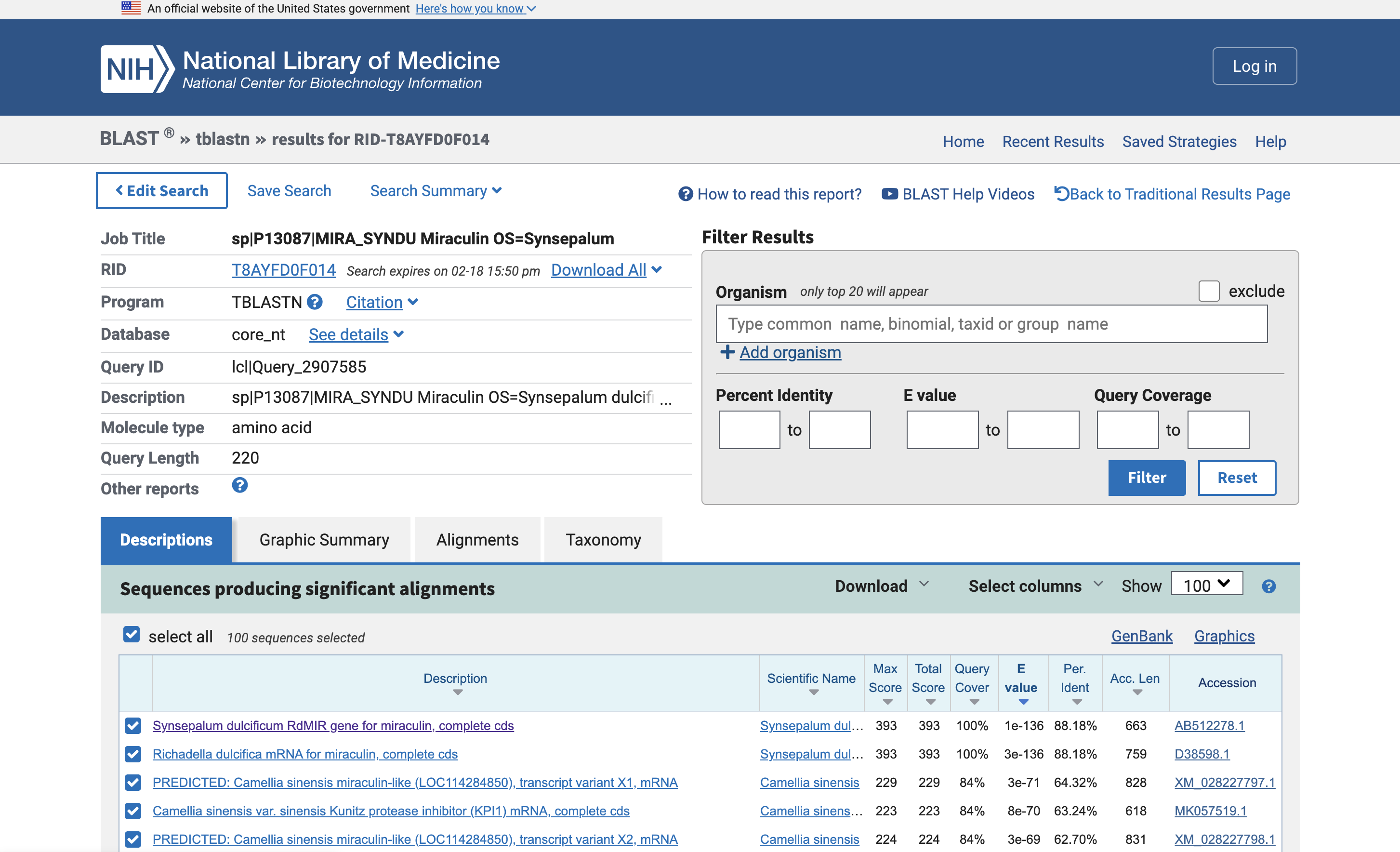
The protein I chose is Miraculin, which is from the miracle berry and famous for temporarily causing sour things to taste sweet. I picked this because I have tried a miracle berry tasting before and it was an interesting experience.
I used tblastn (Translated BLAST) to get the nucleotide sequence that corresponds with Miraculin. This found two nucleotide sequences in a database: AB512278.1 and D38598.1. One appears to be the mRNA rather than the genes.
I used VectorBuilder’s codon optimization tool since Twist’s was down for maintenance. I optimized for E. Coli, so the protein could be mass produced in its “cellular factory”. It gave the following:
Since I chose E. Coli, we can order the gene with a promoter in a plasmid, then use a cell-dependent method of heat shocking the E. Coli to embed the plasmid, then cultivating the E. Coli to produce lots of this protein.
This uses the natural plasmid gene expression mechanisms of E. Coli to transcribe and translate the protein.
Part 4: Prepare a Twist DNA Synthesis Order
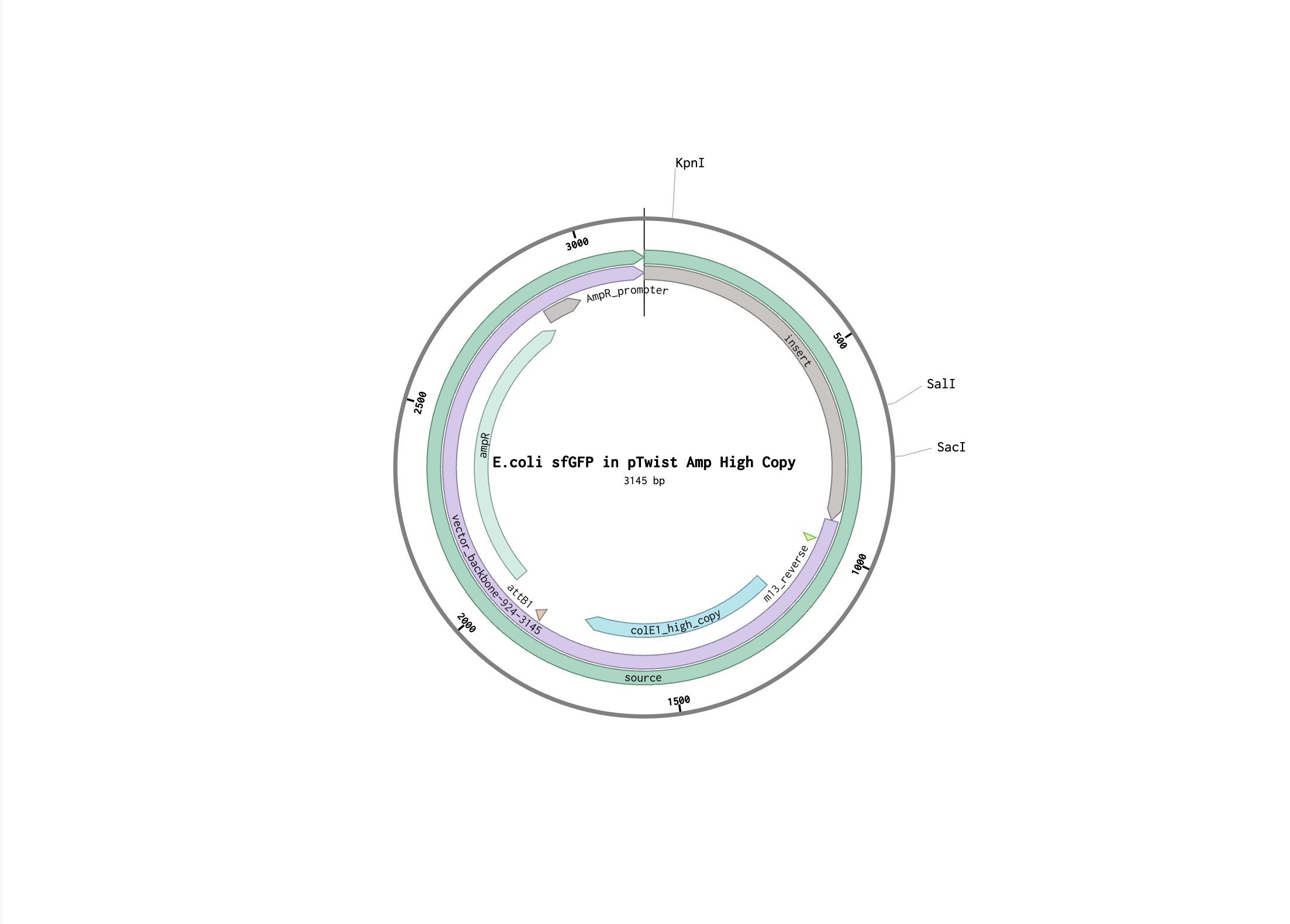
I followed the steps to make provided sfGFP sequence in Benchling. Here is my Benchling project.
Here is the FASTA file of the expression cassette:
(i) What DNA would you want to sequence (e.g., read) and why?
I’d like to sequence DNA of the human microbiome. There’s been recent research about how beneficial flora of the gut and skin microbiome contribute to our health, and there’s already a significant effort to sequence our microbiome in the Human Microbiome Project.
I would also be interested in the widespread sequencing and cataloguing of viruses that make up the common cold. I think it could be useful to detect the geographic spread of these viruses and how they mutate over time, to potentially contribute to a cure.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Sanger Sequencing, since it’s a straightforward and well-tested technique, and I understand it the best.
Is your method first-, second- or third-generation or other? How so?
Sanger Sequencing is a first-generation technique. It’s the earliest and most classic form of sequencing.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input is DNA, regular nucleotides (d*), chain-terminating nucleotides (dd*), primer (like in PCR), DNA-polymerase. You prepare the input by PCRing the sample to have lots of DNA.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
You thermocycle the amplified sample with the nucleotides, primers, and polymerases to build many DNA fragments. These DNA fragments will be different lengths because probabilistically each fragment will incorporate a chain-terminating nucleotide which stops polymerization. Finally, the fragments are run through electrophoresis and imaged one base pair at a time to get the sequence.
What is the output of your chosen sequencing technology?
The result is the electrophoresis imaging data, which can be processed to determine the most likely base pair at each position.
One limitation of the technique is that you need a pure sample of DNA, so it may be inefficient for the volumes of organisms we’d want to sequence.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would want to synthesize DNA origami, as art! I’m curious what it would take to make the smallest art pieces.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use the phosphoramidite method.
What are the essential steps of your chosen sequencing [sic; synthesis?] methods?
Deprotection, coupling, capping, oxidation, and repeat.
What are the limitations of your sequencing [sic; synthesis?] method (if any) in terms of speed, accuracy, scalability?
The limitation, as discussed last homework, is the length of DNA oligos that can be synthesized with this technique. However, this isn’t a problem for DNA origami, which doesn’t need full length DNA.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would edit human DNA, for example to cure hearing loss and tinnitus as a gene therapy. Hearing loss is permanent and affects 5% of the world population. Noise exposure from work and the environment also contribute to increased rates of hearing loss. Birds, unlike humans, can regenerate inner ear cells, and researchers have demonstrated regrowth in cell cultures so there is a theoretical target for gene therapy. There have already been successful gene therapy treatments for deafness in children due to congenital disorders.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-Cas9, since it is the most popular technique today.
How does your technology of choice edit DNA? What are the essential steps?
Cas9 cuts the DNA, then introducing the edits via homology directed repair.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
The guide RNA for Cas9 needs to be designed, as well as the template DNA (for knock-ins).
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Homology-directed repair is not entirely efficient, since the ends that Cas9 break could rejoin without the knock-in sequence. Performing CRISPR in humans (rather than bacteria or cell cultures) requires sophisticated deliveries to get to the target cells or tissues.
I used Ronan’s tool to make this. I uploaded an image of tomatoes but it didn’t render well, so I modified it significantly by hand with the editor.
Then, I attended the Saturday session on Zoom with Ronan, Michelle, and Ice at Ginkgo Bioworks. Here’s the end result:
Post-Lab Questions
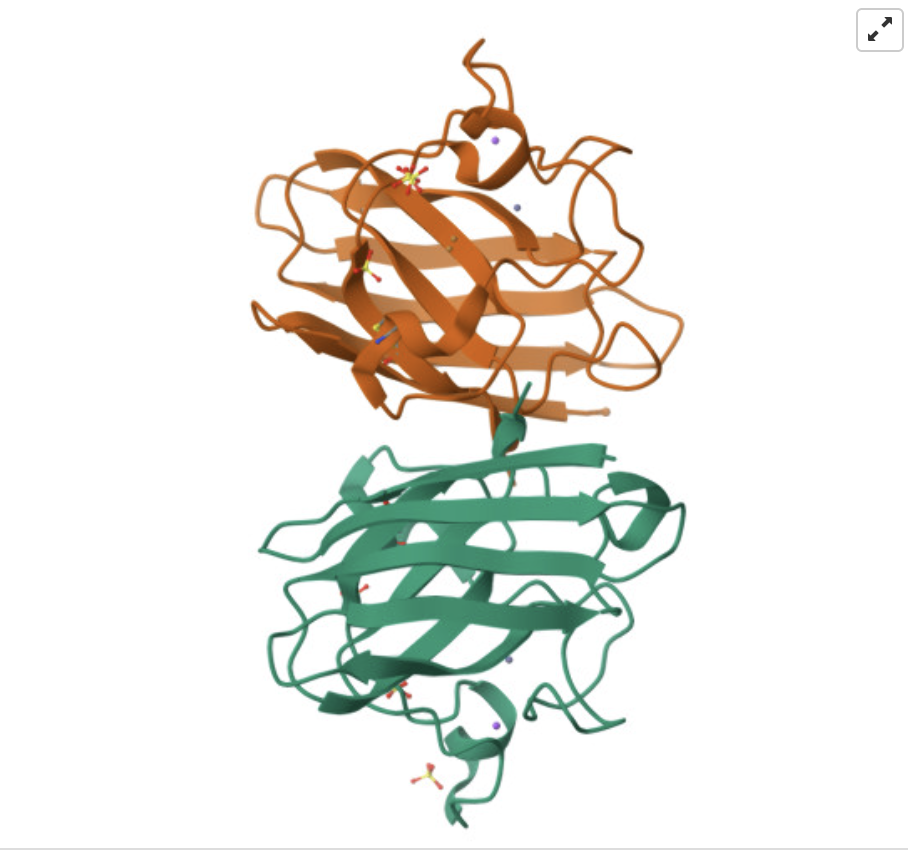
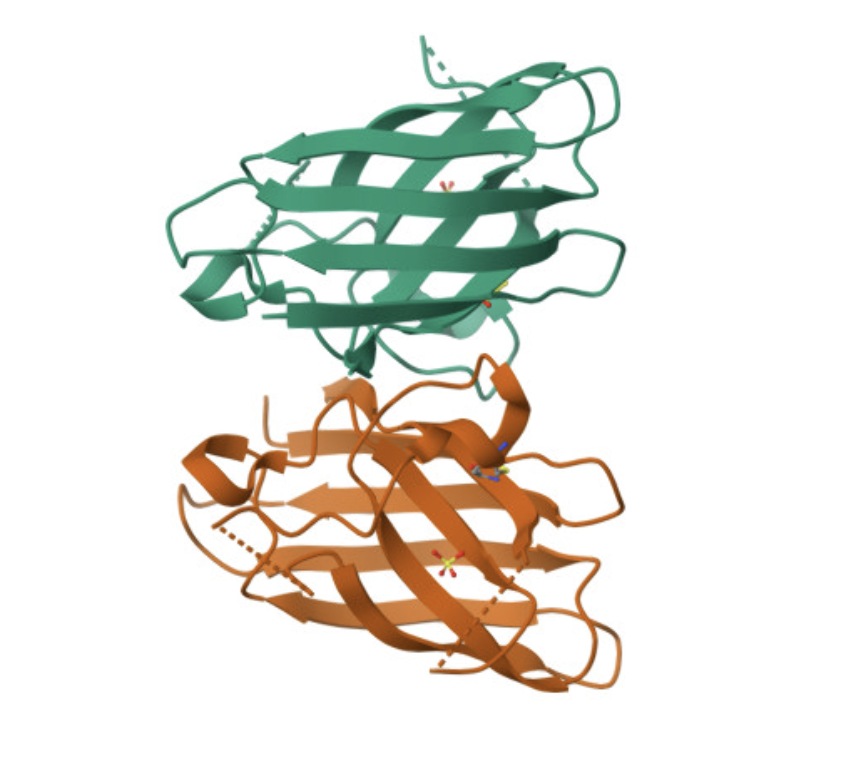
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
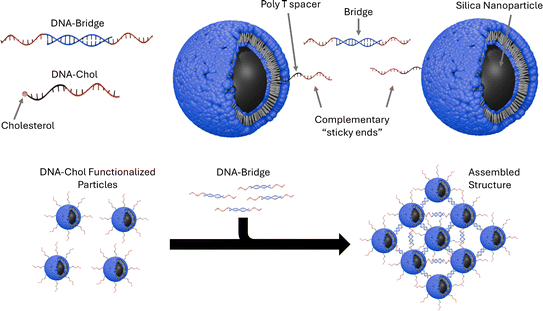
I read Assembly of small silica nanoparticles using lipid-tethered DNA ‘bonds’. This paper used a novel assembly using DNA by by embedding silica nanoparticles in a lipid bilayer, embedding the cholesterol end of a DNA-cholesterol molecule within the bilayer, then assembling the nanoparticles with complementary sticky end “bridge” DNA. This is best explained with the image from the paper below:
They used Opentrons to rapidly iterate on and evalute different concentrations of DNA-Chol, NaCL, and bridge DNA in the assembly mixture. These were then screened with SAXS to determine the structural qualities of each sample.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
One project idea I have is genetically modifying trees for use in urban areas. I’d like to use automation tools to permute different genetic combinations. I envision custom modules that could germinate and monitor an array of seeds for different qualities.
For example, I create a module with grow lights and watering capabilities that can care for the different seed variants and cameras to compare growth rates.
Week 4 HW: Protein Design, Part I
Part A: Conceptual questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
A dalton is 1.66053906892(52)*10−23 g, so 500g = 500 / 1.66053906892(52)*10−23 = 3.0110704e+25 daltons.
If an amino acid averages 100 daltons, then 3e+25 daltons is about 3e+23 amino acids.
~300,000,000,000,000,000,000,000 amino acids!
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We digest the meat and it does not alter our genetics.
Why are there only 20 natural amino acids?
Codon redundancy allows for both more efficient genetic coding and error correction. 20 appears to be enough (as evidenced by life).
Can you make other non-natural amino acids? Design some new amino acids.
Yes, you can make non-natural amino acids (e.g. non-proteinogenic amino acids). Amino acids require an amine, a carboxyl, a central carbon, and a side-chain. You could design one by using a unique side chain that doesn’t exist in the natural amino acids.
Where did amino acids come from before enzymes that make them, and before life started?
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
I would expect it to turn the other way, since D-amino acids are mirrored. Normal alpha helixes are right handed, so I would expect a left handed helix.
Can you discover additional helices in proteins?
Skipped (1/2).
Why are most molecular helices right-handed?
Skipped (2/2).
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
This is because beta sheets can be hydrophillic on one side and hydrophobic on the other, forming a “pleated sheet”.
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Shugang mentions that β-sheets can aggregate to form disease because β-sheets can’t be easily untangled once formed.
Yes, β-sheets give spider silk its unique properties, which inspired the design of materials like Kevlar.
Design a β-sheet motif that forms a well-ordered structure.
We can use the rule from Shugang’s slides: alternate hydrophobic and hydrophillic for every other amino acid.
Part B: Protein Analysis and Visualization
I’m picking the same protein as in week 1: Miraculin, a taste altering protein.
Amino acid sequence:
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
>1ZKR_1|Chains A, B|Major allergen I polypeptide, fused chain 1, chain 2|Felis catus (9685)
MEICPAVKRDVDLFLTGTPDEYVEQVAQYKALPVVLENARILKNCVDAKMTEEDKENALSLLDKIYTSPLCVKMAETCPIFYDVFFAVANGNELLLDLSLTKVNATEPERTAMKKIQDCYVENGLISRVLDGLVMTTISSSKDCMGEHHHHHH
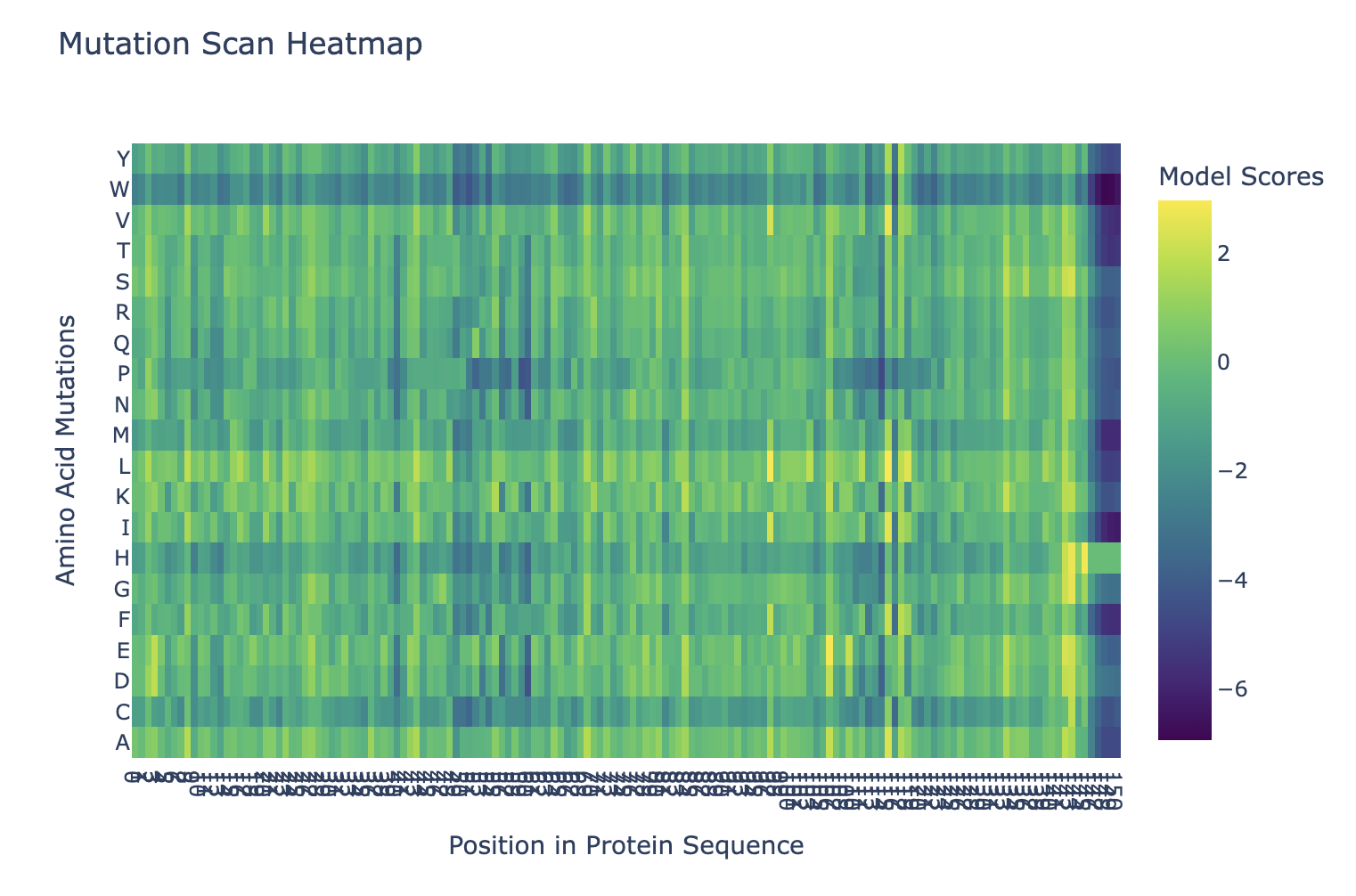
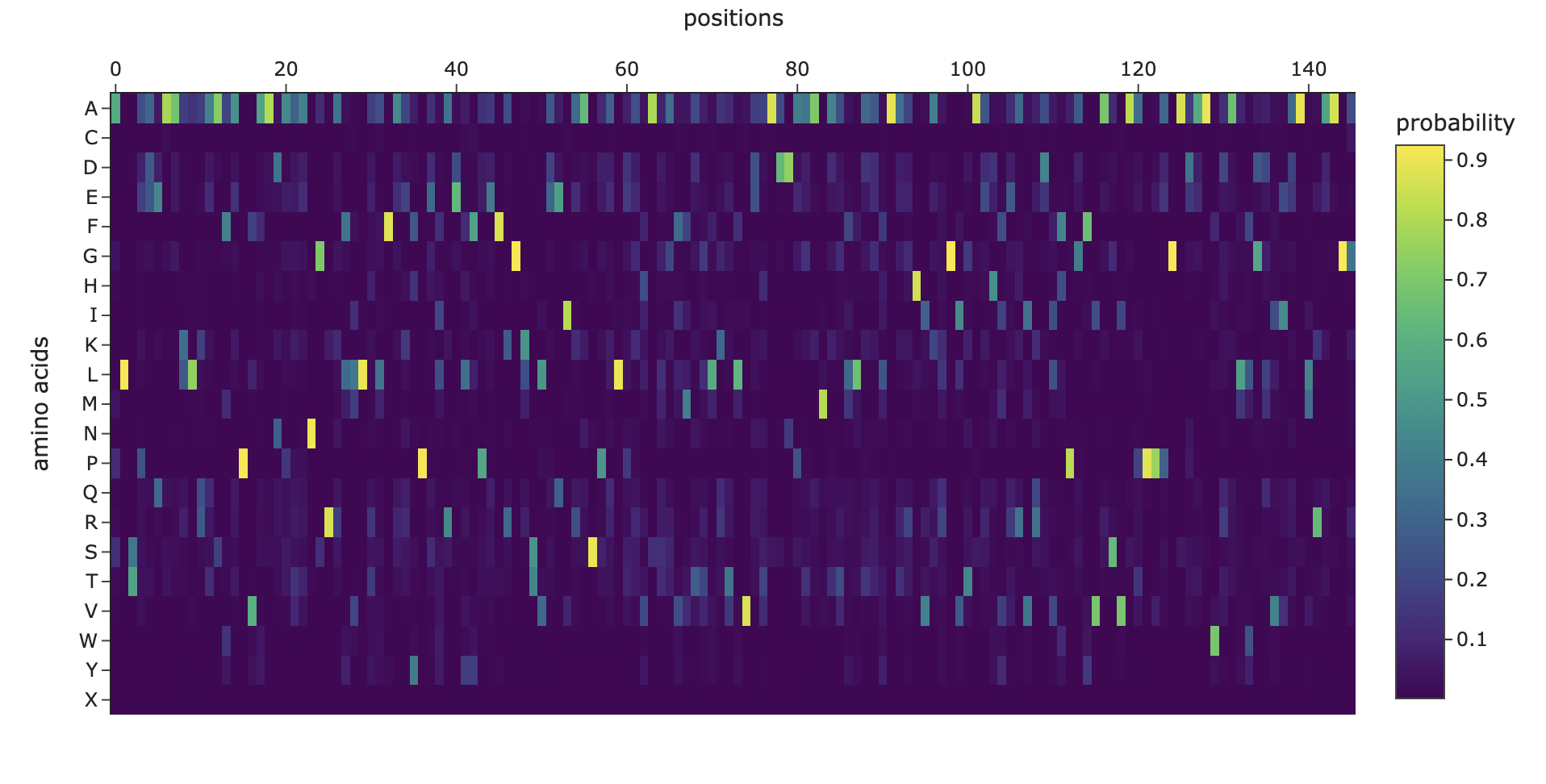
Here’s the mutation scan heatmap. It looks like the amino acids at the end of the protein are most sensitive to mutation. We can also see that “W” tends to be a bad mutation anywhere in the protein.
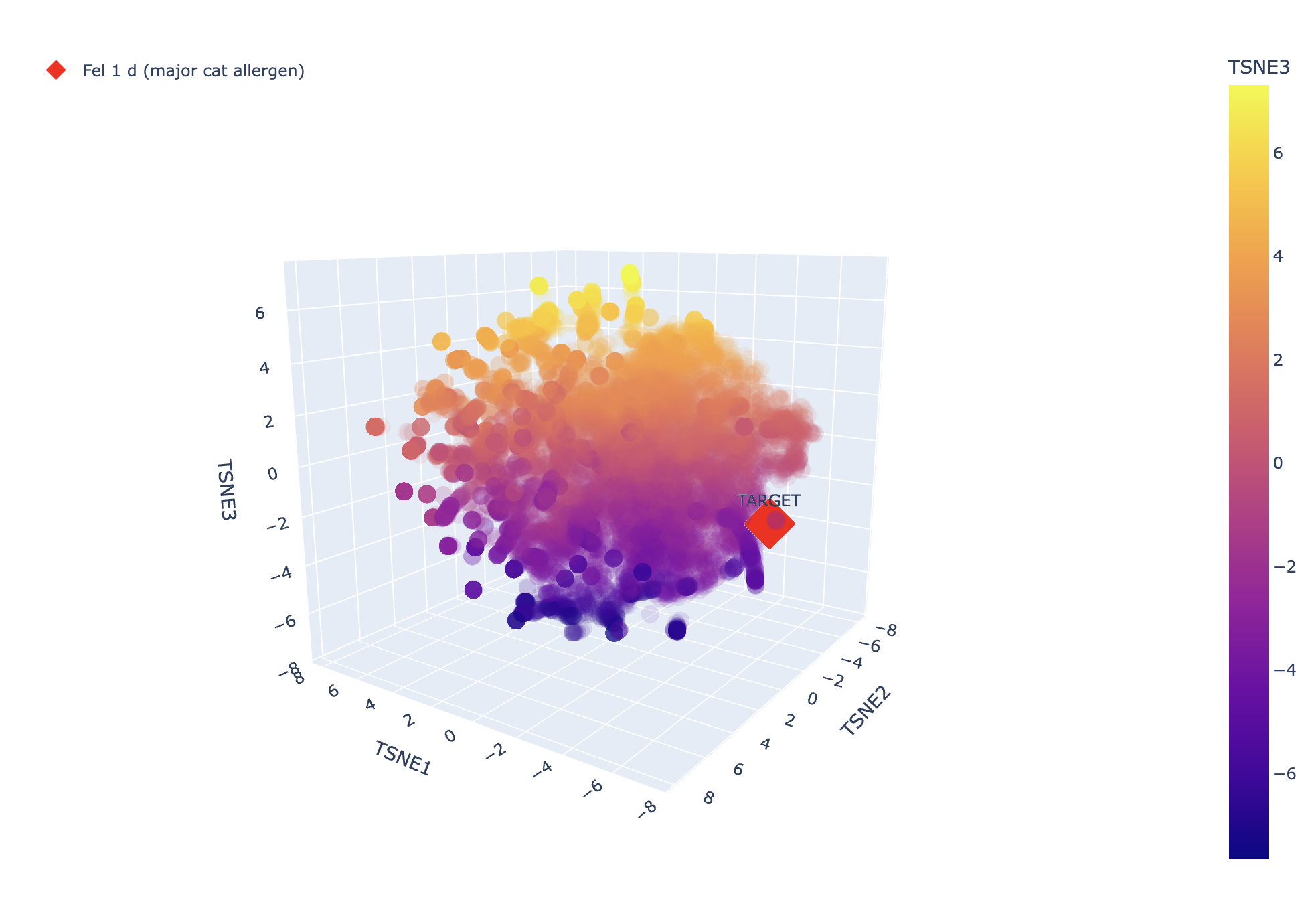
And here’s the TSNE, it looks like it’s not very closely related to the other proteins, which would make sense since it’s a unique allergen (otherwise we might expect cat allergies to correlate with lots of other allergies).
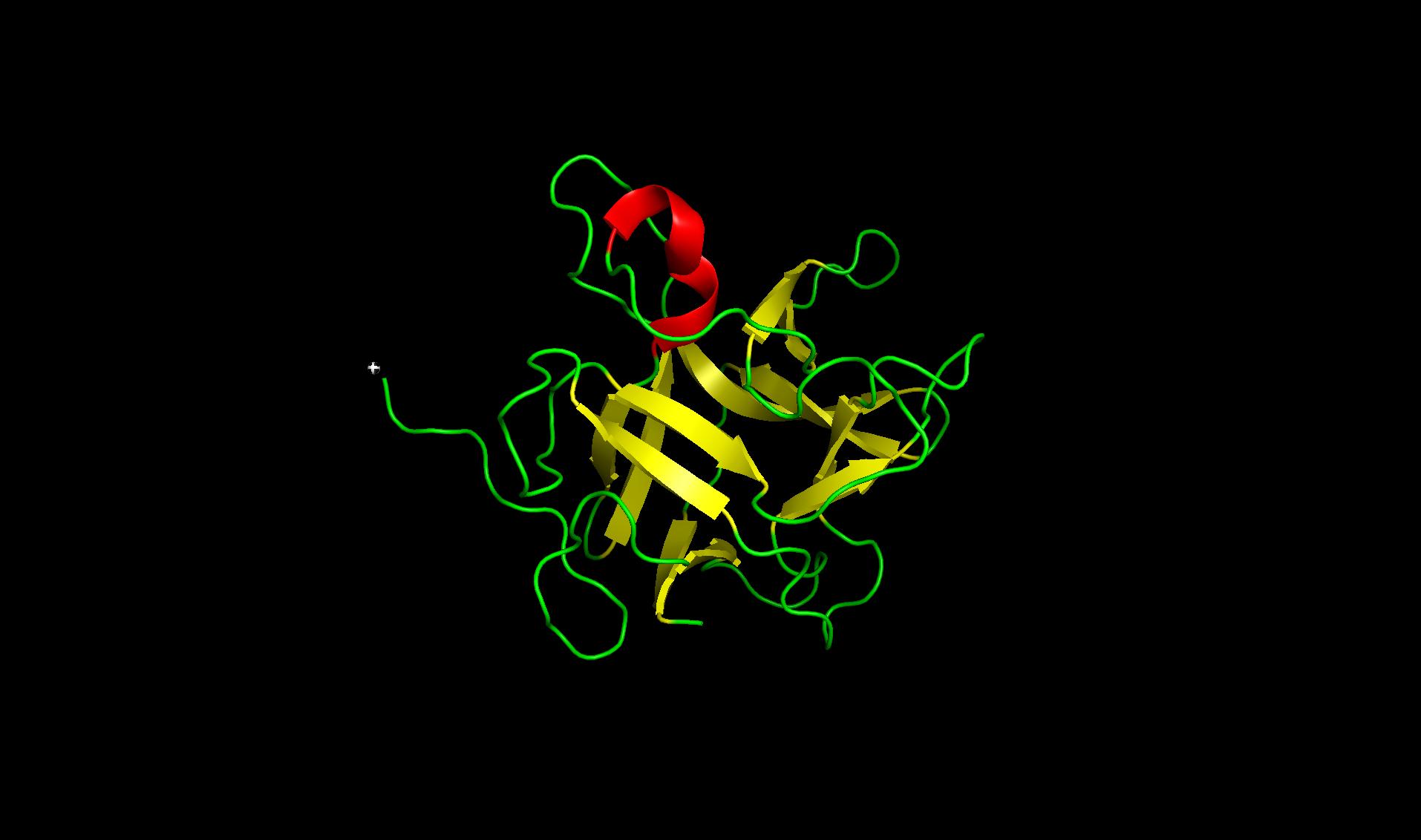
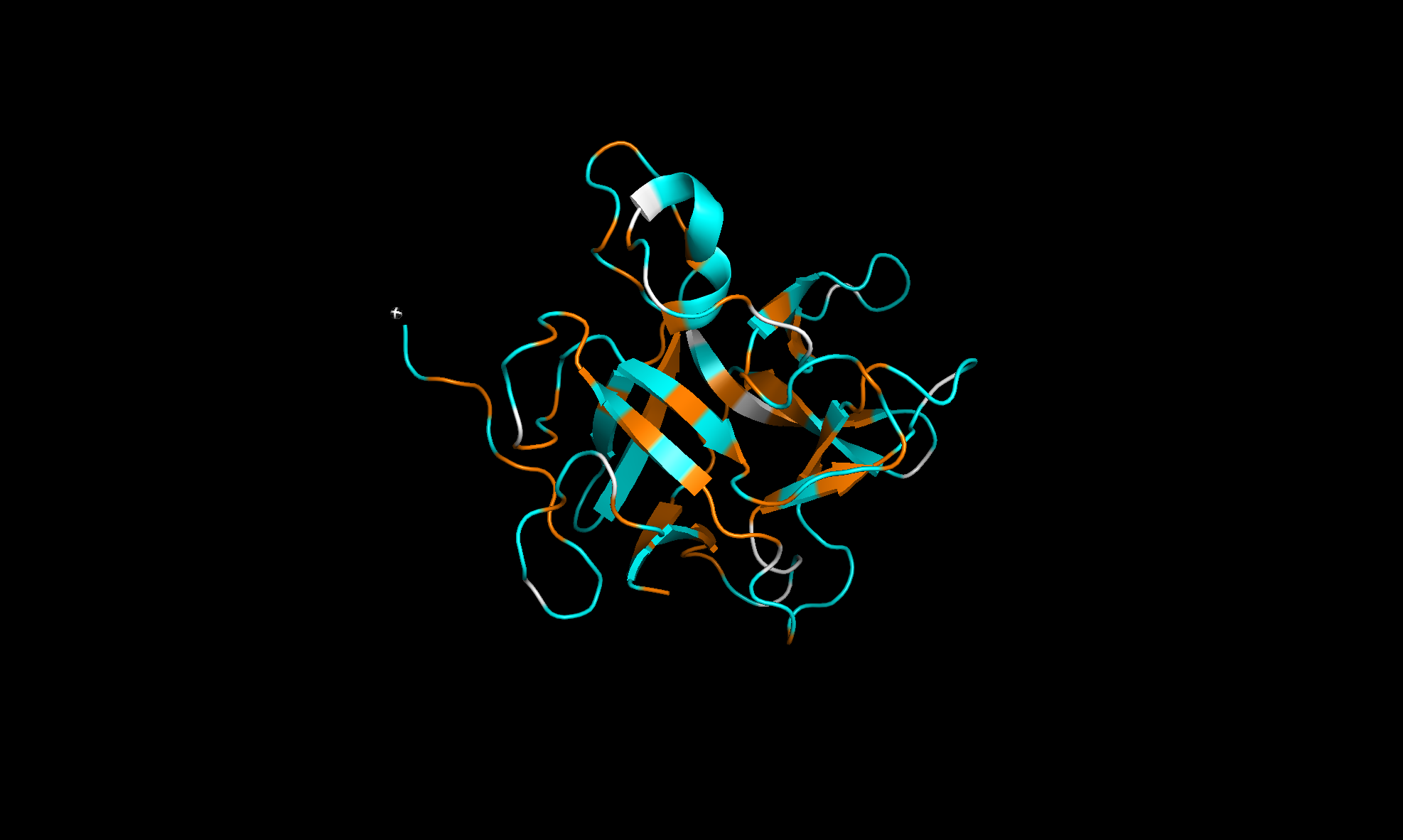
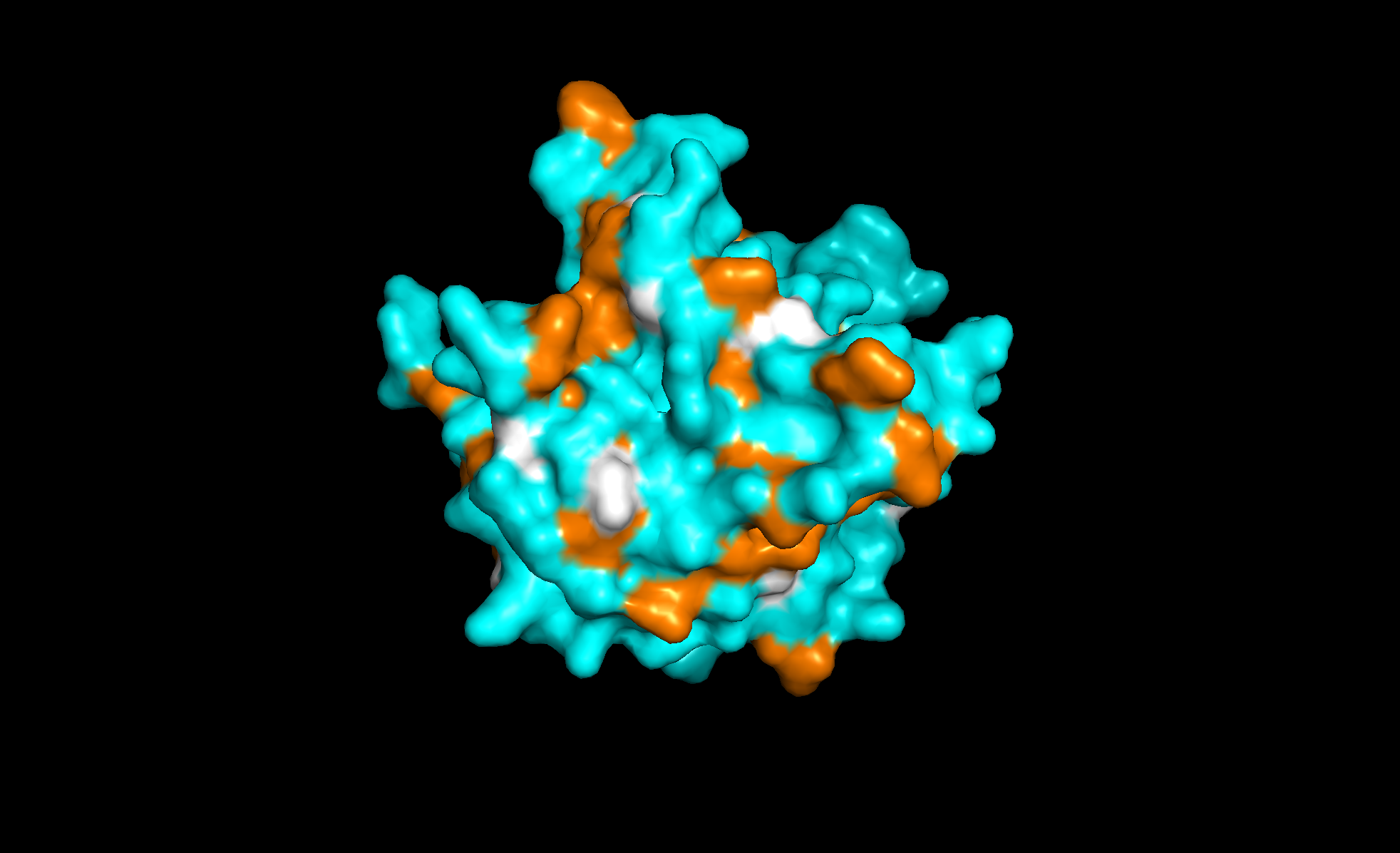
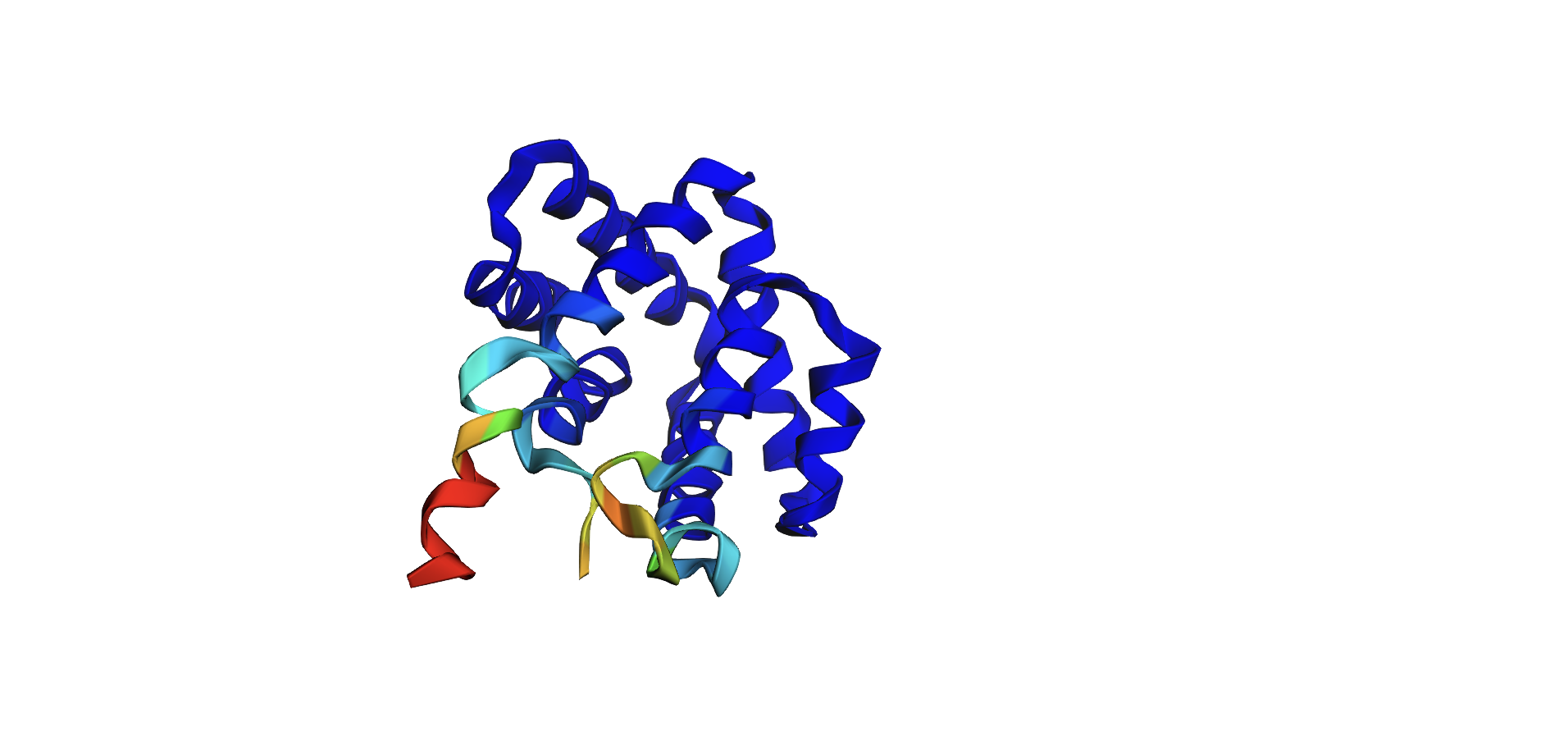
Here’s the predicted fold, colored based on confidence. The red at the end means lower confidence.
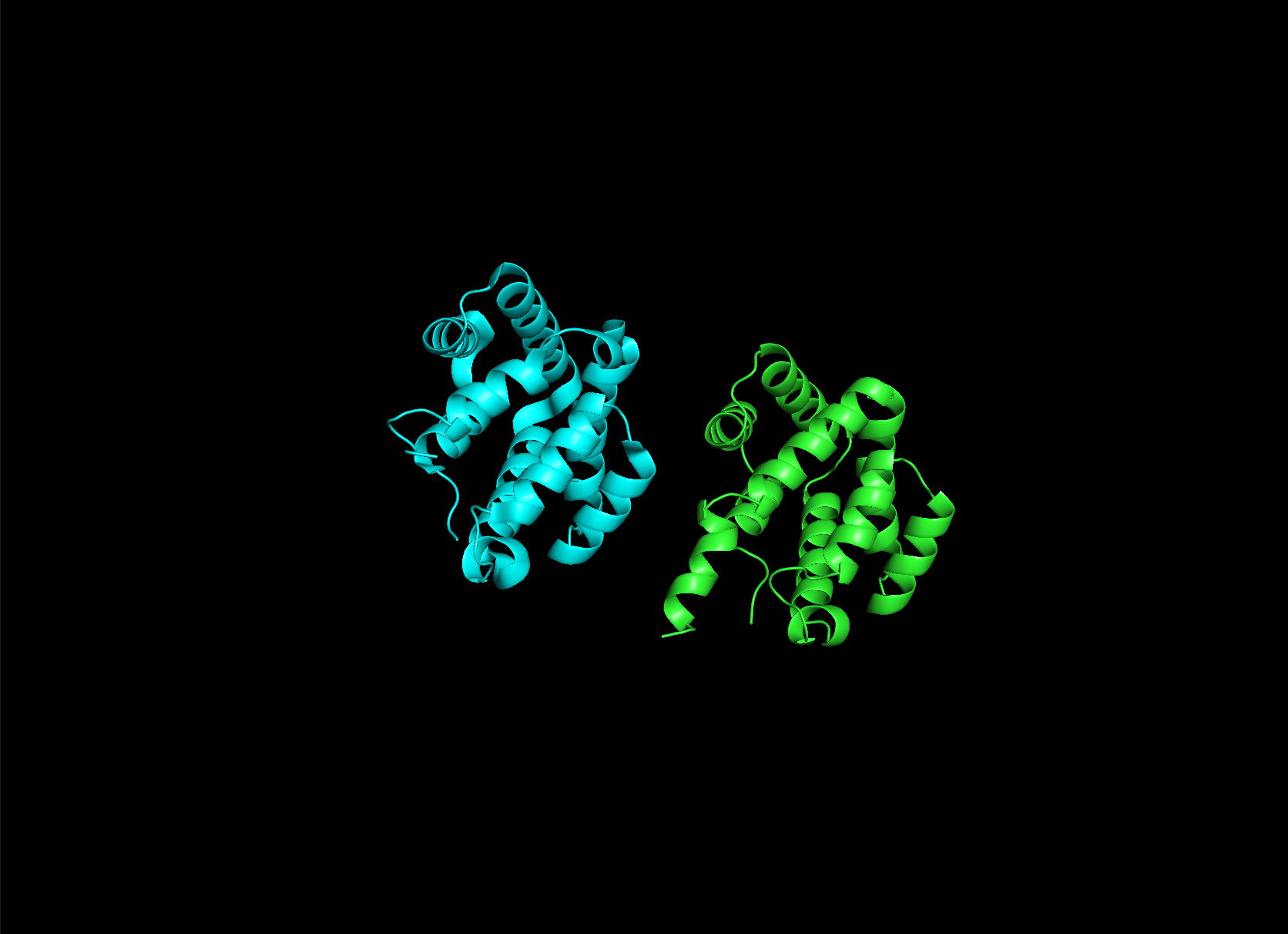
Here it is compared to the actual structure. On the left in cyan is the experimental structure, and on the right is the predicted fold. We can see that the main structure looks similar but the end is totally wrong, in line with the lower confidence. However, this is probably due to the His-tag at the end of the protein:
Note that in PDB data, there were two protein molecules included in the experimental data, apparently as an asymmetric unit, so I removed one for better comparison.
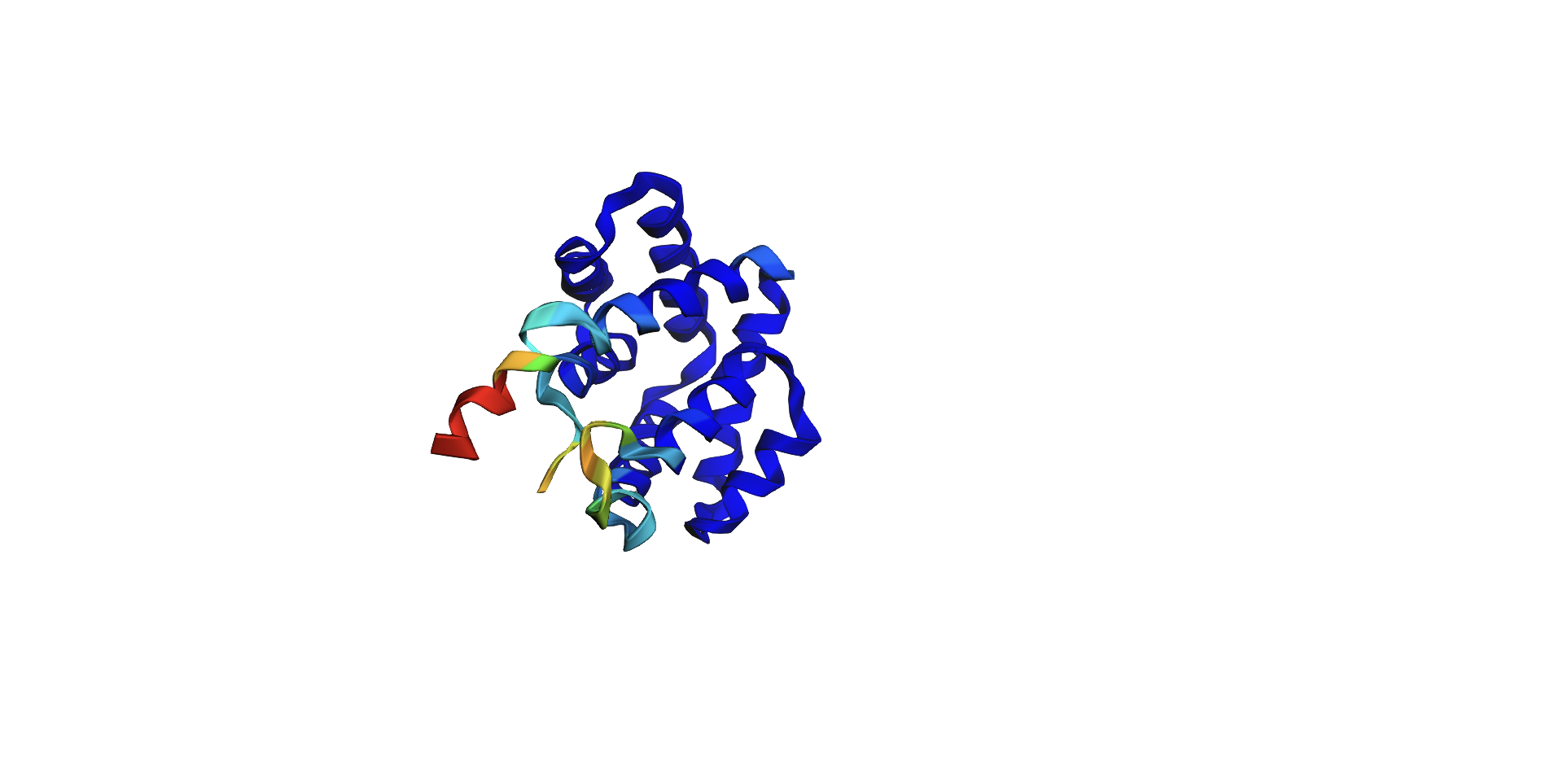
I tried added random pointwise mutations to the protein, and it didn’t appear to affect the fold too much. For example:
Week 5 HW: Protein Design, Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
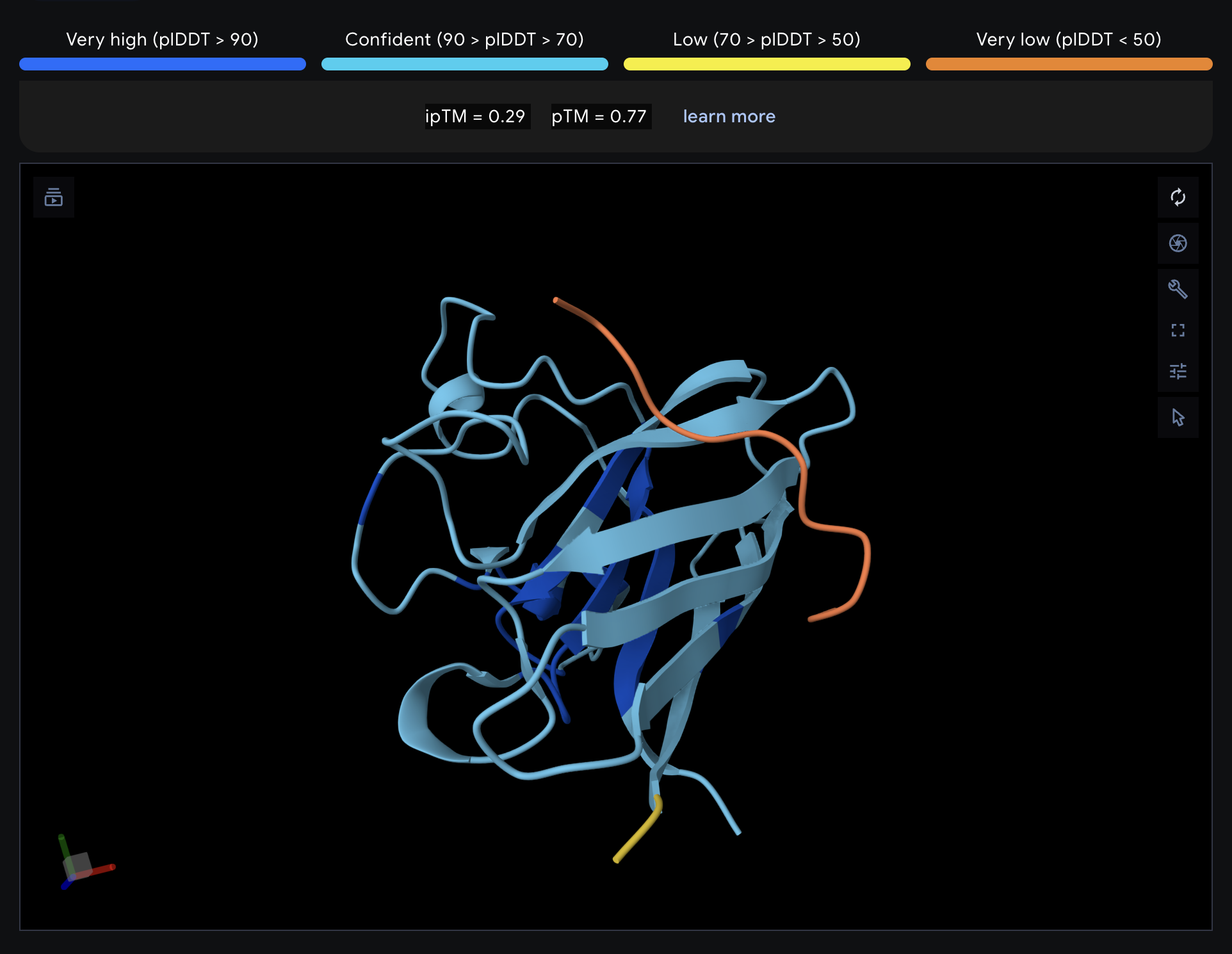
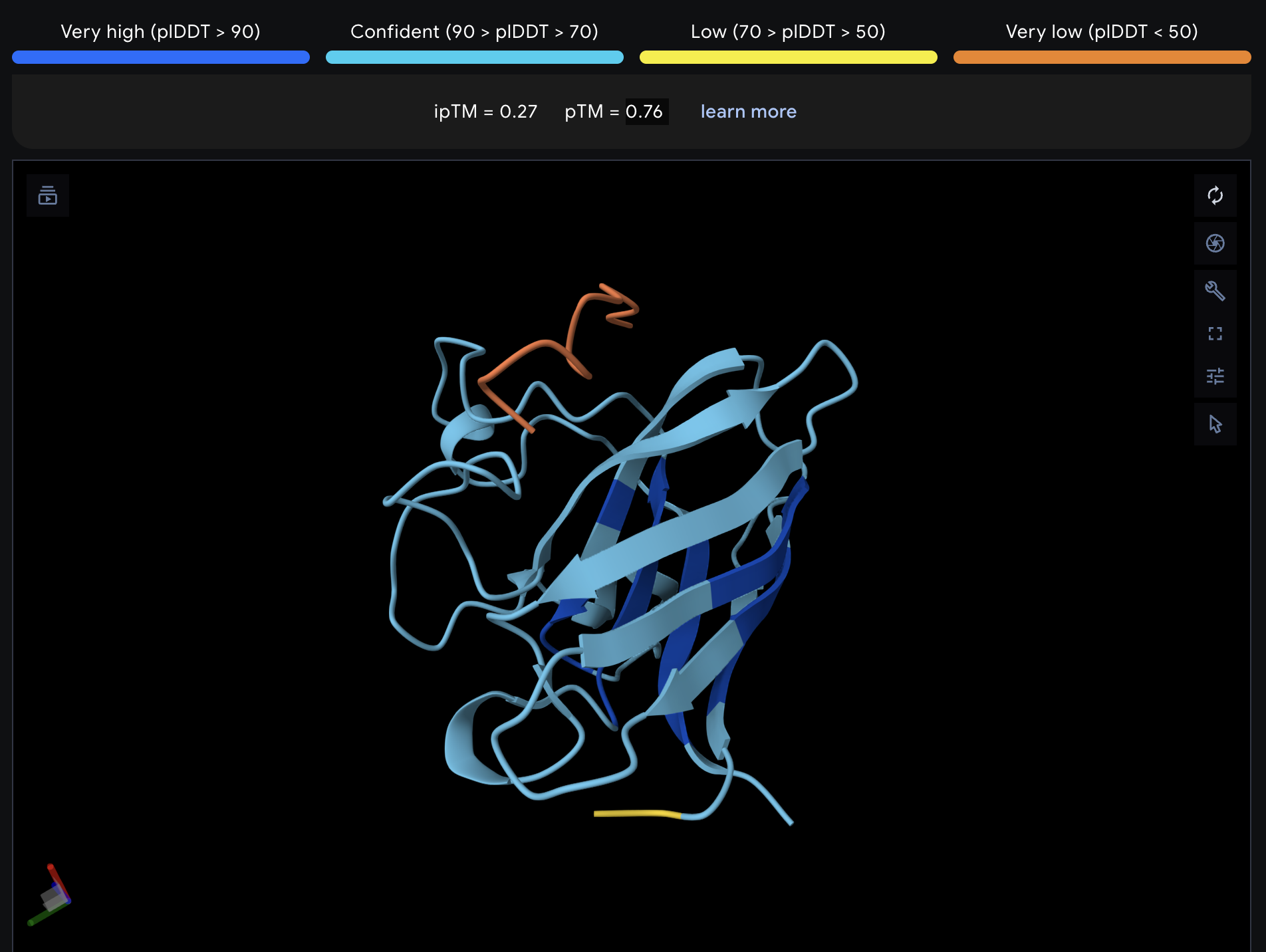
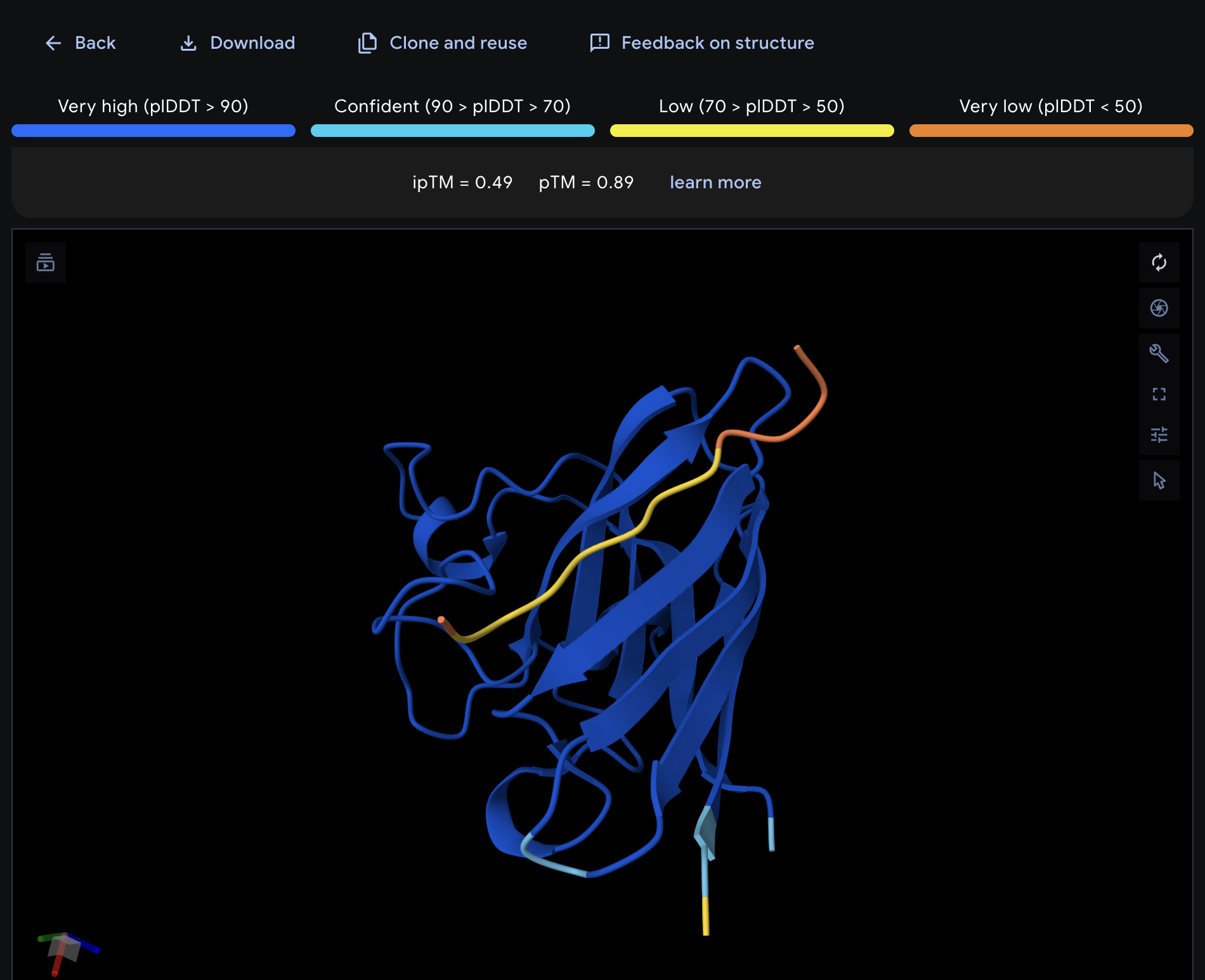
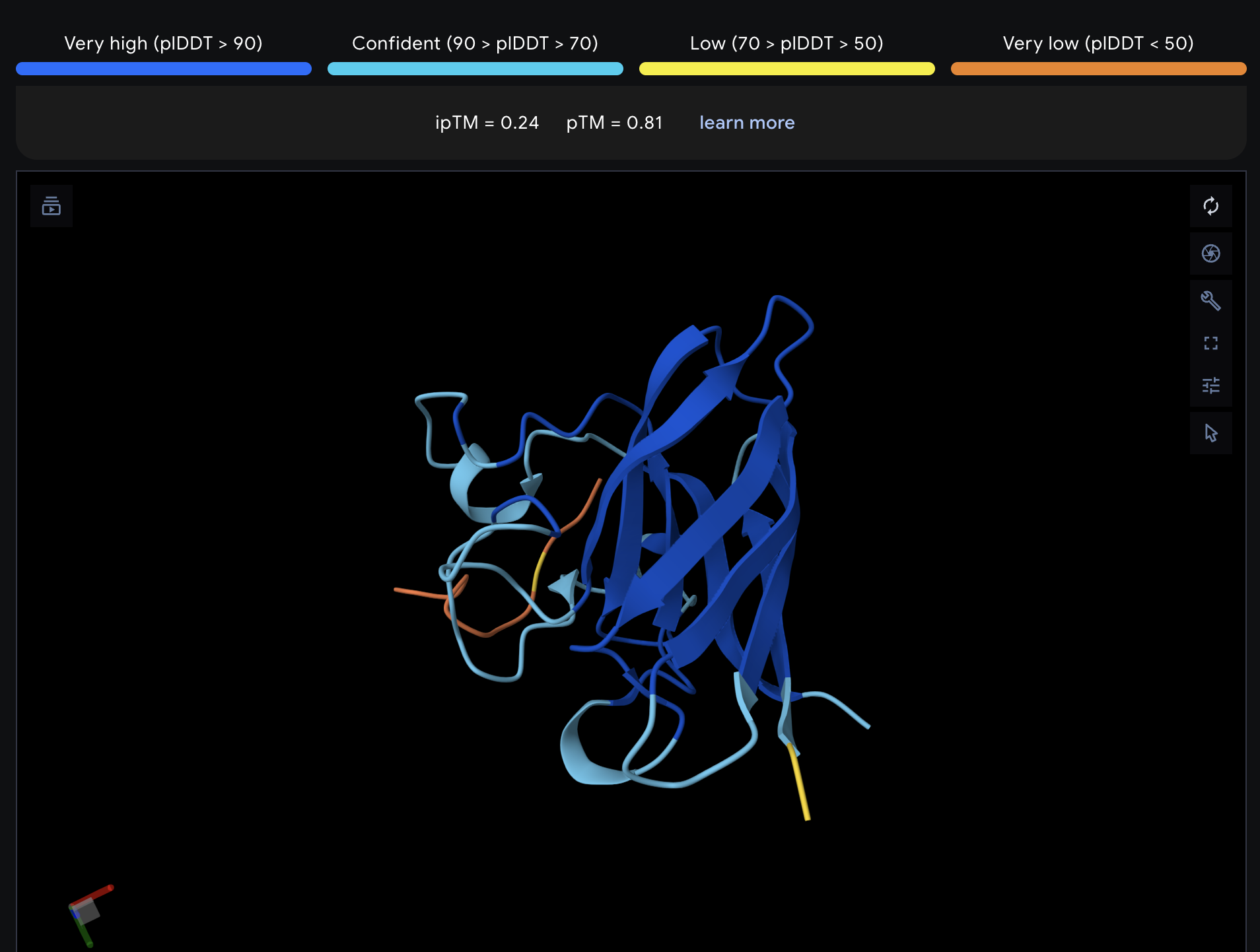
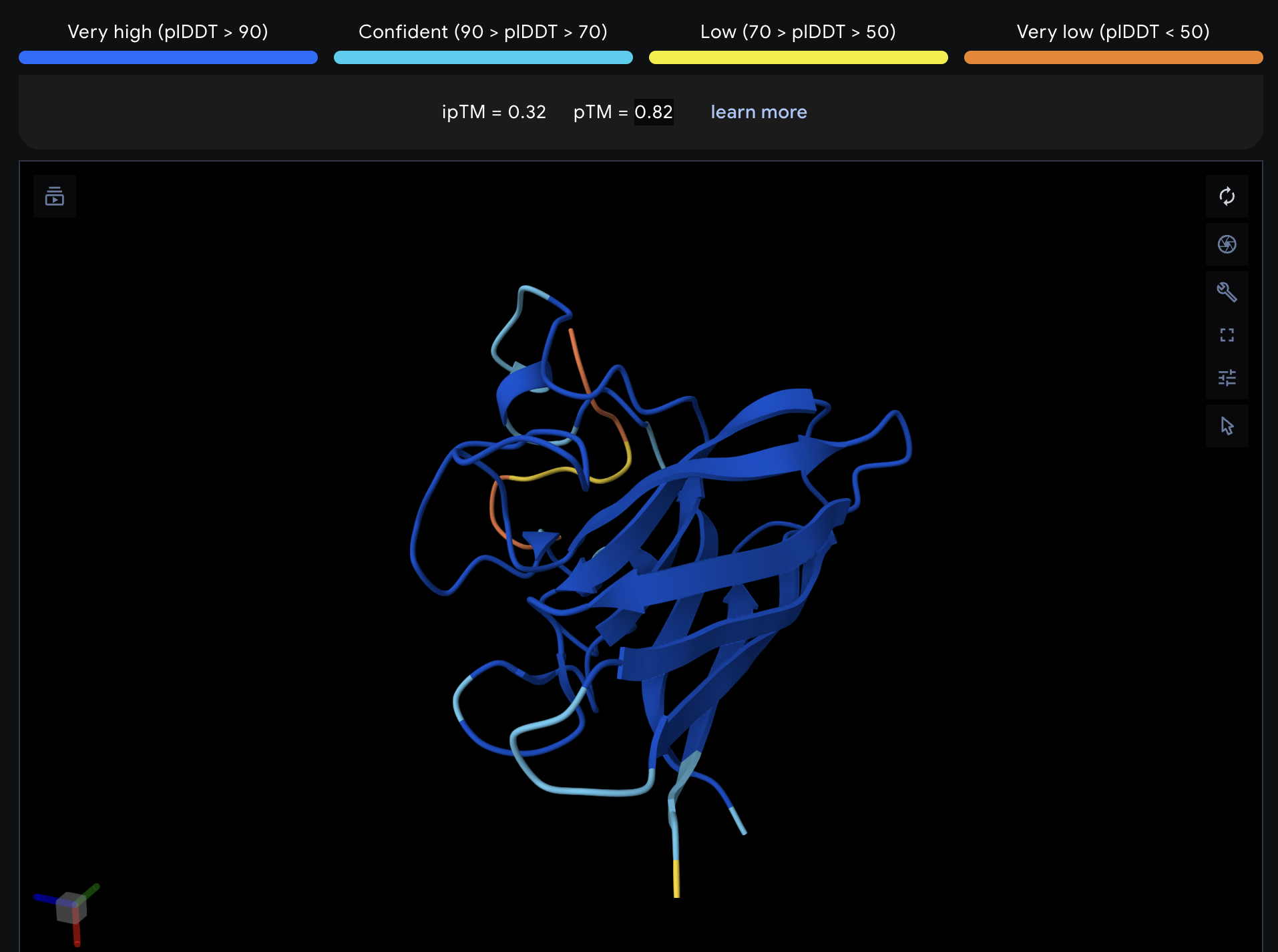
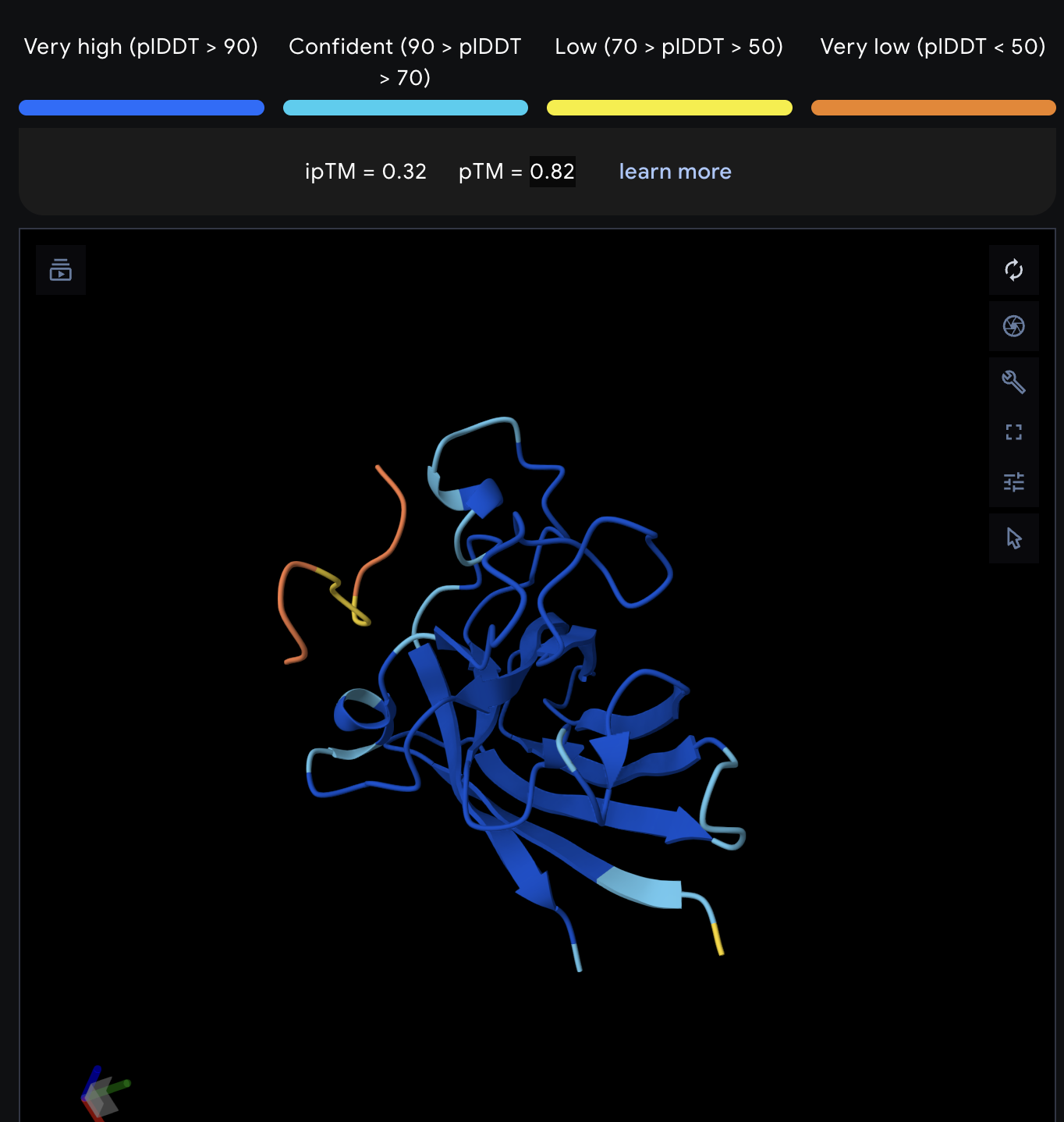
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
All proteins seem largely surface-bound. Binders 0 and 2 seem to engage the beta-barrel region. None seem too close to the dimer interface.
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
One exceeded the known binder: KRYGAVAVRHWA, most are around the same level as the known binder, although the binding sites look wildly different from binder to binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Binder
💧 Solubility
🩸 Hemolysis
🔗 Binding Affinity
📏 Length
⚖️ Molecular Weight
⚡ Net Charge (pH 7)
🎯 Isoelectric Point
💦 Hydrophobicity (GRAVY)
WHYPAVAVALKE
Soluble (1.000)
Non-hemolytic (0.032)
Weak binding (5.591 pKd/pKi)
12 aa
1383.6 Da
-0.15
6.76 pH
0.27 GRAVY
WLYPAVALELKE
Soluble (1.000)
Non-hemolytic (0.052)
Weak binding (5.774 pKd/pKi)
12 aa
1431.7 Da
-1.23
4.86 pH
0.37 GRAVY
KRYGAVAVRHWA
Soluble (1.000)
Non-hemolytic (0.029)
Weak binding (6.059 pKd/pKi)
12 aa
1413.6 Da
2.85
11.00 pH
-0.41 GRAVY
WRYPAAGLELKE
Soluble (1.000)
Non-hemolytic (0.044)
Weak binding (5.565 pKd/pKi)
12 aa
1432.6 Da
-0.23
6.28 pH
-0.70 GRAVY
FLYRWLPSRRGG
Soluble (1.000)
Non-hemolytic (0.044)
Weak binding (5.565 pKd/pKi)
12 aa
1432.6 Da
-0.23
6.28 pH
-0.70 GRAVY
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
The ipTM loosely correlates with the binding affinity. The highest ipTM matches the highest binding affinity. The others hae similar ipTM and they have similar affinities.
All the predicted binders are soluble and none are hemolytic.
KRYGAVAVRHWA looks to be the best based on the criteria, although even it has the best ipTM it is still modest. It bonds to the beta barrel loop.
Choose one peptide you would advance and justify your decision briefly.
I would advance KRYGAVAVRHWA since it has the highest values as predicted by PepMLM and PeptiVerse.
*The “hemolysis” probability is actually non-hemolytic, as per the source, so this heading was updated.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
The peptides from moPPit are more unique and have higher binding affinity. PepMLM peptides share more amino acids with each other, probably because of the encoder model generation.
I would chose the peptides with the highest specificity and use a similar screening as we applied to the PepMLM peptides by checking AlphaFold. Then, I would test in the lab before testing clinically.
Week 6 HW: Genetic Circuits I
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The mix includes: Phusion DNA Polymerase, deoxynucleotides and reaction buffer that has been optimized and includes MgCl2.
DNA Polymerase builds the DNA sequence from deoxynucleotides starting with the template strands and primer. It’s high-fidelity, so it won’t make as many errors as Taq.
Reaction buffer allows the reaction to take place.
What are some factors that determine primer annealing temperature during PCR?
For primer annealing, we want the template to be high enough for the template DNA to be denatured and but low enough that some of the primer is annealed. So the lab protocol suggests to use temperatures near the Tm of the primer (temp where the DNA is 50/50 double/single stranded).
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Restriction enzyme digests are limited to specific sequences in DNA, so we need to make sure that the target site has a existing restriction enzyme cut site nearby. After, we can ligate the strands, but this is non-specific, producing either blunt ends or short sticky ends, so sequences could be joined incorrectly. In PCR we can design short primers to add onto a DNA sequences to create specific sticky ends.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
We can simulate a digest then run a gel through electrophoresis to see if the bands line up.
How does the plasmid DNA enter the E. coli cells during transformation?
From the protocol:
In either of these methods, we shock the cells (either by applying an electrical shock or heat shock) which causes the cell membrane to “open up”. The plasmid now enters the cells by diffusion. After the shock, the cells are fed with SOC growth media and incubated for 1 hour in 37C. That one hour incubation filled with nutrients allows the cells to recover and to start multiplying, some with the plasmid inside. Last, we plate the transformed cells onto a selective media plate, which contains antibiotics.
We heat shock the E. coli to create pores in the bacteria.
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
Golden Gate Assembly is similar to traditional restriction enzyme cloning, but it uses Type IIS instead of classic Type II restriction enzymes. Type IIS enzymes cut outside of the recognition site, unlike classic Type II which cuts in the recognition site. Cutting outside means that the sticky ends can be designed specifically for the joins. It’s similar to Gibson assembly in that the reaction for multiple fragments can take place at the same time, so long as the overhangs are different. The insert can be PCR’ed to add the recognition sites with the overhangs, then ligated with any other fragments to the vector backbone.
Week 7 HW: Genetic Circuits II
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs are analog, instead of digital (e.g. boolean/binary logic), which means they work on numerical values. This means that their behavior can be finely adjusted: they can output specific quantities, and they can take into account the specific/relative magnitudes of inputs. This is well suited for biological applications because biological systems exhibit homeostatic behaviors.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
One example could be releasing a variable dose of therapeutic only if relative proportions of the input signals are out of sync. For example, a theoretical IANN circuit for diabetes could monitor both insulin and glucose levels and only emit insulin if levels are low relative to glucose levels.
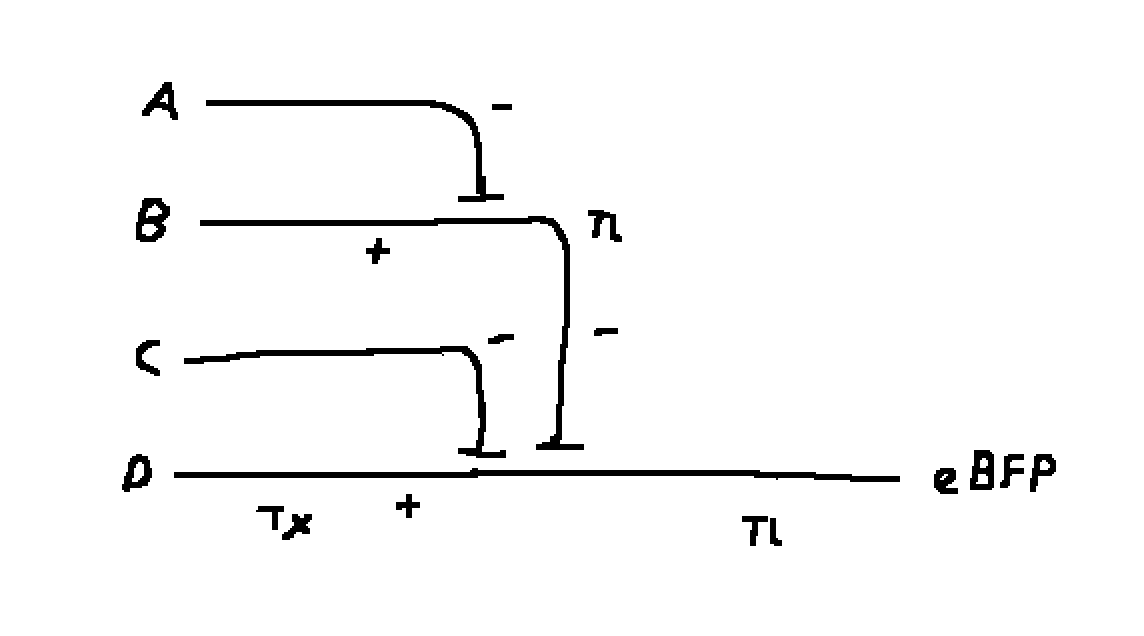
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Layer 1 is A, B, C, D and layer 2 is A - B - C + D.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Some examples include mushroom leather and mycellium packaging. Fungal materials are light, thermally and acoustically insulating and can be grown into moulds. The main disadvantage today is that it is novel so production is currently slow.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
The reason given in lecture by Ren was to better improve the production and quality of fungal biomaterials. Mycellium from fungi is much less fragile than biocement from bacteria, so it if you are interested in engineered biomaterials.
Week 9 HW: Cell-free systems
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell free systems are much simpler to control because the reaction does not have to happen within a cell-wall, so scientists have direct control over concentrations, monitoring, and timing. Cell based production is much harder to control, since you need to control the expression and inputs of the cell. For example, if you need to do experiments to optimize production, or you need to produce proteins that would kill living cells.
Another benefit is that cell free systems can be freeze dried and transported. Living cells require water, which consist of 99% of volume in a biological reaction. Freeze drying allows for much lower cost of transport and storage, allowing for on-demand therapeutics, or rapid manufacturing.
Describe the main components of a cell-free expression system and explain the role of each component.
It is all the parts of the central dogma for protein expression:
DNA: the template DNA for the protein you want to make
DNA polymerase: Copies DNA
RNA polymerase: DNA to RNA
Ribosome: RNA to protein
These are contained in the cell extract.
We also need to add the other things that the cell would have generated for itself, like energy regeneration system.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
ATP is used by the reactions as an energy source, and energy is needed for any of the reactions to take place.
In the recitation, the method presented in purified system diagram uses creatine phosphate to convert ADP to ATP.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotes are much simpler, with plasmid DNA, whereas eukaryotes can produce more complex proteins with post translational modifications. A simple protein like GFP could be a good candidate for prokaryotic systems, whereas eukaryotic systems could be a good candidate for IgG antibodies, which require post translational modification and are native to eukaryotes.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
The challenge for membrane proteins is that they are partially hydrophobic and could misfold in a water solution.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
A few possible reasons could be not enough energy, bottlenecking by transcription, or bottlenecking by translation.
We could measure the system, either by finding the amount of DNA, RNA, or ATP and plotting that over time in order to diagnose where the bottleneck comes from.
Homework question from Kate Adamala
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
My synthetic cell would produce GFP based on the concentration of lactose in the system.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Possibly, but a membrane makes it more ideal if it needs to be used in a biological system, otherwise the chemical concentrations would be diluted.
Could this function be realized by genetically modified natural cell?
Yes, it can be realized in a GMO cell.
Describe the desired outcome of your synthetic cell operation.
It will emit GFP and glow more intensely based on the concentration of lactose.
Design all components that would need to be part of your synthetic cell.
The membrane would be usual materials of phospholipids and cholesterol to make the membrane more stable, with a selective membrane pore to selectively allow lactose through to enable to biosensor.. Bacterial transaction and translation system is okay because the protein and function are simple.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
LacY membrane protein to act as a pore for lactose, GFP expression protein, genes for Lac operon: LacI, LacZ to convert lactose to LacI-binding allolactose, LacO before the GFP gene to be repressed by LacI unless allolactose binds to LacI.
How will you measure the function of your system?
I can measure the amount of light produced in the presence of lactose to see if it is working.
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
How will the idea work, in more detail? Write 3-4 sentences or more.
What societal challenge or market need will this address?
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Homework question from Ally Huang
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
I’m curious about the impact of space on the body’s natural ways of healing. This will go towards understanding how space will affect our bodies and medicine.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
I’m interested in using spatial transcriptomics, which images the expressed proteins in space.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Using the same approach of the FISSEQ paper, I’m interested in seeing what genes are expressed during wound healing, but in the unique environment of space.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
My hypothesis is that gene expression will be modified without gravity and wounds will heal slower as a result of those changes.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
I will simulate wound healing using fibroblasts in space and on Earth, then use FISSEQ and compare the resulting expressed sequences using statistical techniques.