Week 2 HW: DNA Read, Write, and Edit
Part 0: Basics of Gel Electrophoresis
I watched the lecture, recitation, and read the lab. Essentially, we use the negative charge of DNA to pull DNA fragments towards a positive anode in a porous agarose gel. Larger DNA fragments move slower in the agarose gel.
Part 1: Benchling & In-silico Gel Art
I spent some time playing around with Ronan’s gel art site to make a pattern (below on the left). I noticed that some of the restriction enzymes in the gel art tool weren’t on the HTGAA enzyme list, so I didn’t use them.
I think it looks kind of like Darth Vader.
Then, I added the Lambda DNA to Benchling. I made a custom enzyme list with the EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI. Then, I added the restriction enzymes from the gel art tool to make a virtual digest (below on the right). I had some difficulty ordering the digests properly, so I saved them with the names and ordered them by dragging the tabs after.
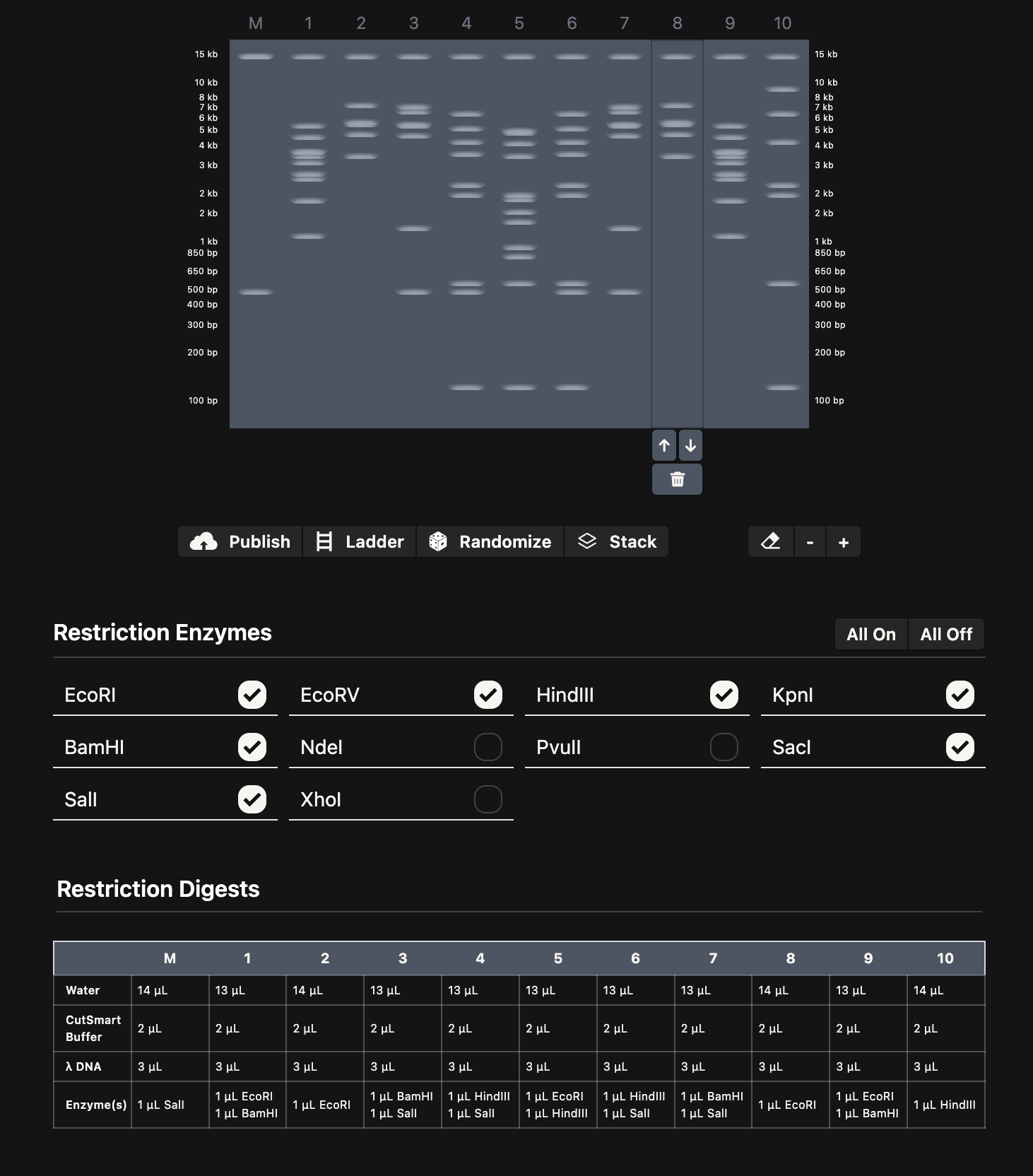
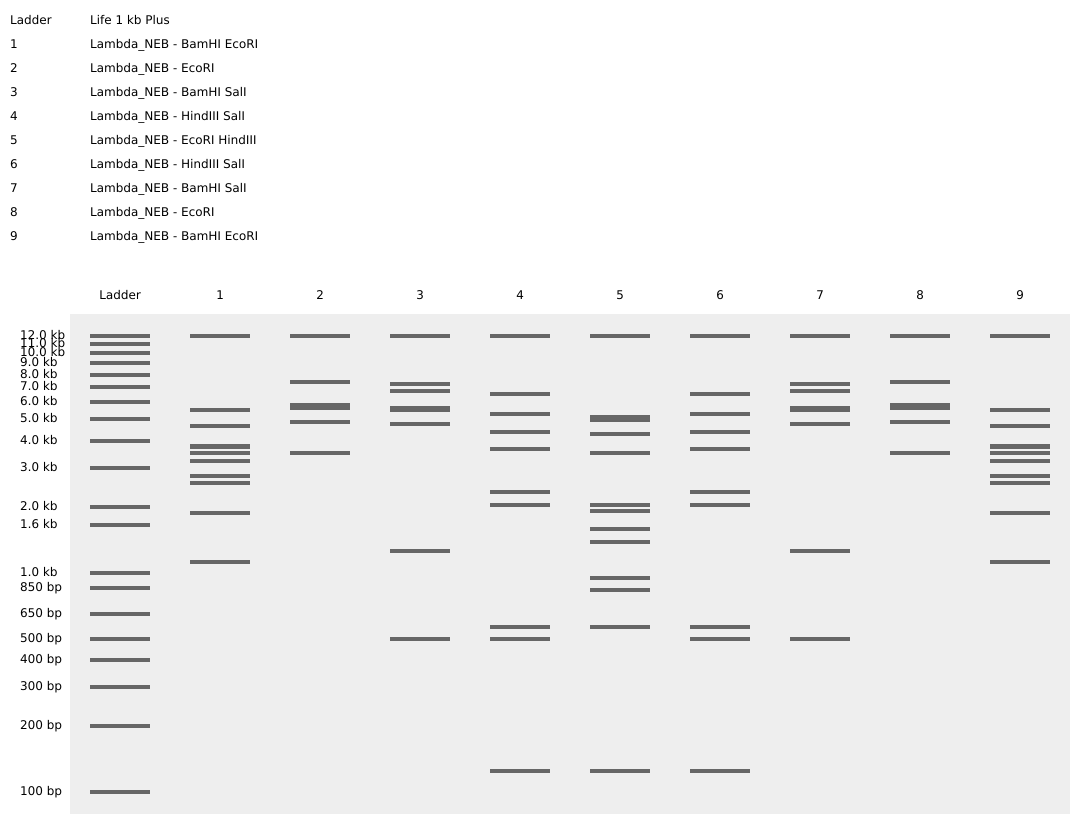
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis (Wet Lab)
N/A: This is optional for committed listeners and I didn’t have access to a wet lab this week.
Part 3: DNA Design Challenge
3.1. Choose your protein
The protein I chose is Miraculin, which is from the miracle berry and famous for temporarily causing sour things to taste sweet. I picked this because I have tried a miracle berry tasting before and it was an interesting experience.
Here is the UniProt entry for Miraculin. UniProt also tags a number of other taste-modifying proteins.
The sequence for Miraculin (in FASTA) is:
3.2. Reverse Translate
I used tblastn (Translated BLAST) to get the nucleotide sequence that corresponds with Miraculin. This found two nucleotide sequences in a database: AB512278.1 and D38598.1. One appears to be the mRNA rather than the genes.
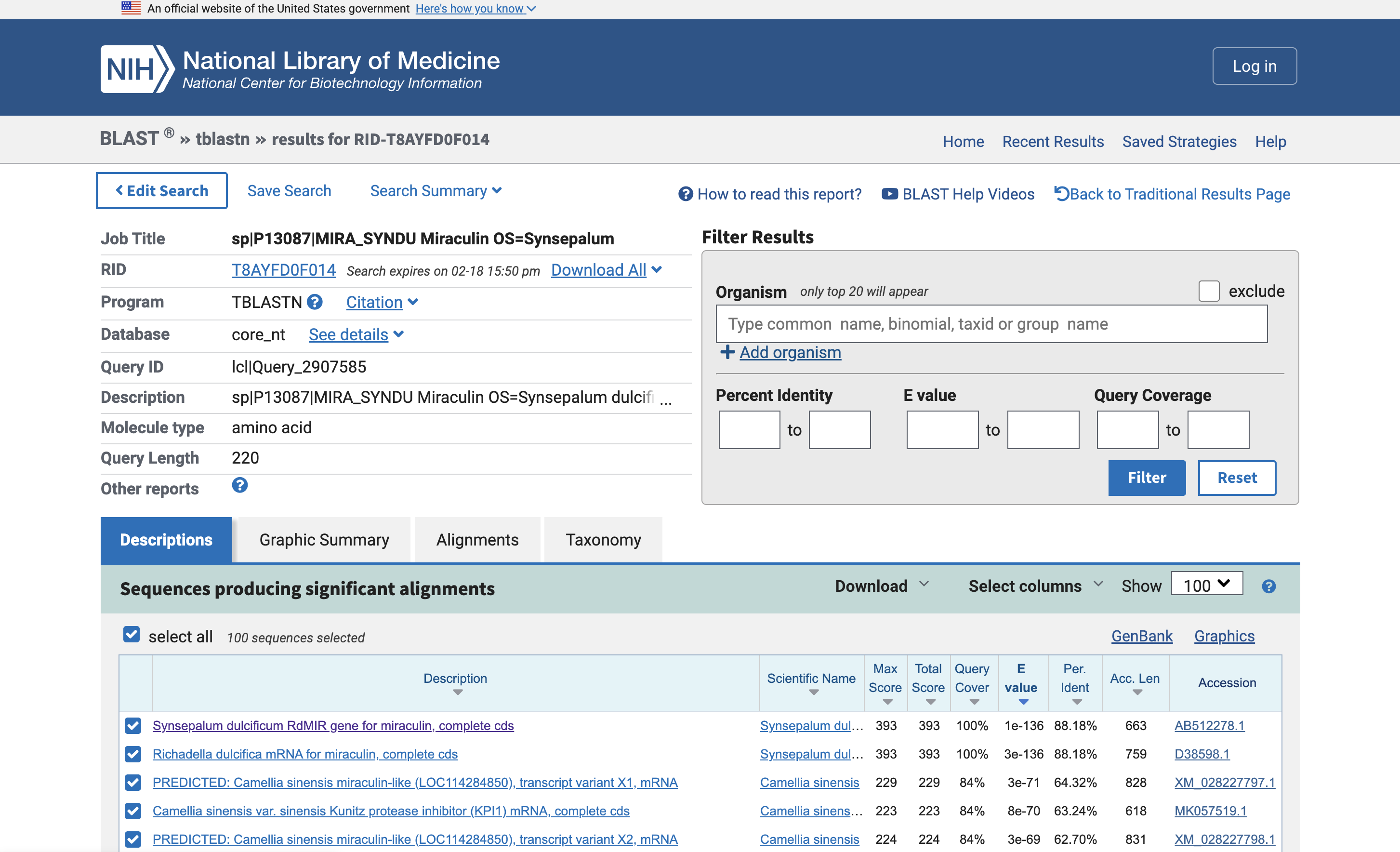
Here is the FASTA for the gene, from AB512278.1:
3.3. Codon optimization
I used VectorBuilder’s codon optimization tool since Twist’s was down for maintenance. I optimized for E. Coli, so the protein could be mass produced in its “cellular factory”. It gave the following:
Pasted Sequence: GC=51.43%, CAI=0.56
Improved DNA[1]: GC=55.81%, CAI=0.94
3.4. You have a sequence! Now what?
Since I chose E. Coli, we can order the gene with a promoter in a plasmid, then use a cell-dependent method of heat shocking the E. Coli to embed the plasmid, then cultivating the E. Coli to produce lots of this protein.
This uses the natural plasmid gene expression mechanisms of E. Coli to transcribe and translate the protein.
Part 4: Prepare a Twist DNA Synthesis Order
I followed the steps to make provided sfGFP sequence in Benchling. Here is my Benchling project.
Here is the FASTA file of the expression cassette:
I imported that into Twist and added the vector. Here is a link to the Benchling project with the Twist draft order.
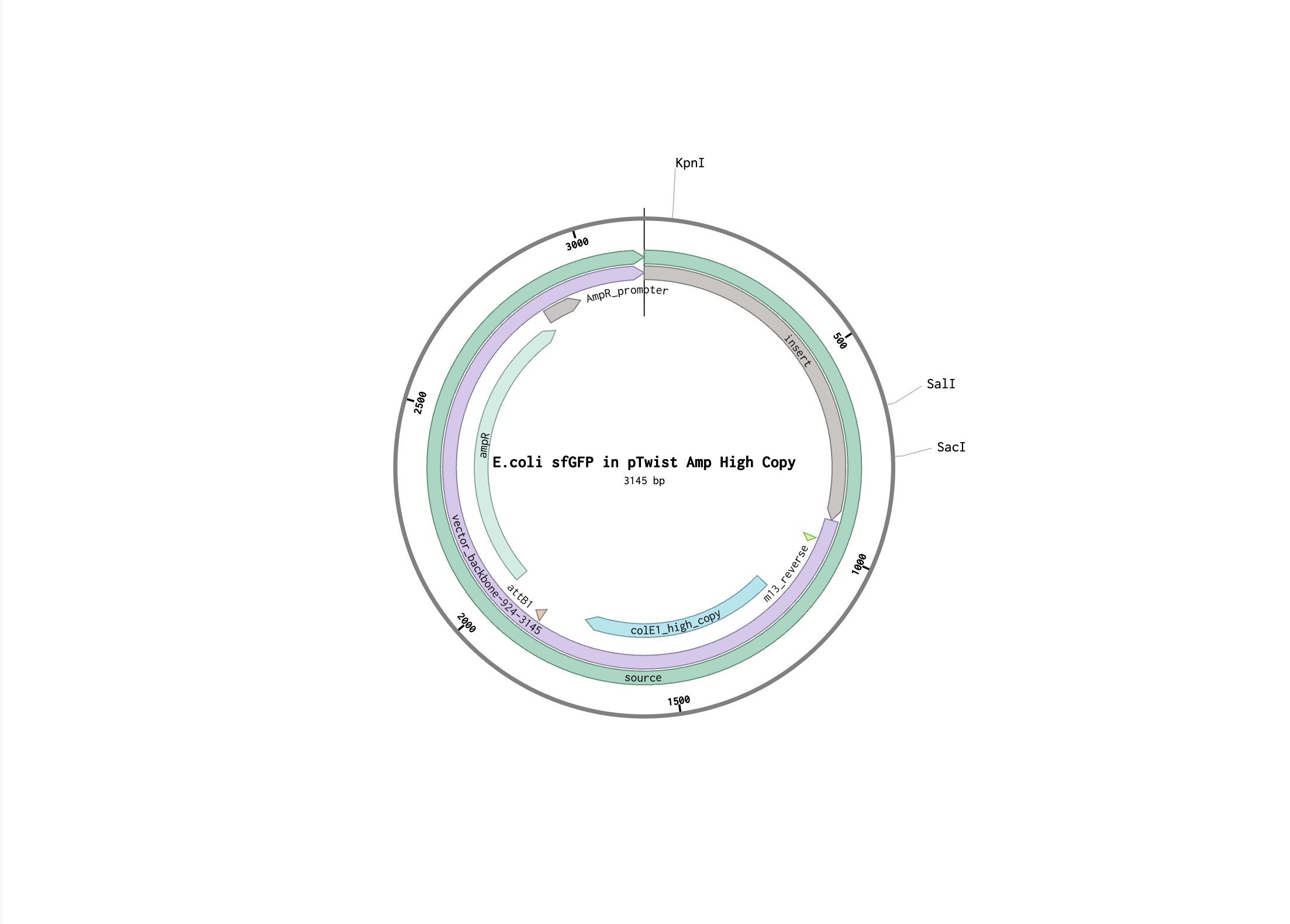
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I’d like to sequence DNA of the human microbiome. There’s been recent research about how beneficial flora of the gut and skin microbiome contribute to our health, and there’s already a significant effort to sequence our microbiome in the Human Microbiome Project.
I would also be interested in the widespread sequencing and cataloguing of viruses that make up the common cold. I think it could be useful to detect the geographic spread of these viruses and how they mutate over time, to potentially contribute to a cure.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Sanger Sequencing, since it’s a straightforward and well-tested technique, and I understand it the best.
- Is your method first-, second- or third-generation or other? How so?
Sanger Sequencing is a first-generation technique. It’s the earliest and most classic form of sequencing.
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input is DNA, regular nucleotides (d*), chain-terminating nucleotides (dd*), primer (like in PCR), DNA-polymerase. You prepare the input by PCRing the sample to have lots of DNA.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
You thermocycle the amplified sample with the nucleotides, primers, and polymerases to build many DNA fragments. These DNA fragments will be different lengths because probabilistically each fragment will incorporate a chain-terminating nucleotide which stops polymerization. Finally, the fragments are run through electrophoresis and imaged one base pair at a time to get the sequence.
- What is the output of your chosen sequencing technology?
The result is the electrophoresis imaging data, which can be processed to determine the most likely base pair at each position.
One limitation of the technique is that you need a pure sample of DNA, so it may be inefficient for the volumes of organisms we’d want to sequence.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would want to synthesize DNA origami, as art! I’m curious what it would take to make the smallest art pieces.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use the phosphoramidite method.
- What are the essential steps of your chosen sequencing [sic; synthesis?] methods?
Deprotection, coupling, capping, oxidation, and repeat.
- What are the limitations of your sequencing [sic; synthesis?] method (if any) in terms of speed, accuracy, scalability?
The limitation, as discussed last homework, is the length of DNA oligos that can be synthesized with this technique. However, this isn’t a problem for DNA origami, which doesn’t need full length DNA.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would edit human DNA, for example to cure hearing loss and tinnitus as a gene therapy. Hearing loss is permanent and affects 5% of the world population. Noise exposure from work and the environment also contribute to increased rates of hearing loss. Birds, unlike humans, can regenerate inner ear cells, and researchers have demonstrated regrowth in cell cultures so there is a theoretical target for gene therapy. There have already been successful gene therapy treatments for deafness in children due to congenital disorders.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-Cas9, since it is the most popular technique today.
- How does your technology of choice edit DNA? What are the essential steps?
Cas9 cuts the DNA, then introducing the edits via homology directed repair.
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
The guide RNA for Cas9 needs to be designed, as well as the template DNA (for knock-ins).
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Homology-directed repair is not entirely efficient, since the ends that Cas9 break could rejoin without the knock-in sequence. Performing CRISPR in humans (rather than bacteria or cell cultures) requires sophisticated deliveries to get to the target cells or tissues.