Week 4 HW: Protein Design, Part I
Part A: Conceptual questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
A dalton is 1.66053906892(52)*10−23 g, so 500g = 500 / 1.66053906892(52)*10−23 = 3.0110704e+25 daltons.
If an amino acid averages 100 daltons, then 3e+25 daltons is about 3e+23 amino acids.
~300,000,000,000,000,000,000,000 amino acids!
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We digest the meat and it does not alter our genetics.
- Why are there only 20 natural amino acids?
Codon redundancy allows for both more efficient genetic coding and error correction. 20 appears to be enough (as evidenced by life).
- Can you make other non-natural amino acids? Design some new amino acids.
Yes, you can make non-natural amino acids (e.g. non-proteinogenic amino acids). Amino acids require an amine, a carboxyl, a central carbon, and a side-chain. You could design one by using a unique side chain that doesn’t exist in the natural amino acids.
- Where did amino acids come from before enzymes that make them, and before life started?
They formed abiotically through natural reactions in the environment. They have even been found on meteorites.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
I would expect it to turn the other way, since D-amino acids are mirrored. Normal alpha helixes are right handed, so I would expect a left handed helix.
- Can you discover additional helices in proteins?
Skipped (1/2).
- Why are most molecular helices right-handed?
Skipped (2/2).
- Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
This is because beta sheets can be hydrophillic on one side and hydrophobic on the other, forming a “pleated sheet”.
- Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Shugang mentions that β-sheets can aggregate to form disease because β-sheets can’t be easily untangled once formed.
Yes, β-sheets give spider silk its unique properties, which inspired the design of materials like Kevlar.
- Design a β-sheet motif that forms a well-ordered structure.
We can use the rule from Shugang’s slides: alternate hydrophobic and hydrophillic for every other amino acid.
Part B: Protein Analysis and Visualization
I’m picking the same protein as in week 1: Miraculin, a taste altering protein.
Amino acid sequence:
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
Using the Colab, I got:
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
BLAST yielded 249 similar proteins with significant identities (~50%), high scores (~200), and low e-values (< 0.05).
These include proteins from tea, fruit, and tobacco.
Does your protein belong to any protein family?
According to UniProt:
Belongs to the protease inhibitor I3 (leguminous Kunitz-type inhibitor) family.
Identify the structure page of your protein in RCSB
Miraculin isn’t in RCSB, so I switched to D7TY99, a homolog found in grapes.
This one does have an RCSB entry.
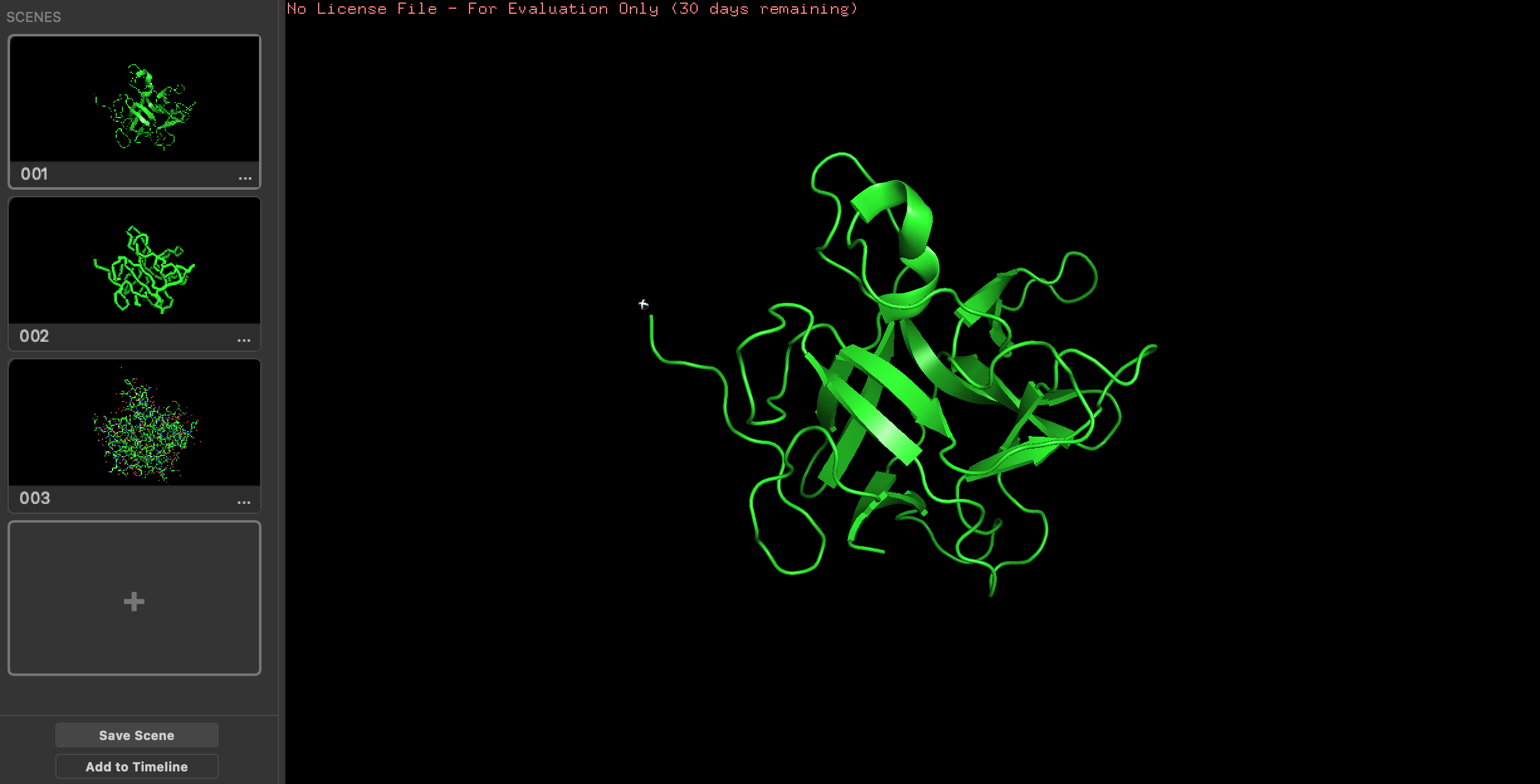
Open the structure of your protein in any 3D molecule visualization software
Here it is visualized in PyMol (different visualization types on the left):
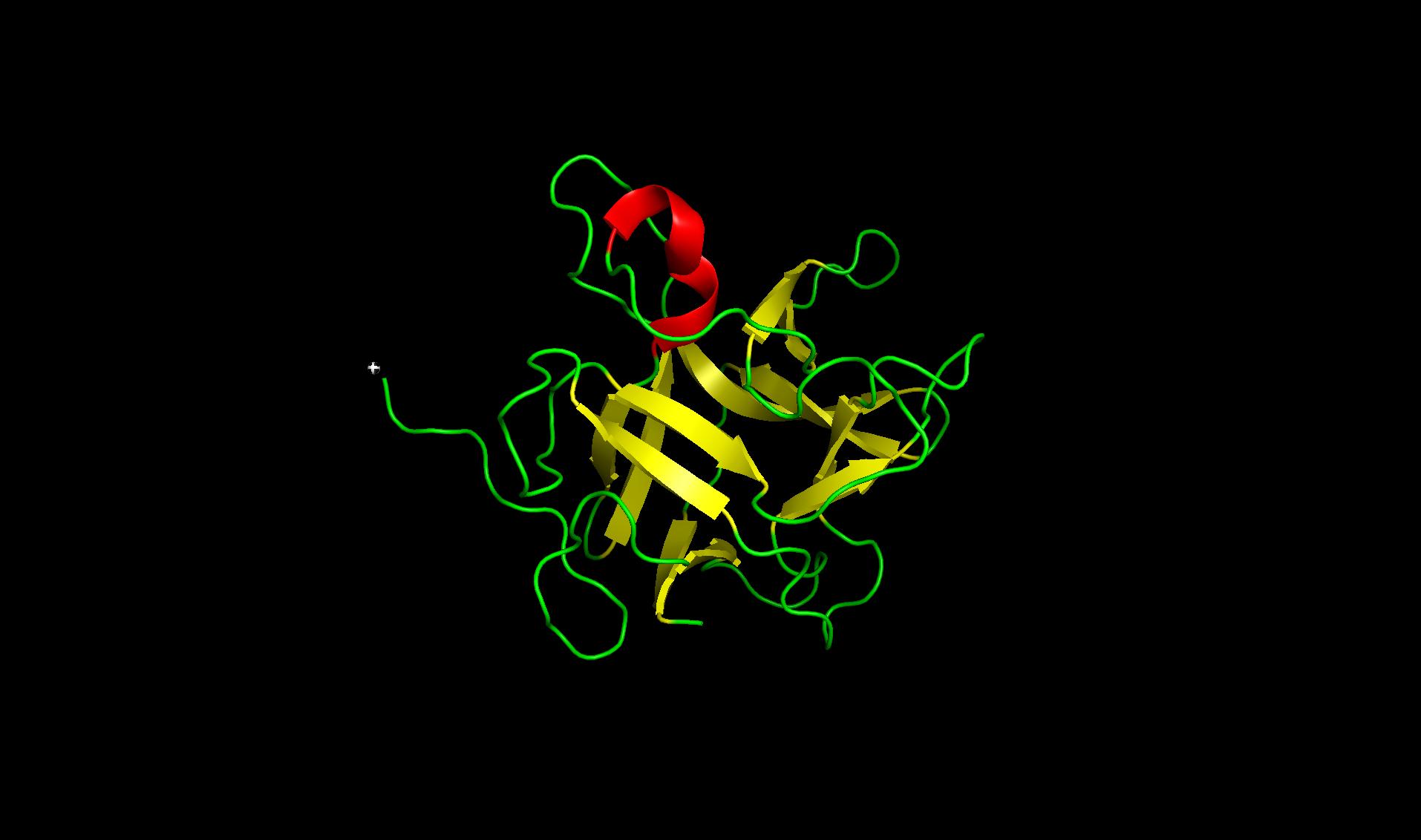
It has more sheets than helixes. Here is the secondary structure sheets colored yellow and helix colored red.
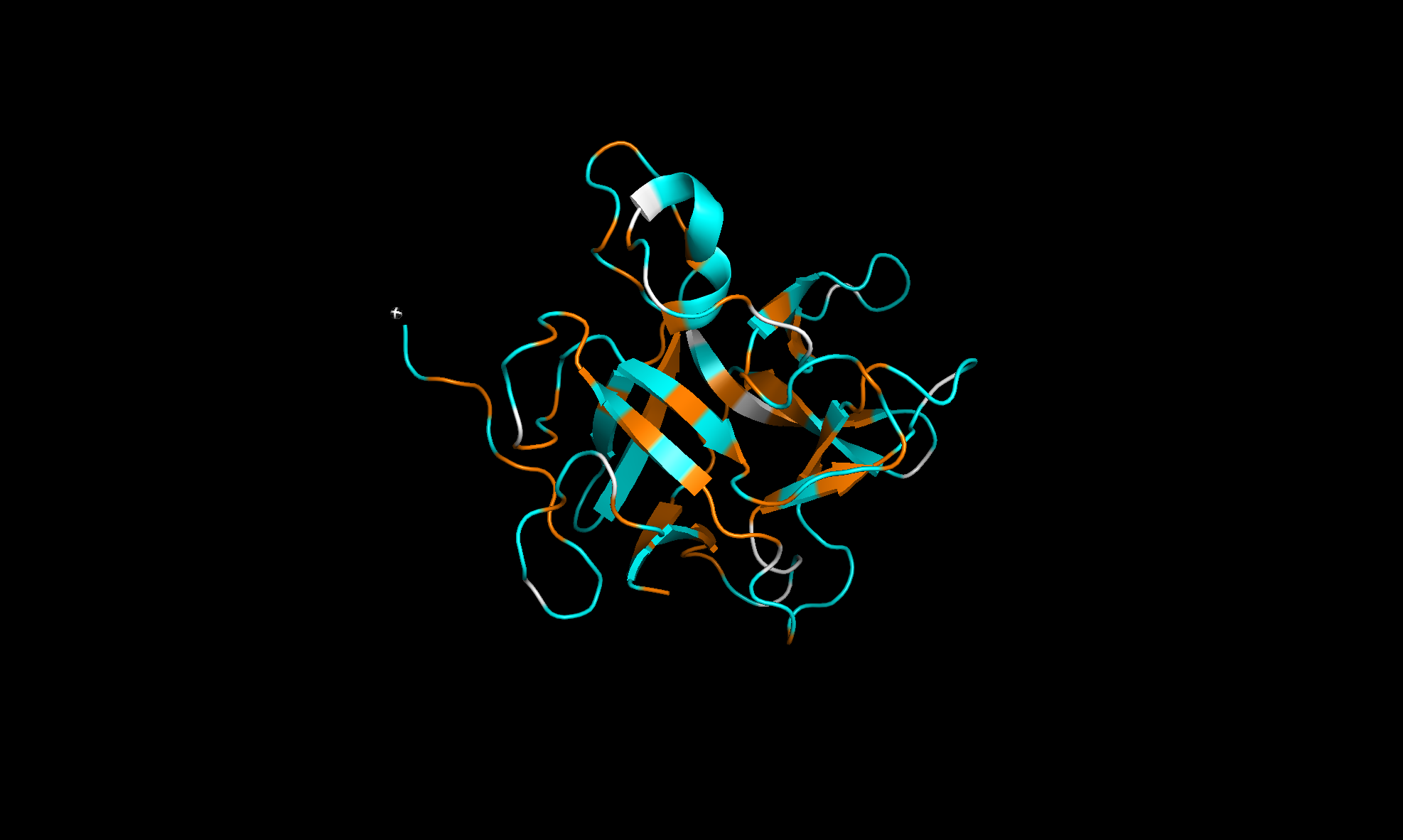
Here with the residue types colored with orange for hydrophobic and cyan for hydrophilic. The regions alternate and looks like more hydrophilic:
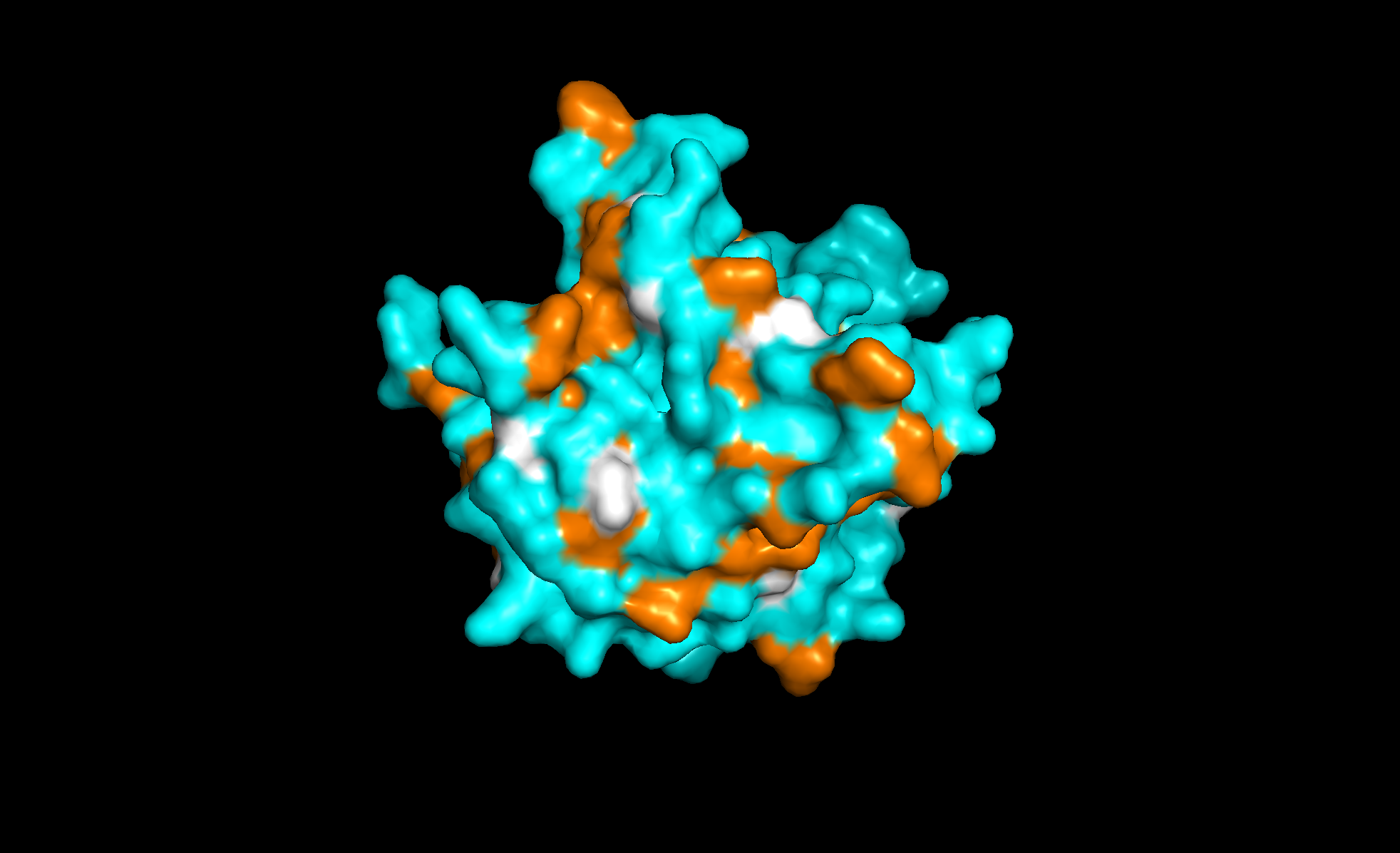
Finally, the surface of the protein. There are at least three “holes” that look like binding sites:
Using ML-based protein design tools
For the ML tools, I’ll be using Fel d 1, which is a major cat allergen protein.
It’s made of two linked peptides: Major allergen I polypeptide chain 1 and Major allergen I polypeptide chain 2 .
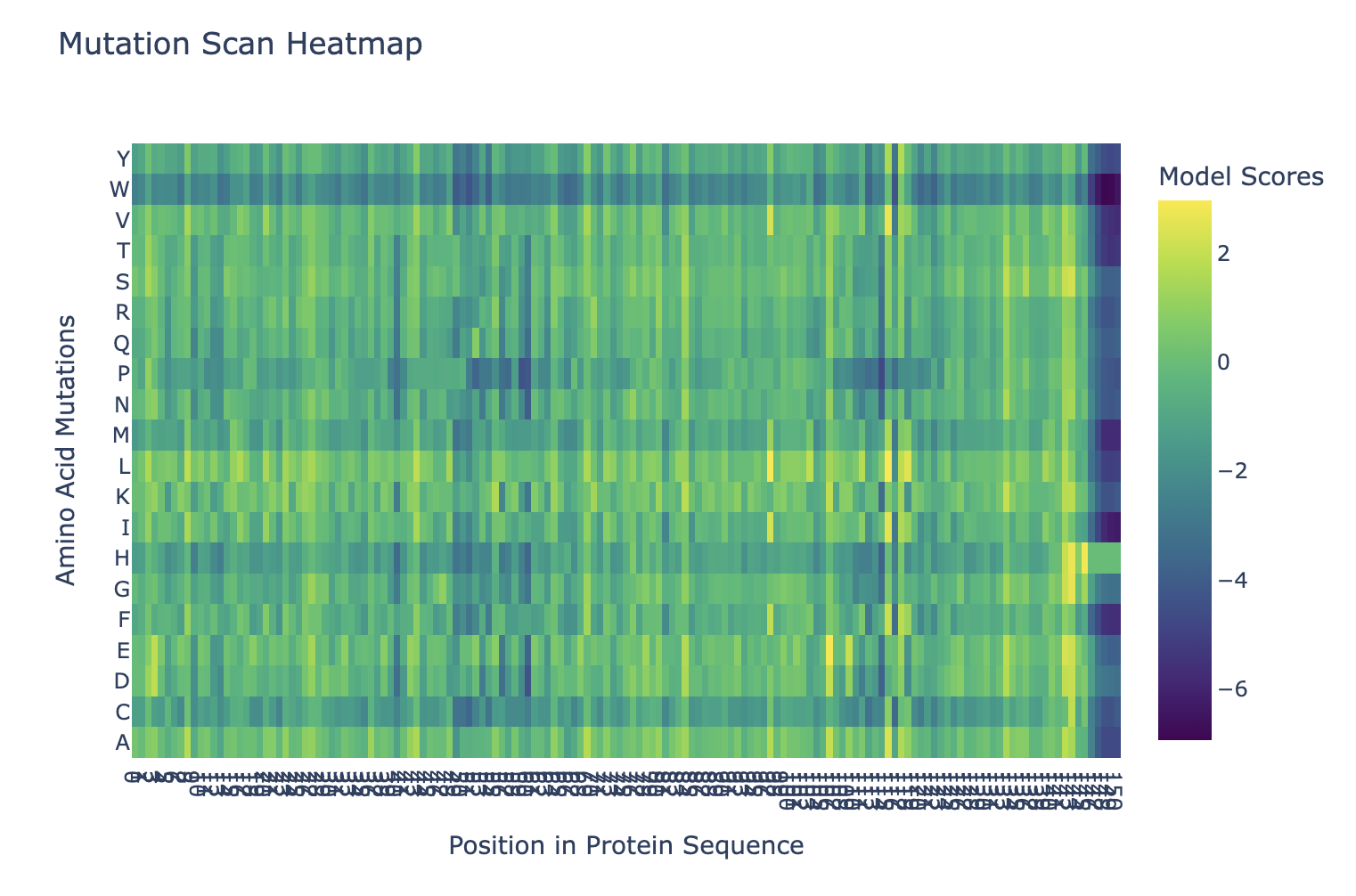
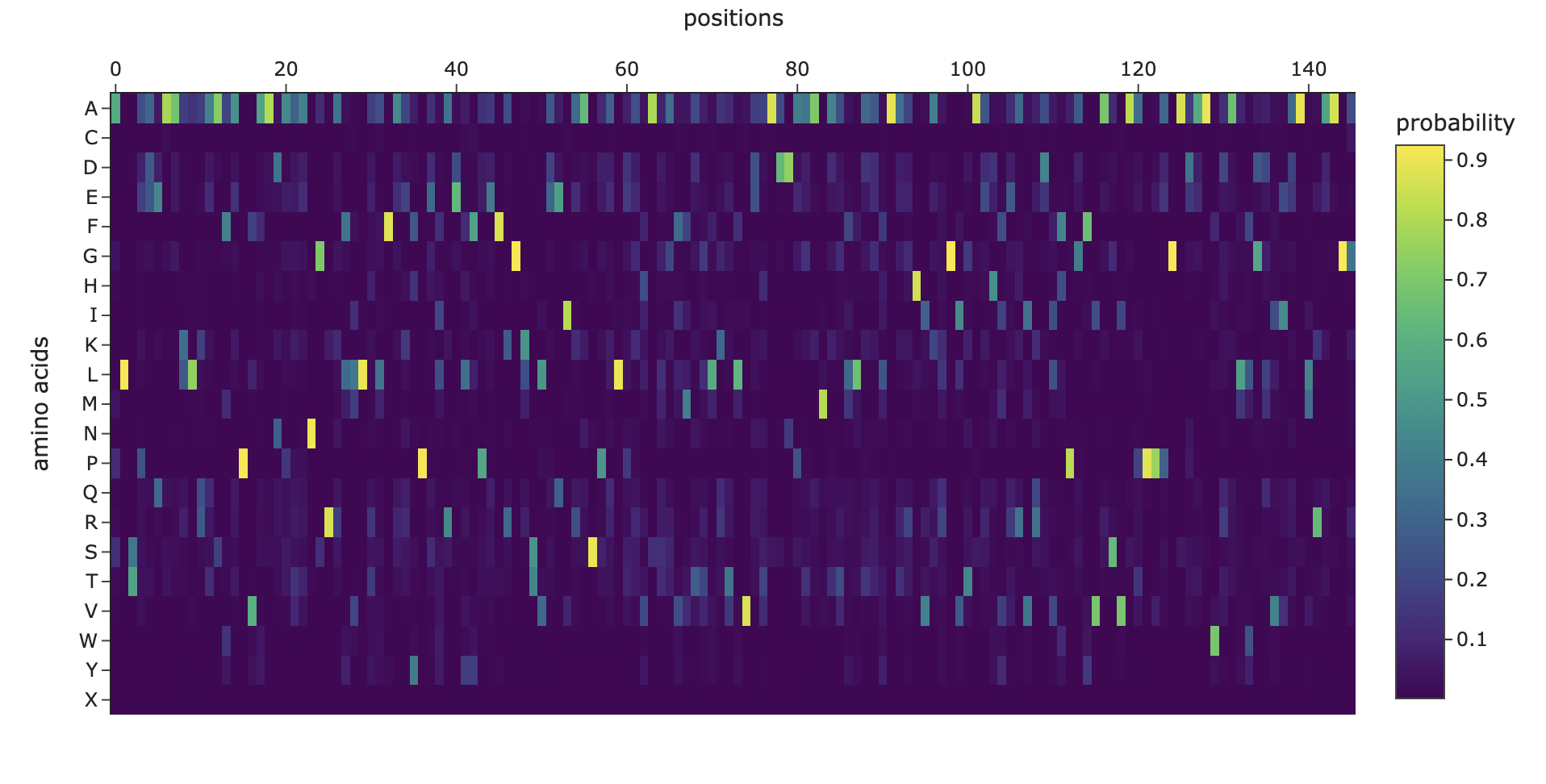
Here’s the mutation scan heatmap. It looks like the amino acids at the end of the protein are most sensitive to mutation. We can also see that “W” tends to be a bad mutation anywhere in the protein.
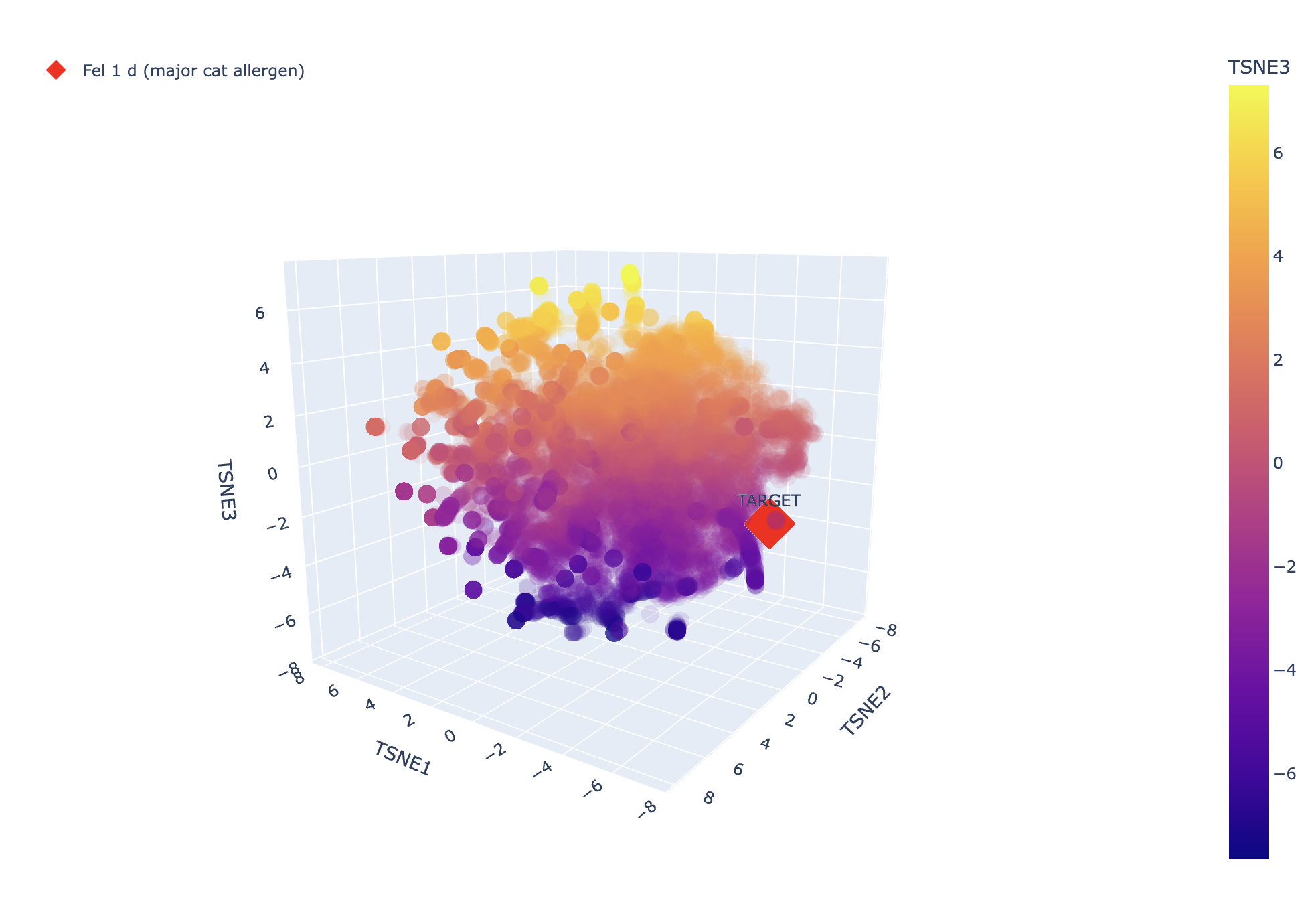
And here’s the TSNE, it looks like it’s not very closely related to the other proteins, which would make sense since it’s a unique allergen (otherwise we might expect cat allergies to correlate with lots of other allergies).
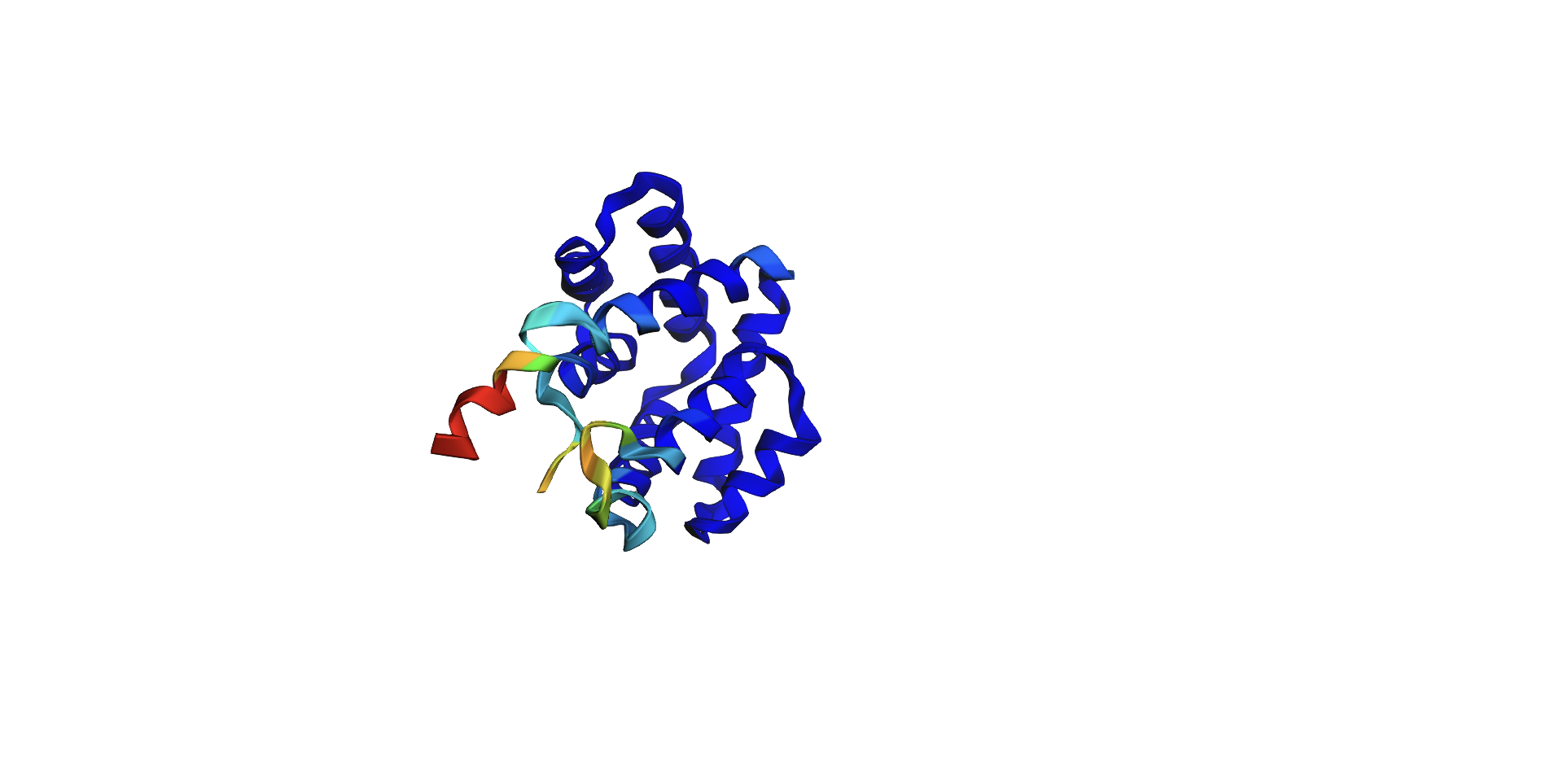
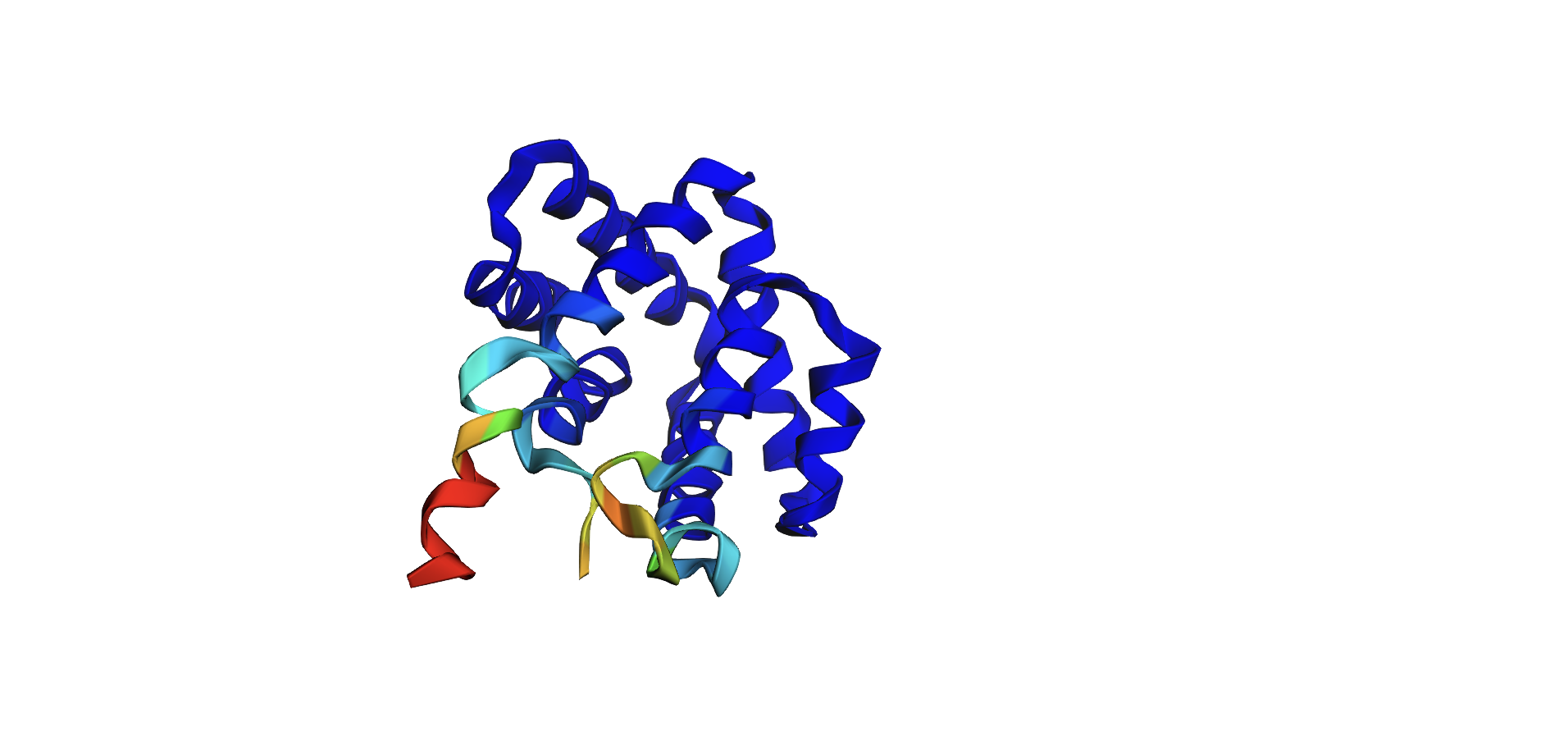
Here’s the predicted fold, colored based on confidence. The red at the end means lower confidence.
Here it is compared to the actual structure. On the left in cyan is the experimental structure, and on the right is the predicted fold. We can see that the main structure looks similar but the end is totally wrong, in line with the lower confidence. However, this is probably due to the His-tag at the end of the protein:
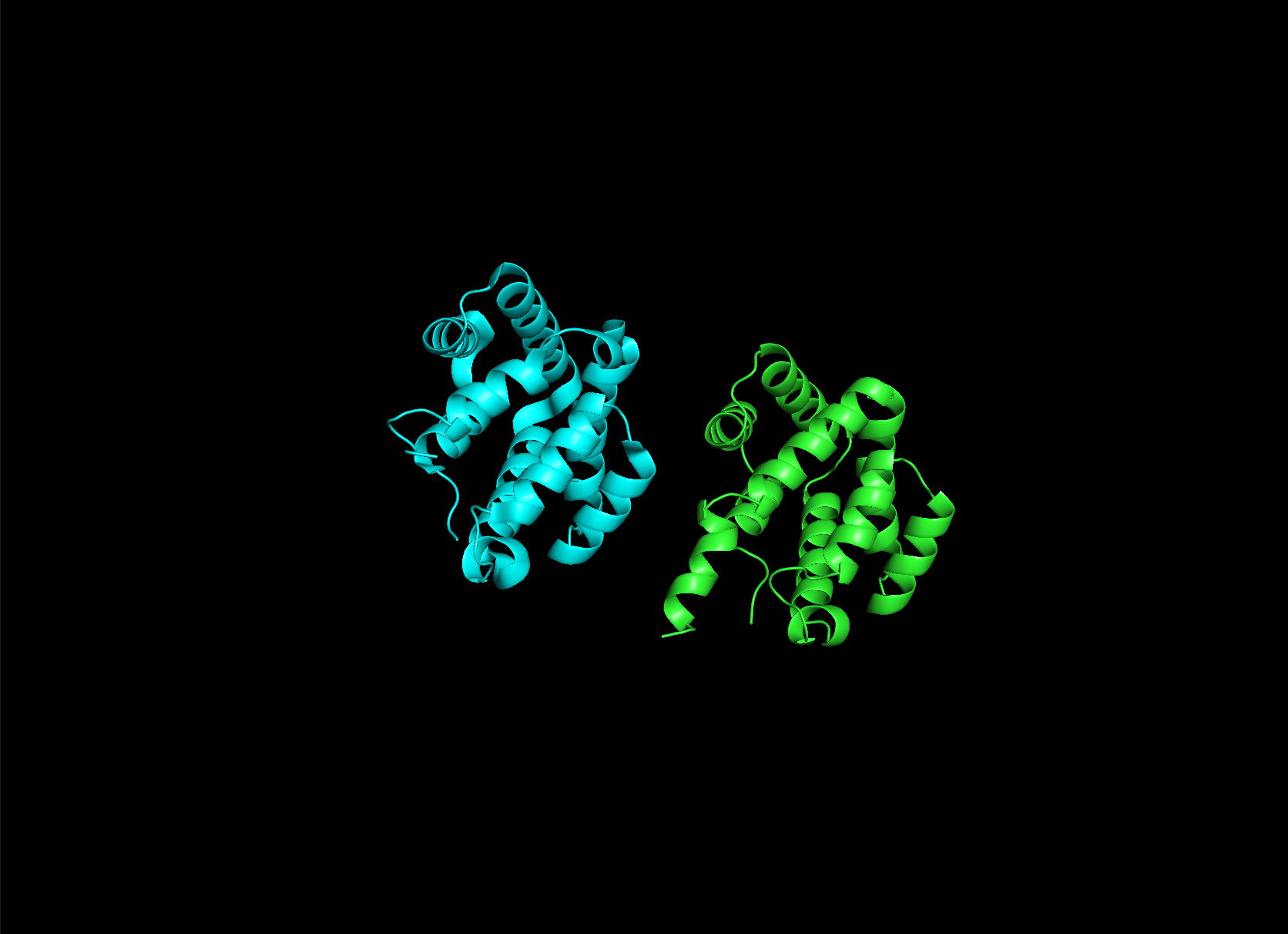
Note that in PDB data, there were two protein molecules included in the experimental data, apparently as an asymmetric unit, so I removed one for better comparison.
Here’s the output of the inverse fold:
I tried added random pointwise mutations to the protein, and it didn’t appear to affect the fold too much. For example: