Week 5 HW: Protein Design, Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Part 1: Generate Binders with PepMLM
I retrieved the UniProt entry (P00441):
There was a UniProt variation entry for the mutation we are targeting: VAR_007131:
ALS1; severe form; reduces structural stability and enzyme activity; increases tendency to form fibrillar aggregate
With the “A4V mutation (Alanine → Valine at residue 4)”:
Note that the start codon’s amino acid Met is removed post translationally, so in the UniProt sequence it’s at residue 5.
Using my Colab notebook, I generated 3 peptides of length 12 and compared it to FLYRWLPSRRGG:
The final result:
| # | Binder | Pseudo Perplexity |
|---|---|---|
| 0 | WHYPAVAVALKE | 9.614430 |
| 1 | WLYPAVALELKE | 14.331262 |
| 2 | KRYGAVAVRHWX | 10.045459 |
| 3 | WRYPAAGLELKE | 14.706538 |
| 4 | FLYRWLPSRRGG | 20.635231 |
Part 2: Evaluate Binders with AlphaFold3
Final results from AlphaFold3:
| # | Binder | PP | ipTM | pTM | Fold |
|---|---|---|---|---|---|
| 0 | WHYPAVAVALKE | 9.6 | 0.29 | 0.77 | 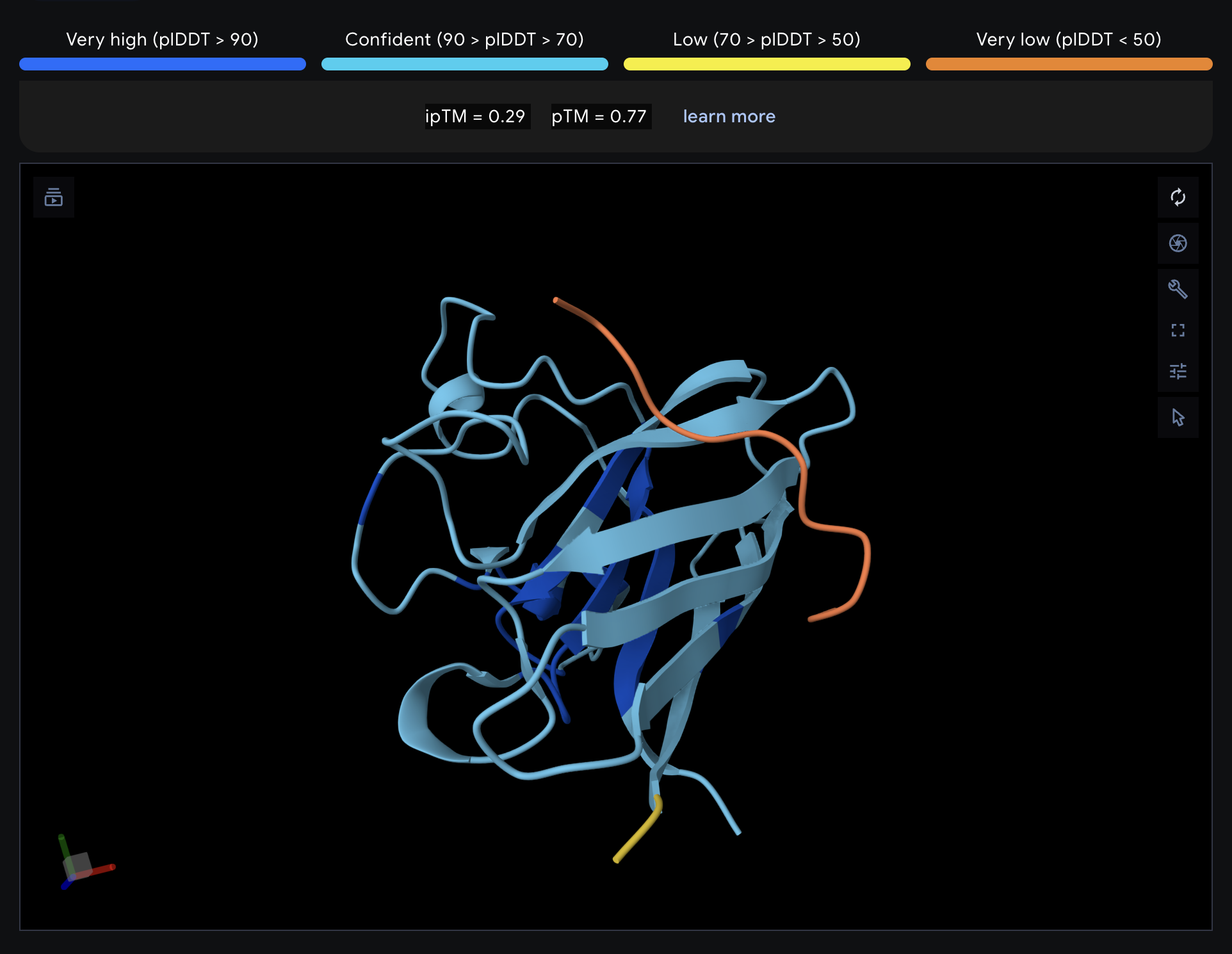
|
| 1 | WLYPAVALELKE | 14.3 | 0.27 | 0.76 | 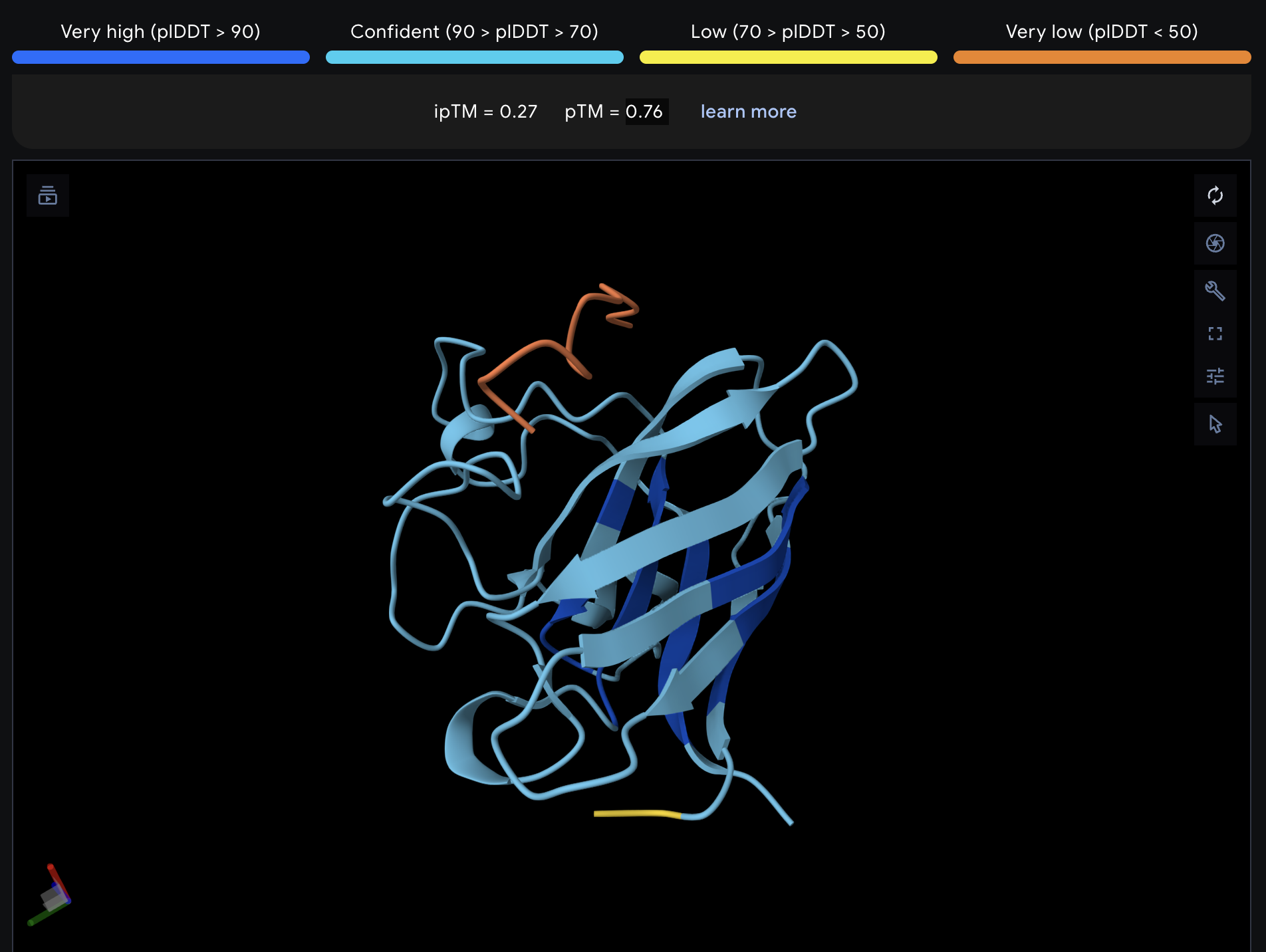
|
| 2 | KRYGAVAVRHWA* | 10.0 | 0.49 | 0.89 | 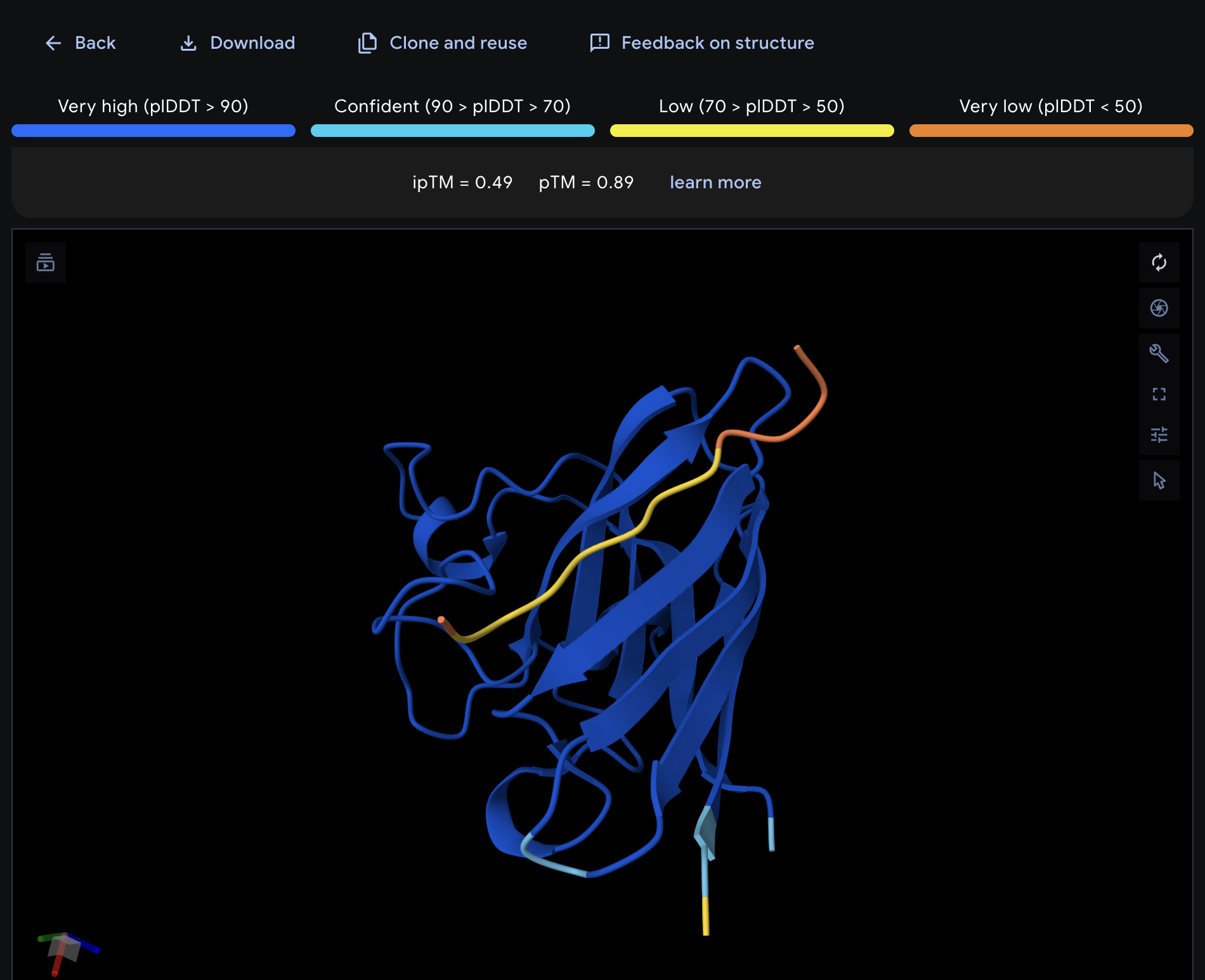
|
| 3 | WRYPAAGLELKE | 14.7 | 0.24 | 0.81 | 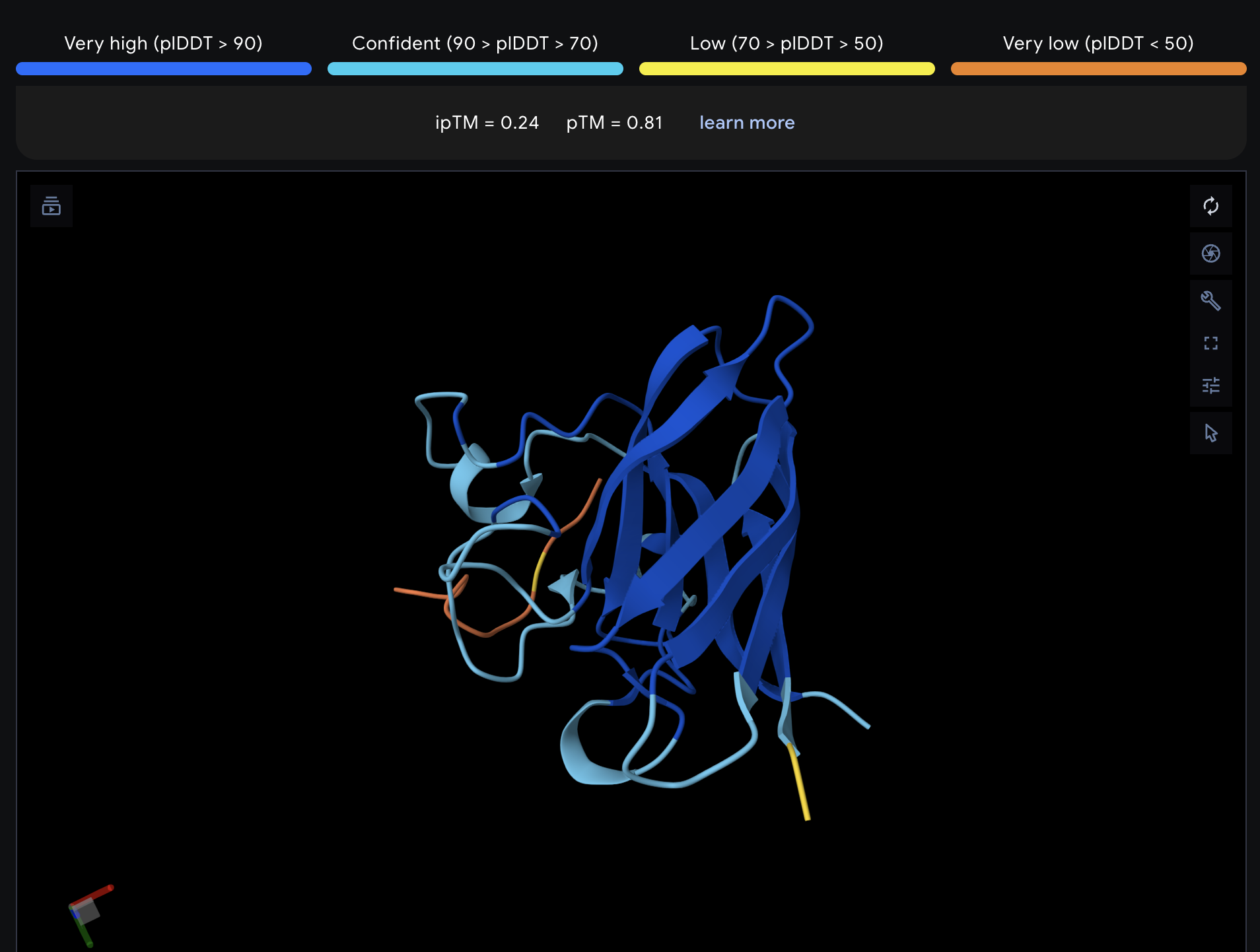
|
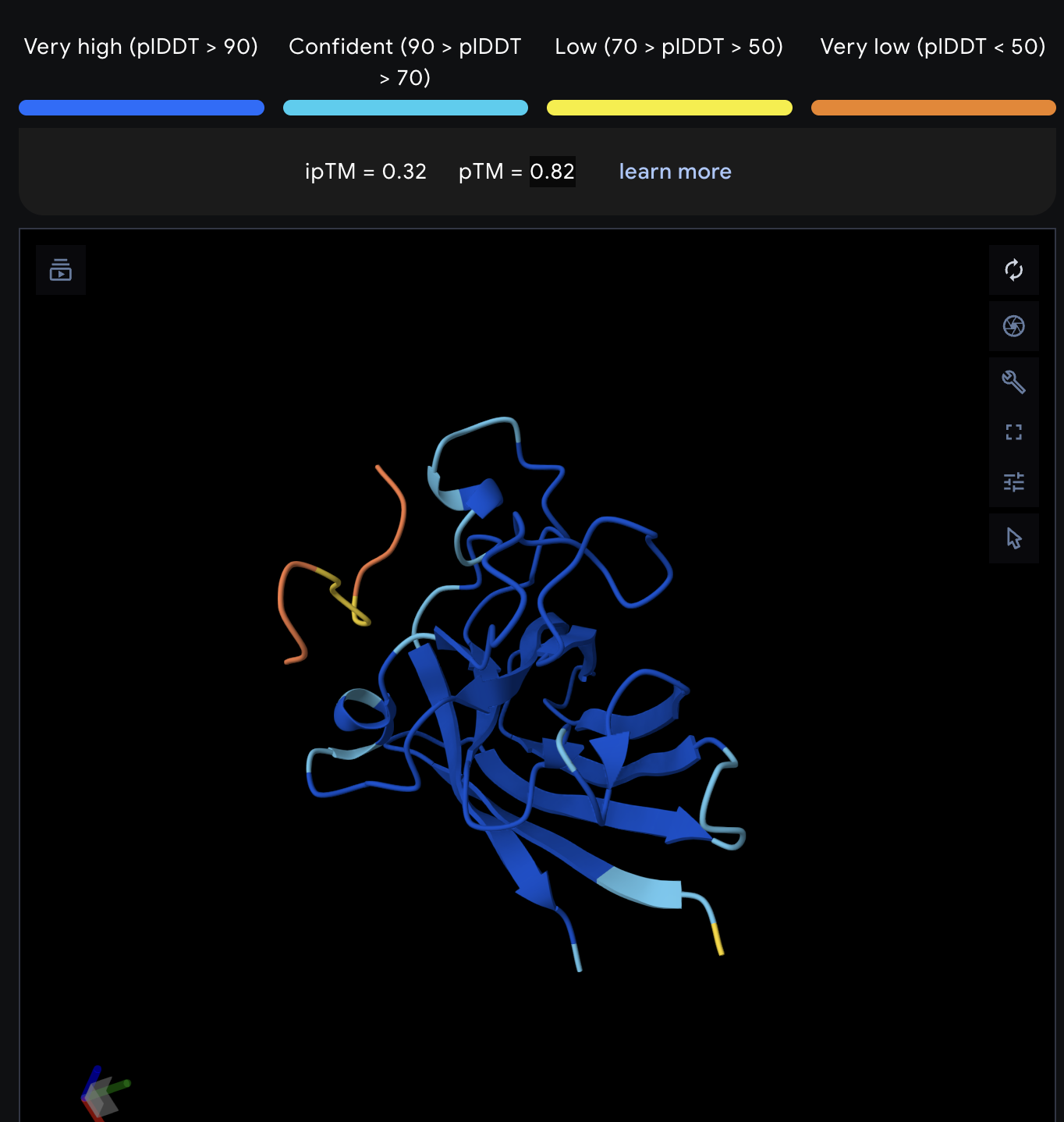
| 4 | FLYRWLPSRRGG | 20.6 | 0.32 | 0.82 | 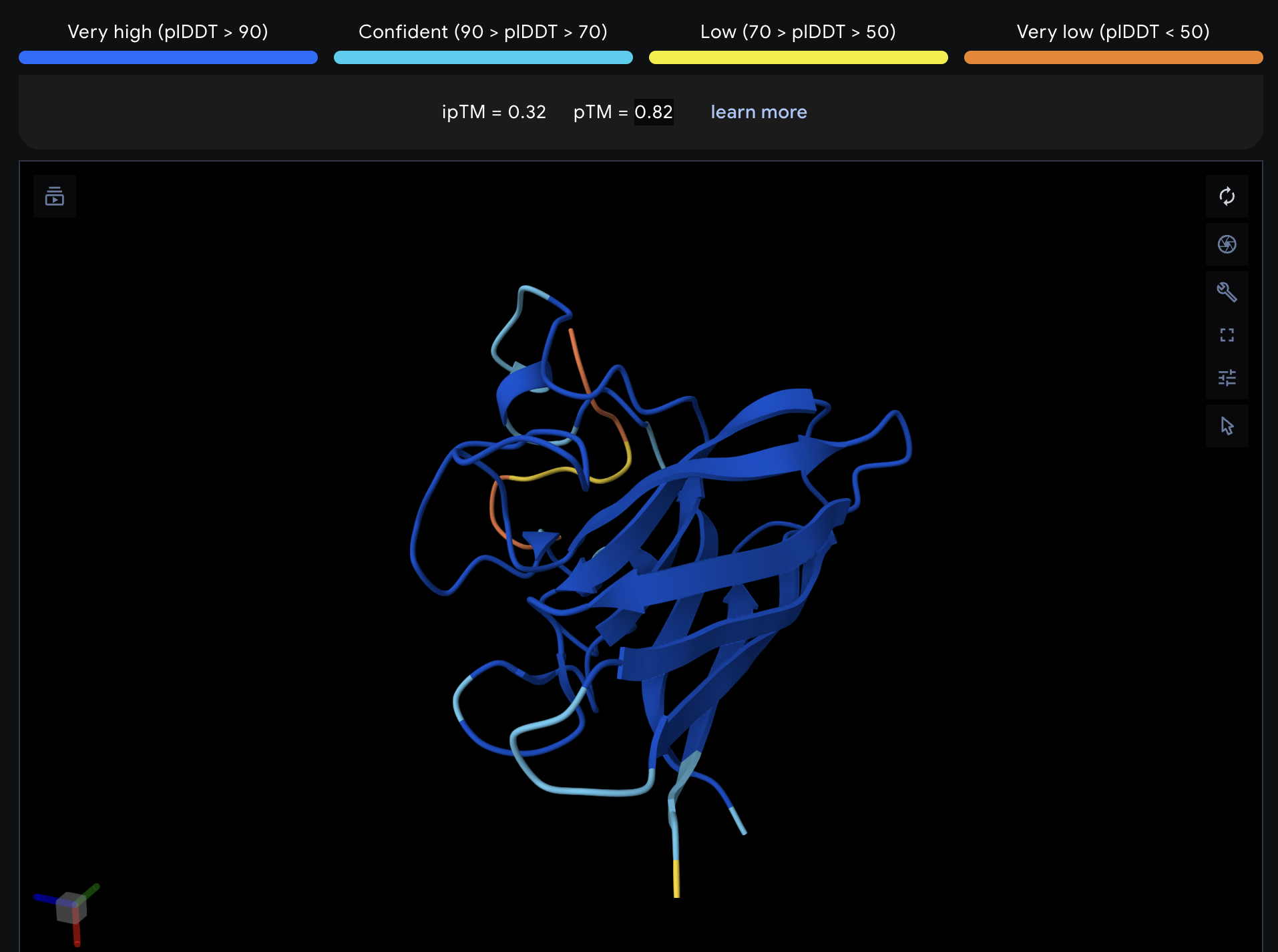
|
*X replaced with A for AlphaFold
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
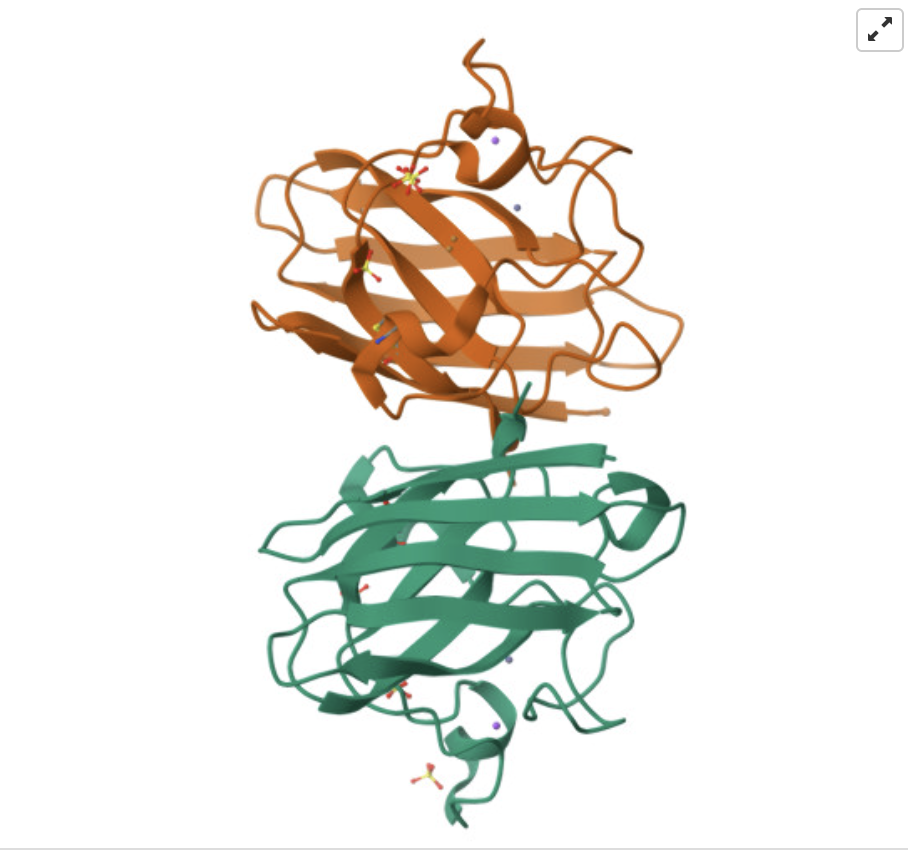
Here is the SODC homodimer structure:
And the structure of the “metal free” A4V SOD1:
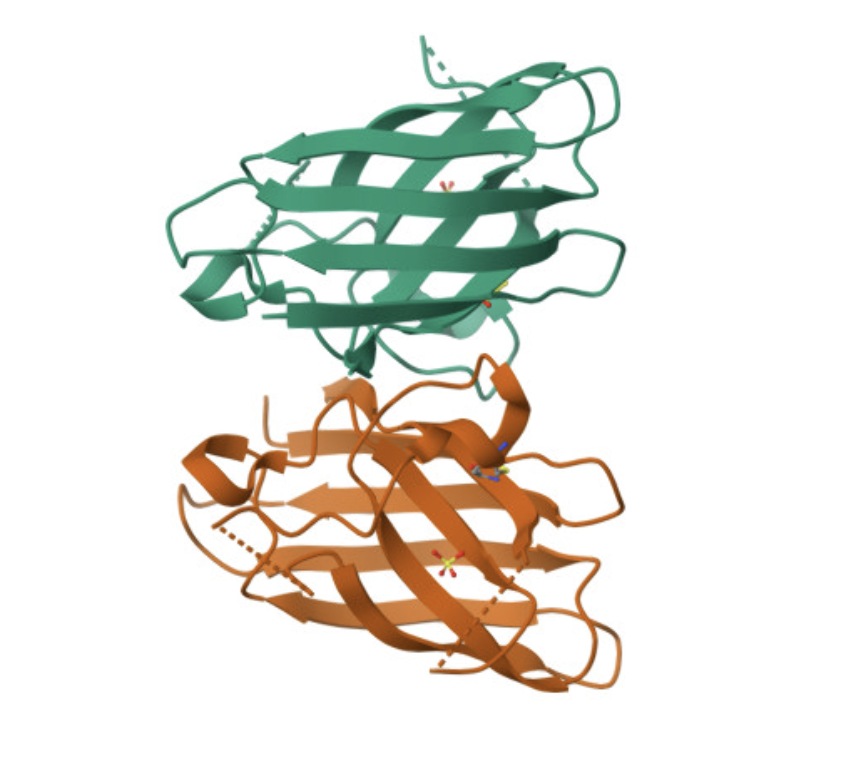
All proteins seem largely surface-bound. Binders 0 and 2 seem to engage the beta-barrel region. None seem too close to the dimer interface.
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
One exceeded the known binder: KRYGAVAVRHWA, most are around the same level as the known binder, although the binding sites look wildly different from binder to binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
| Binder | 💧 Solubility | 🩸 Hemolysis | 🔗 Binding Affinity | 📏 Length | ⚖️ Molecular Weight | ⚡ Net Charge (pH 7) | 🎯 Isoelectric Point | 💦 Hydrophobicity (GRAVY) |
|---|---|---|---|---|---|---|---|---|
| WHYPAVAVALKE | Soluble (1.000) | Non-hemolytic (0.032) | Weak binding (5.591 pKd/pKi) | 12 aa | 1383.6 Da | -0.15 | 6.76 pH | 0.27 GRAVY |
| WLYPAVALELKE | Soluble (1.000) | Non-hemolytic (0.052) | Weak binding (5.774 pKd/pKi) | 12 aa | 1431.7 Da | -1.23 | 4.86 pH | 0.37 GRAVY |
| KRYGAVAVRHWA | Soluble (1.000) | Non-hemolytic (0.029) | Weak binding (6.059 pKd/pKi) | 12 aa | 1413.6 Da | 2.85 | 11.00 pH | -0.41 GRAVY |
| WRYPAAGLELKE | Soluble (1.000) | Non-hemolytic (0.044) | Weak binding (5.565 pKd/pKi) | 12 aa | 1432.6 Da | -0.23 | 6.28 pH | -0.70 GRAVY |
| FLYRWLPSRRGG | Soluble (1.000) | Non-hemolytic (0.044) | Weak binding (5.565 pKd/pKi) | 12 aa | 1432.6 Da | -0.23 | 6.28 pH | -0.70 GRAVY |
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
The ipTM loosely correlates with the binding affinity. The highest ipTM matches the highest binding affinity. The others hae similar ipTM and they have similar affinities.
All the predicted binders are soluble and none are hemolytic.
KRYGAVAVRHWA looks to be the best based on the criteria, although even it has the best ipTM it is still modest. It bonds to the beta barrel loop.
Choose one peptide you would advance and justify your decision briefly.
I would advance KRYGAVAVRHWA since it has the highest values as predicted by PepMLM and PeptiVerse.
Part 4: Generate Optimized Peptides with moPPIt
Here’s my Colab notebook, and the results:
| Binder | Non-hemolytic* | Non-Fouling | Solubility | Affinity | Motif | Specificity |
|---|---|---|---|---|---|---|
| GGSRRGDRPRSV | 0.922 | 0.977 | 0.833 | 7.224 | 0.000 | 0.955 |
| DTCFQQTGEKRY | 0.976 | 0.751 | 0.917 | 6.070 | 0.884 | 0.590 |
| NEKQRTKSDMFH | 0.956 | 0.807 | 0.833 | 5.402 | 0.634 | 0.603 |
| SRKRGSRRDSED | 0.939 | 0.977 | 1.000 | 6.702 | 0.005 | 0.968 |
| YCLQRLNQNSTC | 0.874 | 0.754 | 0.833 | 6.323 | 0.760 | 0.609 |
| DKTDAKGFYQFY | 0.901 | 0.632 | 0.750 | 6.310 | 0.715 | 0.583 |
*The “hemolysis” probability is actually non-hemolytic, as per the source, so this heading was updated.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
The peptides from moPPit are more unique and have higher binding affinity. PepMLM peptides share more amino acids with each other, probably because of the encoder model generation.
I would chose the peptides with the highest specificity and use a similar screening as we applied to the PepMLM peptides by checking AlphaFold. Then, I would test in the lab before testing clinically.