Week 2 HW: DNA Read, Write, & Edit
My Homework
DNA!
Part 1: Benchling & In-silico Gel Art
See this week’s lab protocol “Gel Art: Restriction Digests and Gel Electrophoresis” for details. Overview:
Make a free account at benchling.com Import the Lambda DNA. Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. You might find Ronan’s website a helpful tool for quickly iterating on designs!
My in silico design in Benchling
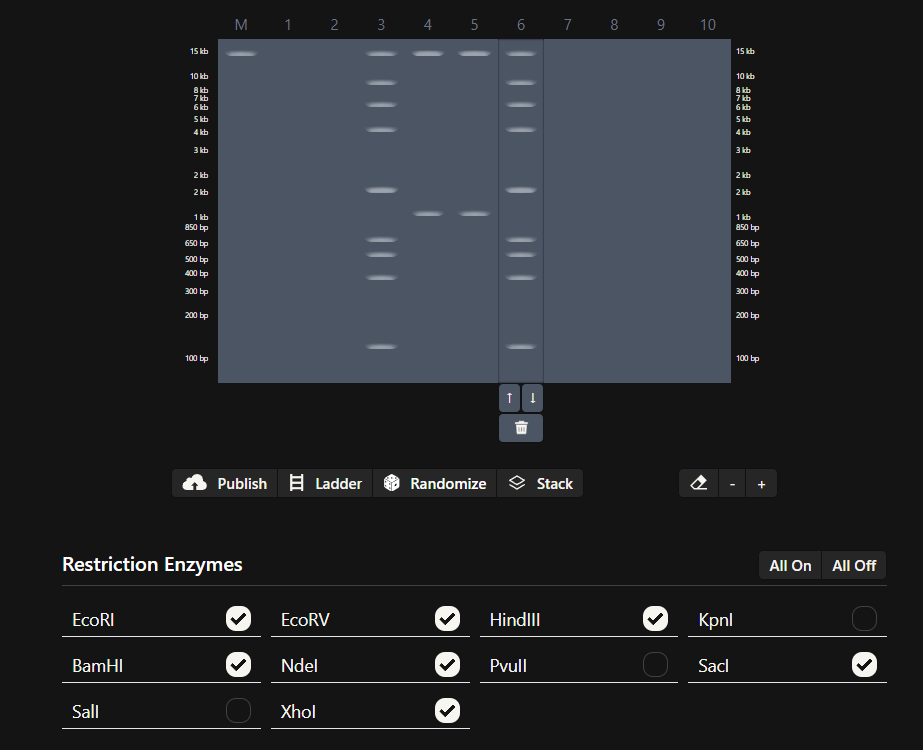
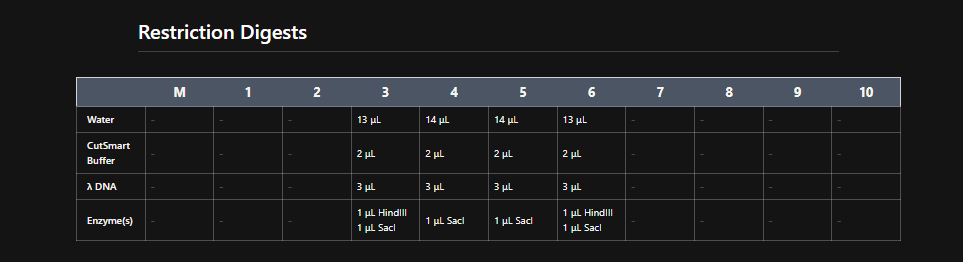
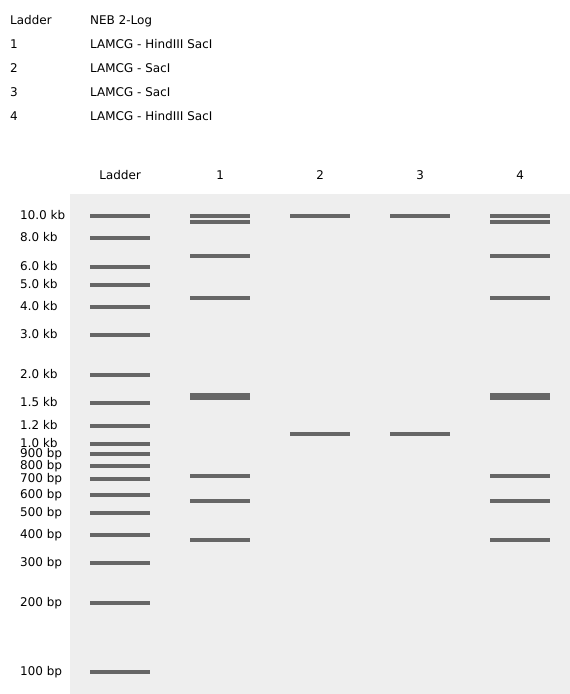
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
In the wet-lab perform the lab experiment you designed in Part 1 and outlined in this week’s lab protocol “Gel Art: Restriction Digests and Gel Electrophoresis”.
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
[Example from our group homework, you may notice the particular format — The example below came from UniProt]
sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1 METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLL EAVIRTVTTLQQLLT
For my final project, I decided to develop a field-deployable diagnostic platform that is user-friendly and low-cost for growers. For this platform, one idea could be to use the CRISPR-Cas12a system; therefore, I chose the enzyme LbCas12a-Ultra. In the article, they improved different components of the CRISPR assay, such as Cas12a and the reporter system. They tested different Cas12a variants, and the best-performing one was LbCas12a-Ultra. This enzyme was generated by IDT, and the sequence is not available in databases or articles. I found this information in UniProt:
tr|A0A5S8WF58|A0A5S8WF58_9FIRM LbCas12a OS=Lachnospiraceae bacterium OX=1898203 PE=1 SV=1 MSKLEKFTNCYSLSKTLRFKAIPVGKTQENIDNKRLLVEDEKRAEDYKGVKKLLDRYYLS FINDVLHSIKLKNLNNYISLFRKKTRTEKENKELENLEINLRKEIAKAFKGNEGYKSLFKKDIIETILPEFLDDKDEIALVNSFNGFTTAFTGFFDNRENMFSEEAKSTSIAFRCINENL TRYISNMDIFEKVDAIFDKHEVQEIKEKILNSDYDVEDFFEGEFFNFVLTQEGIDVYNAIIGGFVTESGEKIKGLNEYINLYNQKTKQKLPKFKPLYKQVLSDRESLSFYGEGYTSDEEV LEVFRNTLNKNSEIFSSIKKLEKLFKNFDEYSSAGIFVKNGPAISTISKDIFGEWNVIRDKWNAEYDDIHLKKKAVVTEKYEDDRRKSFKKIGSFSLEQLQEYADADLSVVEKLKEIIIQ KVDEIYKVYGSSEKLFDADFVLEKSLKKNDAVVAIMKDLLDSVKSFENYIKAFFGEGKETNRDESFYGDFVLAYDILLKVDHIYDAIRNYVTQKPYSKDKFKLYFQNPQFMGGWDKDKET DYRATILRYGSKYYLAIMDKKYAKCLQKIDKDDVNGNYEKINYKLLPGPNKMLPKVFFSKKWMAYYNPSEDIQKIYKNGTFKKGDMFNLNDCHKLIDFFKDSISRYPKWSNAYDFNFSET EKYKDIAGFYREVEEQGYKVSFESASKKEVDKLVEEGKLYMFQIYNKDFSDKSHGTPNLHTMYFKLLFDENNHGQIRLSGGAELFMRRASLKKEELVVHPANSPIANKNPDNPKKTTTLS YDVYKDKRFSEDQYELHIPIAINKCPKNIFKINTEVRVLLKHDDNPYVIGIDRGERNLLYIVVVDGKGNIVEQYSLNEIINNFNGIRIKTDYHSLLDKKEKERFEARQNWTSIENIKELK AGYISQVVHKICELVEKYDAVIALEDLNSGFKNSRVKVEKQVYQKFEKMLIDKLNYMVDKKSNPCATGGALKGYQITNKFESFKSMSTQNGFIFYIPAWLTSKIDPSTGFVNLLKTKYTS IADSKKFISSFDRIMYVPEEDLFEFALDYKNFSRTDADYIKKWKLYSYGNRIRIFRNPKKNNVFDWEEVCLTSAYKELFNKYGINYQQGDIRALLCEQSDKAFYSSFMALMSLMLQMRNS ITGRTDVDFLISPVKNSDGIFYDSRNYEAQENAILPKNADANGAYNIARKVLWAIGQFKKAEDEKLDKVKIAISNKEWLEYAQTSVKH
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
[Example: Get to the original sequence of phage MS2 L-protein from its genome phage MS2 genome - Nucleotide - NCBI]
Lysis protein DNA sequence atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttaccaatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
LbCas12a from NCBI Genbank: OK557998.1 atgagcaagctggagaagtttacaaactgctactccctgtctaagaccctgaggttcaaggccatccctgtgggcaagacccaggagaacatcgacaataagcggctgctggtggaggacgagaagagagccgaggattataagggcgtgaagaagctgctggatcgctactatctgtcttttatcaacgacgtgctgcacagcatcaagctgaagaatctgaacaattacatcagcctgttccggaagaaaaccagaaccgagaaggagaataaggagctggagaacctggagatcaatctgcggaaggagatcgccaaggccttcaagggcaacgagggctacaagtccctgtttaagaaggatatcatcgagacaatcctgccagagttcctggacgataaggacgagatcgccctggtgaacagcttcaatggctttaccacagccttcaccggcttctttgataacagagagaatatgttttccgaggaggccaagagcacatccatcgccttcaggtgtatcaacgagaatctgacccgctacatctctaatatggacatcttcgagaaggtggacgccatctttgataagcacgaggtgcaggagatcaaggagaagatcctgaacagcgactatgatgtggaggatttctttgagggcgagttctttaactttgtgctgacacaggagggcatcgacgtgtataacgccatcatcggcggcttcgtgaccgagagcggcgagaagatcaagggcctgaacgagtacatcaacctgtataatcagaaaaccaagcagaagctgcctaagtttaagccactgtataagcaggtgctgagcgatcgggagtctctgagcttctacggcgagggctatacatccgatgaggaggtgctggaggtgtttagaaacaccctgaacaagaacagcgagatcttcagctccatcaagaagctggagaagctgttcaagaattttgacgagtactctagcgccggcatctttgtgaagaacggccccgccatcagcacaatctccaaggatatcttcggcgagtggaacgtgatccgggacaagtggaatgccgagtatgacgatatccacctgaagaagaaggccgtggtgaccgagaagtacgaggacgatcggagaaagtccttcaagaagatcggctccttttctctggagcagctgcaggagtacgccgacgccgatctgtctgtggtggagaagctgaaggagatcatcatccagaaggtggatgagatctacaaggtgtatggctcctctgagaagctgttcgacgccgattttgtgctggagaagagcctgaagaagaacgacgccgtggtggccatcatgaaggacctgctggattctgtgaagagcttcgagaattacatcaaggccttctttggcgagggcaaggagacaaacagggacgagtccttctatggcgattttgtgctggcctacgacatcctgctgaaggtggaccacatctacgatgccatccgcaattatgtgacccagaagccctactctaaggataagttcaagctgtattttcagaaccctcagttcatgggcggctgggacaaggataaggagacagactatcgggccaccatcctgagatacggctccaagtactatctggccatcatggataagaagtacgccaagtgcctgcagaagatcgacaaggacgatgtgaacggcaattacgagaagatcaactataagctgctgcccggccctaataagatgctgccaaaggtgttcttttctaagaagtggatggcctactataaccccagcgaggacatccagaagatctacaagaatggcacattcaagaagggcgatatgtttaacctgaatgactgtcacaagctgatcgacttctttaaggatagcatctcccggtatccaaagtggtccaatgcctacgatttcaacttttctgagacagagaagtataaggacatcgccggcttttacagagaggtggaggagcagggctataaggtgagcttcgagtctgccagcaagaaggaggtggataagctggtggaggagggcaagctgtatatgttccagatctataacaaggacttttccgataagtctcacggcacacccaatctgcacaccatgtacttcaagctgctgtttgacgagaacaatcacggacagatcaggctgagcggaggagcagagctgttcatgaggcgcgcctccctgaagaaggaggagctggtggtgcacccagccaactcccctatcgccaacaagaatccagataatcccaagaaaaccacaaccctgtcctacgacgtgtataaggataagaggttttctgaggaccagtacgagctgcacatcccaatcgccatcaataagtgccccaagaacatcttcaagatcaatacagaggtgcgcgtgctgctgaagcacgacgataacccctatgtgatcggcatcgataggggcgagcgcaatctgctgtatatcgtggtggtggacggcaagggcaacatcgtggagcagtattccctgaacgagatcatcaacaacttcaacggcatcaggatcaagacagattaccactctctgctggacaagaaggagaaggagaggttcgaggcccgccagaactggacctccatcgagaatatcaaggagctgaaggccggctatatctctcaggtggtgcacaagatctgcgagctggtggagaagtacgatgccgtgatcgccctggaggacctgaactctggctttaagaatagccgcgtgaaggtggagaagcaggtgtatcagaagttcgagaagatgctgatcgataagctgaactacatggtggacaagaagtctaatccttgtgcaacaggcggcgccctgaagggctatcagatcaccaataagttcgagagctttaagtccatgtctacccagaacggcttcatcttttacatccctgcctggctgacatccaagatcgatccatctaccggctttgtgaacctgctgaaaaccaagtataccagcatcgccgattccaagaagttcatcagctcctttgacaggatcatgtacgtgcccgaggaggatctgttcgagtttgccctggactataagaacttctctcgcacagacgccgattacatcaagaagtggaagctgtactcctacggcaaccggatcagaatcttccggaatcctaagaagaacaacgtgttcgactgggaggaggtgtgcctgaccagcgcctataaggagctgttcaacaagtacggcatcaattatcagcagggcgatatcagagccctgctgtgcgagcagtccgacaaggccttctactctagctttatggccctgatgagcctgatgctgcagatgcggaacagcatcacaggccgcaccgacgtggattttctgatcagccctgtgaagaactccgacggcatcttctacgatagccggaactatgaggcccaggagaatgccatcctgccaaagaacgccgacgccaatggcgcctataacatcgccagaaaggtgctgtgggccatcggccagttcaagaaggccgaggacgagaagctggataaggtgaagatcgccatctctaacaaggagtggctggagtacgcccagaccagcgtgaagcac
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
[Example from Codon Optimization Tool | Twist Bioscience while avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI]
Lysis protein DNA sequence with Codon-Optimization ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCACCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
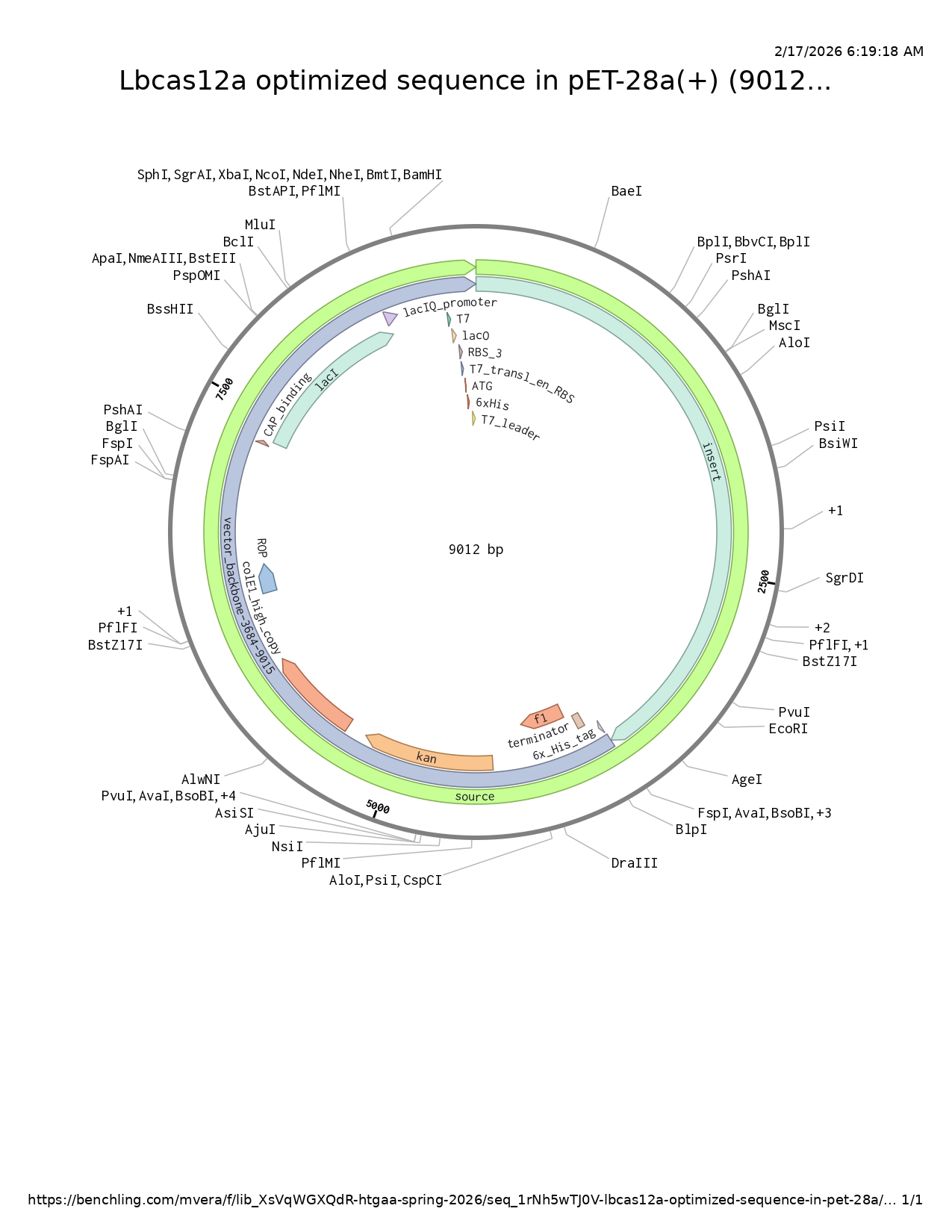
I used the codon optimization tool from Twist Bioscience because I plan to produce LbCas12a in E. coli, purify it, and prepare it for use in a cell-free system. E. coli is a well-characterized organism and is relatively simple to handle for protein expression and purification. LbCas12a is an enzyme originally derived from Lachnospiraceae bacterium. Since I chose the expression vector pET-28a(+), I selected the option in the program to optimize codon usage for E. coli and to avoid introducing restriction sites for BamHI and XhoI, as I will use these enzymes for cloning.
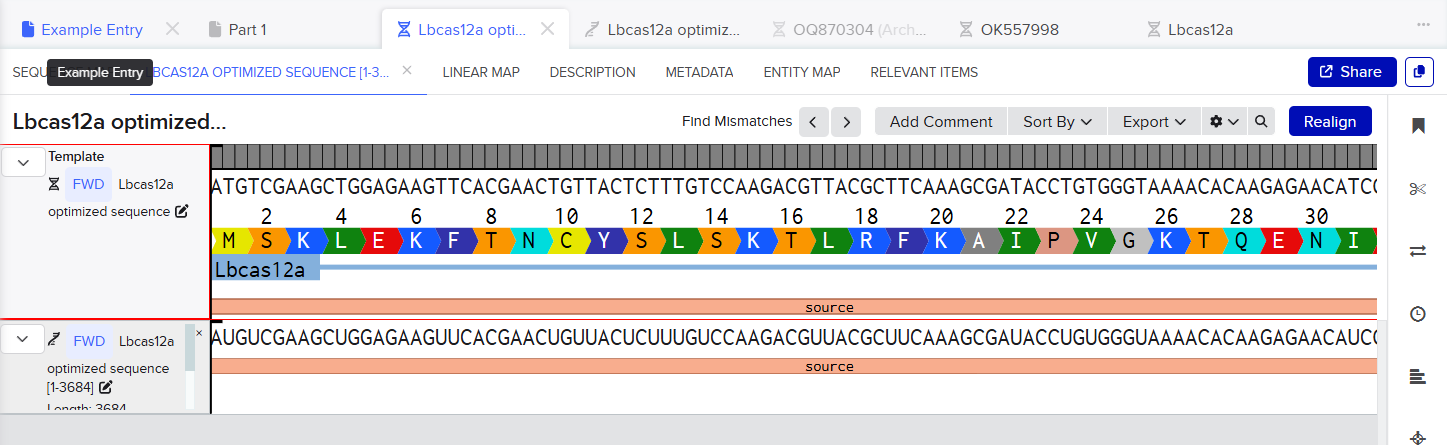
LbCas12a DNA sequence with Codon-optimization ATGTCGAAGCTGGAGAAGTTCACGAACTGTTACTCTTTGTCCAAGACGTTACGCTTCAAAGCGATACCTGTGGGTAAAACACAAGAGAACATCGATAACAAGAGACTGCTGGTGGAAGACGAGAAACGAGCTGAAGATTACAAGGGCGTCAAGAAGTTATTGGATCGATACTATCTTTCCTTTATCAACGATGTTCTGCACTCGATTAAATTGAAGAACCTCAATAACTATATATCTCTGTTCCGCAAGAAGACTCGAACCGAAAAGGAAAACAAGGAGCTTGAGAACTTAGAGATTAACCTCAGAAAGGAGATAGCCAAAGCTTTCAAGGGAAACGAGGGGTACAAATCCTTGTTCAAGAAGGATATAATCGAAACGATACTGCCCGAATTTCTGGACGATAAAGATGAAATTGCTCTGGTCAACTCGTTCAATGGCTTCACGACTGCATTTACGGGTTTCTTCGACAACCGTGAGAACATGTTCAGCGAAGAGGCTAAAAGCACGAGCATTGCCTTTCGTTGCATTAACGAGAACCTGACCCGGTACATAAGTAATATGGACATATTTGAGAAAGTAGACGCTATCTTTGATAAGCACGAAGTCCAAGAGATTAAAGAGAAGATCCTTAATTCAGACTACGATGTAGAGGACTTCTTTGAAGGGGAGTTCTTCAACTTCGTACTGACCCAAGAGGGCATAGATGTCTACAATGCAATTATAGGCGGCTTCGTCACCGAGTCGGGCGAAAAGATAAAAGGCTTGAACGAATACATTAACCTGTACAACCAGAAGACAAAACAAAAGCTCCCGAAGTTCAAGCCCCTGTATAAACAAGTTCTCTCAGACCGCGAGAGTTTGTCTTTCTACGGTGAGGGTTACACTTCGGATGAAGAGGTCCTCGAAGTATTTCGCAATACGTTAAACAAGAACTCAGAGATATTTTCTTCCATTAAGAAATTAGAGAAGCTCTTCAAGAACTTTGATGAGTACTCCTCAGCTGGTATATTCGTGAAGAATGGGCCAGCAATCTCCACCATTAGTAAGGATATATTTGGCGAATGGAACGTAATCCGTGACAAATGGAACGCTGAATACGATGATATCCATCTGAAGAAGAAGGCCGTAGTGACGGAGAAGTACGAAGACGACCGTCGGAAGTCCTTTAAGAAGATAGGAAGCTTCAGTCTCGAACAGCTCCAGGAGTACGCAGATGCGGATTTAAGTGTAGTCGAGAAATTGAAGGAGATAATAATTCAAAAGGTTGATGAGATCTACAAGGTCTATGGGTCTAGTGAGAAACTCTTCGACGCCGACTTCGTGTTGGAAAAGTCACTGAAGAAGAATGATGCCGTTGTGGCCATTATGAAGGACCTGCTGGATTCAGTGAAGAGCTTTGAGAACTACATTAAAGCATTCTTCGGAGAAGGTAAGGAAACTAATCGCGACGAGTCTTTCTACGGAGACTTCGTTCTTGCGTATGACATCCTTTTGAAGGTGGACCACATTTACGATGCAATTAGAAACTACGTCACTCAGAAGCCTTACTCCAAGGACAAGTTTAAGCTTTACTTCCAAAACCCTCAATTTATGGGCGGTTGGGACAAAGACAAAGAGACTGACTATCGCGCCACGATCTTACGCTATGGAAGCAAGTACTACTTAGCGATTATGGACAAGAAGTACGCAAAGTGCCTGCAAAAGATAGACAAGGATGATGTTAACGGCAACTACGAGAAAATTAATTACAAGCTGCTTCCTGGCCCAAATAAGATGCTGCCAAAGGTCTTCTTTTCTAAGAAGTGGATGGCTTACTACAACCCATCAGAGGACATACAAAAGATTTATAAGAACGGCACTTTCAAGAAGGGTGACATGTTTAACCTCAATGACTGTCATAAGTTGATTGACTTCTTCAAGGACAGTATATCCCGATACCCTAAGTGGTCCAATGCGTACGACTTCAATTTCTCAGAGACAGAAAAGTACAAAGACATCGCGGGTTTCTATCGCGAGGTGGAAGAACAAGGATACAAGGTTTCTTTCGAGTCAGCTTCTAAGAAAGAGGTCGACAAGCTGGTGGAAGAGGGTAAGCTGTACATGTTTCAGATTTACAATAAGGATTTCTCAGATAAGTCACACGGAACTCCCAACCTGCACACCATGTACTTCAAACTGCTGTTCGATGAGAACAACCACGGTCAAATTCGCTTGTCTGGCGGCGCAGAACTTTTCATGCGGCGCGCTTCTCTGAAGAAAGAGGAACTTGTTGTTCATCCAGCCAACAGCCCTATCGCTAACAAGAATCCTGACAACCCCAAGAAAACTACTACGTTGAGTTACGACGTTTACAAGGATAAGAGATTTTCCGAGGACCAGTACGAGTTGCATATTCCGATAGCCATTAATAAATGCCCTAAGAACATTTTCAAGATCAATACGGAAGTACGTGTCCTTTTGAAACACGACGACAACCCATACGTGATTGGGATTGACAGAGGAGAGCGTAACTTATTATATATCGTCGTCGTCGACGGCAAGGGAAACATAGTTGAACAGTATAGCTTAAATGAGATAATCAACAACTTCAACGGTATCCGTATCAAAACAGATTACCACTCTTTACTTGACAAGAAGGAGAAGGAGCGCTTCGAGGCTCGTCAAAATTGGACTAGCATTGAGAATATTAAAGAGCTCAAGGCTGGGTATATCAGCCAGGTTGTACATAAGATCTGCGAGCTGGTTGAAAAGTACGACGCGGTCATCGCACTTGAGGACCTGAACAGTGGTTTTAAGAACAGCCGTGTGAAGGTCGAGAAGCAGGTATACCAAAAGTTTGAGAAGATGCTGATAGATAAGCTGAACTACATGGTAGACAAGAAATCTAATCCATGTGCGACTGGTGGGGCCTTAAAGGGATATCAGATAACCAACAAATTTGAGTCCTTCAAGAGCATGTCGACCCAGAACGGATTCATATTTTACATCCCGGCTTGGTTGACATCCAAGATCGATCCTAGCACTGGTTTCGTTAACCTCTTGAAGACTAAGTACACGAGTATAGCTGACAGTAAGAAGTTTATCTCATCTTTCGATCGTATTATGTACGTTCCAGAGGAAGACCTTTTCGAGTTTGCGTTAGATTACAAGAACTTCAGCCGAACTGACGCAGACTACATTAAGAAGTGGAAATTGTATAGCTATGGGAACCGAATTCGCATCTTCCGGAATCCAAAGAAGAATAACGTTTTCGACTGGGAAGAGGTGTGTTTAACTTCAGCGTATAAAGAGCTGTTTAATAAGTACGGAATAAATTACCAACAAGGTGACATCCGCGCCCTTCTGTGCGAGCAATCGGATAAAGCATTTTACTCCTCGTTCATGGCATTGATGTCATTAATGTTGCAAATGCGGAACTCCATAACTGGTAGAACCGATGTAGACTTTCTTATCTCACCGGTTAAGAACAGTGATGGTATCTTCTACGATTCTCGGAATTATGAGGCTCAAGAGAACGCAATCCTGCCCAAGAACGCTGACGCGAATGGAGCCTATAATATCGCACGTAAAGTACTGTGGGCTATAGGTCAATTTAAGAAGGCCGAAGACGAGAAATTGGACAAAGTTAAAATCGCAATTTCTAACAAAGAGTGGCTGGAATATGCGCAAACAAGTGTGAAGCAT
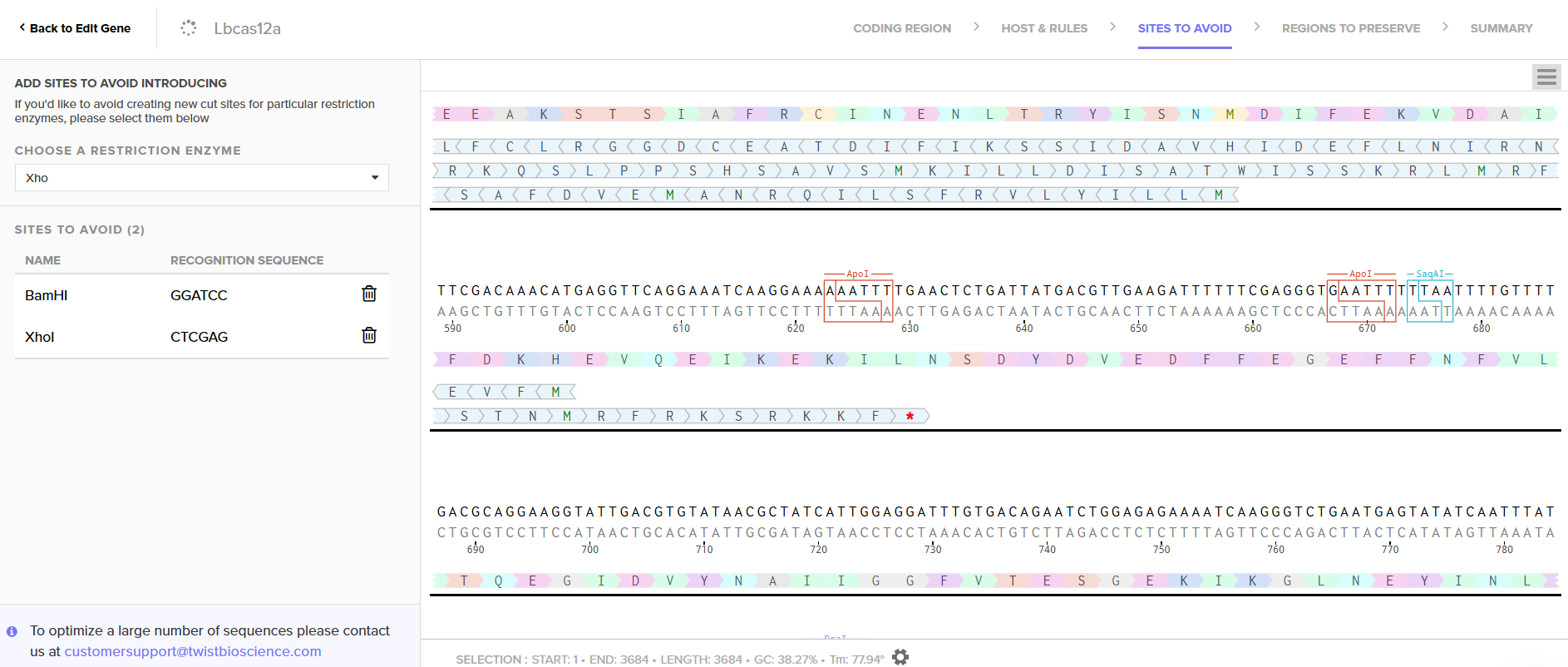
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
It could be used both tecnologies cell-dependent system and cell-free system. In a cell-dependent system, the optimized LbCas12a gene will be cloned into an expression vector such as pET-28a(+) and transformed into E. coli expression strains (for example, BL21(DE3)). Once inside the bacteria, the DNA is first transcribed into messenger RNA (mRNA) by RNA polymerase. In the pET system, transcription is typically driven by the T7 promoter, which is activated after induction (commonly with IPTG). The mRNA is then translated by ribosomes in the cytoplasm, where transfer RNAs (tRNAs) recognize codons and incorporate the corresponding amino acids to synthesize the LbCas12a protein. After translation, the protein can be purified, often using Nickle affinity and cation exchange chromatography.
In a cell-free expression system, the same DNA template (plasmid or linear DNA) is added to a reaction mixture containing the molecular machinery required for transcription and translation. This mixture typically includes RNA polymerase, ribosomes, tRNAs, amino acids, nucleotides, enzymes, and energy sources. The DNA is transcribed into mRNA in vitro, and ribosomes translate the mRNA into protein directly in the reaction tube. Cell-free systems allow faster protein production and easier control of reaction conditions, which is useful for rapid prototyping or diagnostic applications.
3.5. [Optional] How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level. Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below. [Example shows the biomolecular flow in central dogma from DNA to RNA to Protein] Special note that all “T” were transcribed into “U” and that the 3-nt codon represents 1-AA.
Splicing can generate different mRNA variants with different exon organization, resulting in the production of different proteins. Alternative promoters within a single gene or different transcription start sites can also generate different protein isoforms.
Part 4: Prepare a Twist DNA Synthesis Order
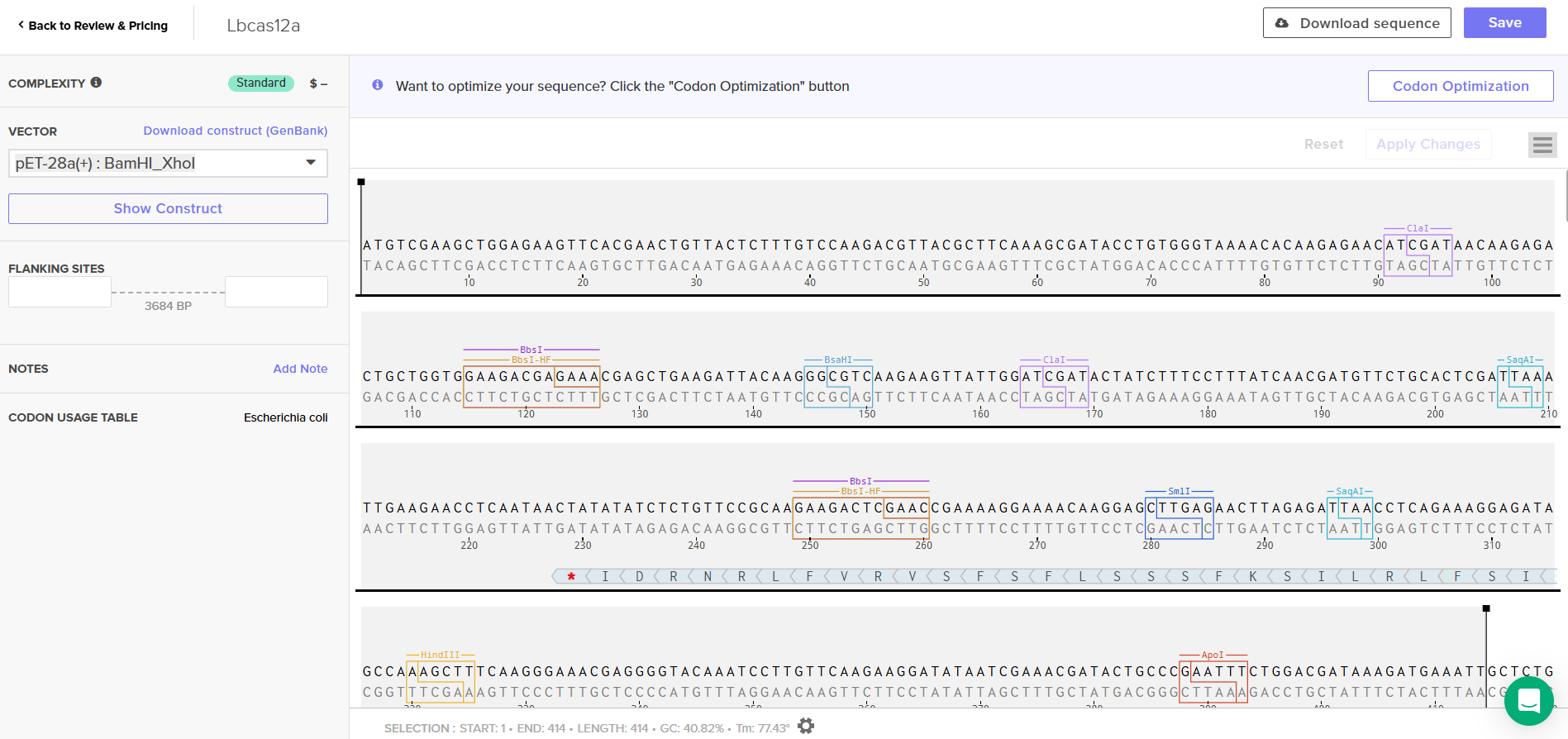
In this case, I can order the synthetic fragment (LbCas12a gene) containing the restriction sites and clone it into the expression vector pET-28a(+) using the restriction enzymes BamHI and XhoI.
Order from Twist
Insert and desired expression vector
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank). DNA-based digital data storage technology. Source: Archives in DNA: Workshop Exploring Implications of an Emerging Bio-Digital Technology through Design Fiction - Scientific Figure on ResearchGate. Available from: https://www.researchgate.net/figure/DNA-based-digital-data-storage-technology_fig1_353128454 [accessed 11 Feb 2025]
For the development of a field-deployable CRISPR platform to detect Aster Yellows Phytoplasma (AYP), I would sequence DNA from symptomatic garlic plants that test positive for AYP. I plan to do this in two ways: (1) cloning and sequencing three molecular marker genes to confirm AYP identity and compare strains, and (2) whole-genome sequencing to fully characterize the entire AYP genome from field isolates. These sequencing approaches are important because they reveal conserved and variable regions of the pathogen, which helps me design guide RNAs that are specific, sensitive, and robust across different AYP strains found in real agricultural samples.
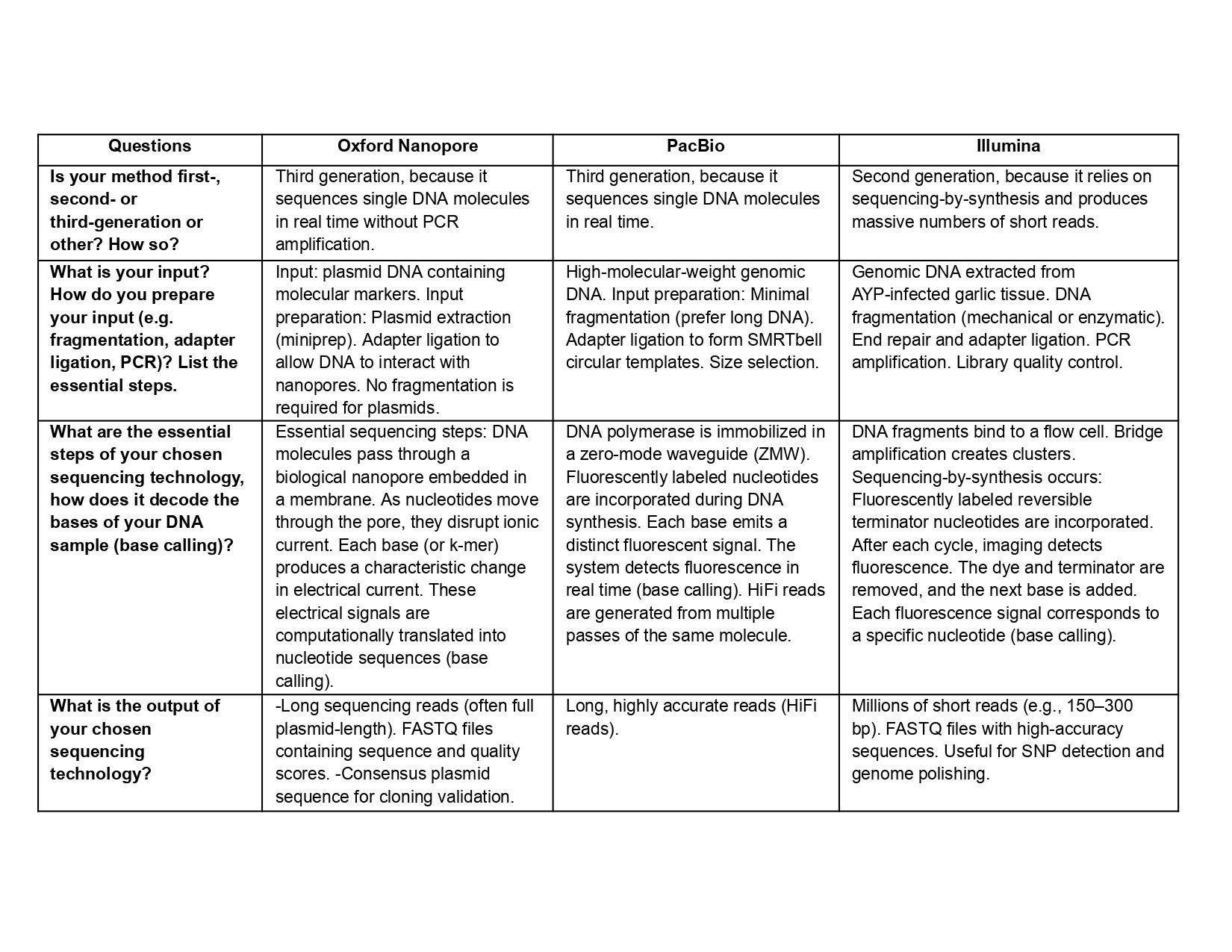
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions: Is your method first-, second- or third-generation or other? How so? What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps. What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)? What is the output of your chosen sequencing technology?
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :) See some famous examples of DNA design DNA origami by Paul W. K. Rothemund, California Institute of Technology, 2004. 100 nanometers in diameter.
For now I want to synthetize the individual gene LbCas12a to clone in E.coli and produce and purify it. Also, it could be a genetic circuit in a cell free-system where LbCas12a generates itself or by using Toehold switch system for detection. (ii) What technology or technologies would you use to perform this DNA synthesis and why? Also answer the following questions: What are the essential steps of your chosen sequencing methods? What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
(i) For this project, I would like to synthesize the individual gene encoding LbCas12a (from Lachnospiraceae bacterium) for heterologous expression in E. coli. The goal is to clone the codon-optimized gene into an expression vector (such as pET-28a(+)), produce the recombinant protein, and purify it for CRISPR-based detection of AYP. Synthesizing this gene allows me to optimize codon usage for E. coli and remove unwanted restriction sites. In addition to expressing LbCas12a as a standalone protein, I am also interested in synthesizing a genetic circuit for a cell-free system. This system could integrate Cas12a or toehold switch technology. In this design, the DNA construct could encode LbCas12a under a T7 promoter, enabling in vitro transcription and translation directly within a cell-free reaction. This would allow on-demand production of the CRISPR effector enzyme during the diagnostic reaction itself, reducing the need for pre-purified protein. I do not yet have a fully defined design, but I plan to refine and optimize the system as the project progresses.
(ii) To synthesize the LbCas12a gene and the genetic circuit constructs, I would use commercial gene synthesis services, such as those provided by Twist Bioscience (synthetic fragments or clonal genes). Twist uses high-throughput, phosphoramidite silicon-based DNA synthesis technology to chemically synthesize DNA fragments with high accuracy and scalability.
Gene synthesis typically follows these essential steps:
Oligonucleotide synthesis Short DNA oligonucleotides (~60–200 nt) are chemically synthesized using phosphoramidite chemistry.
Oligo assembly Overlapping oligos are assembled into longer double-stranded DNA (dsDNA) fragments.
Error correction and amplification Gene fragments are purified and subjected to quality control, including size and length verification. Sequence verification may be performed by next-generation sequencing (NGS), and enzymatic error correction combined with PCR amplification improves sequence fidelity. The validated gene fragments are then ready to be shipped. For clonal genes, additional cloning and bacterial amplification steps may be required.
Cloning into a vector The synthesized gene is inserted into a plasmid backbone for propagation and expression.
Sequence verification The final construct is validated by Sanger sequencing or next-generation sequencing to confirm sequence accuracy.
Although commercial DNA synthesis is highly efficient and accurate, it has several limitations. Errors can still occur during oligonucleotide synthesis and assembly, particularly in repetitive sequences or regions with very high GC content, which may require additional verification and correction. The synthesis of very long DNA constructs can be technically challenging and more expensive, as longer sequences increase the probability of errors and assembly difficulties. Turnaround time may also vary depending on the complexity and size of the construct, sometimes requiring several days to weeks.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why? Colossal Biosciences Inc., a biotechnology company using genetic engineering to de-extinct various historic animals such as the woolly mammoth, dodo, and dire wolf.
(ii) What technology or technologies would you use to perform these DNA edits and why? Also answer the following questions: How does your technology of choice edit DNA? What are the essential steps? What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing? What are the limitations of your editing methods (if any) in terms of efficiency or precision?
(i) I am interested in editing plant genomes to improve agriculturally important traits such as disease resistance, stress tolerance (drought, heat), and yield stability. Specifically, I would target genes involved in plant-pathogen interactions, immune signaling pathways, and susceptibility (S) genes that facilitate pathogen infection. (ii) I would use CRISPR-Cas systems, delivered through engineered plant viruses. CRISPR-Cas is highly programmable, efficient, and relatively simple to design compared to earlier genome-editing tools (e.g., ZFNs or TALENs). Using plant viruses as delivery vectors allows systemic spread of guide RNAs throughout the plant, enabling editing directly in planta. First, the target gene sequence must be identified and analyzed to select an appropriate editing site adjacent to a compatible PAM sequence. Guide RNAs (gRNAs) are then computationally designed to ensure high specificity and minimal off-target effects. The selected guide sequence is cloned into a plant viral vector capable of systemic infection. The input materials include the guide RNA construct, a Cas nuclease (either stably expressed in the plant or delivered separately), the engineered viral vector, and the target plant tissue. Once delivered into plant cells, the Cas protein and guide RNA form a ribonucleoprotein complex that recognizes the target DNA sequence and introduces a double-strand break, which is subsequently repaired by the plant’s endogenous DNA repair mechanisms, resulting in the desired mutation. Editing efficiency can vary depending on plant species, en vironemtnal conditions and viral delivery efficiency. Off-target mutations may occur if guide RNAs are not carefully designed. Precise edits using HDR are typically inefficient in plants compared to knockouts generated through NHEJ. Additionally, viral vectors may have cargo size limitations and may not efficiently infect all plant tissues or germline cells, which can affect heritability of edits.