Week 4 HW: Protein Design Part I
My homework
DNA!
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)

- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans do not become cows or fish after eating them because the proteins we consume are first broken down into their basic components during digestion. Although all living organisms use the same 20 standard amino acids as building blocks of proteins, these proteins are digested into individual amino acids by enzymes such as pepsin in the stomach and trypsin in the small intestine. These amino acids are then absorbed and reused by our cells to synthesize new proteins according to the instructions encoded in human DNA. Therefore, the amino acids from beef or fish are simply raw materials that our bodies use to build human proteins.
- Why are there only 20 natural amino acids?
According to Doig (2016), the amino acids were selected during the early “RNA world” because they allow proteins to form stable, soluble structures with tightly packed hydrophobic cores and functional binding pockets. The chosen amino acids cover a wide range of properties such as size, charge, hydrophobicity, and functional groups, which enable proteins to have diverse and stable 3D structures. Other possible amino acids were likely excluded because they would not improve protein stability, solubility, or biosynthetic efficiency and cost more conformational entropy to fold than their branched isomers.
- Where did amino acids come from before enzymes that make them, and before life started?
Before life began and before enzymes existed, amino acids likely formed through prebiotic chemical reactions under early Earth conditions. According to Kirschning (2022), amino acids have been detected in meteorites, suggesting that some were synthesized in space and delivered to Earth through extraterrestrial material. In addition, amino acids could be produced on the early Earth through abiotic reactions involving simple molecules such as CO₂, NH₃, H₂O, and other small compounds under energy sources like lightning, UV radiation, or hydrothermal activity.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Natural proteins are made of L-amino acids, which typically form right-handed α-helices. If the chirality is reversed and D-amino acids are used, the geometry is mirrored, producing a left-handed α-helix.
- Can you discover additional helices in proteins?
Yes, additional helices in proteins can be identified using experimental structural techniques such as X-ray crystallography and cryo-electron microscopy, as well as computational prediction tools like AlphaFold and ESMFold.
- Why are most molecular helices right-handed?
Most molecular helices in proteins are right-handed because proteins are composed of L-amino acids. The stereochemistry of L-amino acids favors backbone conformations that form stable hydrogen bonds between the carbonyl oxygen of residue i and the amide hydrogen of residue i+4, which stabilizes a right-handed α-helix. A left-handed helix would create unfavorable steric interactions for L-amino acids, so the right-handed α-helix is energetically preferred.
- Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because they form hydrogen-bonding networks that can interact with neighboring β-strands from other protein molecules. The main driving forces for β-sheet aggregation are hydrogen bonding and hydrophobic interactions, which allow β-strands from different proteins to stack together into stable sheet-like structures.
- Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Many amyloid diseases involve β-sheet structures because misfolded proteins can reorganize into highly stable cross-β sheet fibrils. In these structures, β-strands align perpendicular to the fibril axis and form repetitive hydrogen-bond networks between protein molecules. Amyloid β-sheet structures can be used as biomaterials because they form extremely stable, self-assembling nanofibers with high mechanical strength.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
- Briefly describe the protein you selected and why you selected it.
I chose the enzyme LbCas12a because I am going to develop a field-deployable diagnostic platform that is user-friendly and low-cost for growers using the CRISPR-Cas12a system.
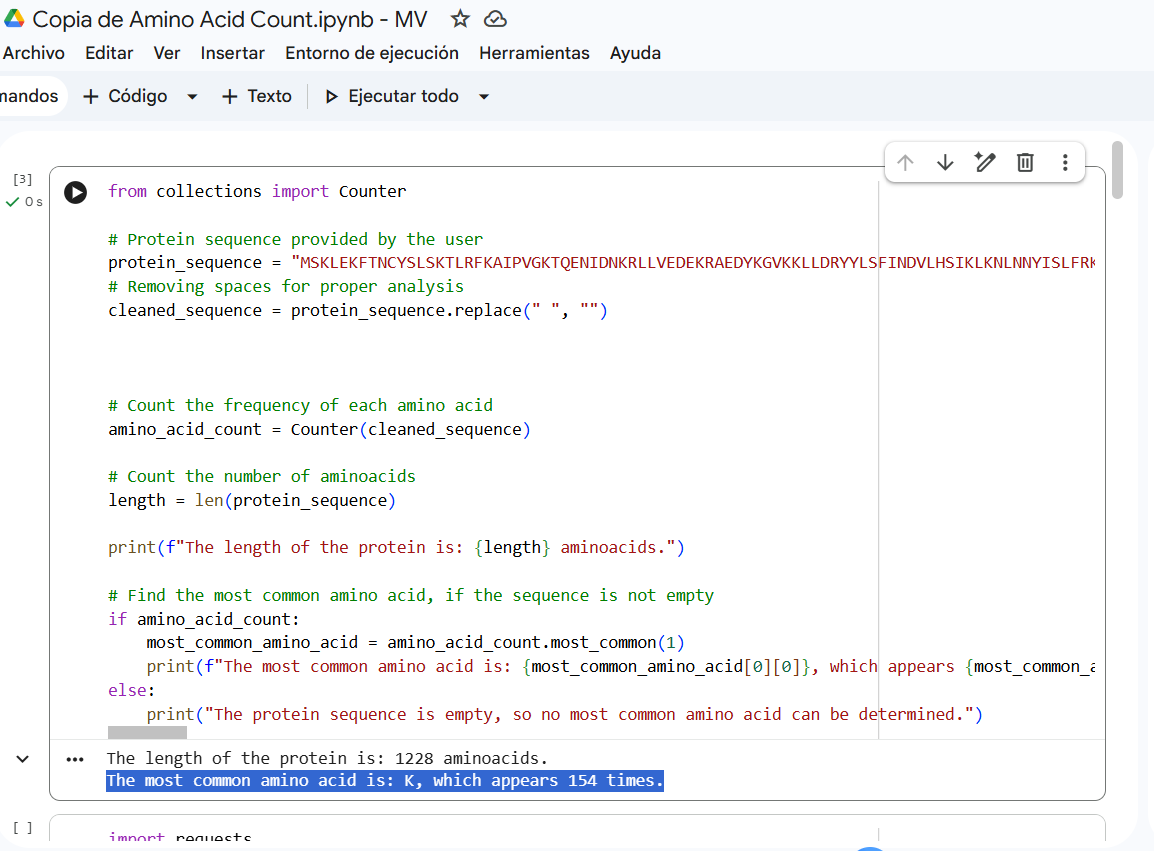
- Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
Sequence length: 1,228 AA. According to google Colab script, the most common amino acid is: K, which appears 154 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Does your protein belong to any protein family? According to Interpro, family membership: CRISPR-associated endonuclease Cas12a.
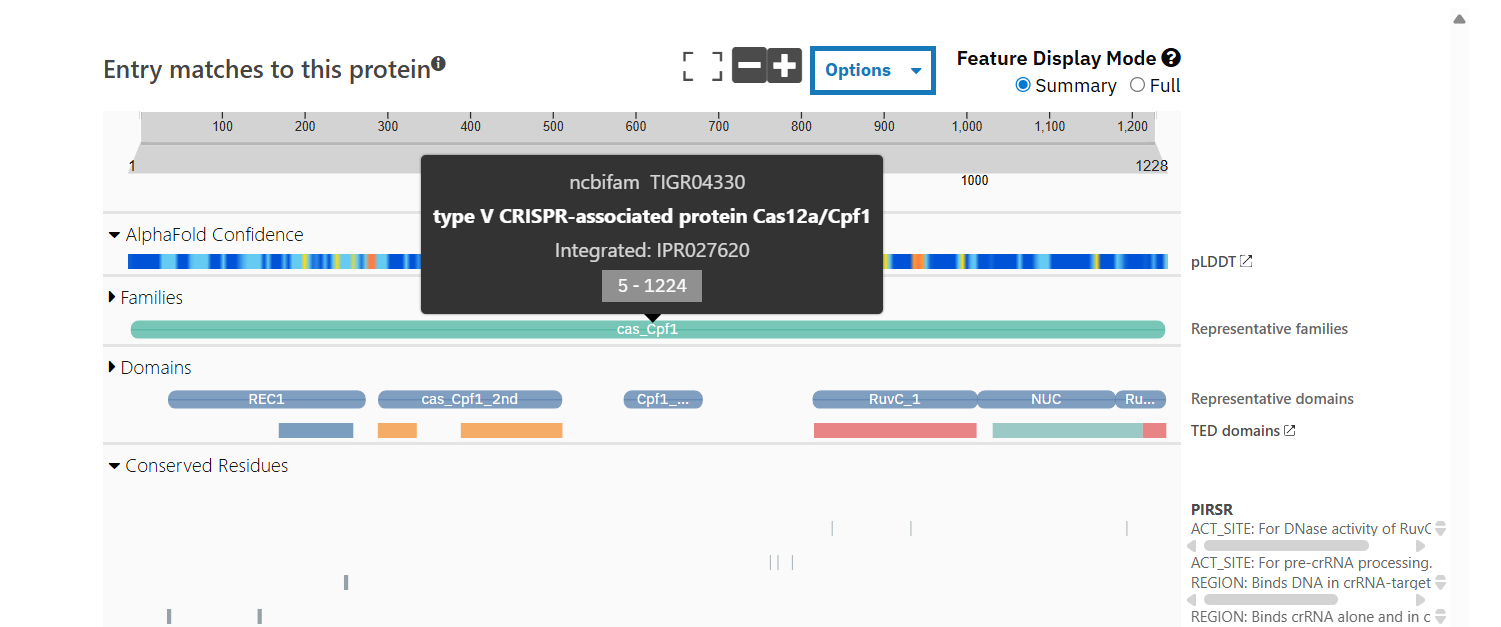
NCBIFam: type V CRISPR-associated protein Cas12a/Cpf1
- Identify the structure page of your protein in RCSB When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å) The structure was deposited in July 2019 and solved at 3.25 Å resolution by cryo-EM. This is considered a good-quality structure for electron microscopy.
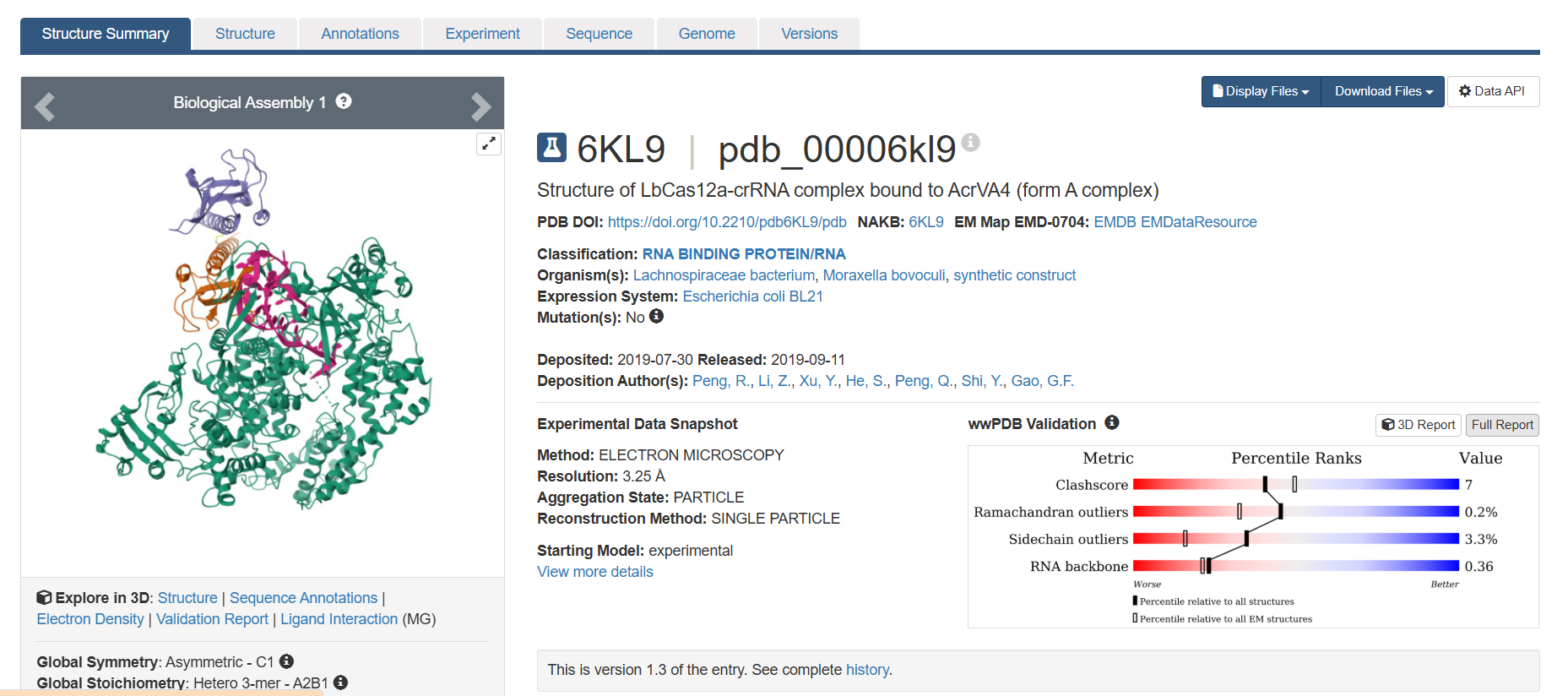
Are there any other molecules in the solved structure apart from protein? Yes, it is a complex. There are several molecules such as AcrVA4 protein, Mg+2 ion and RNA (42-MER) (crRNA).
Does your protein belong to any structure classification family? When I enter the PDB ID in the search bar, no results appear.
- Open the structure of your protein in any 3D molecule visualization software: PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands) Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Cartoon
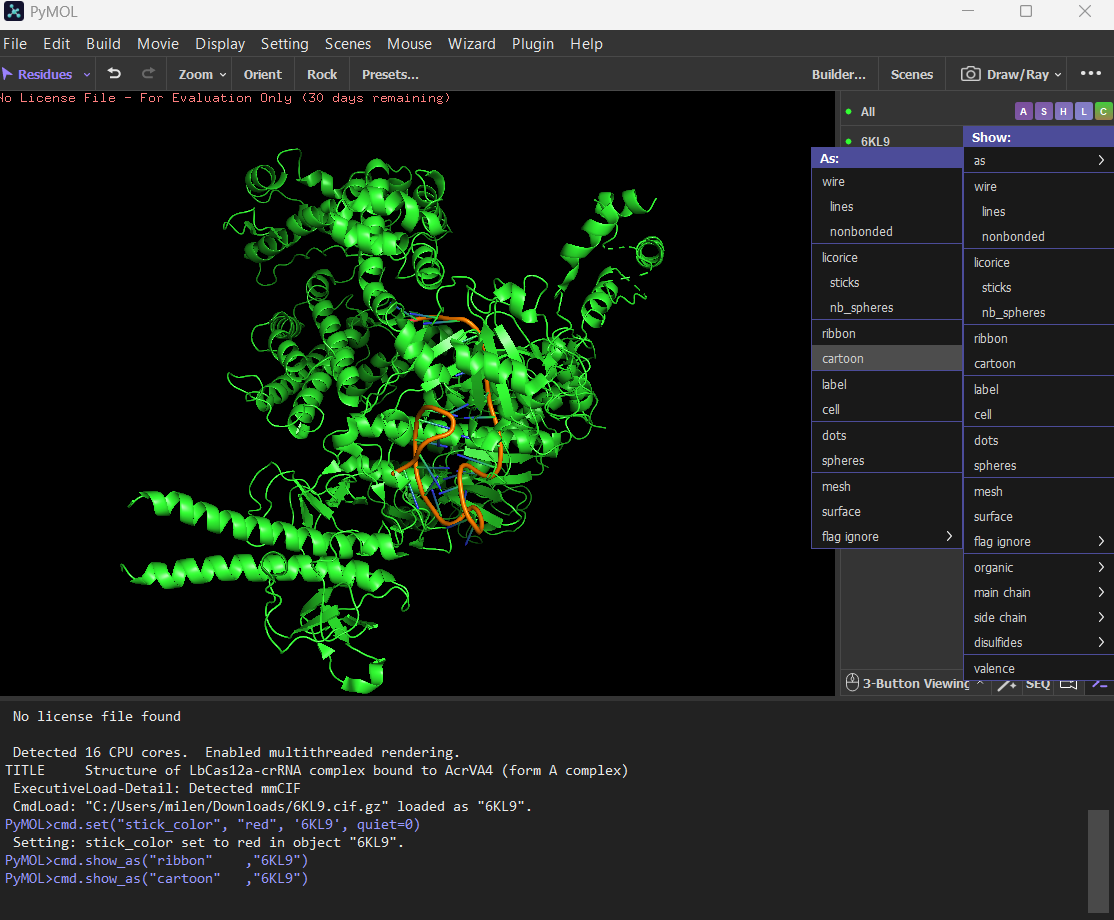
Ribbon
Ball and stick
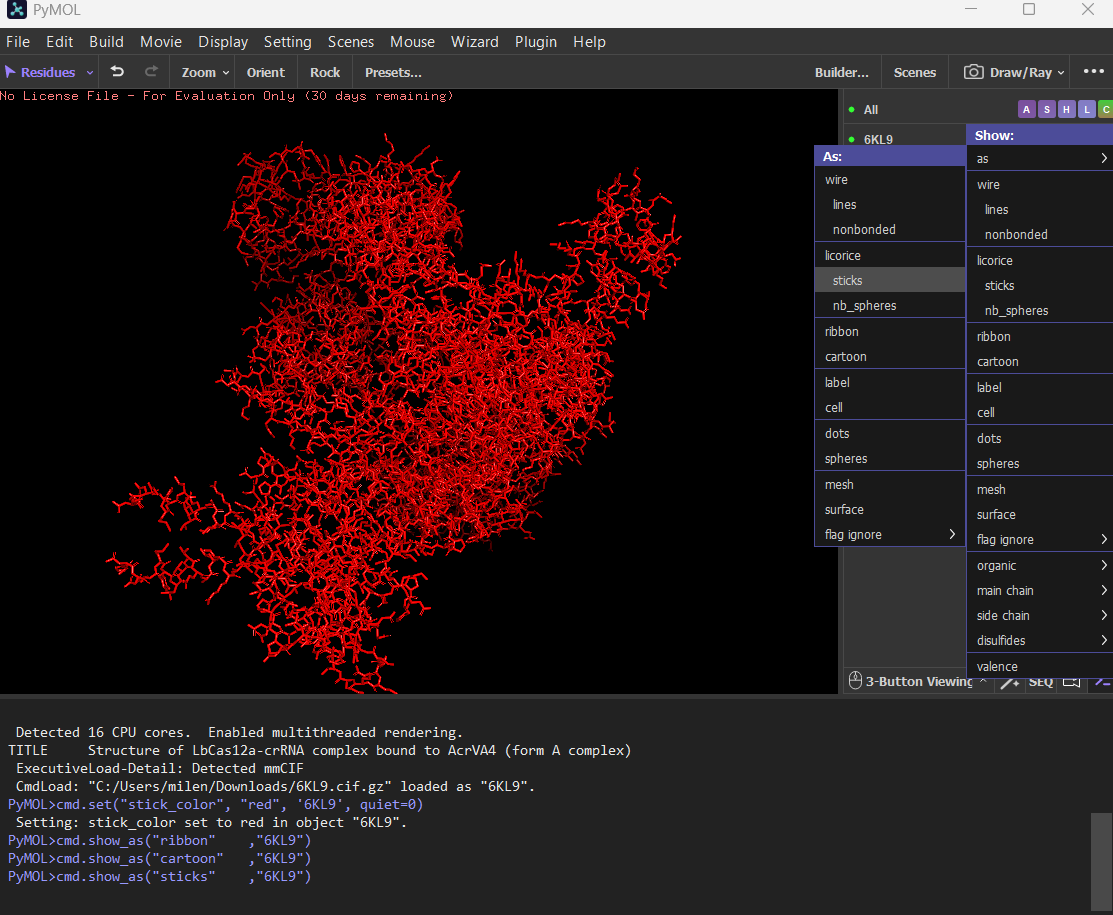
Color the protein by secondary structure. Does it have more helices or sheets? The protein has more helices (red) than sheets (yellow). It is predominantly alpha-helical, with fewer beta-sheet regions.
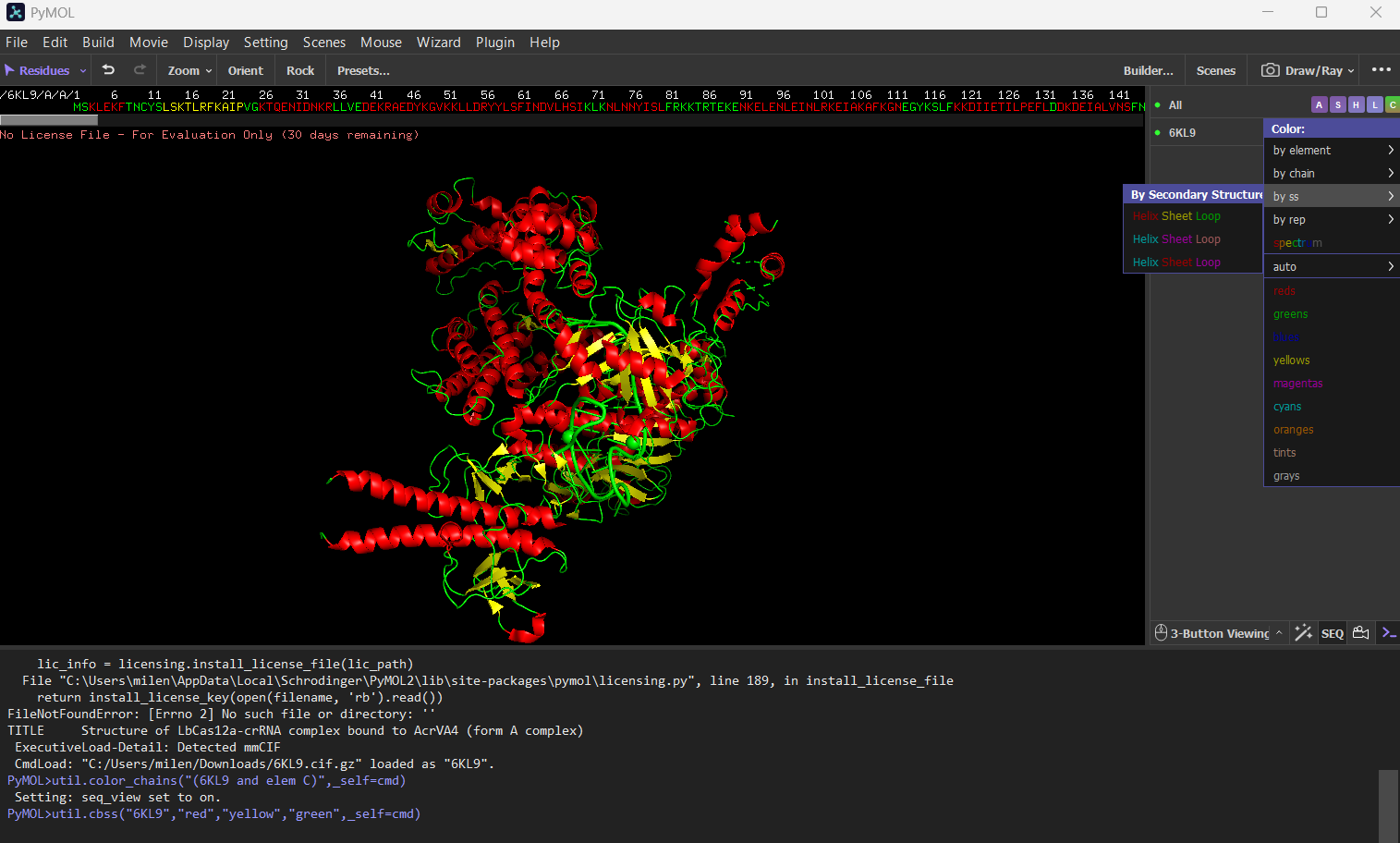
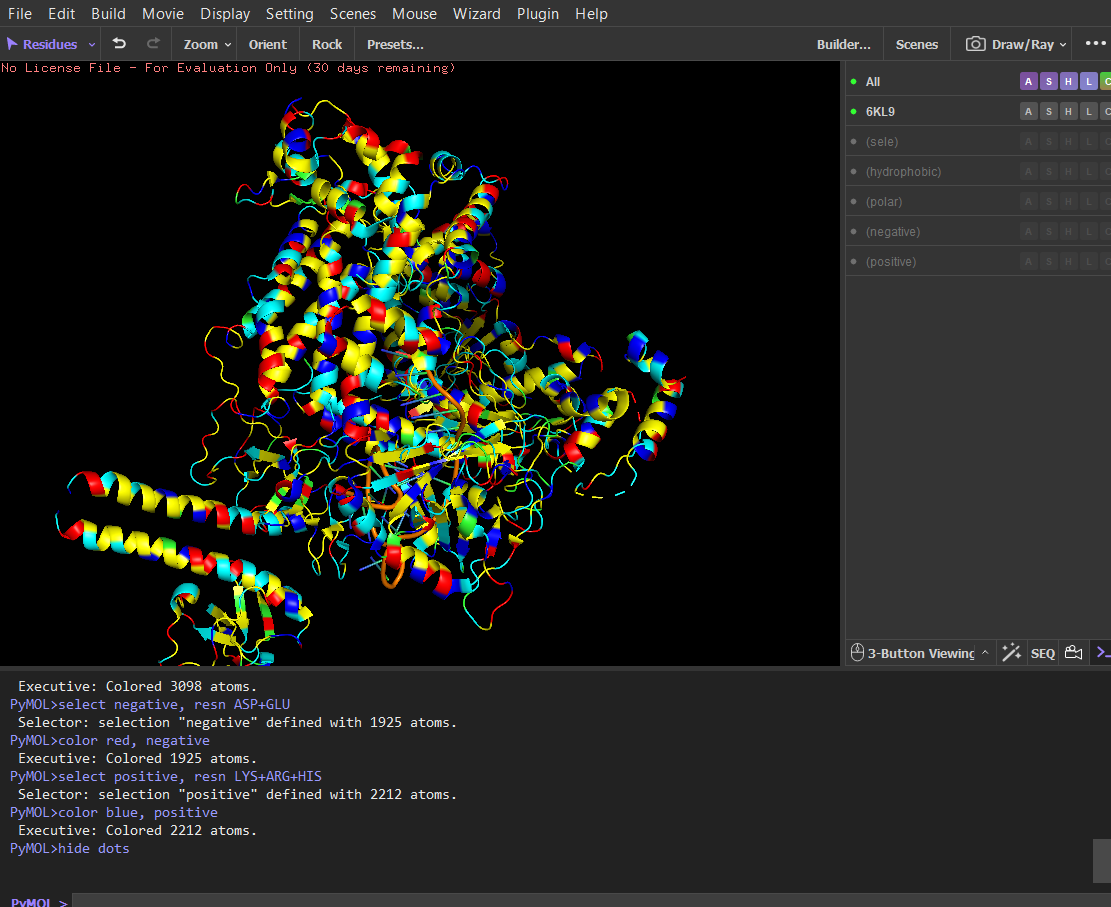
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Script
Hydrophobic (yellow) select hydrophobic, resn ALA+VAL+LEU+ILE+MET+PHE+TRP+PRO color yellow, hydrophobic
Polar without charge (cyan) select polar, resn SER+THR+ASN+GLN+TYR color cyan, polar
Negatives (red) select negative, resn ASP+GLU color red, negative
Positives (blue) select positive, resn LYS+ARG+HIS color blue, positive
After coloring the protein by residue type, hydrophobic residues (ALA, VAL, LEU, ILE, MET, PHE, TRP, PRO) are mainly located in the interior of the protein structure. These residues form the structural core and help stabilize the protein through hydrophobic interactions. In contrast, hydrophilic and charged residues (SER, THR, ASN, GLN, TYR, ASP, GLU, LYS, ARG, HIS) are predominantly exposed on the surface. Many positively charged residues are concentrated in the nucleic acid-binding region, which is consistent with the protein’s function in binding negatively charged RNA and DNA molecules. Overall, the distribution shows a typical soluble protein organization: a hydrophobic core and a hydrophilic, charged surface.
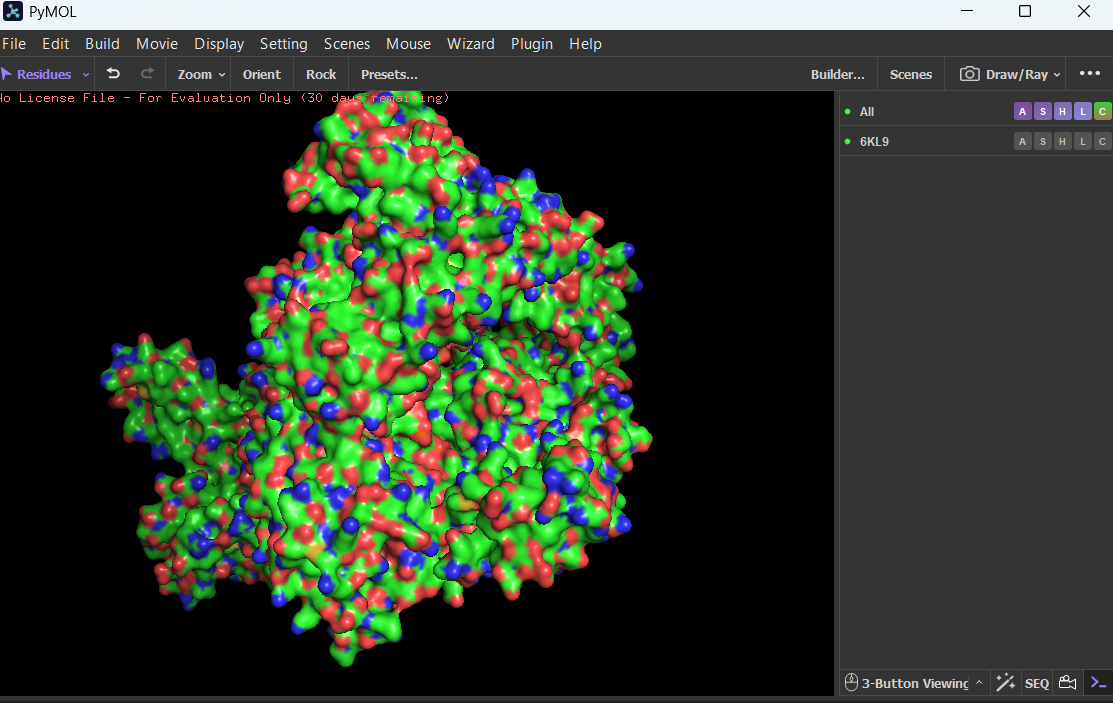
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes, the protein surface shows several cavities and grooves. There is a prominent cleft that likely corresponds to the nucleic acid-binding pocket of Cas12a. These surface indentations are consistent with functional binding sites.
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein. Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU. Choose your favorite protein from the PDB. We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
- Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
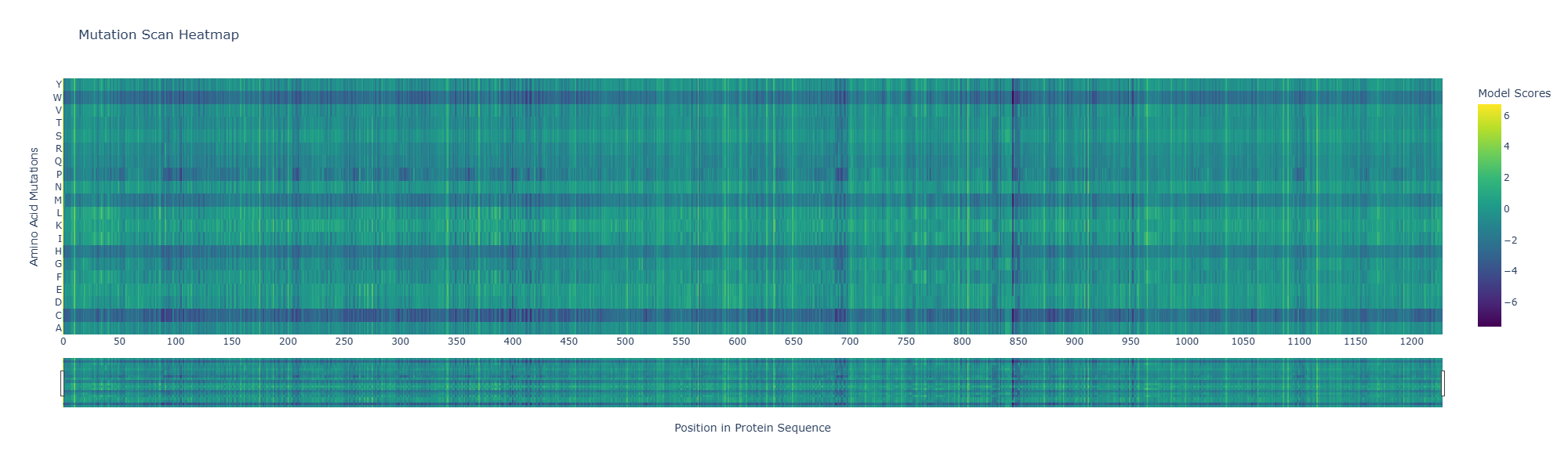
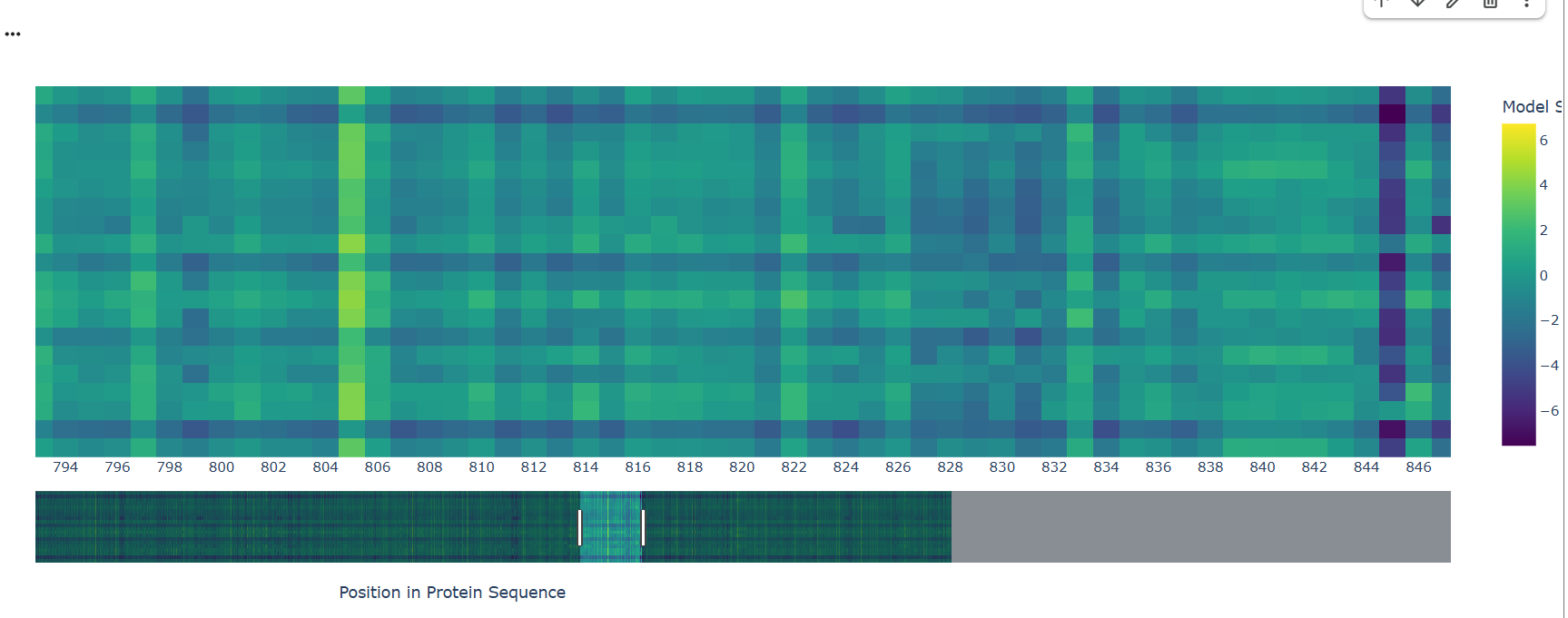
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
A notable pattern is observed around position 845, where most substitutions show strongly negative scores, indicating high intolerance to mutation. This suggests that the residue at this position is under strong evolutionary constraint, likely contributing to structural stability or functional activity. In contrast, other regions display more neutral scores, suggesting mutational flexibility.
- (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
Database with experimental deep mutational scans datasets: https://www.mavedb.org/ and https://proteingym.org/
I used the GB1 protein deep mutational scanning (DMS) dataset from Olson et al. (2014), available in MaveDB. This dataset contains experimental measurements of how single amino acid mutations affect the function of a 55 amino acid protein. I used this protein sequence established in the article: protein_sequence = “QYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTE” I generated mutation scores for the same protein using the ESM2 language model. For each possible single mutation, I calculated how likely the mutation is compared to the wild-type amino acid. Then, I compared the model scores with the experimental functional scores using Spearman correlation. Results: Spearman ρ = -0.104 p-value = 8 × 10⁻⁴
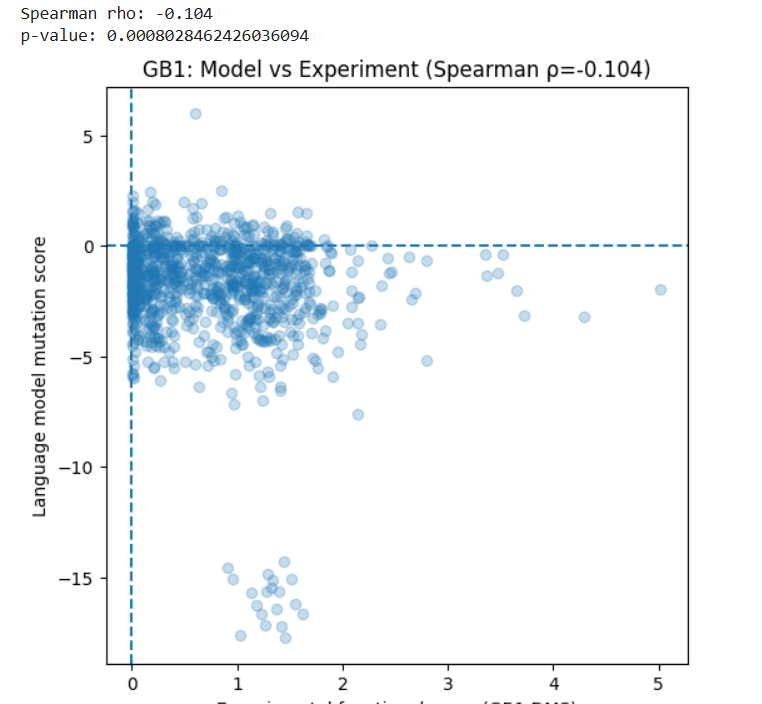
This shows a very weak negative correlation between the model predictions and the experimental data. In simple terms, the language model does not strongly predict which mutations improve or reduce protein function in this case. This may be because: The model is trained to predict natural sequence patterns, not experimental fitness. Protein function depends on structural and biophysical effects that may not be fully captured by sequence likelihood.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
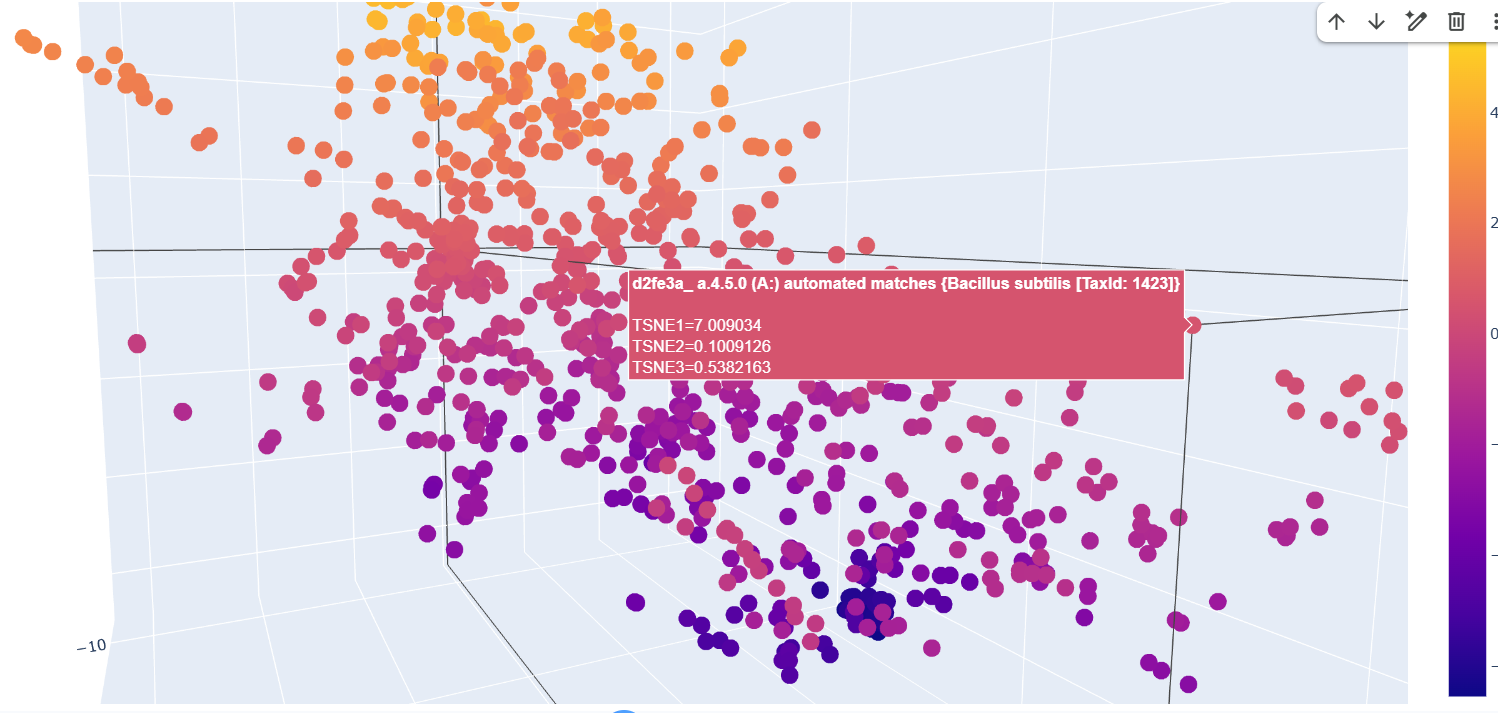
The formed neighborhoods approximate structurally similar proteins rather than taxonomically related ones. For example, sequences from Francisella tularensis, Campylobacter jejuni, Streptococcus pyogenes, Vibrio cholerae, and Bacillus subtilis cluster together despite belonging to different bacterial species. All share the SCOP classification a.4.5.0, indicating a common structural fold. This suggests that the embedding captures structural similarity across evolutionary distance.
- Place your protein in the resulting map and explain its position and similarity to its neighbors.
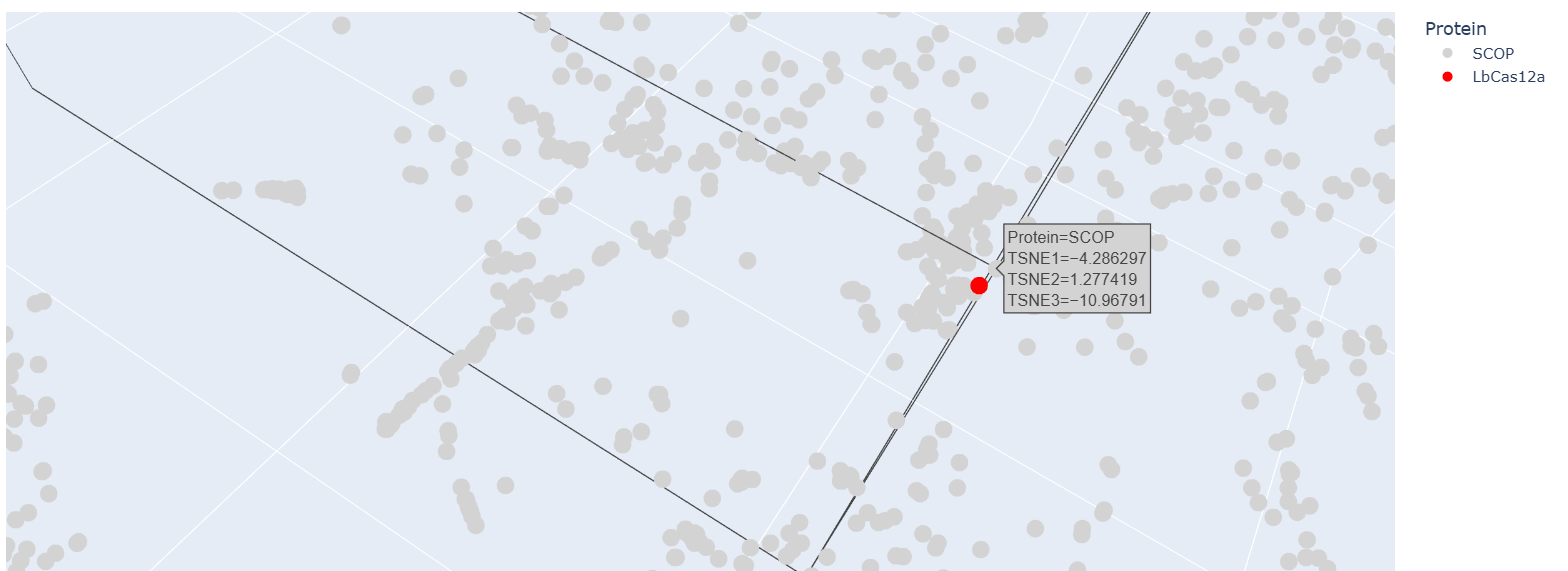
After embedding SCOP domain proteins using ESM2 and reducing dimensionality with t-SNE, I placed LbCas12a into the same latent space. The SCOP proteins form a broad, continuous distribution rather than sharply separated clusters, reflecting the diversity of protein folds in the dataset. LbCas12a appears within the overall distribution rather than being completely isolated. It lies near a local group of SCOP proteins, suggesting that the language model identifies some similarity in sequence patterns or structural features between LbCas12a and those neighboring proteins. Because embeddings were generated using mean pooling over the entire sequence, the model likely captures global sequence properties rather than specific catalytic functions. Therefore, proximity in latent space reflects general evolutionary and structural similarity rather than precise functional classification.
C2. Protein Folding
Folding a protein
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Since LbCas12a is a very large protein, running the ESMfold and inverse folding analyses would be computationally demanding. For this reason, I chose TEM-1 β-lactamase for parts C2 and C3 of the assignment, as its smaller size makes the analysis faster and easier to perform. The structure predicted by ESMFold closely matches the experimentally determined structure. After structural alignment in PyMOL, the RMSD between the predicted and experimental coordinates was 0.44 Å across 252 aligned atoms, indicating a very high structural similarity. The overall fold and arrangement of secondary structure elements are almost identical. Minor deviations occur mainly in flexible terminal regions, which are typically less structured and more difficult to predict accurately. The ESMFold prediction had a high average pLDDT score (~95.9), indicating very high confidence in the predicted structure.
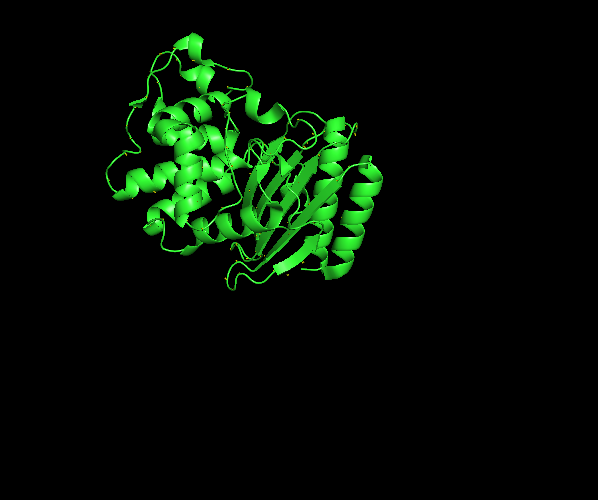 β-lactamase sequence structure from RCSB PDB visualized in Pymol
β-lactamase sequence structure from RCSB PDB visualized in Pymol
 β-lactamase sequence structure from ESMFold visualized in Pymol
β-lactamase sequence structure from ESMFold visualized in Pymol
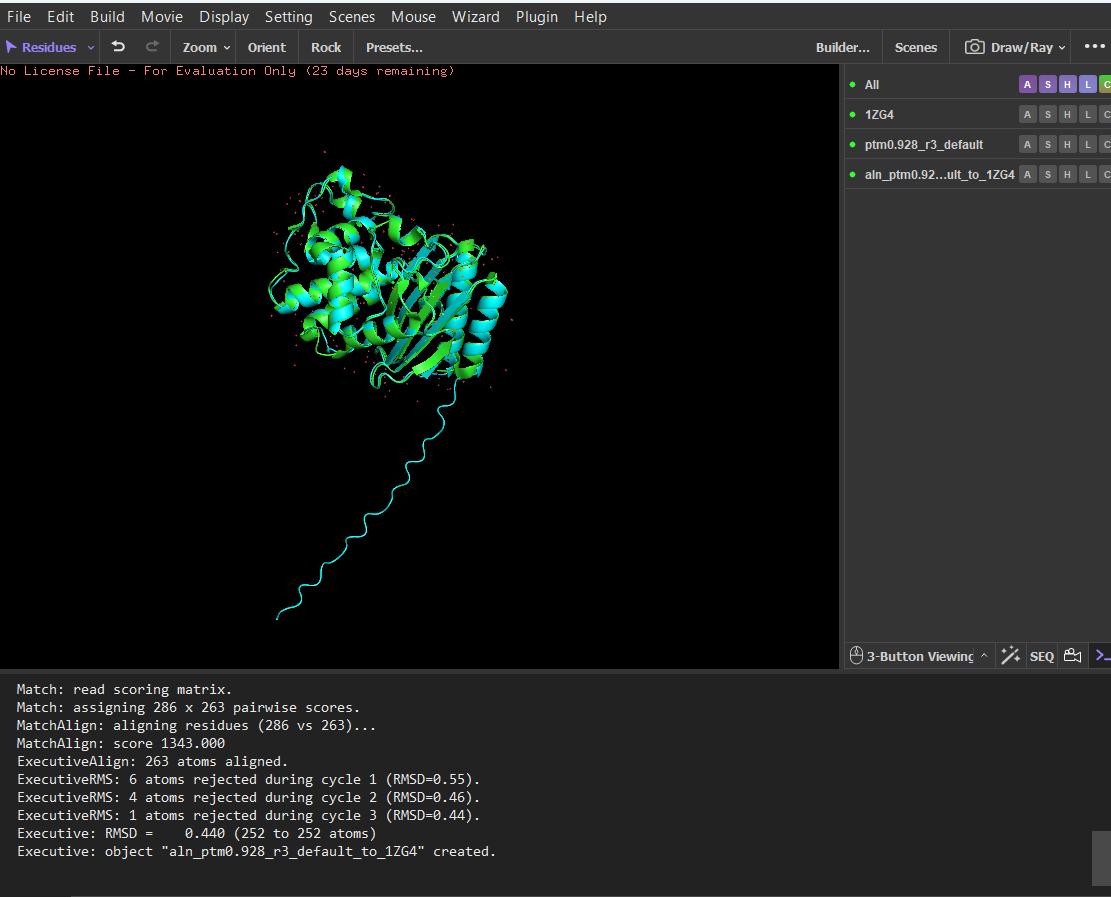 Structural alignment between the experimental β-lactamase structure and the structure predicted by ESMFold in PyMOL
Structural alignment between the experimental β-lactamase structure and the structure predicted by ESMFold in PyMOL
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I tested the structural resilience of the protein by introducing point mutations predicted by the mutational scan. I first mutated residue 68 to glutamate (E), which was predicted to be unfavorable (blue), and then mutated residue 59 to aspartate (D), which was predicted to be tolerated (green). For each mutant sequence, I predicted the structure using ESMFold and aligned it with the original predicted structure in PyMOL. The alignments produced extremely low RMSD values (0.057 Å for the mutation at position 68 and 0.038 Å for the mutation at position 59), indicating that the global protein fold remained essentially unchanged. These results suggest that the protein structure is highly resilient to single amino-acid substitutions, as these mutations do not significantly alter the overall structure.
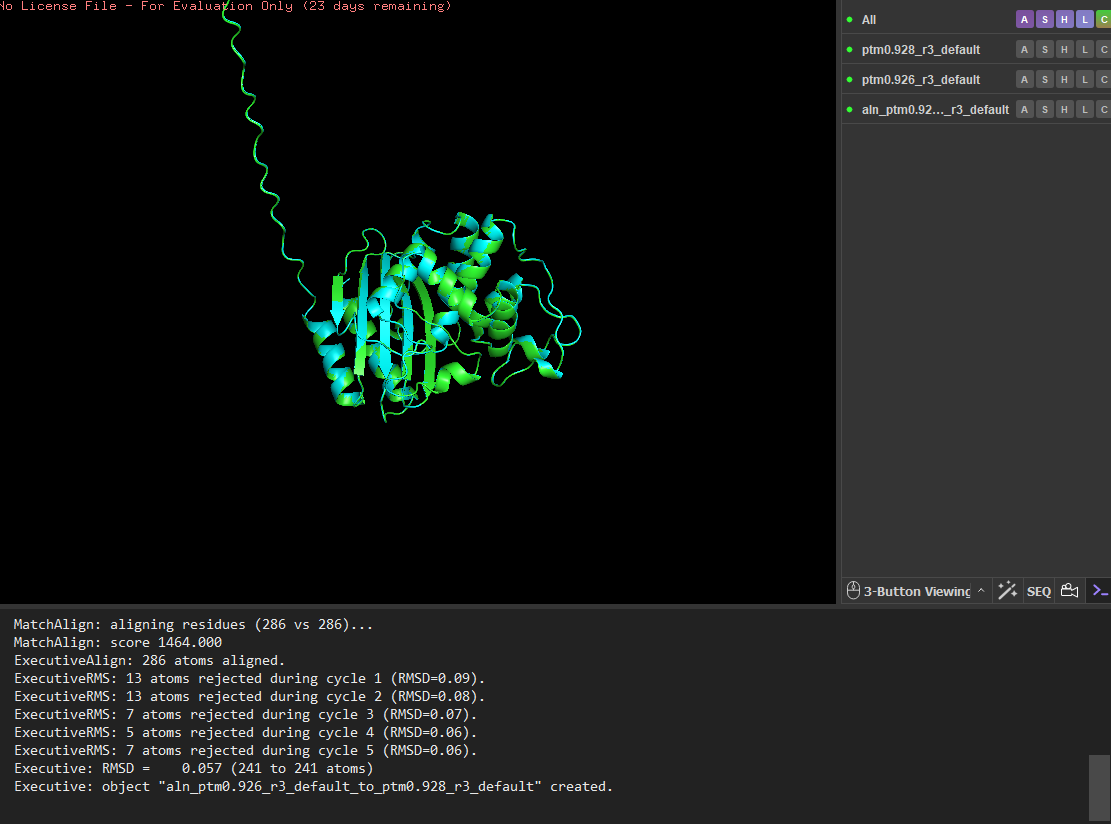 Residue 68 was mutated to glutamate (E), and the mutated structure was aligned with the original ESMFold-predicted structure using PyMOL.
Residue 68 was mutated to glutamate (E), and the mutated structure was aligned with the original ESMFold-predicted structure using PyMOL.
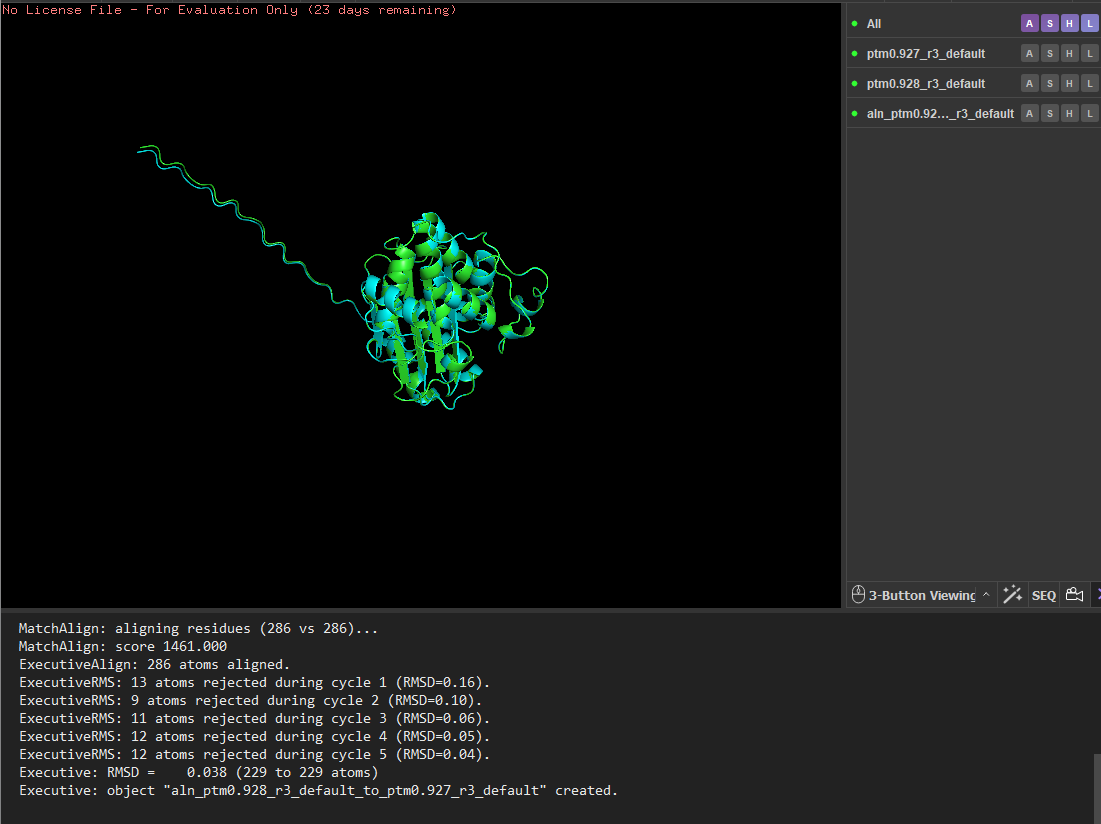 Residue 59 was mutated to aspartate (D), and the mutated structure was aligned with the original ESMFold-predicted structure using PyMOL.
Residue 59 was mutated to aspartate (D), and the mutated structure was aligned with the original ESMFold-predicted structure using PyMOL.
I then mutated a larger segment (residues 8-14) by replacing them with glutamate residues. The initial alignment showed larger deviations, suggesting local structural changes. However, after alignment refinement, the core structure still aligned well with a low RMSD (~0.056 Å), indicating that the overall fold remained stable. These results suggest that the protein structure is highly resilient to both point mutations and small segment mutations, with most structural changes occurring in flexible regions rather than the stable core.
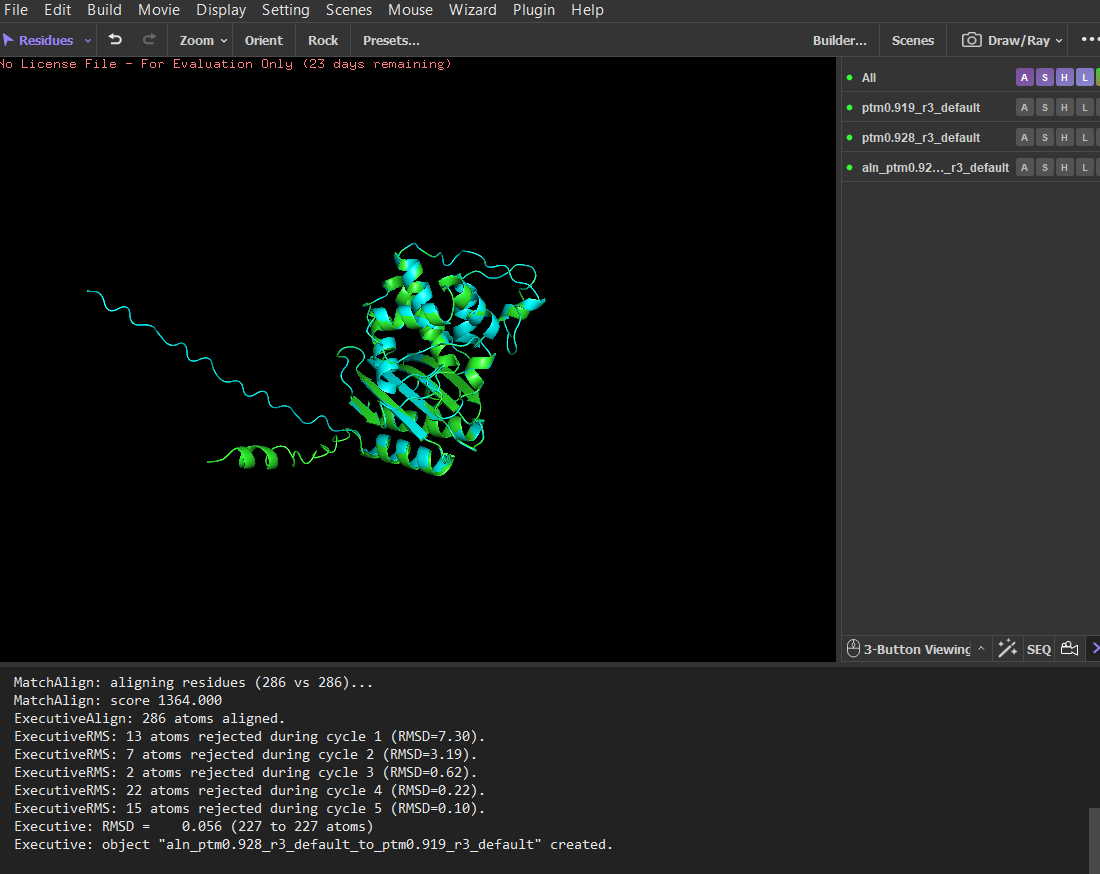 14 residues were mutated to glutamate (E), and the mutated structure was aligned with the original ESMFold-predicted structure using PyMOL.
14 residues were mutated to glutamate (E), and the mutated structure was aligned with the original ESMFold-predicted structure using PyMOL.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Below is the sequence generated by ProteinMPNN.
1ZG4, score=1.4111, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020 HPETLVKVKDAEDQLGARVGYIELDLNSGKILESFRPEERFPMMSTFKVLLCGAVLSRIDAGQEQLGRRIHYSQNDLVEYSPVTEKHLTDGMTVRELCSAAITMSDNTAANLLLTTIGGPKELTAFLHNMGDHVTRLDRWEPELNEAIPNDERDTTMPVAMATTLRKLLTGELLTLASRQQLIDWMEADKVAGPLLRSALPAGWFIADKSGAGXERGSRGIIAALGPXDGKPSRIVVIYTTGSQATMDERNRQIAEIGASLIKHW T=0.1, sample=0, score=0.7625, seq_recovery=0.4867 SAAVLDVVRAAEERLGAPVGLVVMDLATGEVLAEYNANALFPLNSTSKVFLAAYVLHLVDRGELSLDQLVKYDESDIVPNSPVTKKHIERGMTVRELMEAAIQHSDNTAYNLLLKLIGGPEALTAWLRSIGDTVTRLTHYEPELKANVPGDTADTGTPLATATTLRTILTGDVLSPESRALLREFMAANTVCGARFPSALPAGWALYCKCGSGXKNGSMNIVAAFGPXNGAPTHIVVIFTSGSKASLAEIEAAIAEIAAAIIAHL
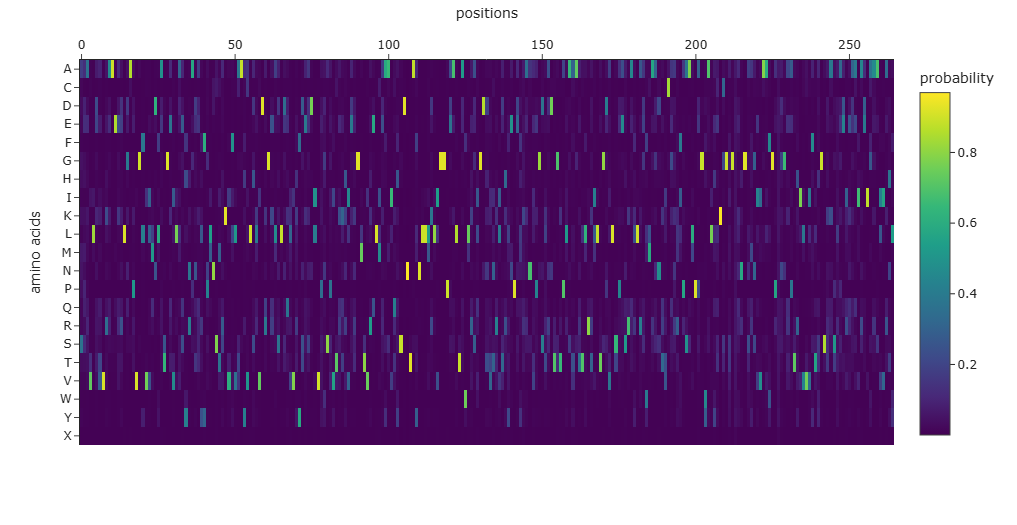
The amino-acid probability heatmap shows the probability of each residue at every position of the protein backbone. Positions with strong probability peaks correspond to residues that are strongly preferred by the model, indicating structurally constrained sites such as buried residues or catalytic positions. Positions with lower probabilities across many amino acids correspond to more flexible or surface-exposed regions. The designed sequence generated by ProteinMPNN has a score of 0.7625, which represents the negative log-likelihood of the sequence given the backbone structure, with lower scores indicating better structural compatibility. The sequence recovery of 0.4867 indicates that approximately 48.7% of the residues match the native sequence, demonstrating that multiple sequences can adopt the same protein fold.
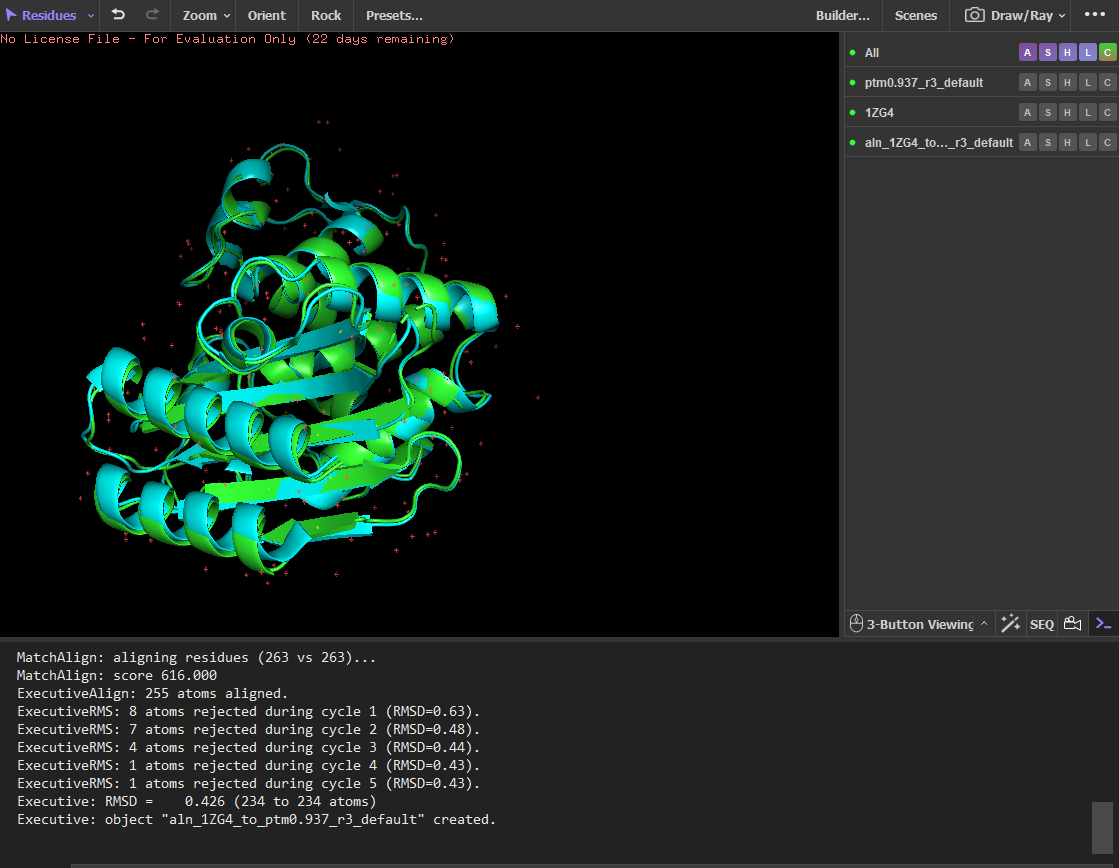
- Input this sequence into ESMFold and compare the predicted structure to your original.
The sequence generated by the inverse folding model (ProteinMPNN) was used as input for ESMFold to predict its structure. The predicted structure was then aligned with the original crystal structure (PDB: 1ZG4) using PyMOL. Despite the sequence recovery being approximately 48%, the structural alignment shows a very low RMSD value of 0.426 Å, indicating that the two structures are nearly identical. This result demonstrates that different amino-acid sequences can adopt very similar three-dimensional folds, highlighting that protein structure is often more conserved than sequence.
References
Balasco, N., Diaferia, C., Morelli, G., Vitagliano, L., & Accardo, A. (2021). Amyloid-like aggregation in diseases and biomaterials: Osmosis of structural information. Frontiers in Bioengineering and Biotechnology, 9. https://doi.org/10.3389/fbioe.2021.641372
Cheng, P.-N., Liu, C., Zhao, M., Eisenberg, D., & Nowick, J. S. (2012). Amyloid β-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nature Chemistry, 4(11), 927–933. https://doi.org/10.1038/nchem.1433
Doig, A. J. (2017). Frozen, but no accident – why the 20 standard amino acids were selected. The FEBS Journal, 284(9), 1296–1305. https://doi.org/10.1111/febs.13982
Kirschning, A. (2022). On the evolutionary history of the twenty encoded amino acids. Chemistry – A European Journal, 28(55). https://doi.org/10.1002/chem.202201419