Week 5 HW: Protein Design part II
My Homework
Part 1: Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
- Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
- Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
- To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
- Record the perplexity scores that indicate PepMLM’s confidence in the binders.
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Mutated sequence_SODC_HUMAN MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Pseudo-perplexity values represent PepMLM’s confidence in each peptide sequence. Lower values indicate sequences that are more consistent with patterns learned by the model. In our results, the generated peptides showed lower perplexity scores (6.2-15.1) compared with the known SOD1-binding peptide FLYRWLPSRRGG (20.6), suggesting the model considers the generated sequences more probable under its learned sequence distribution.
In the PepMLM study, most experimentally validated peptide binders exhibit pseudo-perplexity values below ~40, with many falling between approximately 5 and 20. Lower PPL values indicate higher model confidence in the peptide-protein interaction. In our results, the generated peptides showed PPL values between 6.2 and 15.1, which fall within the range reported in the paper, while the known SOD1-binding peptide had a higher PPL of 20.6, suggesting lower confidence by the model.
Part 2: Evaluate Binders with AlphaFold3
- Navigate to the AlphaFold Server: alphafoldserver.com
- For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
- Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
- In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
The amino acid X represents an unknown or unspecified residue predicted by PepMLM. Since AlphaFold does not accept ambiguous residues, alanine (A) was used as a replacement. Alanine is commonly used in protein modeling because it has a small, non-reactive side chain, which minimizes steric and chemical effects on the predicted structure. This makes it a reasonable neutral placeholder that is unlikely to strongly bias peptide-protein interactions in the model.
| Index | Peptide | iPTM | Binding description |
|---|---|---|---|
| 0 | WRYGATGVAHKK | 0.80 | Peptide binds on the surface of the β-barrel of one SOD1 monomer, away from the N-terminus. The interaction appears surface-bound and does not strongly approach the dimer interface. |
| 1 | WRYPATALRHKX | 0.87 | Peptide binds along the β-barrel surface of one monomer, distant from the N-terminal region. The peptide remains solvent-exposed and mostly surface-associated and it is away from the dimer interface. |
| 2 | WHYPAAGVEHGX | 0.65 | Peptide interacts with loop regions on the protein surface, away from the N-terminus, β-barrel core and dimer interface. Binding appears weak and loosely surface-bound. |
| 3 | WHYYATGAAHGX | 0.86 | Peptide binds near the β-barrel surface, not close to the N-terminal A4V region and dimer interface. Interaction occurs on the outer protein surface. |
| 4 | FLYRWLPSRRGG (known binder) | 0.89 | Peptide binds close to the N-terminus where the A4V mutation is located and lies along the β-barrel surface near the dimer interface. |
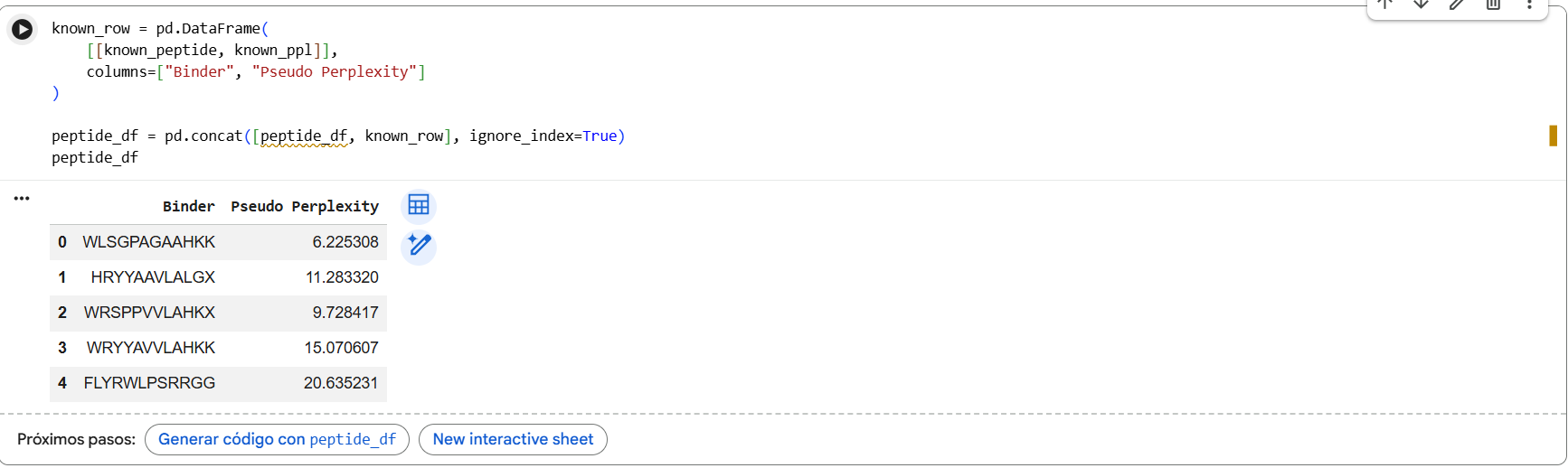
0-WRYGATGVAHKK & SOD1 Mutant
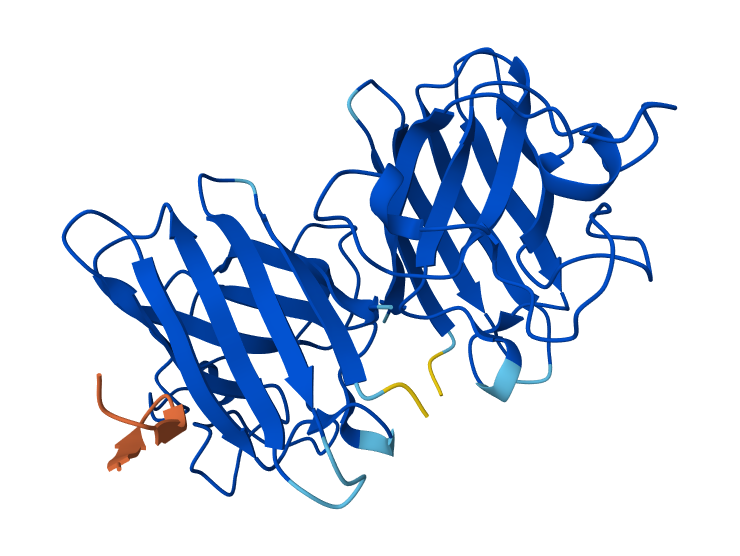
1-WRYPATALRHKX & SOD1 Mutant
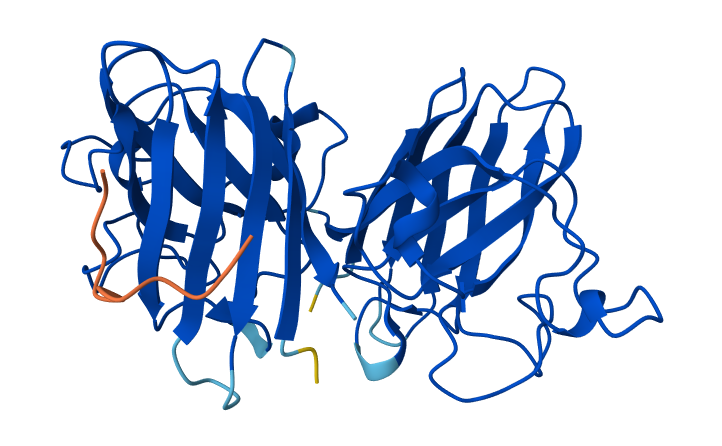
2-WHYPAAGVEHGX & SOD1 Mutant
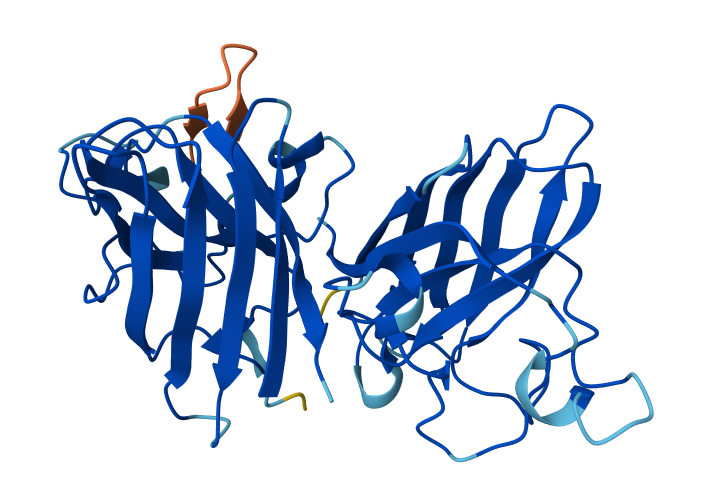
3-WHYYATGAAHGX & SOD1 Mutant
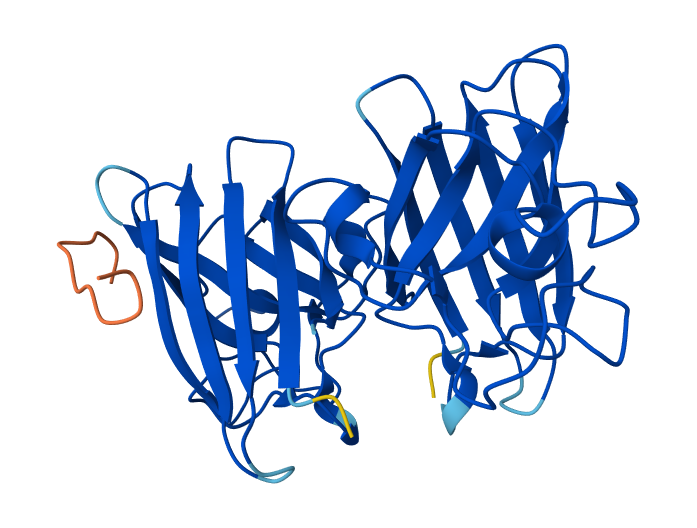
4-FLYRWLPSRRGG & SOD1 Mutant
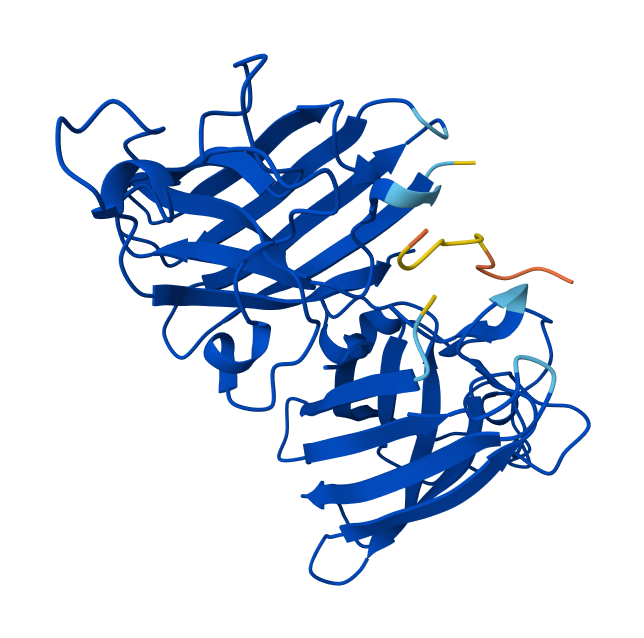
In AlphaFold predictions, pTM reflects confidence in the overall fold of the protein complex, while ipTM measures the confidence of the interface between interacting chains. ipTM values above ~0.8 indicate high-confidence interfaces, values between 0.6-0.8 represent moderate confidence, and values below 0.6 suggest unreliable interactions. The predicted complexes show ipTM values between 0.65 and 0.89, indicating mostly moderate to high confidence. Most PepMLM-generated peptides bind to surface regions of the SOD1 β-barrel, but do not localize near the N-terminal A4V region and dimer interface. Only the known binder FLYRWLPSRRGG binds close to the N-terminus and near the dimer interface, and it also produces the highest ipTM score (0.89). Although some generated peptides show relatively high ipTM values (0.87 and 0.86), none match or exceed the known binder in both binding location and interface confidence.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
- Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
- Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
- Choose one peptide you would advance and justify your decision briefly.
0-WRYGATGVAHKK
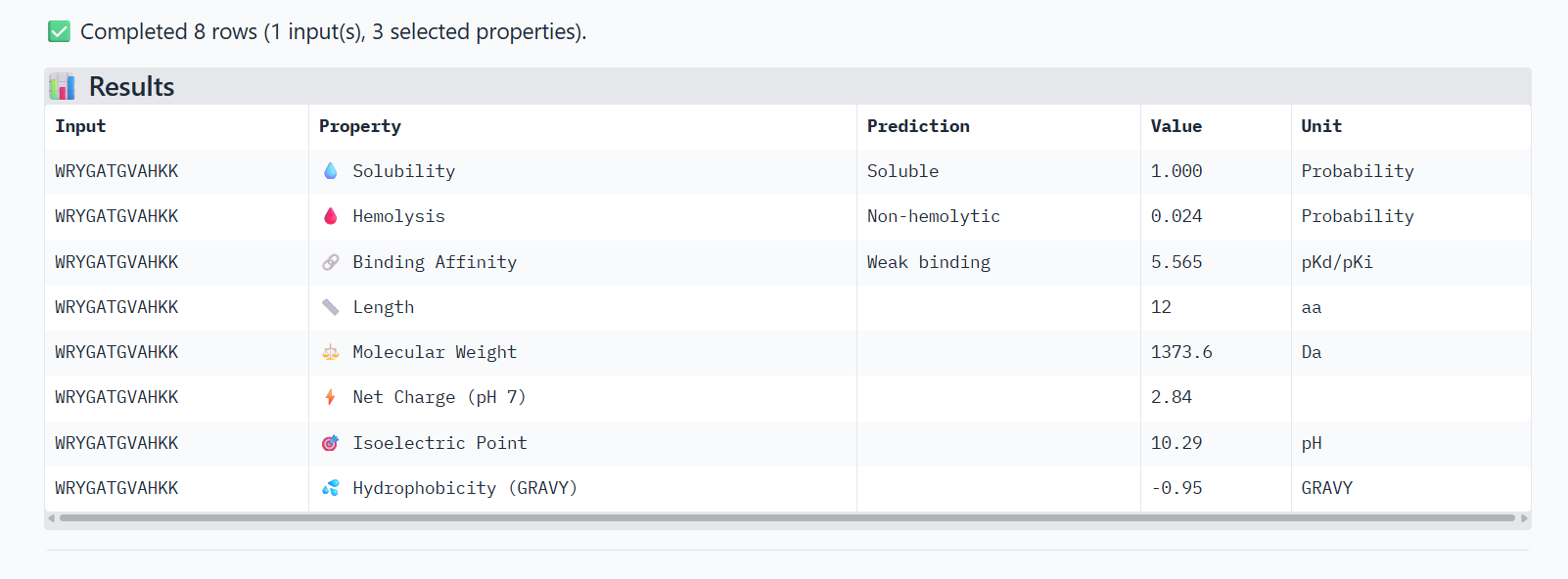
1-WRYPATALRHKX X→ Alanine
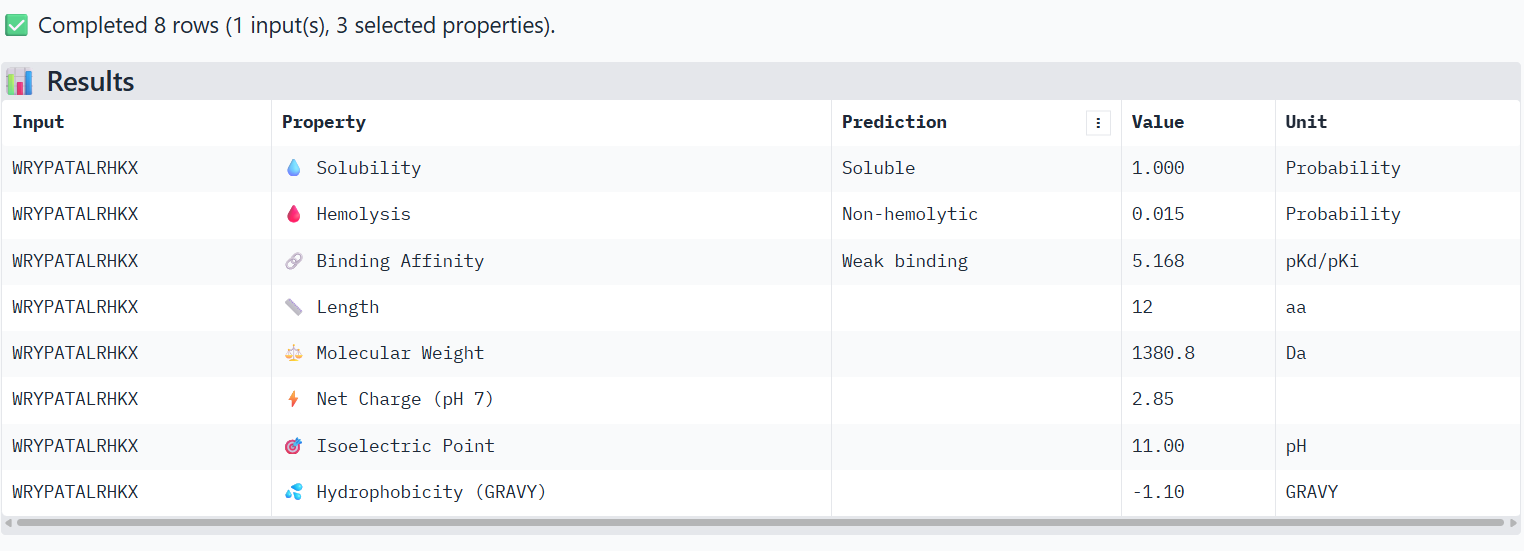
2-WHYPAAGVEHGX X→ Alanine
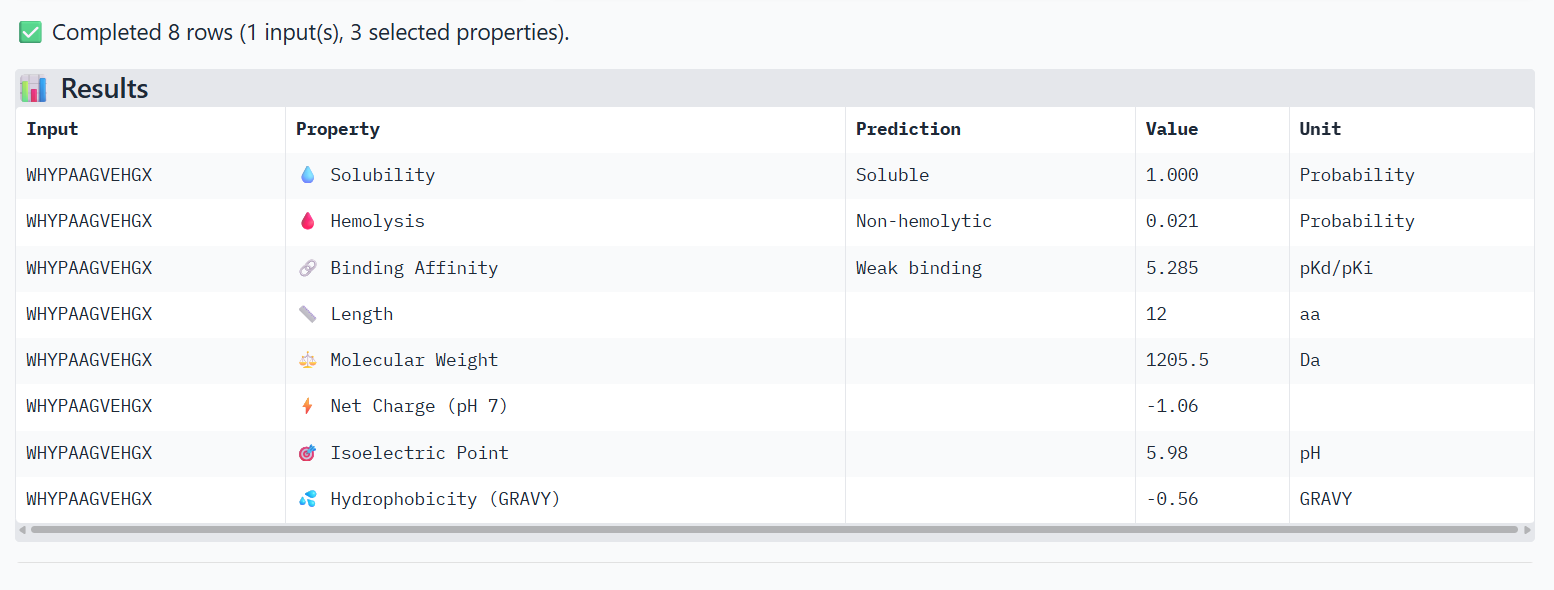
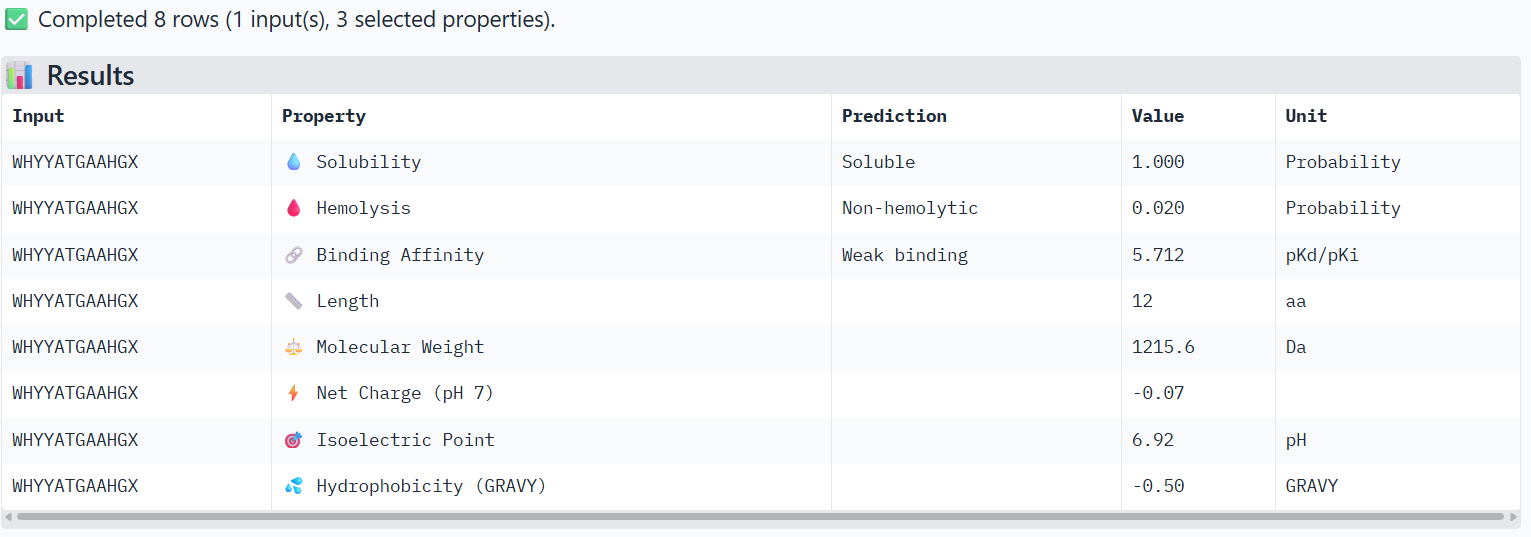
3-WHYYATGAAHGX X→ Alanine
4-FLYRWLPSRRGG (known binder)
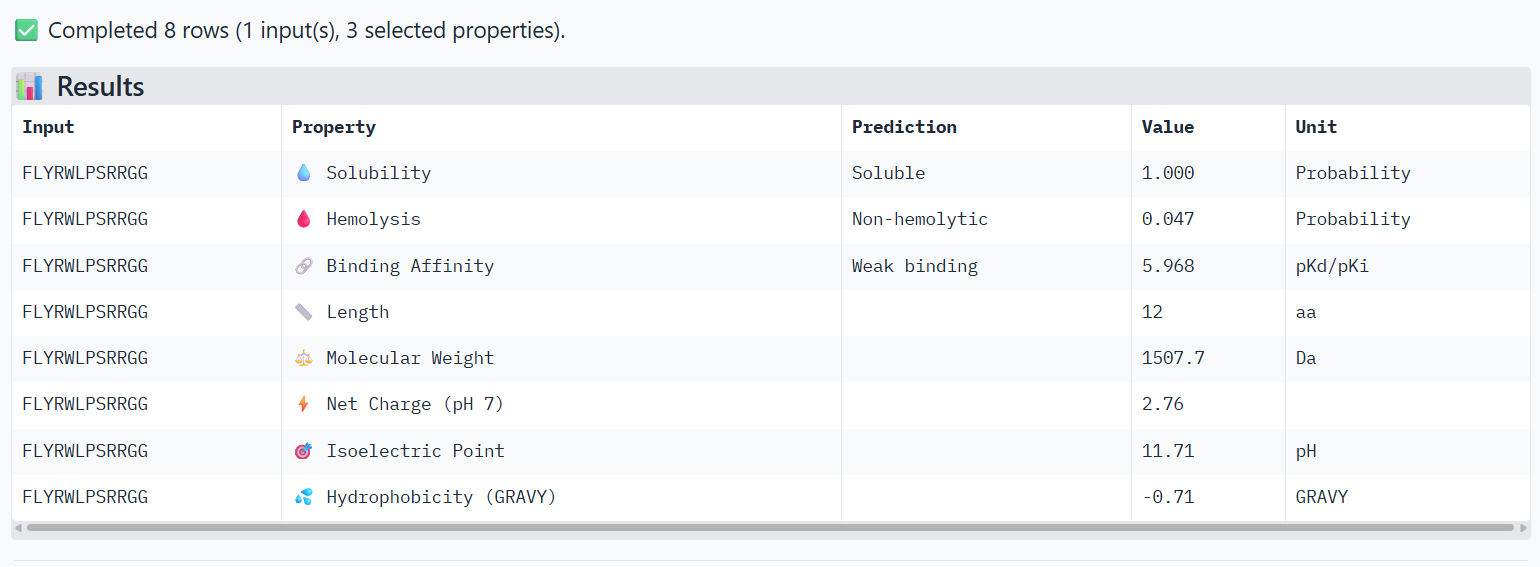
The PeptiVerse analysis shows that all peptides are predicted to be highly soluble (probability = 1.0) and non-hemolytic, indicating favorable safety and formulation properties for therapeutic development. The predicted binding affinities for all peptides fall within a similar range (pKd/pKi ≈ 5.1-5.7), suggesting relatively weak binding interactions overall. These affinity predictions do not perfectly correlate with the structural confidence observed in AlphaFold3. For example, peptide WRYPATALRHKX (index 1) showed a high structural interface score in AlphaFold (ipTM ≈ 0.87) but has the lowest predicted affinity (5.168) in PeptiVerse. Conversely, the known binder FLYRWLPSRRGG shows the highest predicted affinity (5.968) and also had the highest AlphaFold interface confidence. The peptides also fall within a similar molecular weight range (~1200-1500 Da) and most carry a moderate positive charge, which may favor protein-protein interactions. Although several PepMLM-generated peptides show similar therapeutic properties, peptide WHYYATGAAHGX (index 3) provides the best balance between structural and biochemical predictions. It shows a relatively high AlphaFold interface confidence (ipTM ≈ 0.86) together with weaker predicted binding affinity (pKd/pKi ≈ 5.71, but higher than the other ones). In addition, it has favorable therapeutic properties including high solubility, low hemolysis probability, and a smaller molecular weight. Its near-neutral net charge may also reduce nonspecific interactions compared with more positively charged peptides.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
- Open the moPPit Colab linked from the HuggingFace moPPIt model card
- Make a copy and switch to a GPU runtime.
- In the notebook:
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
- After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
The peptides generated with moPPIt differ from the PepMLM peptides because they are not just sampled based on sequence likelihood, but are actively guided toward multiple objectives, including binding affinity, motif constraints, solubility, and hemolysis. As a result, moPPIt peptides (e.g., GHSYFRGCGTYW, YYTMTCYTTIPY) tend to show higher predicted affinity scores (~6-7.3) compared to PepMLM peptides (~5-6), and also include specific sequence features (motifs) that target selected regions of SOD1. However, this optimization comes with trade-offs. Some moPPIt peptides show high hemolysis probabilities (~0.89-0.97) and only moderate solubility, indicating that improving binding and motif targeting may negatively impact therapeutic safety. In contrast, PepMLM peptides were generally non-hemolytic and highly soluble, but had weaker predicted binding. Before advancing these peptides to clinical studies, structural validation using AlphaFold or docking should confirm that the peptides bind to the intended region on SOD1. Second, in vitro assays (e.g., binding affinity measurements such as SPR or ITC) should verify the predicted interactions. Third, toxicity and stability assays should be performed to assess hemolysis, aggregation, and degradation. Finally, promising candidates would need to be optimized to balance binding strength with safety and pharmacological properties.
| Peptide | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|
| GHSYFRGCGTYW | 0.95 | 0.83 | 7.13 | 0.64 |
| TDSQMRKFGPFY | 0.89 | 0.66 | 6.05 | 0.69 |
| YYTMTCYTTIPY | 0.91 | 0.75 | 7.28 | 0.82 |
| SFGKTCVKTEQV | 0.98 | 0.75 | 6.77 | 0.90 |