FIELD-DEPLOYABLE CRISPR-CAS12A DETECTION DEVICE FOR ASTER YELLOWS PHYTOPLASMA

SECTION 1: ABSTRACT
Aster yellows phytoplasma (AYP) is a phloem-limited bacterial plant pathogen affecting garlic production in Minnesota. Because phytoplasmas are difficult to culture, diagnosis relies mainly on laboratory-based PCR, qPCR, and sequencing, which limits rapid field decision-making for growers. The overall goal of this project is to develop a proof-of-concept, field-deployable CRISPR-Cas12a diagnostic workflow for detecting phytoplasma and AYP in garlic. I hypothesize that combining simplified sample preparation, RPA isothermal amplification, CRISPR-Cas12a detection, and lateral-flow or fluorescence readout can enable faster plant disease detection while maintaining sensitivity comparable to qPCR. The first aim is to design and computationally evaluate RPA primers and Cas12a crRNAs targeting cpn60 and ribosomal protein (RP) sequences generated from phytoplasma-positive garlic samples. The second aim is to validate the CRISPR-Cas12a detection workflow using amplified targets and extracted nucleic acids, including preliminary testing with three literature-derived crRNAs that produced detectable fluorescence signal in phytoplasma-positive targets. The third aim is to translate the assay toward a portable detection platform using multiplex lateral flow and future probe-invasion logic. Methods include multilocus sequence analysis, crRNA design in Google Colab, BLAST/NUPACK screening, RPA, Cas12a reporter assays, LOD estimation, qPCR comparison, and field-compatible sample preparation optimization.
SECTION 2: PROJECT AIMS
Aim 1: Design CRISPR-Cas12a crRNAs for cpn60 and RP targets to establish a proof-of-concept nucleic-acid detection workflow for phytoplasma-positive plant samples.
Aim 2: Test crRNA and RPA primers, evaluate invasion probes and determine assay specificity (off-targets effects), sensitivity and limit of detection.
Aim 3: Develop a low-cost, field-deployable CRISPR diagnostic for AYP detection.
SECTION 3: BACKGROUND
Background and Literature Context
Phytoplasmas are wall-less, phloem-limited bacterial plant pathogens that cannot be cultured in vitro, making biological characterization and disease management very challenging. They are transmitted primarily by sap-feeding insect vectors such as leafhoppers and psyllids, but they can also spread through vegetative propagation, which is relevant for garlic because cloves are used as planting material (Weintraub, & Beanland, 2006). Early and accurate detection prevents entering infected planting stock to production systems and spreading disease in the field. Current phytoplasma detection relies on nested PCR, qPCR, and sequencing, but these methods require laboratory infrastructure, trained personnel, and may not provide immediate results (Zao et al., 2021). AYP was first detected in garlic in Minnesota in 2012, with outbreaks recorded in 2017 and 2021, and spread to planting material in 2018 and 2022 (Mollov et al., 2014). In 2024, infestations were detected throughout Minnesota. However, there are no precise data on its incidence. AYP is a concern for production, because there are not available effective treatments for this emergent pathogen in Minnesota’s garlic crops, and current diagnostic methods are time-consuming and costly. There is a need to develop biotechnological tools for the detection and management of this plant pathogen. There are some studies that use the CrisprCas12a system for detection of phytoplasma. Wei, Yang & Shih, (2026) describes a phytoplasma detection protocol using RPA-Cas12a system in which RPA first amplifies phytoplasma DNA and Cas12a/crRNA then recognizes the target and activates collateral cleavage of a reporter for fluorescence or lateral-flow readout. Lagner et al., (2025) develop a CRISPR-Cas12a phytoplasma detection system improving assay sensitivity by testing engineered Cas12a variants and modified reporters. The authors found that LbCas12a-Ultra combined with a 7-nt stem-loop reporter improved sensitivity by approximately 10-fold compared with standard wild-type LbCas12a and linear reporters. They also used multiple conserved 16S rRNA target sites to increase detection coverage across diverse phytoplasma groups. However, the study also recognized some limitations. Without a pre-amplification step, very low-titer phytoplasma samples may be missed increasing the risk of false negatives. Lateral flow (LFA) had an LOD between 10 pM and 100 pM, which is less sensitive than fluorescence-based readouts. The authors highlighted that additional validation with more phytoplasma samples, off-target screening, and further LOD improvement in LFA are needed before translating it to the field (Lagner et al., 2025). Many CRISPR detection platforms rely on Cas12a trans-cleavage. In trans-cleavage assays, once Cas12a is activated by target recognition, it indiscriminately cleaves single-stranded DNA reporters. This produces strong signal amplification, but it also makes multiplexing difficult because all targets activate the same reporter pool. It can also create specificity problems because trans-cleavage does not always require perfect crRNA-target pairing, especially in PAM-distal regions. Many studies used lab-on-chip and orthogonal Cas for the multiplexing, however these strategies depend on complex chip design and construction and different Cas proteins according to the number of genes detected (Jain et al., 2023; Shen et al., 2023; Ding et al., 2020). The study of Lin et al., (2025) addressed this limitation by developing a Cas12a cis-cleavage-mediated lateral flow assay, called cc-LFA. Instead of relying only on collateral trans-cleavage, this method uses a “double-key” recognition system: first, Cas12a recognizes and cleaves the target DNA; second, the released PAM-distal sticky-end DNA fragment is recognized by a specific invasion probe. This design improves specificity and enables multiplexed detection because different crRNA/invasion-probe pairs can generate different target-specific outputs. The authors demonstrated multiplex detection of respiratory pathogens and HPV subtypes, supporting the idea that probe-invasion logic could be adapted to distinguish phytoplasma strains. However, the authors also reported that cc-LFA still requires further optimization for low-copy samples, especially samples with high Ct values, which is highly relevant for phytoplasma detection because phytoplasmas often occur at low and uneven titers in plant tissues.
Key Technology Limitations Identified
There are several limitations such as
i) sensitivity,
ii) specificity,
iii) multiplexing and
iv) field deployment.
i) Phytoplasmas are often present at low concentration in plant tissue, and their distribution can be uneven because they are phloem-limited. A field assay must have a comparable sensitivity, efficiency and LOD to qPCR (gold standard).
ii) Aster yellows phytoplasma belongs to a genetically diverse group, and closely related phytoplasmas may share conserved regions in 16S rRNA or other molecular markers. A test that only detects “phytoplasma positive” may not be sufficient for epidemiology or management if the goal is to distinguish AYP from other phytoplasma groups such as blueberry-associated phytoplasmas. Trans-cleavage-based Cas12a assays may generate signals even when target pairing is imperfect, which can increase the risk of false positives when closely related sequences are present.
iii) Many CRISPR-Cas12a assays are easy to design for one target, but become difficult when multiple targets such as phytoplasma strains and internal controls must be detected in the same reaction. This diagnostic tool should ideally include at least three layers of information: a plant/sample quality control, a universal phytoplasma target, and a specific AYP target using probe-invasion.
iv) A field-detection system should minimize sample processing, reduce contamination risk, integrate amplification and detection, and provide simple interpretation for growers. Current methods may require DNA extraction, pipetting, incubation, fluorescence readers, or interpretation of faint lateral-flow bands. This creates a gap between laboratory validation and real-world use by growers, extension specialists, or field researchers.
Novelty and Innovation of This Project
The final output of the project is to develop a CRISPR-Cas12a field-detection device with a sensitivity and LOD similar to qPCR, which can be used easily by the growers and able to detect phytoplasma and AYP from simple sample preparation. This field-detection device combines three steps simple sample preparation, isothermal amplification with RPA to increase sensitivity and Cas12a detection with probe-invasion logic for higher specificity and multiplexed output. A major innovative direction is the use of double-key recognition for plant pathogen diagnostics. A standard Cas12a trans-cleavage assay asks only one main question: “Did Cas12a become activated?” In contrast, the proposed probe-invasion strategy asks two questions: “Did Cas12a recognize and cleave the correct target?” and “Does the released DNA fragment match the correct invasion probe?” This additional recognition step could reduce false positives from closely related phytoplasmas and increase specificity. LFA assay will be improved by adding a line for plant endogenous control which is an internal extraction/amplification control line that can identify inhibited samples from negative results.
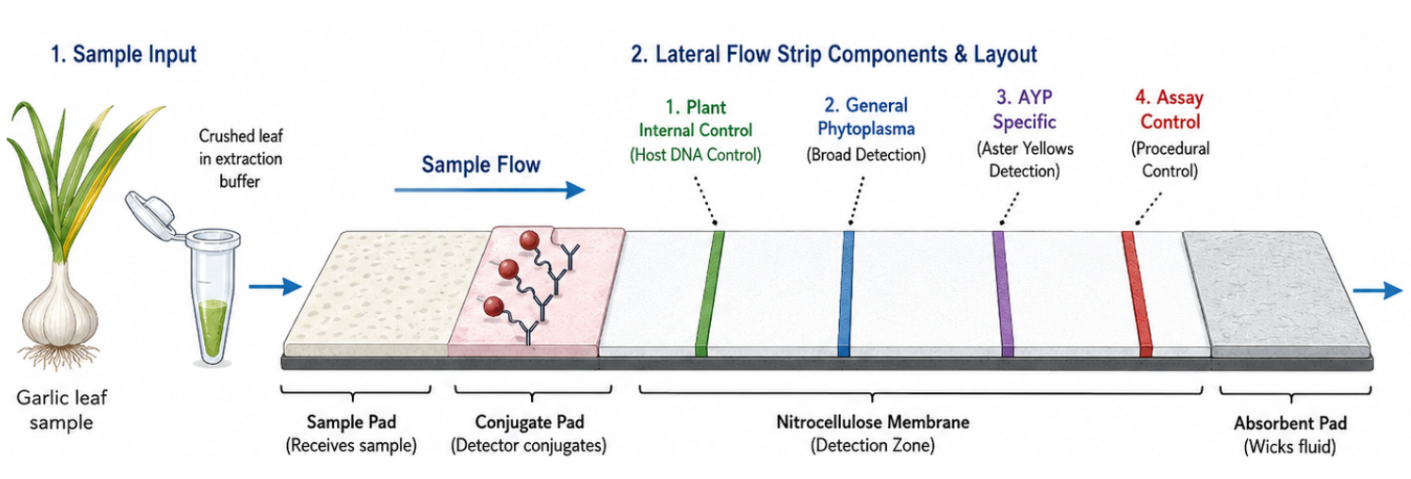 Figure 1. Multiplex lateral flow assay for simultaneous detection of phytoplasma, AYP, an assay control line, and an endogenous plant internal extraction/amplification control. The endogenous control helps identify inhibited or low-quality samples, reducing false-negative interpretation.
Figure 1. Multiplex lateral flow assay for simultaneous detection of phytoplasma, AYP, an assay control line, and an endogenous plant internal extraction/amplification control. The endogenous control helps identify inhibited or low-quality samples, reducing false-negative interpretation.
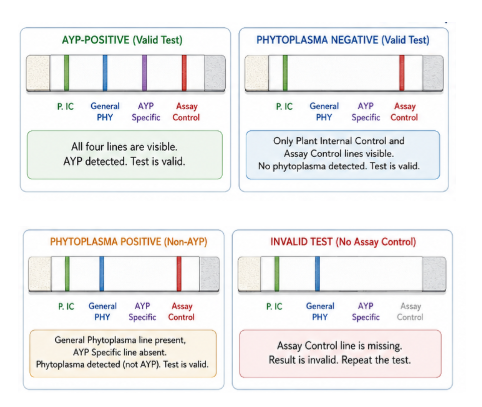 Figure 2. The strip includes four lines: plant internal control, general phytoplasma, AYP-specific, and assay control. An AYP-positive sample shows all four lines; a phytoplasma-negative sample shows only the plant internal control and assay control; and a non-AYP phytoplasma sample shows the general phytoplasma line without the AYP-specific line. If the assay control line is missing, the test is invalid and should be repeated.
Figure 2. The strip includes four lines: plant internal control, general phytoplasma, AYP-specific, and assay control. An AYP-positive sample shows all four lines; a phytoplasma-negative sample shows only the plant internal control and assay control; and a non-AYP phytoplasma sample shows the general phytoplasma line without the AYP-specific line. If the assay control line is missing, the test is invalid and should be repeated.
Why This Project Matters and Potential Impact
Aster yellows phytoplasma is an important agricultural pathogen because it can affect a wide range of hosts and can be difficult to manage once established in a field. In garlic, the problem is especially important because infected bulbils may be asymptomatic or show unclear symptoms, yet still contribute to disease spread through planting material. A field-deployable diagnostic tool could help growers, researchers, and extension programs make faster decisions about seed garlic quality, disease monitoring, and vector-associated risk. This project matters because current phytoplasma diagnostics are often centralized in laboratories, which can delay management decisions. A rapid field test could allow earlier detection of infected plants or planting stock before the disease spreads further. The ability to distinguish general phytoplasma infection from AYP-specific infection would also improve epidemiological studies by helping researchers understand which phytoplasma groups are present in garlic production systems. If successful, the platform could be adapted to other crops and other phloem-limited pathogens, like some plant viruses creating a broader diagnostic framework for plant disease surveillance. The project also has potential scientific impact because it connects plant pathology, CRISPR diagnostics, synthetic biology, and AI-assisted interpretation. Most current plant disease tests provide a simple positive/negative result. In contrast, this project aims to move toward a more informative diagnostic device that can classify pathogen identity, reduce ambiguous visual interpretation, and eventually connect results with a mobile app for data recording and disease mapping. This could shift phytoplasma detection from a purely laboratory-based process to a decentralized, data-connected surveillance system.
Ethical Implications
This project involves ethical considerations related to accuracy, responsible deployment, and agricultural decision-making. A false negative could lead a grower to plant infected garlic seed or fail to remove infected plants, allowing disease spread. A false positive could cause unnecessary economic loss if healthy planting material is discarded or if a farm is incorrectly associated with disease. Therefore, the ethical principles of non-maleficence and responsibility are central to this project: the technology should not harm growers through inaccurate results, overconfident interpretation, or premature deployment before validation. Another ethical issue is justice. Field-deployable diagnostics should benefit small and medium growers, not only large operations with access to advanced laboratory testing. If the device is developed successfully, it should be affordable, easy to use, and accompanied by clear instructions so that non-specialists can interpret results appropriately. The project should also avoid overclaiming what the device can do. For example, an early prototype may be useful for research screening, but not yet validated for regulatory or commercial certification decisions. To ensure ethical development, this project should include strong validation against qPCR, sequencing, and known positive and negative controls. The assay should be tested with multiple garlic tissues, different phytoplasma titers, healthy plant controls, non-target phytoplasmas, and plant-associated microbes to evaluate sensitivity and specificity. The mobile-app output should clearly report uncertainty, such as “positive,” “negative,” or “inconclusive,” rather than forcing a binary result when the signal is weak. In addition, any field deployment should include training, data privacy considerations, and recommendations that diagnostic results be confirmed by laboratory methods before major management or economic decisions are made.
Summary Gap Addressed by This Project
The current gap is that phytoplasma diagnostics need a system that is sensitive enough for low-titer plant samples, specific enough to distinguish closely related phytoplasmas, multiplexed enough to provide more than one diagnostic answer, and simple enough for field deployment. Existing RPA-Cas12a and LFA assays provide a strong foundation, but they still face limitations in sample preparation, false negative, visual interpretation, and multiplexed specificity. This project addresses that gap by proposing a next-generation field-deployable platform that integrates AIOD-style CRISPR-Cas12a detection in a simple sample preparation for reducing false negatives, engineered reporters, probe-invasion specificity, and future microfluidic/app-based readout for Aster yellows phytoplasma detection in garlic.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Experimental design overview
This final project is a continuation of my ongoing research on Aster yellows phytoplasma (AYP) in cultivated garlic in Minnesota. The broader research project focuses on understanding the genetic diversity, detection, epidemiology, and persistence of AYP in garlic. The HTGAA component extends this work toward the design and proof-of-concept validation of a field-deployable CRISPR-Cas12a diagnostic device for AYP detection. This device is envisioned as a portable system that combines simple sample preparation, isothermal DNA amplification using RPA, CRISPR-Cas12a detection, and lateral-flow or fluorescence-based readout. The current AYP project is generating the genetic information needed to design the diagnostic assay. Specifically, multilocus sequence analysis using markers such as 16S rRNA, cpn60, and ribosomal protein (RP) genes will identify conserved and variable regions across AYP strains from garlic. In parallel, phytoplasma enrichment and whole-genome sequencing will provide additional genomic information to improve target selection. This is important because phytoplasmas are difficult to sequence directly from infected plant material due to their low abundance, dependence on the host plant matrix, and the overwhelming amount of host DNA in infected samples. The DNA design component of this project will include the design of RPA primers, Cas12a crRNAs, fluorescent reporters, and probe-invasion oligonucleotides. These designed DNA/RNA components will be used to detect AYP specifically, distinguish AYP from other phytoplasma groups when possible, and reduce false-positive or false-negative results. This approach builds on recent CRISPR-Cas12a diagnostic systems for phytoplasma detection and on newer Cas12a cis-cleavage/probe-invasion strategies for multiplexed and highly specific nucleic acid detection (Wei et al., 2026; Lagner et al., 2025; Lin et al., 2025). Previous CRISPR-Cas12a phytoplasma work showed that LbCas12a-Ultra, stem-loop reporter design, and multiplexed target sites can improve detection, but the lateral-flow format still had a higher LOD than fluorescence-based detection, with reported LFA detection between approximately 10 pM and 100 pM target DNA (Lagner et al., 2025). The proposed project aims to improve this limitation by integrating RPA amplification, internal controls, and a double-recognition probe-invasion strategy.
Hypothesis
I hypothesize that a field-deployable AYP detection system can be made more sensitive and specific by combining: RPA amplification of conserved AYP genomic regions, CRISPR-Cas12a target recognition, engineered reporter/probe design, and probe-invasion-based confirmation of Cas12a cis-cleavage products. This system should reduce false negatives by including an internal plant control and reduce false positives by requiring two recognition steps: Cas12a/crRNA recognition of the target sequence and invasion-probe recognition of the released PAM-distal DNA fragment.
Detailed Experimental Plan and Timeline
1. Compile and curate AYP sequence data from the ongoing garlic project
Timeline: 4 months
The first step will be to organize the sequence information already generated from the AYP garlic project. This will include 16S rRNA, cpn60, and ribosomal protein sequences obtained from positive garlic samples. Also, whole genome sequencing of around ten positive samples allow to characterize the phytoplasma strains and to design crRNAs more specific to AYP detection. Additional reference sequences from GenBank will be included to represent AYP subgroups, closely related phytoplasmas, blueberry stunt phytoplasma, and other relevant phytoplasma groups. This sequence dataset will serve as the foundation for diagnostic target selection.
Tools and techniques: GenBank, BLASTn, Geneious prime, MAFFT, MEGA, iPhyClassifier, cpnClassiPhyR, and multilocus sequence analysis.
Expected result: A curated sequence alignment showing conserved AYP regions suitable for broad detection and variable regions suitable for subgroup or strain-level discrimination.
Protocols
Nucleic acid extraction and detection using qPCR
Whole-genome sequencing phytoplasma enrichment
3. Identify candidate genomic regions for CRISPR-Cas12a and RPA assay design
Timeline: Month five
Candidate diagnostic regions will be selected based on three criteria: conservation among garlic-associated AYP sequences, divergence from non-target phytoplasmas, and compatibility with RPA and Cas12a recognition. The goal is to identify at least two types of targets: one broad phytoplasma target and one AYP-specific target. A third optional target may be designed for a related phytoplasma group, such as blueberry stunt phytoplasma, to test whether the platform can discriminate among phytoplasma groups.
Tools and techniques: Multiple sequence alignment, BLAST specificity screening, PAM identification for Cas12a, and in silico off-target analysis.
Expected result: A short list of candidate target regions that can support RPA primer design and Cas12a crRNA design.
Design CRISPR-Cas12a crRNAs using a custom bioinformatic pipeline
I developed with help to ChatGPT a Google Colab pipeline to scan the curated cpn60 and RP sequences for Cas12a PAM sites, including TTTA, TTTC, and TTTG, on both forward and reverse strands. The pipeline extracts the downstream spacer sequence, scores candidate crRNAs based on conservation in target sequences, mismatch patterns in non-target sequences, GC content, and position in the amplicon. This computational design is the main DNA/RNA design component of my project.
Screen candidate crRNAs for specificity
The top crRNA candidates will be checked by BLAST against NCBI databases and against garlic-related sequences to identify possible off-target matches. Candidates that match garlic or non-target phytoplasmas too strongly will be deprioritized. Based on my preliminary results, some cpn60 and RP crRNAs appear conserved across multiple phytoplasmas rather than AYP-specific, so these may be useful for general phytoplasma detection, while future whole-genome sequencing data will be needed to design more AYP-specific crRNAs.
Evaluate crRNA secondary structure using NUPACK
The top crRNA spacers will be evaluated for predicted secondary structure. Strong hairpins or structures that interfere with crRNA-target binding will be avoided. The expected result is to prioritize crRNAs that are both computationally specific and structurally accessible.
5. Design RPA primers for isothermal amplification
Timeline: month six
RPA primers will be designed to amplify short diagnostic fragments containing the selected Cas12a target sites. Because RPA works under isothermal conditions and is compatible with portable detection, it is appropriate for a field-deployable device. Several primer pairs will be designed for each target region, and each primer set will be screened in silico for specificity, primer-dimer formation, secondary structure, and compatibility with the downstream Cas12a assay.
DNA design component: RPA forward and reverse primers.
Tools and techniques: Primer3, NCBI Primer-BLAST, IDT OligoAnalyzer, Multiple Primer Analyzer, and sequence alignment.
Expected result: At least 2–3 candidate RPA primer sets for each diagnostic target.
6. Validate CRISPR-Cas12a detection system using amplified targets
Timeline: month six
Each amplicon (generated by PCR) will be tested with the designed Cas12a/crRNA combinations. Fluorescence detection will be used first because it allows quantitative measurement of signal intensity over time. The best crRNA will be selected based on high signal with the AYP target, low background in negative controls, and minimal cross-reactivity with non-target phytoplasmas.
Tools and techniques: Cas12a reaction, crRNA screening, fluorescence plate reader, endpoint fluorescence measurement, and signal-to-noise analysis.
Expected result: At least one crRNA should generate a strong Cas12a signal for AYP-positive samples with low background in negative controls.

7. Validate CRISPR-Cas12a detection system using extracted nucleic acids
Timeline: month six
After validating the CRISPR-Cas12a system with amplified targets, the assay will be tested using total nucleic acids extracted directly from plant samples. This step is important because field samples contain plant DNA, possible PCR/RPA inhibitors, and variable phytoplasma concentrations. DNA extracted from phytoplasma-positive garlic samples, healthy garlic controls, and non-target plant/pathogen samples will be used as input for the RPA-Cas12a workflow. This validation will determine whether the assay can detect phytoplasma targets in a realistic sample matrix, not only in purified amplicons.
Tools and techniques: Total DNA extraction from garlic tissue, phytoplasma-positive garlic DNA, healthy garlic DNA, non-target plant DNA, RPA amplification from extracted nucleic acids, Cas12a/crRNA detection, fluorescent ssDNA reporter, fluorescence plate reader, endpoint fluorescence analysis, signal-to-noise analysis, inhibition assessment.
Expected result: Phytoplasma-positive extracted DNA samples should produce a detectable CRISPR-Cas12a signal after RPA amplification, while healthy garlic DNA and no-template controls should remain close to background. Some samples with low phytoplasma titer or high inhibitor content may produce weak or delayed signal, which will help define the practical performance of the assay in real plant matrices.
8. Validate RPA amplification using phytoplasma-positive and negative samples
Timeline: month seven
The selected RPA primers will be tested using DNA from phytoplasma-positive garlic samples and healthy garlic controls. The first validation will confirm whether the designed primers can amplify the expected target region. Amplicons will be checked by gel electrophoresis, and selected products may be sequenced to confirm target identity.
Tools and techniques: RPA amplification, total DNA from garlic samples, positive AYP/phytoplasma DNA, healthy garlic DNA, no-template control, gel electrophoresis, DNA purification, Sanger sequencing or plasmid sequencing if needed.
Expected result: Successful amplification of the expected cpn60, RP target or other region in phytoplasma-positive samples, with no amplification or minimal nonspecific amplification in healthy garlic and no-template controls.
9. Optimize simplified sample preparation for field deployment
Timeline: month seven and eight
After validating the CRISPR-Cas12a system with extracted nucleic acids, I will optimize a simplified sample preparation method that does not require a conventional DNA extraction workflow. This step is essential because growers would not be able to perform column-based or CTAB-based nucleic acid extraction in the field. Instead, the goal is to develop a rapid sample preparation method that releases phytoplasma DNA from garlic tissue while minimizing inhibitors that could affect RPA amplification and Cas12a detection. Different garlic tissues, such as leaves, cloves, basal plate, and roots, will be tested because phytoplasma distribution may vary across the plant. Simple preparation conditions may include crude tissue maceration, dilution, heat treatment, short incubation in extraction buffer, filtration, or direct use of clarified plant sap. These crude-prep inputs will then be tested in the RPA-Cas12a workflow and compared with purified DNA from the same samples.
Tools and techniques: Simple plant tissue maceration, crude sap preparation, rapid lysis buffer testing, heat-assisted sample preparation, dilution series to reduce inhibitors, filtration or clarification, RPA-Cas12a detection, fluorescence plate reader, lateral flow compatibility testing, qPCR comparison using purified DNA as reference.
Expected result: A simplified sample preparation method should allow detection of phytoplasma-positive garlic samples without full nucleic acid extraction. The expected signal may be lower than with purified DNA, but the optimized method should still produce a clear positive signal above background. The best preparation method will be selected based on signal intensity, reproducibility, speed, ease of use, and compatibility with field conditions.
10. Determine analytical sensitivity and limit of detection
Timeline: month nine
The limit of detection will be evaluated using serial dilutions of target DNA, purified amplicons, plasmid standards, or positive sample DNA. The goal is to identify the lowest target concentration that can be consistently detected by the RPA-Cas12a workflow. Fluorescence signal will be measured over time and compared with negative controls to define a positivity threshold.
Tools and techniques: Serial dilution, purified target amplicon or plasmid standard, RPA-Cas12a detection, fluorescence kinetics, endpoint RFU analysis, limit-of-detection estimation, positive/negative threshold calculation.
Expected result: A preliminary LOD value for the proof-of-concept assay. The expected result is that higher target concentrations will produce faster and stronger fluorescence signals, while low concentrations near the LOD will show delayed or weaker signal.
11. Compare CRISPR-Cas12a detection with qPCR
Timeline: month ten
The CRISPR-Cas12a assay will be compared with the current qPCR detection method used for phytoplasma detection. The same DNA samples will be tested by qPCR and by RPA-Cas12a to evaluate agreement between methods. This comparison will help determine whether the CRISPR assay can detect samples with high, medium, and low phytoplasma titers.
Tools and techniques: qPCR, CRISPR-Cas12a fluorescence assay, Cq-value comparison, positive/negative agreement analysis, sensitivity and specificity estimation, signal-to-noise comparison.
Expected result: Samples with strong qPCR amplification are expected to produce strong CRISPR-Cas12a signal. Samples with high Cq values or low phytoplasma concentration may show weaker or inconsistent CRISPR signal, helping define the practical sensitivity of the assay.
12. Test the assay across different garlic sample matrices
Timeline: month eleven
The assay will be tested using DNA extracted from different garlic tissues, such as leaves, cloves, basal plate, roots, and bulbils. This is important because phytoplasmas are phloem-limited and may be unevenly distributed in plant tissues. Testing different matrices will help identify which tissue type gives the most reliable detection signal.
Tools and techniques: Garlic tissue sampling, total DNA extraction, RPA amplification, CRISPR-Cas12a detection, qPCR comparison, tissue-specific detection analysis.
Expected result: Some tissues are expected to produce stronger and more consistent detection than others. The result will help define the best sample type for future field detection.
13. Design invasion probes after validating RPA primers and crRNAs
Timeline: month 12 and 13
Invasion probes will be designed only after the best RPA amplicon and crRNA combination has been selected. This is because the invasion probe depends on the Cas12a cis-cleavage product generated from a specific target. According to the probe-invasion strategy, Cas12a first cuts the target DNA and releases a PAM-distal fragment with a short sticky-end region. The invasion probe then binds to this exposed sticky end, creating a second specificity checkpoint.
Tools and techniques: Cas12a cis-cleavage site analysis, PAM-distal fragment prediction, invasion probe design, probe hybridization design, sequence complementarity analysis, NUPACK or similar structure prediction, literature-guided design from the cc-LFA paper.
Expected result: A candidate invasion probe that specifically recognizes the Cas12a-generated cleavage product. This should improve specificity because the assay would require both crRNA recognition and invasion probe binding.
14. Translate fluorescence detection into a lateral flow assay concept
Timeline: month 14
After fluorescence-based detection is validated, the assay will be adapted conceptually to a lateral flow assay format. The proposed LFA will include an assay control line, a plant internal control line, a general phytoplasma line, and an AYP-enriched or AYP-specific line. This format will help distinguish valid negatives, phytoplasma positives, AYP-like positives, and invalid tests.
Tools and techniques: Lateral flow assay design, labeled reporter probes, biotin/FAM or other tag-based detection, multiplex strip design, plant internal control design, assay control design, visual readout interpretation.
Expected result: A conceptual LFA design capable of reducing false negatives and improving interpretation. A valid negative result should show the plant internal control and assay control lines. A broad phytoplasma-positive sample should show the general phytoplasma line, and an AYP-positive sample should show both the phytoplasma and AYP-specific lines.
15. Use liquid handling automation to optimize assay conditions
Timeline: Throughout experimental optimization
A liquid handling robot can be used to automate the preparation of multiple RPA and CRISPR-Cas12a reaction conditions. This will help test different concentrations of Cas12a, crRNA, reporter, RPA input, magnesium concentration, incubation time, and temperature. Automation will reduce pipetting variability and allow more systematic optimization of the assay.
Tools and techniques: Liquid handling robot, automated reaction setup, multi-condition assay optimization, fluorescence plate reader, replicate testing, signal-to-noise analysis.
Expected result: More reproducible and quantitative optimization of the CRISPR-Cas12a assay. The robot should help identify conditions that maximize signal in positive samples while minimizing background in negative controls.
16. Final proof-of-concept implementation
Timeline: 1.5 years
The final proof-of-concept workflow will combine sample preparation, RPA amplification, CRISPR-Cas12a detection, and fluorescence or lateral flow readout. The initial version will likely detect broad phytoplasma or AYP-enriched targets using cpn60 and RP. Future whole-genome sequencing data will be used to identify more variable AYP-specific regions and improve diagnostic specificity.
Tools and techniques: Sample preparation, RPA, CRISPR-Cas12a, fluorescence readout, LFA concept, qPCR comparison, DNA design iteration, whole-genome-guided target discovery.
Expected result: A feasible workflow for field-deployable phytoplasma detection in garlic. The expected output is a validated proof-of-concept assay and a clear roadmap for improving specificity using WGS-derived crRNAs.

I will use cell-free reactions as the core detection system for my final project. In this project, the reaction is cell-free because it does not require living cells; instead, purified or pre-assembled components such as RPA reagents, Cas12a, crRNA, target DNA, and reporter probes are combined in vitro. The crRNA will guide Cas12a to the phytoplasma target sequence, and if the target is present, Cas12a will become activated and cleave the reporter to generate a fluorescence or lateral flow signal. This approach is useful for a field-deployable diagnostic because the reaction can potentially be simplified, miniaturized, and eventually freeze-dried for portable testing. I will also use a liquid handling robot to help automate and optimize the CRISPR-Cas12a assay. The robot can prepare multiple reaction conditions in parallel, including different Cas12a concentrations, crRNA concentrations, reporter concentrations, RPA input volumes, incubation times, and controls. This will reduce pipetting variability and make it easier to compare conditions quantitatively. Using automation will help identify the reaction setup that gives the highest signal in phytoplasma-positive samples and the lowest background in negative controls.
SECTION 5: Results & Quantitative Expectations
1. What aspect of your final project did you choose to validate? (min. 2 sentences)
To validate a key aspect of my final project, I developed and tested a proof-of-concept fluorescence-based CRISPR-Cas12a detection assay for Aster yellows phytoplasma (AYP) using published phytoplasma 16S rRNA crRNAs. The validation focused on determining whether the Cas12a system could successfully recognize phytoplasma DNA targets and generate measurable fluorescence signals across a dilution series of amplified target DNA. Multiple optimization plates were performed to evaluate assay performance, including comparison of different crRNAs (Cas site 1, Cas site 3, and Cas site 9), fluorescence gain settings, dilution ranges, and assay reproducibility. These experiments allowed preliminary determination of assay sensitivity, dynamic range, and analytical limit of detection (LOD). Also, I validated the DNA design component of my final project. Specifically, I designed preliminary CRISPR-Cas12a crRNA candidates targeting the cpn60 and ribosomal protein (RP) molecular markers from phytoplasma-positive garlic samples and reference phytoplasma sequences. I chose to validate this aim computationally because crRNA design is a critical first step before experimental CRISPR-Cas12a testing. The goal was to determine whether cpn60 and RP contain suitable Cas12a target sites that could be used for a proof-of-concept phytoplasma detection assay.
2. Write down a detailed protocol of how you validated this aspect of your final project. (Numbered list or paragraph is fine)
Validation of Crisprcas12 system using crRNAs from literature
- A 1.2 kb PCR product amplified from an AYP-positive garlic sample was used as the target DNA.
- The amplicon concentration was estimated using Nanodrop and gel visualization.
- Serial dilution series were prepared using nuclease-free water.
- CRISPR-Cas12a reactions were assembled using IDT Alt-R LbCas12a Ultra, crRNA, fluorescence reporter, reaction buffer, and target DNA.
- Three phytoplasma crRNAs were tested: Cas site 1, Cas site 3, and Cas site 9.
- Negative controls included NTC (no target DNA), No-crRNA controls, and No-Cas controls.
- Each reaction was prepared in a final volume of 50 µL containing 45 µL master mix and 5 µL target DNA.
- Reactions were loaded into black Greiner 96-well half-area plates and sealed.
- Plates were pre-heated at 37°C before kinetic fluorescence reading.
- Fluorescence measurements were performed in a BioTek Synergy H1 plate reader using Ex/Em 485/528 nm with top optics.
- Kinetic fluorescence was monitored every 2 minutes for 2 hours.
- Plate 1 and Plate 2 initially used automatic gain settings, Plate 3 used fixed gain 35, and Plate 4 used fixed gain 90.
- Data were normalized as ΔRFU = RFU(t) - RFU(0).
- Mean ΔRFU values and standard deviations were calculated for each concentration and condition.
- The preliminary LOD was estimated using the endpoint threshold formula: mean NTC + 3 * standard deviation of the NTC.
Design of crRNAs for phytoplasma and AYP detection
- I curated available cpn60 and RP sequence datasets from my ongoing phytoplasma characterization work in garlic and from selected reference sequences. These sequences came from phytoplasma-positive garlic samples and reference genomes/sequences relevant to Aster yellows phytoplasma and related phytoplasmas.
- I organized the sequences into target and non-target datasets. The target dataset included phytoplasma sequences relevant to the current AYP detection project, while the non-target or priority off-target dataset included phytoplasmas that should not be detected by an AYP-specific assay, such as blueberry/Vaccinium-associated phytoplasmas and other non-AYP phytoplasmas.
- I used a custom Google Colab pipeline to clean the FASTA files, remove exact duplicate sequences, and prepare the sequences for crRNA design. The pipeline scanned both forward and reverse-complement strands for LbCas12a-compatible PAM sequences. The PAM motifs used were: TTTA TTTC TTTG These represent real versions of the LbCas12a PAM motif TTTV, where V can be A, C, or G. After each PAM site, the pipeline extracted candidate spacer sequences of 20–23 nucleotides. Each candidate crRNA spacer was evaluated for basic design properties, including spacer length, GC content, homopolymer content, and target coverage. The pipeline exported two types of BLAST-ready sequences: spacer only PAM + spacer The spacer-only BLAST was used to evaluate general sequence similarity. The PAM + spacer BLAST was used to evaluate more realistic Cas12a off-target risk because Cas12a recognition depends on both the PAM and the spacer sequence.
- I performed BLAST screening for candidate crRNAs from both cpn60 and RP.
- I first evaluated whether the candidate crRNAs matched multiple phytoplasmas.
- I then performed additional BLAST searches excluding phytoplasmas to evaluate whether candidate crRNAs had potential off-target matches outside the phytoplasma group.
- I also performed BLAST searches against garlic / Allium sativum-associated sequences to evaluate whether the candidates had similarity to the host plant or garlic-associated organisms.
The preliminary results showed that the pipeline successfully generated candidate crRNAs for both cpn60 and RP. However, the BLAST results showed that many candidate crRNAs matched multiple phytoplasmas, indicating that these target sites are conserved across phytoplasma taxa. Additionally, when screening against garlic-associated sequences, the top crRNA candidates also showed partial similarity to garlic or garlic-associated sequences. Based on these results, I classified the current cpn60 and RP crRNAs as preliminary proof-of-concept candidates, not final AYP-specific crRNAs. These candidates are still useful for validating the CRISPR-Cas12a detection workflow, but deeper specificity screening and future whole-genome-guided design will be needed to identify more specific AYP targets.
3. What synthetic biology techniques did you utilize in validating this aspect of your final project? You can refer to the list of techniques in question 8. (min. 4 sentences)
This validation incorporated several synthetic biology techniques discussed during HTGAA 2026. DNA design and sequence analysis were used to identify phytoplasma target regions suitable for Cas12a recognition and crRNA design. PCR amplification was used to generate target DNA templates for CRISPR-Cas12a detection and dilution series experiments. Cell-free CRISPR-Cas12a reactions were performed using purified Cas12a enzyme, crRNA, and a fluorescent reporter system. Finally, quantitative fluorescence analysis and computational data analysis were used to evaluate kinetic behavior, dynamic range, and preliminary analytical LOD.
For designing crRNAs, I used sequence-based DNA design to identify candidate nucleic-acid recognition elements for a CRISPR-Cas12a diagnostic assay. The designed biological components include Cas12a crRNA spacer sequences and future RPA primer targets. Second, I used CRISPR guide RNA design principles, including PAM identification, spacer selection, GC-content evaluation, and off-target screening. This is a synthetic biology design process because the crRNA is a programmable nucleic-acid component that determines the specificity of the diagnostic system. Third, I used computational screening and design automation through a Google Colab pipeline. The pipeline scans molecular marker sequences, identifies possible Cas12a target sites, ranks candidates, and exports sequences for downstream BLAST and NUPACK analysis. Fourth, I used a design-build-test-learn framework. In this first design cycle, I designed crRNAs from available cpn60 and RP markers, tested them computationally by BLAST, learned that many target sites are conserved across phytoplasmas or have host-associated similarities, and identified the need to redesign future crRNAs using whole-genome sequencing data.
4. You must present data as part of your final project and include some analysis of that data. The data may be collected experimentally in the lab or generated as simulated data (e.g., using the Asimov Kernel or another simulation method). (min. 2 sentences)
Four optimization plates were performed to evaluate the CRISPR-Cas12a fluorescence assay. Plate 1 demonstrated that the assay chemistry was functional because positive reactions generated increasing fluorescence over time while controls remained close to baseline. Cas site 1 and Cas site 3 generated stronger fluorescence signals compared with Cas site 9 (Figures 3-6). Plate 2 evaluated a finer dilution range using Cas site 3. Several high-concentration wells produced detector overflow (OVRFLW), indicating that fluorescence exceeded the dynamic range of the plate reader when using automatic gain settings (Figures 7 and 8). Plate 3 tested a fixed gain of 35, but fluorescence signals became weak and difficult to separate from controls. Plate 4 used a fixed gain of 90 and generated the best kinetic separation between positive reactions and controls (Figures 9 and 10)). Strong concentration-dependent fluorescence activation was observed from 30 pM down to approximately 1.875 pM final target concentration in the reaction. Using a conservative threshold rule (mean NTC + 3 SD), the preliminary reliable LOD was estimated at approximately 1.875 pM final target concentration (Figure 11).
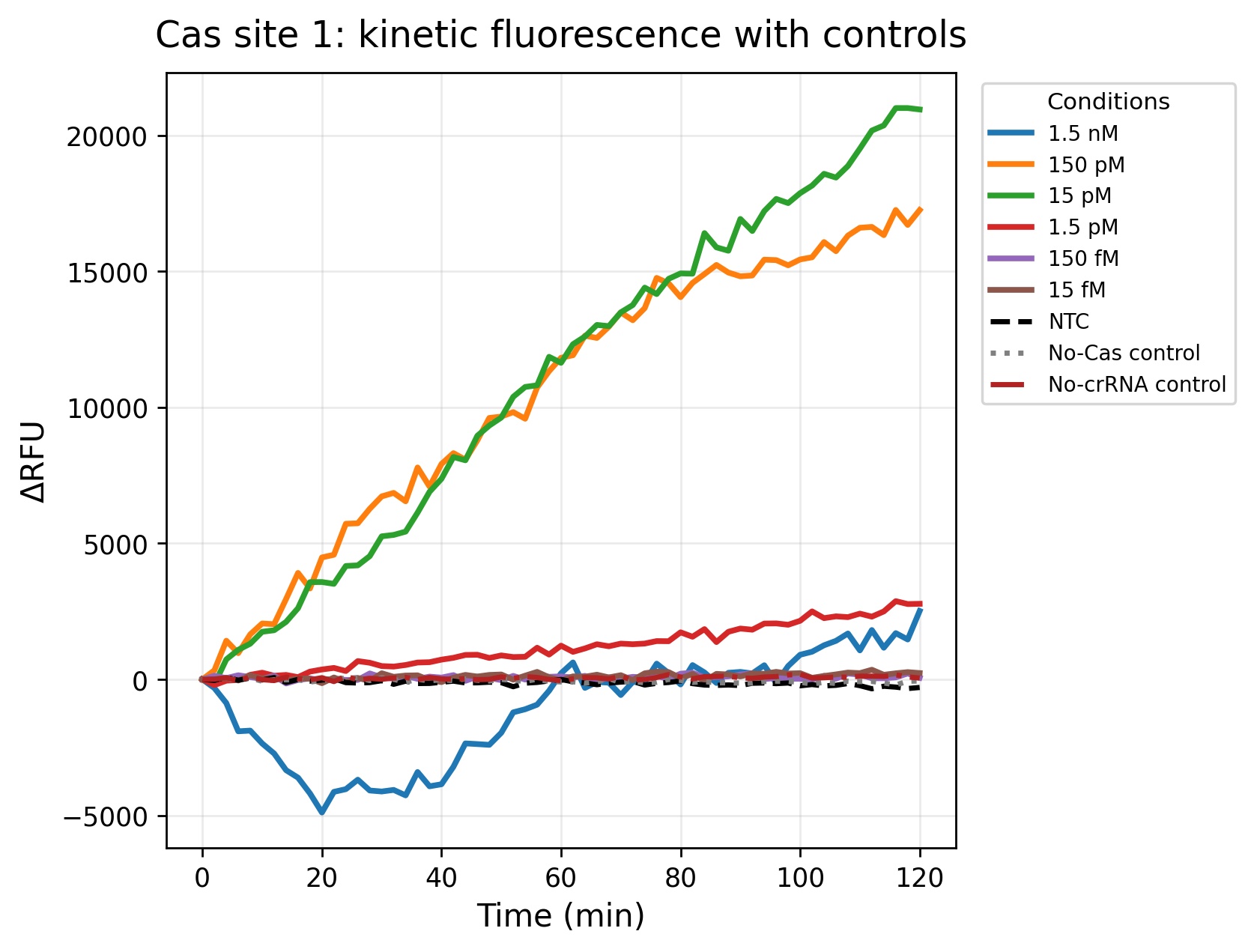 Figure 3. Kinetic fluorescence response of Cas site 1 across the corrected final target concentrations during the 120-minute CRISPR-Cas12a assay. The 150 pM and 15 pM conditions generated the strongest fluorescence accumulation over time, indicating efficient Cas12a collateral cleavage activity and robust target detection. In contrast, lower concentrations such as 150 fM and 15 fM remained close to the baseline signal observed for the controls. The NTC, No-Cas control, and No-crRNA control showed minimal fluorescence variation throughout the experiment, supporting the specificity of the assay and demonstrating that fluorescence activation was dependent on both the target DNA and the complete CRISPR-Cas12a reaction components.
Figure 3. Kinetic fluorescence response of Cas site 1 across the corrected final target concentrations during the 120-minute CRISPR-Cas12a assay. The 150 pM and 15 pM conditions generated the strongest fluorescence accumulation over time, indicating efficient Cas12a collateral cleavage activity and robust target detection. In contrast, lower concentrations such as 150 fM and 15 fM remained close to the baseline signal observed for the controls. The NTC, No-Cas control, and No-crRNA control showed minimal fluorescence variation throughout the experiment, supporting the specificity of the assay and demonstrating that fluorescence activation was dependent on both the target DNA and the complete CRISPR-Cas12a reaction components.
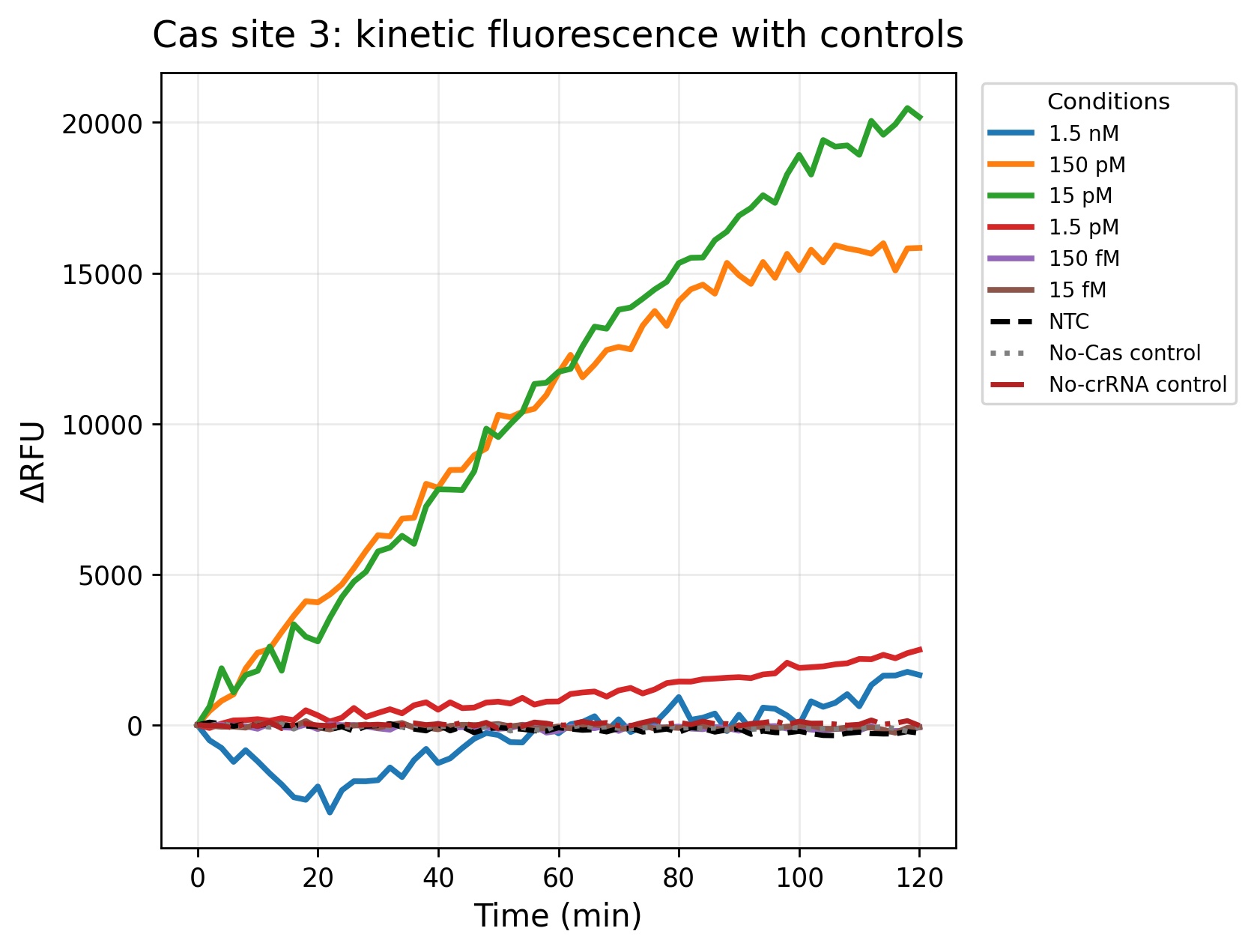 Figure 4. Fluorescence kinetics obtained for Cas site 3 during the 2-hour incubation period. Similar to Cas site 1, the intermediate concentrations (150 pM and 15 pM) produced the highest ΔRFU values, demonstrating strong concentration-dependent fluorescence activation. The 1.5 pM concentration generated detectable but lower fluorescence accumulation, whereas femtomolar concentrations remained close to the control baseline. Overall, Cas site 3 showed robust assay performance and reproducible signal separation from the negative controls, supporting its potential utility as a sensitive crRNA target site for phytoplasma detection.
Figure 4. Fluorescence kinetics obtained for Cas site 3 during the 2-hour incubation period. Similar to Cas site 1, the intermediate concentrations (150 pM and 15 pM) produced the highest ΔRFU values, demonstrating strong concentration-dependent fluorescence activation. The 1.5 pM concentration generated detectable but lower fluorescence accumulation, whereas femtomolar concentrations remained close to the control baseline. Overall, Cas site 3 showed robust assay performance and reproducible signal separation from the negative controls, supporting its potential utility as a sensitive crRNA target site for phytoplasma detection.
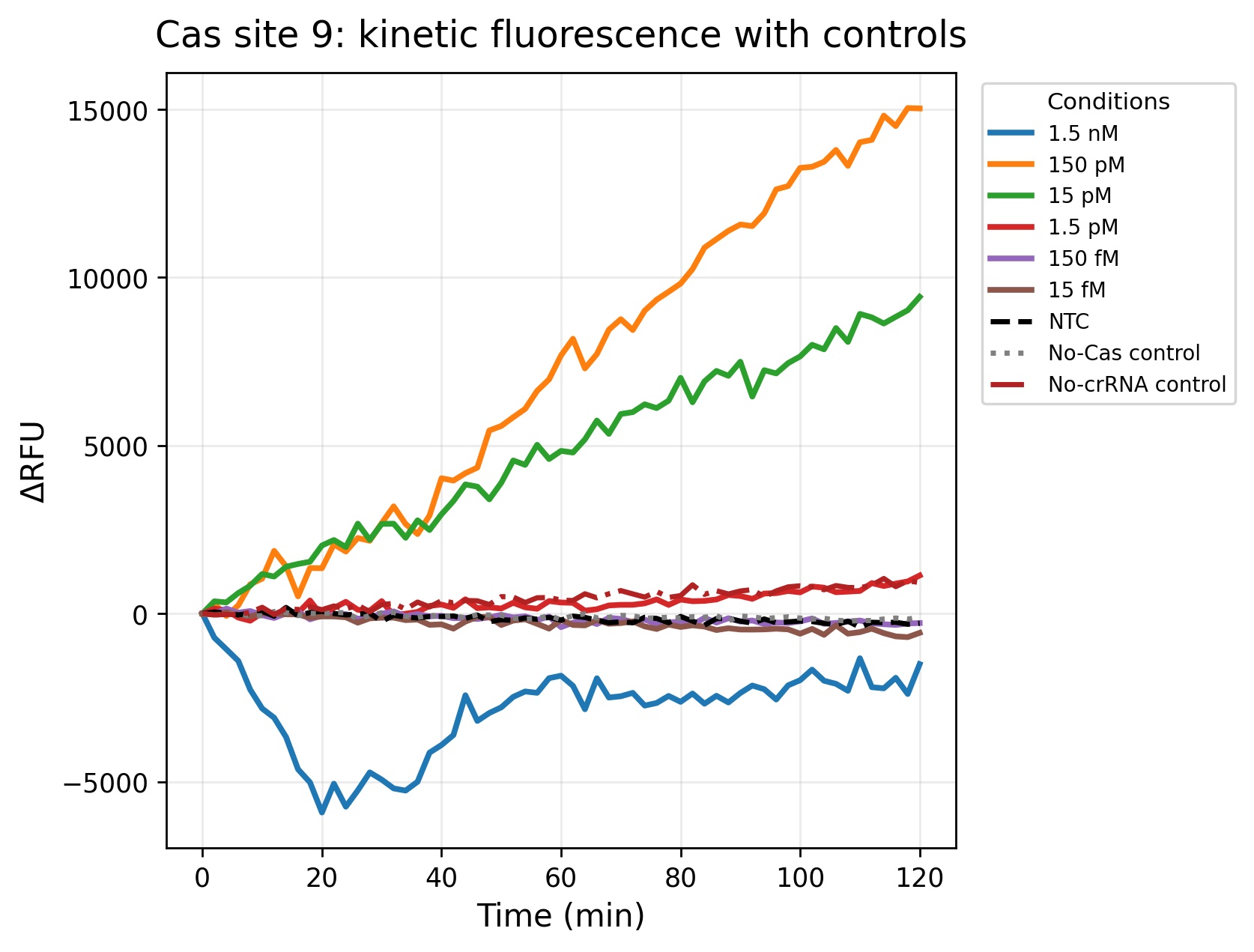 Figure 5. Fluorescence kinetics for Cas site 9. Compared to Cas site 1 and Cas site 3, Cas site 9 produced lower overall fluorescence intensity and slower signal accumulation, suggesting reduced crRNA efficiency or lower target accessibility. Nevertheless, the 150 pM and 15 pM conditions still generated clear fluorescence separation from the controls during the assay. Lower concentrations remained close to baseline fluorescence, indicating reduced sensitivity at the lower detection range. The controls remained relatively stable throughout the experiment, supporting target-dependent fluorescence activation mediated by Cas12a collateral cleavage.
Figure 5. Fluorescence kinetics for Cas site 9. Compared to Cas site 1 and Cas site 3, Cas site 9 produced lower overall fluorescence intensity and slower signal accumulation, suggesting reduced crRNA efficiency or lower target accessibility. Nevertheless, the 150 pM and 15 pM conditions still generated clear fluorescence separation from the controls during the assay. Lower concentrations remained close to baseline fluorescence, indicating reduced sensitivity at the lower detection range. The controls remained relatively stable throughout the experiment, supporting target-dependent fluorescence activation mediated by Cas12a collateral cleavage.
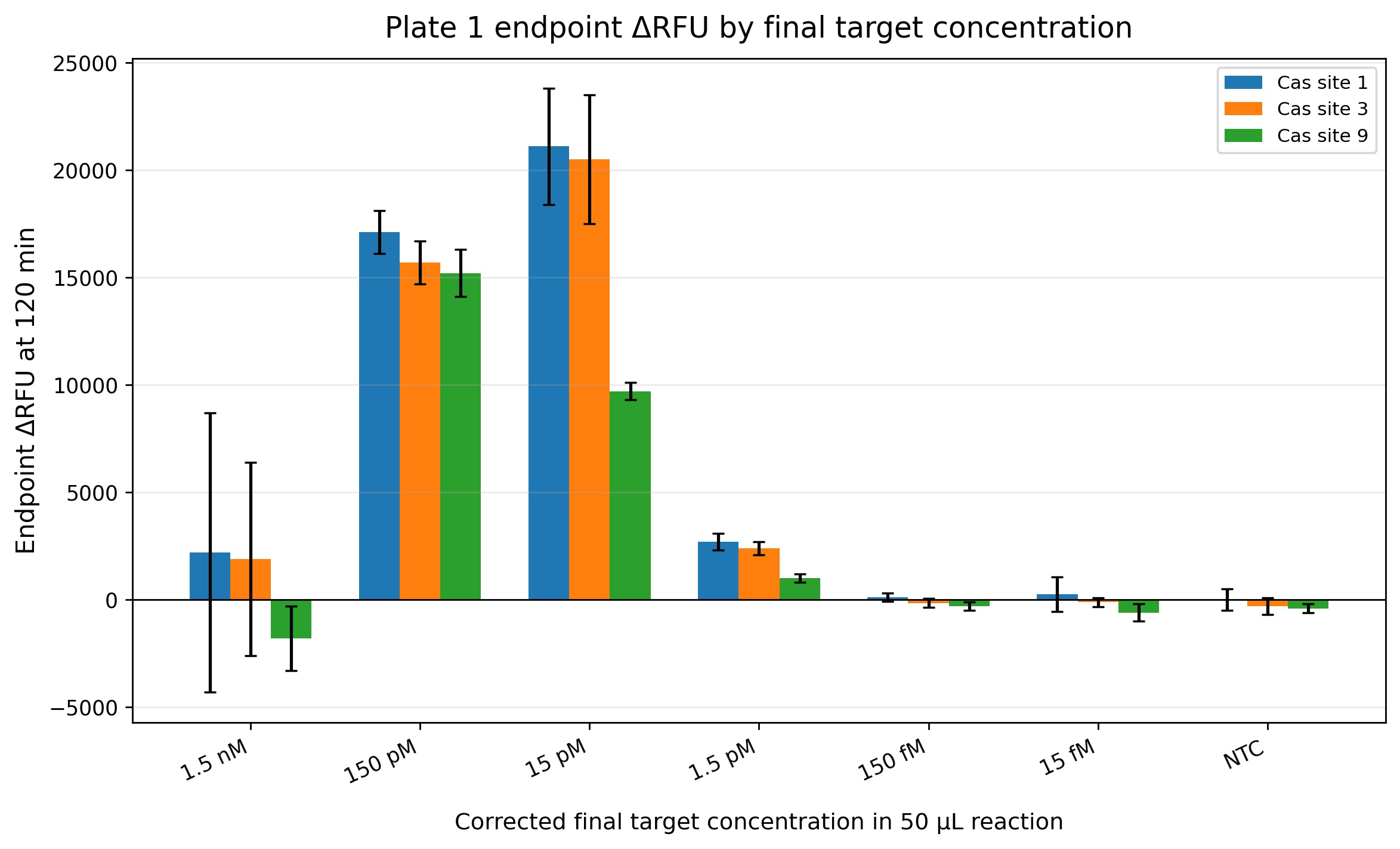 Figure 6. This endpoint analysis summarizes the fluorescence signal measured after 120 minutes of incubation for the three evaluated crRNA target sites. Cas site 1 and Cas site 3 produced the strongest endpoint fluorescence values at intermediate concentrations (150 pM and 15 pM), while Cas site 9 generated lower overall endpoint fluorescence. Interestingly, the highest concentration (1.5 nM) did not produce the strongest fluorescence response, which may suggest partial reaction inhibition, signal saturation effects, or suboptimal enzyme-to-target stoichiometry at high target abundance. The lower concentrations (150 fM and 15 fM) remained near the control baseline, indicating that these concentrations are close to or below the preliminary analytical limit of detection of the assay under these experimental conditions.
Figure 6. This endpoint analysis summarizes the fluorescence signal measured after 120 minutes of incubation for the three evaluated crRNA target sites. Cas site 1 and Cas site 3 produced the strongest endpoint fluorescence values at intermediate concentrations (150 pM and 15 pM), while Cas site 9 generated lower overall endpoint fluorescence. Interestingly, the highest concentration (1.5 nM) did not produce the strongest fluorescence response, which may suggest partial reaction inhibition, signal saturation effects, or suboptimal enzyme-to-target stoichiometry at high target abundance. The lower concentrations (150 fM and 15 fM) remained near the control baseline, indicating that these concentrations are close to or below the preliminary analytical limit of detection of the assay under these experimental conditions.
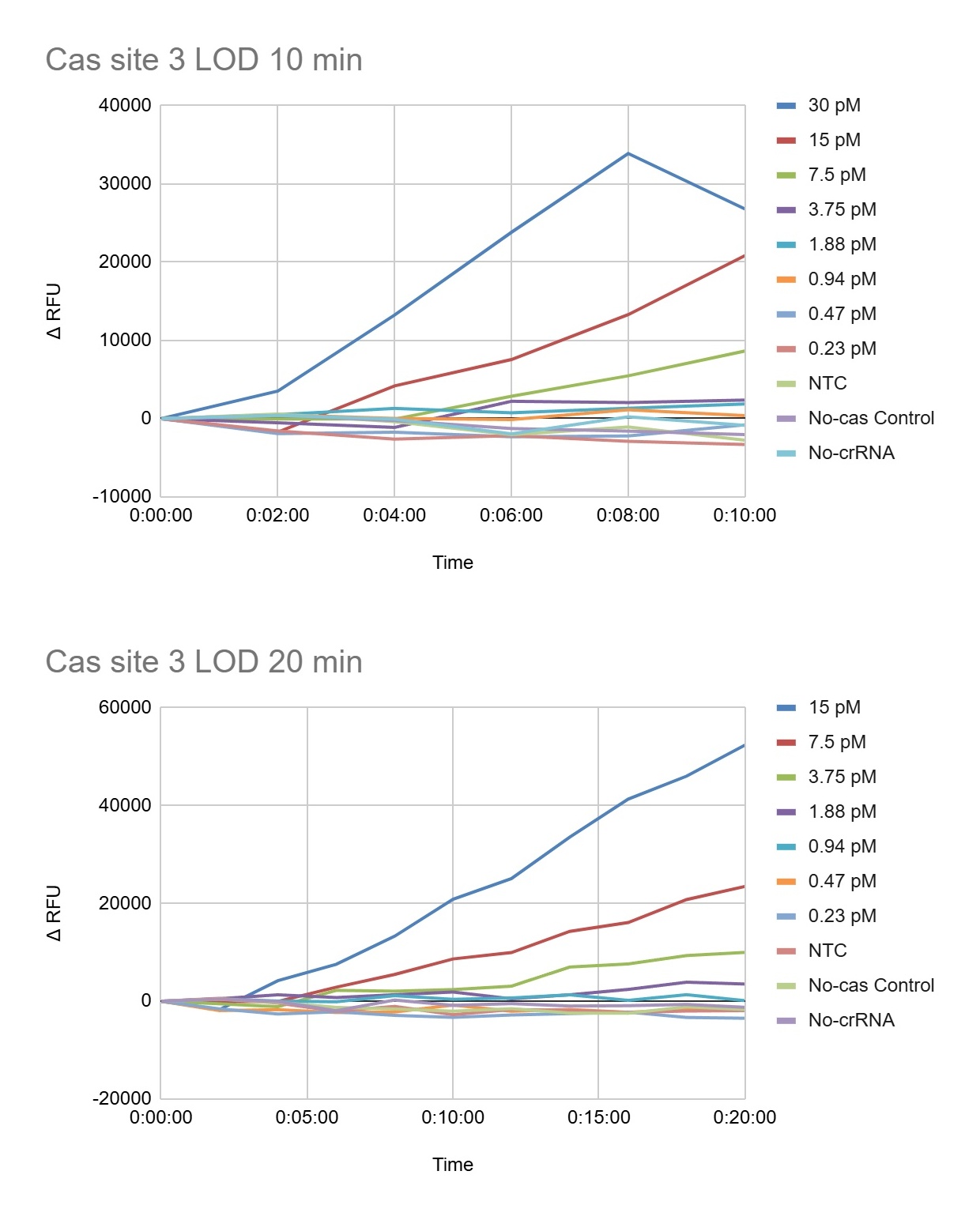 Figure 7. Illustrates the early fluorescence kinetics of the CRISPR-Cas12a assay plate 2 during the first A) 10 and B) 20 minutes of the reaction. At 10 minutes, the highest target concentrations (30 pM and 15 pM final concentration) already showed strong fluorescence separation from the controls, indicating rapid Cas12a activation. By 20 minutes, intermediate concentrations such as 7.5 pM and 3.75 pM also became clearly distinguishable from the NTC, No-Cas, and No-crRNA controls. These data demonstrate that the assay is capable of generating detectable signals within a short incubation period for higher phytoplasma target concentrations.
Figure 7. Illustrates the early fluorescence kinetics of the CRISPR-Cas12a assay plate 2 during the first A) 10 and B) 20 minutes of the reaction. At 10 minutes, the highest target concentrations (30 pM and 15 pM final concentration) already showed strong fluorescence separation from the controls, indicating rapid Cas12a activation. By 20 minutes, intermediate concentrations such as 7.5 pM and 3.75 pM also became clearly distinguishable from the NTC, No-Cas, and No-crRNA controls. These data demonstrate that the assay is capable of generating detectable signals within a short incubation period for higher phytoplasma target concentrations.
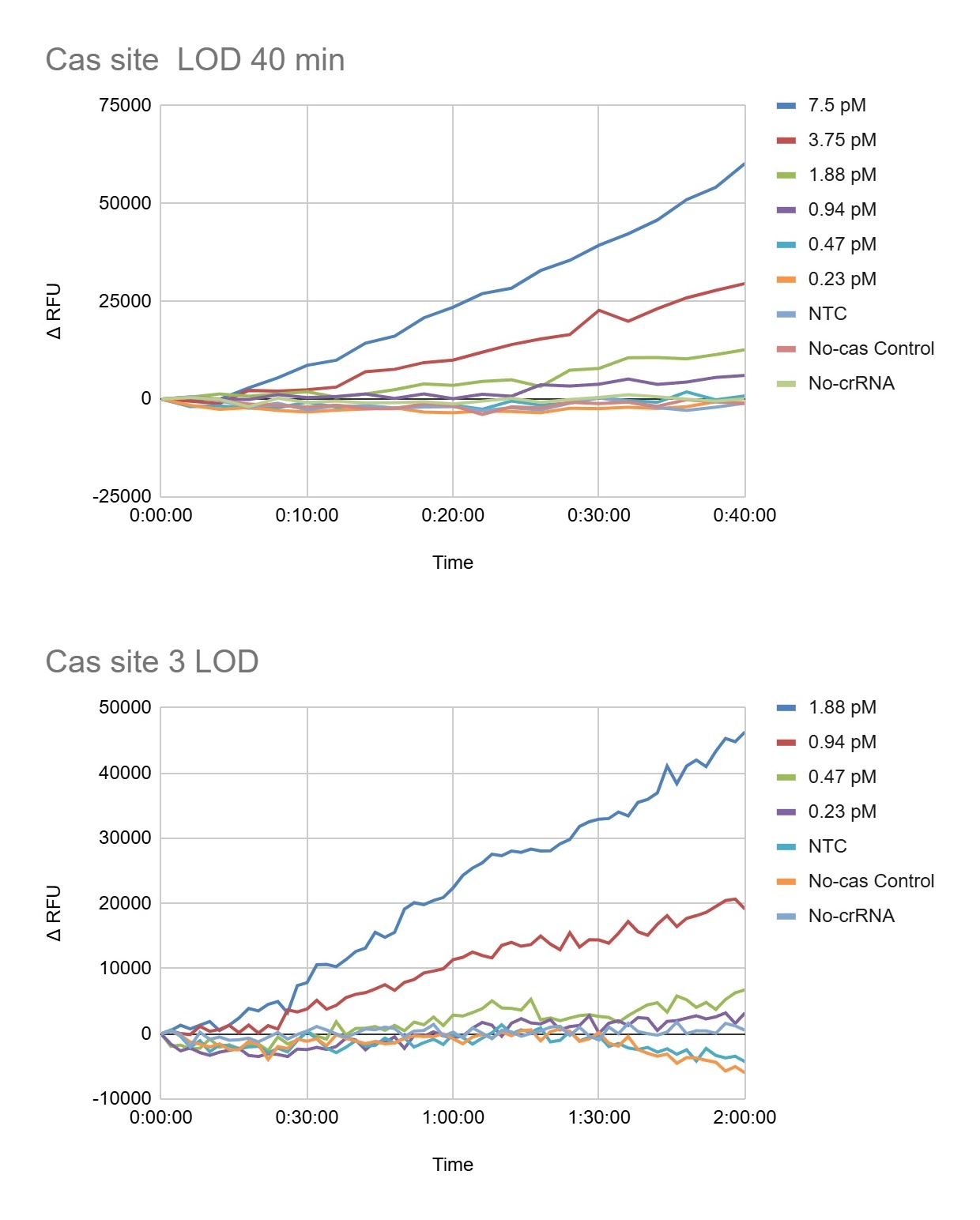 Figure 8. Presents the extended kinetic analysis of plate 2 at 40 minutes and 2 hours. The highest target concentrations produced detector saturation (OVRFLW), indicating that fluorescence exceeded the dynamic range of the plate reader under those conditions. The 7.5 pM and 3.75 pM conditions produced strong fluorescence accumulation over time, while lower concentrations such as 1.88 pM generated slower but still detectable increases in ΔRFU. In contrast, concentrations of 0.94 pM and below remained close to the control baseline, suggesting that these concentrations are near or below the preliminary analytical limit of detection (LOD). The controls remained relatively stable throughout the experiment, demonstrating that fluorescence activation was target-dependent and associated with Cas12a collateral cleavage activity.
Figure 8. Presents the extended kinetic analysis of plate 2 at 40 minutes and 2 hours. The highest target concentrations produced detector saturation (OVRFLW), indicating that fluorescence exceeded the dynamic range of the plate reader under those conditions. The 7.5 pM and 3.75 pM conditions produced strong fluorescence accumulation over time, while lower concentrations such as 1.88 pM generated slower but still detectable increases in ΔRFU. In contrast, concentrations of 0.94 pM and below remained close to the control baseline, suggesting that these concentrations are near or below the preliminary analytical limit of detection (LOD). The controls remained relatively stable throughout the experiment, demonstrating that fluorescence activation was target-dependent and associated with Cas12a collateral cleavage activity.
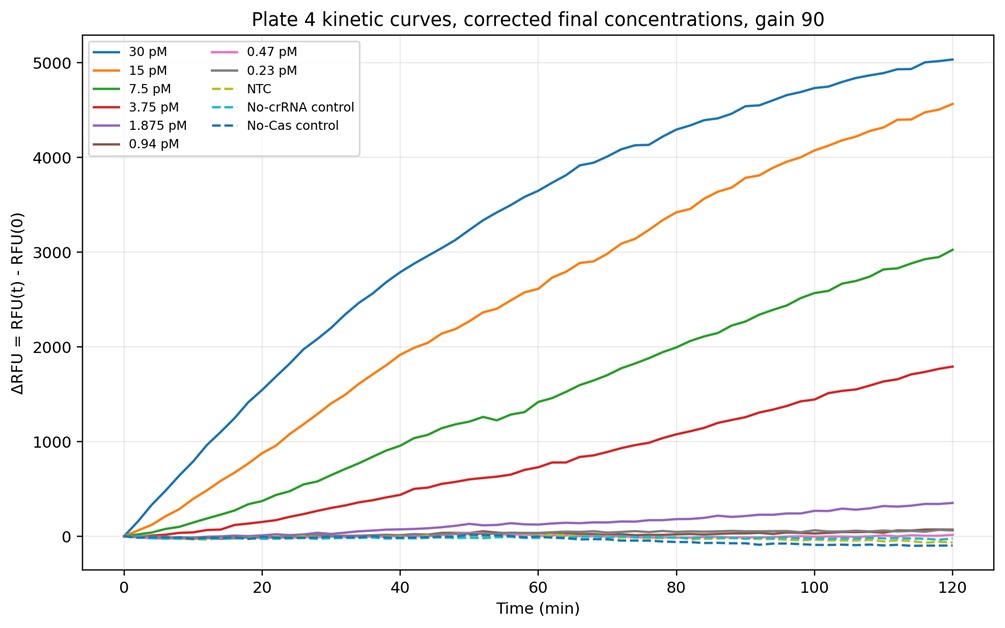 Figure 9. Complete kinetic fluorescence profiles obtained for Plate 4 using fixed gain 90. Strong concentration-dependent Cas12a activation was observed for the highest target concentrations (30 pM, 15 pM, 7.5 pM, and 3.75 pM), which generated rapid and continuous increases in ΔRFU over the 2-hour incubation period. Lower concentrations such as 1.875 pM produced slower but still detectable fluorescence accumulation, while concentrations below 0.94 pM remained close to the control baseline. Importantly, the NTC, No-crRNA, and No-Cas controls remained relatively stable throughout the experiment, indicating that fluorescence activation was target-dependent and associated with Cas12a collateral cleavage activity rather than nonspecific background fluorescence.
Figure 9. Complete kinetic fluorescence profiles obtained for Plate 4 using fixed gain 90. Strong concentration-dependent Cas12a activation was observed for the highest target concentrations (30 pM, 15 pM, 7.5 pM, and 3.75 pM), which generated rapid and continuous increases in ΔRFU over the 2-hour incubation period. Lower concentrations such as 1.875 pM produced slower but still detectable fluorescence accumulation, while concentrations below 0.94 pM remained close to the control baseline. Importantly, the NTC, No-crRNA, and No-Cas controls remained relatively stable throughout the experiment, indicating that fluorescence activation was target-dependent and associated with Cas12a collateral cleavage activity rather than nonspecific background fluorescence.
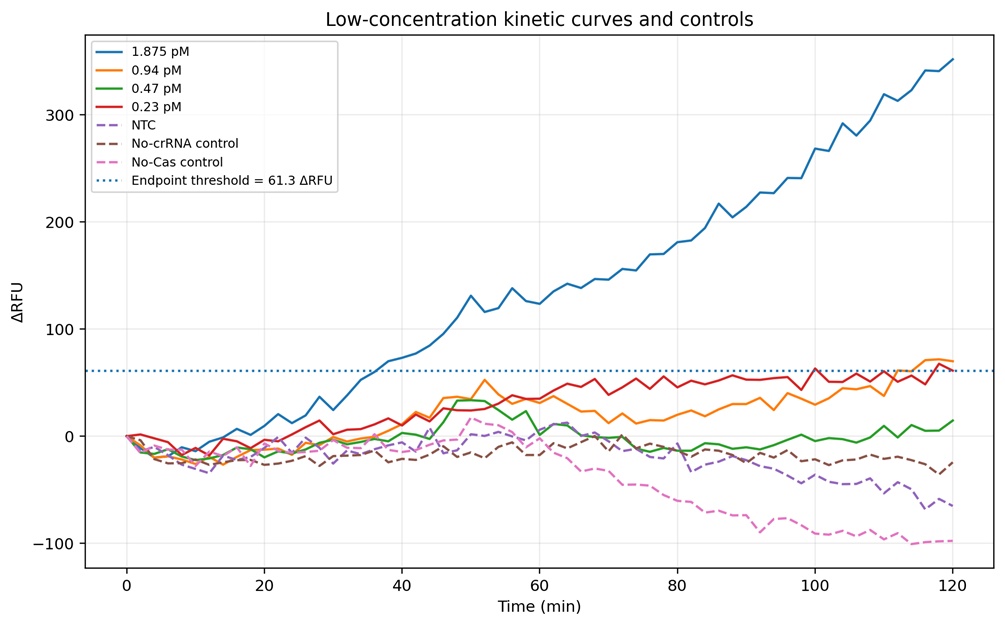 Figure 10. Low-concentration region of the assay to better visualize the preliminary analytical limit of detection (LOD) of the plate 4. The 1.875 pM condition consistently crossed the endpoint threshold (61.3 ΔRFU), indicating reliable target detection under these assay conditions. In contrast, the 0.94 pM condition produced weaker and more variable fluorescence accumulation that remained near the threshold boundary, suggesting borderline detection. Concentrations of 0.47 pM and 0.23 pM largely overlapped with the controls and did not show reproducible signal separation. The NTC, No-crRNA, and No-Cas controls remained close to baseline throughout the experiment, supporting the specificity of the fluorescence activation observed at higher target concentrations
Figure 10. Low-concentration region of the assay to better visualize the preliminary analytical limit of detection (LOD) of the plate 4. The 1.875 pM condition consistently crossed the endpoint threshold (61.3 ΔRFU), indicating reliable target detection under these assay conditions. In contrast, the 0.94 pM condition produced weaker and more variable fluorescence accumulation that remained near the threshold boundary, suggesting borderline detection. Concentrations of 0.47 pM and 0.23 pM largely overlapped with the controls and did not show reproducible signal separation. The NTC, No-crRNA, and No-Cas controls remained close to baseline throughout the experiment, supporting the specificity of the fluorescence activation observed at higher target concentrations
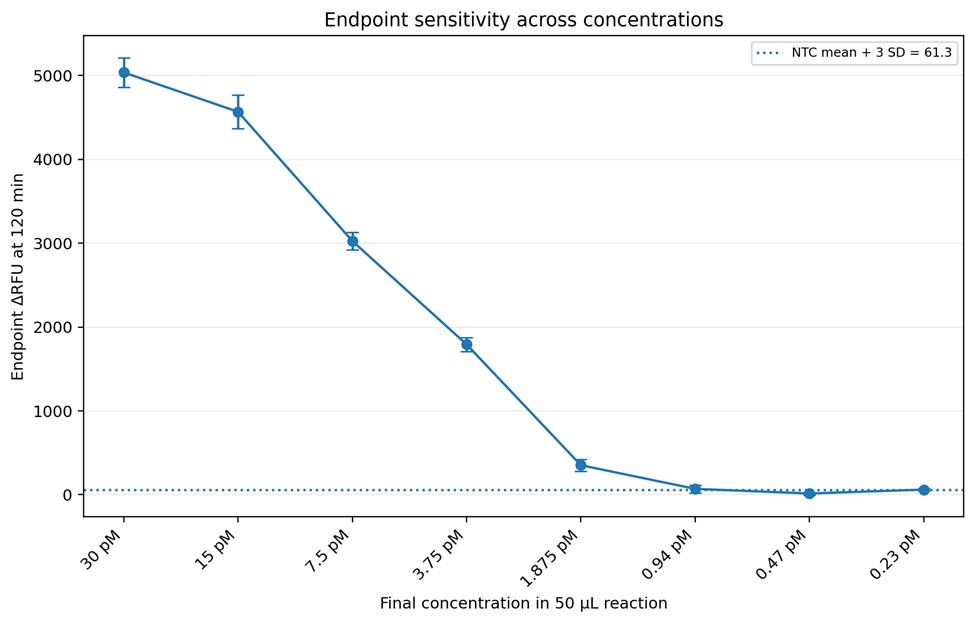 Figure 11. Endpoint sensitivity analysis measured after 120 minutes of incubation of the plate 4. A strong inverse relationship between target dilution and endpoint ΔRFU was observed, demonstrating the quantitative behavior of the CRISPR-Cas12a assay across the tested concentration range. The dotted horizontal line represents the preliminary detection threshold calculated as the mean NTC signal plus three standard deviations (NTC + 3 SD = 61.3 ΔRFU). Based on this criteria, 1.875 pM was identified as the preliminary reliable analytical limit of detection because it consistently produced endpoint fluorescence above the threshold. Lower concentrations (0.94 pM and below) generated signals near or below the detection threshold, indicating reduced assay reliability at these concentrations.
Figure 11. Endpoint sensitivity analysis measured after 120 minutes of incubation of the plate 4. A strong inverse relationship between target dilution and endpoint ΔRFU was observed, demonstrating the quantitative behavior of the CRISPR-Cas12a assay across the tested concentration range. The dotted horizontal line represents the preliminary detection threshold calculated as the mean NTC signal plus three standard deviations (NTC + 3 SD = 61.3 ΔRFU). Based on this criteria, 1.875 pM was identified as the preliminary reliable analytical limit of detection because it consistently produced endpoint fluorescence above the threshold. Lower concentrations (0.94 pM and below) generated signals near or below the detection threshold, indicating reduced assay reliability at these concentrations.
Table 1. Summary statistics of plate 4
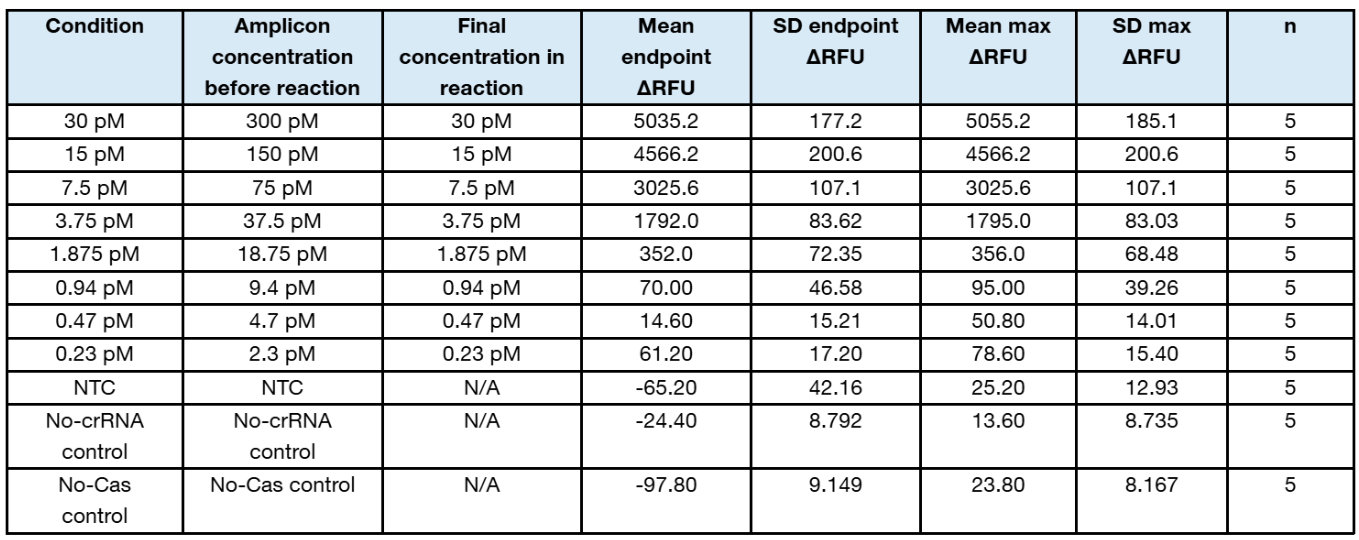
- Gain 90 produced strong kinetic separation without the very low signal observed at gain 35.
- The assay shows robust detection down to 1.875 pM final target concentration using the conservative 5/5 replicate threshold rule.
- The 0.94 pM final concentration is borderline and should be repeated with more replicates or additional intermediate concentrations before calling it positive.
- For a formal analytical LOD, repeat the low range around 3.75, 1.875, 0.94, and 0.47 pM with at least 8 to 20 replicates and calculate the concentration detected in ≥95% of replicates.
Graphs were generated with ChatGPT and Excel using real data.
The computational pipeline successfully generated preliminary CRISPR-Cas12a crRNA candidates for both cpn60 and RP targets. These candidates were exported as spacer-only and PAM+spacer sequences for BLAST-based specificity screening. The main result was that candidate crRNAs from both cpn60 and RP matched multiple phytoplasmas. This suggests that the available cpn60 and RP regions contain short Cas12a target sites that are conserved across phytoplasma taxa. Therefore, these markers may be more appropriate for broad phytoplasma detection or AYP-enriched proof-of-concept detection, rather than strict AYP-specific diagnosis. A second important result was that the top candidate crRNAs also showed partial BLAST hits to garlic-associated sequences when the search was limited to Allium sativum. This does not automatically mean that Cas12a would be activated by garlic DNA, because Cas12a requires proper PAM context and sufficient spacer complementarity. However, it is an important design warning because a diagnostic assay for garlic should avoid crRNAs with host-associated similarity whenever possible.
Table 2. Summary of preliminary crRNA design results for cpn60 and RP markers.
| Marker | Pipeline result | BLAST specificity result | Current interpretation | Next step |
|---|---|---|---|---|
| cpn60 | Candidate crRNAs were generated successfully. | Matches multiple phytoplasmas. | Useful as a broad phytoplasma proof-of-concept marker. | Deprioritize for AYP-specific detection. |
| RP | Candidate crRNAs were generated successfully. | Also matches multiple phytoplasmas in current candidates. | Potentially useful as an AYP-enriched/discriminatory marker, but not final. | Continue screening and compare with WGS targets. |
At this stage, the most important result is not that I found a final AYP-specific crRNA, but that I validated the crRNA design workflow and identified a key biological limitation: cpn60 and RP contain conserved short Cas12a target sites that may not be sufficient for strict AYP specificity.
5. Did you encounter any unexpected challenge(s) when performing your validation? If so, describe the challenge(s) and strategies to overcome it. If not, discuss potential problems, difficulties, limitations, and/or alternative strategies to overcome challenges in your final project. (min. 4 sentences).
One major issue was fluorescence detector overflow caused by automatic gain settings in the plate reader. In Plate 2, some high-concentration wells saturated the detector and produced OVRFLW values that could not be used for quantitative analysis. To overcome this problem, subsequent experiments used fixed gain values to improve comparability between experiments. Another challenge was balancing assay sensitivity and background noise. Plate 3 used fixed gain 35, but the fluorescence signals were weak and difficult to separate from controls. Increasing the gain to 90 in Plate 4 significantly improved signal separation while still avoiding detector overflow at most concentrations. Additional limitations include fluorescence baseline drift, possible secondary structure effects from the long 1.2 kb amplicon, and limited sample availability for repeated optimization experiments. Future experiments will focus on testing shorter amplicons, engineered reporters, and larger replicate numbers near the LOD range. One unexpected challenge was that many of the top cpn60 and RP crRNA candidates matched multiple phytoplasmas in BLAST. This means that the short Cas12a target regions within these molecular markers are more conserved than expected. As a result, these markers may not be ideal for designing a strictly AYP-specific assay. A second challenge was that some candidates also showed partial similarity to garlic-associated sequences. This is important because the final diagnostic test will be used with garlic tissue, so crRNAs must be screened not only against other phytoplasmas, but also against the host plant and organisms commonly associated with garlic samples. To overcome these limitations, I will classify the current cpn60 and RP candidates as proof-of-concept crRNAs rather than final diagnostic crRNAs. They can be used to test the CRISPR-Cas12a detection chemistry, optimize RPA amplification, evaluate reporter readout, and determine the general feasibility of the workflow. For the next design cycle, I will use enriched phytoplasma whole-genome sequencing data to identify more variable or unique AYP genomic regions. These WGS-derived regions should provide better target specificity than cpn60 and RP alone. I will also expand the off-target screening pipeline to include non-target phytoplasmas, garlic host sequences, garlic-associated viruses, and relevant plant-associated microbes.
SECTION 6: ADDITIONAL INFORMATION
12. References
Ding, X., Yin, K., Li, Z., Lalla, R. V., Ballesteros, E., Sfeir, M. M., & Liu, C. (2020a). Ultrasensitive and visual detection of SARS-COV-2 using all-in-one dual CRISPR-CAS12A assay. Nature Communications, 11(1). https://doi.org/10.1038/s41467-020-18575-6
Jain, P., Rananaware, S., Vesco, E., Shoemaker, G., Anekar, S., Sandoval, L. S., Meister, K., Macaluso, N., & Nguyen, L. (2023). Programmable RNA Detection with CRISPR-CAS12A. https://doi.org/10.21203/rs.3.rs-2549171/v1
Lagner, J. R., Newberry, E. A., Rivera, Y., Zhang, L., Vakulskas, C. A., & Qi, Y. (2025). Amplification-free detection of plant pathogens by improved CRISPR-CAS12A systems: A case study on Phytoplasma. Frontiers in Plant Science, 16. https://doi.org/10.3389/fpls.2025.1544513
Lin, M., Qiu, Z., Hao, M., Qi, W., Zhang, T., Shen, Y., Xiao, H., Liang, C., Xie, L., Jiang, Y., Cheng, M., Tian, T., & Zhou, X. (2025). CAS12A cis-cleavage mediated lateral flow assay enables multiplex and ultra-specific Nucleic acid detection. Nature Communications, 16(1). https://doi.org/10.1038/s41467-025-60917-9
Mollov, D., Lockhart, B., Saalau-Rojas, E., & Rosen, C. (2014). First report of a 16sri (Aster yellows) group phytoplasma on garlic (allium sativum) in the United States. Plant Disease, 98(3), 419–419. https://doi.org/10.1094/pdis-07-13-0689-pdn
Shen, J., Chen, Z., Xie, R., Li, J., Liu, C., He, Y., Ma, X., Yang, H., & Xie, Z. (2023). CRISPR/CAS12A-assisted isothermal amplification for rapid and specific diagnosis of respiratory virus on an microfluidic platform. Biosensors and Bioelectronics, 237, 115523. https://doi.org/10.1016/j.bios.2023.115523
Weintraub, P. G., & Beanland, L. (2006). Insect vectors of Phytoplasmas. Annual Review of Entomology, 51(1), 91–111. https://doi.org/10.1146/annurev.ento.51.110104.151039
Wei, W., Yang, Y., Shih, J. (2026). Rapid and Sensitive Detection of Phytoplasma Diseases Using a CRISPR/Cas12a DETECTR Assay Combined with Isothermal Recombinase Polymerase Amplification. In: Janik, K., Tabarelli, M. (eds) Phytoplasma. Methods in Molecular Biology, vol 3008. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-5104-9_6
Zhao, Y., Wei, W., Davis, R. E., Lee, I.-M., & Bottner-Parker, K. D. (2021). The agent associated with blue dwarf disease in wheat represents a new phytoplasma taxon, ‘candidatus phytoplasma tritici.’ International Journal of Systematic and Evolutionary Microbiology, 71(1). https://doi.org/10.1099/ijsem.0.004604
13. Create a supply list and budget for your project (bullet-point list)
- What supplies, equipment, and budget is needed for your project to work?
Sample collection and preparation
Garlic tissue samples: leaves, cloves, basal plate, roots, and bulbils
Disposable sampling bags, tubes, pestles, gloves, wipes, and labels - $500
Simple field-compatible extraction/lysis buffer components for crude sample preparation - $1000
Filtration or clarification materials, such as syringe filters, spin columns, or paper-based sample pads - $1000
Nucleic acid extraction and quality control
DNA extraction reagents or commercial plant DNA extraction kits - $1000
Nuclease-free water, low-bind tubes, strip tubes, and PCR plates - $1000
DNA quantification reagents for Qubit - $500
RPA amplification
RPA reagent kits or isothermal amplification reagents - $500–1,200
Synthesis of RPA primers for cpn60, RP, plant internal control, and future AYP-specific targets - $500
CRISPR-Cas12a detection
LbCas12a or LbCas12a-Ultra enzyme - $2000
Synthetic crRNAs for cpn60, RP, phytoplasma, AYP, and internal control targets - $1000
Fluorescent ssDNA reporters - $1000
Black 96-well fluorescence plates and optical seals - $500
Probe-invasion and lateral flow development
Invasion probe oligonucleotides with required modifications - $800
Lateral flow strips or custom LFA development materials: sample pads, conjugate pads, nitrocellulose membrane, absorbent pads, backing cards - $1,000
Labeled probes or reporter tags, such as biotin, FAM, digoxigenin, or other capture labels - $900
Streptavidin-gold nanoparticles or equivalent LFA conjugates - $800
Validation and controls
qPCR reagents for comparison with the CRISPR assay - $1000
Replicate testing materials for LOD, sensitivity, and specificity experiments - $500
Automation and equipment access
- Liquid handling robot materials and supplies - $1000
Bioinformatics and design tools
Google Colab for crRNA design pipeline - $0
NCBI BLAST, GenBank, NUPACK, Primer-BLAST, and IDT OligoAnalyzer - $0
Geneious Prime for sequence analysis - existing institutional license
Estimated total budget
Minimum proof-of-concept budget: approximately $8,000
This would cover primer/crRNA design, RPA testing, fluorescence-based CRISPR-Cas12a validation, and basic LOD testing using existing lab equipment.
Expanded validation budget: approximately $12,000
This would include more crRNAs, more RPA primer sets, qPCR comparison, multiple garlic tissues, non-target controls, and better LOD/sensitivity testing. Advanced LFA/probe-invasion prototype budget: approximately $20,000
This would include custom invasion probes, modified reporters, lateral flow materials, multiplex strip design, and optimization of a field-compatible readout.