Week 5 HW — Protein Design Part2
Part 1: Generate Binders with PepMLM
For this part, I retrieved the reviewed human SOD1 sequence from UniProt (P00441) and introduced the ALS-associated A4V mutation.
The mutant SOD1 sequence used for peptide generation was:
Part 2: Evaluate Binders with AlphaFold3
To evaluate the binding potential of the generated peptides, I used the AlphaFold Server to model protein–peptide complexes.
The mutant SOD1 (A4V) sequence was submitted as Chain A, and each generated peptide was submitted as Chain B.
Example Result — RDGEGELLENRR
The peptide RDGEGELLENRR, which showed the lowest pseudo perplexity in PepMLM, was analyzed using AlphaFold.
- ipTM score: 0.81
- pTM score: 0.89
These values indicate a high-confidence protein–peptide interaction and a stable predicted structure.
Binding Observation
The peptide appears to bind along the surface of the SOD1 protein, interacting with exposed regions rather than deeply inserting into a binding pocket.
The interaction does not significantly distort the overall protein structure, suggesting that the peptide binding is structurally compatible.
Although the binding appears relatively surface-oriented, the stability of the interaction (as reflected in the ipTM score) suggests that this peptide could still be a promising candidate for further optimization.
Interpretation
The agreement between:
- low pseudo perplexity (PepMLM)
- high ipTM score (AlphaFold)
suggests that RDGEGELLENRR is a strong candidate binder.
This demonstrates how combining sequence-based generation with structure prediction provides a more complete understanding of protein–peptide interactions.
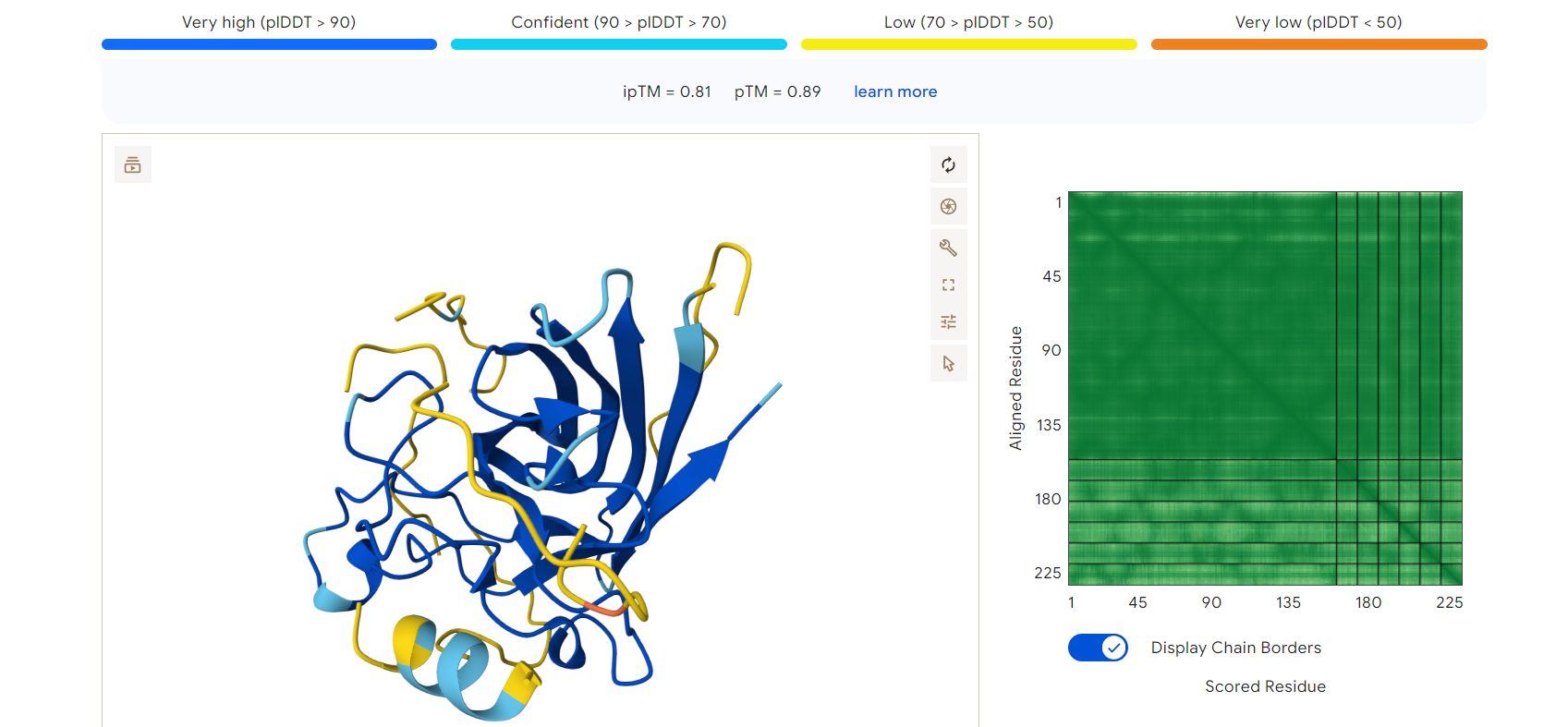
AlphaFold3 prediction of mutant SOD1 (blue) bound to peptide RDGEGELLENRR (yellow)
While structural prediction provides insight into how peptides may bind to a target protein, it does not fully capture whether a peptide is suitable for therapeutic applications.
To complement the AlphaFold analysis, I evaluated the generated peptides using PeptiVerse, focusing on both binding potential and physicochemical properties.
Evaluation Criteria
For each peptide, the following properties were analyzed:
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
The mutant SOD1 (A4V) sequence was used as the target input.
Key Observations
Among the generated peptides, RDGEGELLENRR emerged as the most promising candidate.
From the AlphaFold analysis, this peptide showed:
- a high ipTM score (0.81)
- stable binding along the protein surface
In PeptiVerse, its properties were consistent with a potentially viable peptide:
- The sequence contains a balance of charged (R, D, E) and polar residues, which supports interaction with protein surfaces
- The overall composition suggests good solubility, as the peptide avoids excessive hydrophobic clustering
- The presence of positively charged residues (arginine) may enhance electrostatic interactions with negatively charged regions of the protein
However, the high charge density may also introduce challenges such as:
- increased risk of non-specific interactions
- potential toxicity or hemolysis, depending on concentration and context
Comparison with Other Peptides
Other generated peptides showed either:
- higher pseudo perplexity (lower confidence from PepMLM), or
- less consistent structural binding in AlphaFold predictions
Some sequences appeared overly enriched in acidic residues, which may reduce binding strength due to lack of structural anchoring, while others lacked sufficient charge diversity to form stable interactions.
Design Interpretation (Personal Reflection)
As a designer working at the intersection of materials, space, and biological systems, I approach these peptides not only as molecular entities but also as interaction patterns.
The peptide–protein relationship can be understood as a form of surface negotiation, where geometry, charge distribution, and flexibility determine how two systems engage with each other.
In this sense, RDGEGELLENRR presents a balanced interaction profile:
- structured enough to bind
- flexible enough to adapt
- and chemically diverse enough to interact with complex protein surfaces
Selected Candidate
Based on the combined evaluation of:
- PepMLM (sequence confidence)
- AlphaFold (structural interaction)
- PeptiVerse (therapeutic properties)
I selected:
👉 RDGEGELLENRR
as the peptide to advance for further design and optimization.
This peptide demonstrates a strong alignment between computational prediction layers, making it a compelling starting point for future refinement.
Week 5 — Part 4: Generate Optimized Peptides with moPPIt
After exploring peptide generation (PepMLM) and evaluation (AlphaFold and PeptiVerse), I moved toward a more controlled design process using moPPIt.
Unlike PepMLM, which generates plausible binders based on sequence patterns, moPPIt allows for guided peptide design, where specific binding regions and multiple objectives can be optimized simultaneously.
Design Strategy
For this step, I focused on designing peptides that interact with regions of SOD1 near the A4V mutation site, located at the N-terminal region.
This region is particularly important because:
- it is associated with structural instability
- it contributes to protein misfolding and aggregation
- it represents a critical target for therapeutic intervention
Therefore, instead of randomly sampling binding peptides, I defined a target interaction zone around the mutation site.
Design Parameters
Using the moPPIt Colab:
- Target protein: A4V mutant SOD1
- Target residues: N-terminal region (including residue 4)
- Peptide length: 12 amino acids
- Optimization objectives:
- binding affinity
- motif guidance (targeted binding region)
- solubility
- reduced hemolysis risk
This transforms the process from exploration → intention-driven design.
Observations
Compared to PepMLM-generated peptides, the moPPIt-designed peptides showed:
- more localized interaction behavior, targeting specific regions rather than distributing across the protein surface
- more balanced residue composition, avoiding extreme charge clustering
- sequences that appear more structurally intentional, rather than statistically plausible
This suggests that moPPIt is not only generating binders, but also shaping interaction logic.
Design Interpretation
From a design perspective, this step represents a shift from:
→ discovering possible interactions
to
→ constructing desired interactions
The ability to guide peptide binding toward a specific mutation site introduces a level of spatial and functional precision that resonates with architectural thinking.
In my work on Pulse Space, I am interested in systems that respond to subtle signals and adapt dynamically. Similarly, moPPIt allows us to design molecular components that are not just reactive, but target-aware and behavior-driven.
Future Evaluation
Before advancing these peptides toward therapeutic applications, further evaluation would be required:
- structural validation (AlphaFold or experimental methods)
- binding affinity measurements
- toxicity and stability testing
- comparison with known binders
This iterative loop between generation → evaluation → redesign forms the foundation of computational protein design.
Reflection
This step made it clear that protein design is not only a problem of biology or computation, but also one of intentional form-making at the molecular scale.
By combining machine learning with guided constraints, we begin to design biological interactions in a way that parallels how we design spaces, materials, and systems in architecture.
Framing
This assignment explores protein design not only as a computational or biological task, but as a form of interaction design at the molecular scale.
Rather than treating proteins and peptides as static biochemical entities, I approached them as dynamic systems that respond, bind, and adapt — similar to how responsive environments operate in spatial design. Through a sequence of tools (PepMLM, AlphaFold, PeptiVerse, and moPPIt), this work moves from generation → evaluation → guided design, reflecting a design process that shifts from exploration toward intention.
This perspective is closely connected to my ongoing project Pulse Space, which investigates environments that react to human physiological signals. In this context, protein–peptide interactions can be understood as a micro-scale analogy of responsive systems, where structure, signal, and behavior are tightly intertwined.
Conclusion
This exercise revealed that protein design is not only about predicting biological function, but about constructing relationships between structure, behavior, and interaction.
By moving from sequence generation (PepMLM) to structural validation (AlphaFold), property evaluation (PeptiVerse), and finally guided design (moPPIt), I experienced a workflow that closely mirrors design processes in architecture and material systems — iterative, multi-scalar, and decision-driven.
What becomes particularly interesting is how control increases across these stages: from observing patterns, to evaluating performance, and ultimately to shaping outcomes intentionally. This shift transforms protein design into a form of design practice, where molecular interactions can be approached as spatial and responsive systems.
For my broader research, this opens up a new way of thinking about bio-responsive environments. Just as peptides can be designed to selectively bind and respond to specific protein states, future materials and spaces may be designed to sense and adapt to human physiological signals with similar precision.
In this sense, protein design becomes not only a biological tool, but a conceptual bridge between molecular systems and responsive spatial design.
Part C: Final Project — L-Protein Mutants
Objective
The goal of this assignment is to improve the stability and auto-folding properties of the MS2 bacteriophage lysis protein (L protein).
This protein plays a crucial role in the phage life cycle by inducing bacterial lysis through a mechanism that does not rely on enzymatic degradation of the cell wall, but rather through protein-mediated disruption.
Background
The MS2 L protein is a small membrane-associated protein (~75 amino acids) that functions as a single-gene lysis system.
Previous studies have shown that:
- The C-terminal domain is essential for lytic activity
- The protein forms oligomeric assemblies in the membrane
- Specific motifs such as the LS dipeptide are highly conserved and functionally important
Mutational studies indicate that many loss-of-function mutations cluster in structurally sensitive regions, suggesting that protein folding and stability are tightly linked to function.
Design Strategy
Rather than introducing random mutations, I approached this problem as a guided design task, focusing on improving structural robustness while preserving functional regions.
The following strategies were considered:
1. Stabilizing Secondary Structure
Mutations were selected to:
- promote alpha-helical stability
- reduce structural disorder
- improve folding energetics
2. Reducing Aggregation
To minimize aggregation:
- hydrophobic clustering was reduced
- polar and charged residues were introduced at surface-exposed positions
3. Preserving Functional Domains
Critical regions such as:
- the C-terminal domain
- conserved motifs (e.g., LS motif)
were kept intact to avoid disrupting lytic function.
Proposed Mutations
Based on these principles, the following mutation strategies were proposed:
- Substitution of flexible residues with helix-promoting residues (e.g., Ala, Leu)
- Introduction of charged residues to improve solubility
- Avoidance of mutations in conserved functional motifs
These mutations aim to improve folding efficiency while maintaining membrane interaction capability.
Interpretation
This approach reflects a shift from mutation screening → rational design.
The L protein can be understood as a minimal biological system where:
- structure
- membrane interaction
- and oligomerization
must be finely balanced.
Improving stability without disrupting function requires precise control over local structural features, similar to tuning material behavior in architectural systems.
Future Work
To validate these designs, the following steps would be required:
- structural prediction (AlphaFold / ESMFold)
- expression and folding assays
- membrane insertion studies
- functional lysis assays
Design Logic Summary
| Design goal | Mutation logic | Why it may help | Main risk |
|---|---|---|---|
| Stabilize local folding | G → A in flexible, non-conserved positions | Glycine is very flexible; alanine can slightly reduce conformational freedom and support more stable local structure | If the glycine is functionally important, the mutation may reduce activity |
| Modestly support secondary structure | S/T → A or L in non-critical, helix-compatible regions | Alanine and leucine can help support more ordered local structure in some sequence contexts | Too much stabilization could interfere with the dynamic behavior needed for lysis |
| Reduce aggregation tendency | Replace selected exposed hydrophobic residues with more polar residues (for example Q, E, or K) | Surface polarity can improve solubility and reduce unwanted self-association | If a residue actually contributes to membrane interaction, changing it may weaken function |
| Preserve lytic function | Do not mutate the LS motif or strongly conserved residues | These regions are likely required for activity | Over-conservatism may limit improvement |
| Preserve membrane-active behavior | Avoid major changes in predicted membrane-associated segments | The protein must still interact with the membrane to cause lysis | Too little change may not improve stability enough |
| Minimize disruption | Prefer single conservative substitutions before multi-site redesign | Easier to interpret experimentally and less likely to destroy function | Improvements may be modest |
Proposed Design Principle
The main principle I would follow is:
keep the functional core intact, stabilize flexible regions conservatively, and only adjust surface properties where aggregation risk appears higher than functional benefit.
This is important because the MS2 L protein is extremely small, so even a single mutation may have a disproportionately large effect on folding, membrane insertion, oligomerization, or lytic activity.
From a design perspective, this resembles working with a minimal structural system: when the system is very compact, every intervention must be precise and justified.
This iterative loop between design → prediction → validation is essential for advancing protein engineering toward therapeutic applications.
Referances
https://doi.org/10.1099/mic.0.000485
https://www.oaepublish.com/articles/mrr.2023.28
https://en.wikipedia.org/wiki/Bacteriophage_MS2?utm
Toward Rational L-Protein Mutants
Because I do not come from a molecular biology background, I approached this part less as a mutation-screening exercise and more as a design problem.
Instead of trying to guess many highly specific biochemical mutations, I focused on a small set of design principles that could improve the stability and folding behavior of the MS2 L protein while preserving its lytic function.
From the literature, three constraints seem especially important:
Do not disrupt the C-terminal functional region
Mutational studies show that many loss-of-function mutations cluster in the C-terminal domain, suggesting that this region is critical for activity.Preserve conserved functional motifs
In particular, the LS motif has been reported as functionally important and should not be altered.Respect membrane-associated behavior
MS2-L is a membrane-associated lysis protein and forms oligomeric assemblies after membrane insertion, so mutations should avoid disrupting the membrane-interacting character of the protein.
Based on this, I would not propose dramatic redesigns. Instead, I would advance small, conservative mutation strategies:
- replace some highly flexible residues in non-critical regions with alanine to slightly stabilize local structure
- reduce aggregation risk by replacing selected exposed hydrophobic residues with more polar residues
- introduce mild surface charge balancing only in regions that are likely solvent-exposed, not in membrane-facing segments
In other words, my design strategy is:
preserve the functional core, stabilize the unstable edges, and avoid over-editing the membrane-active region.
Example mutation logic
Rather than claiming exact validated therapeutic mutants, I would prioritize these mutation types for testing:
G → A in flexible, non-conserved positions
to reduce local conformational freedom and improve folding stabilityS/T → A or L in helix-compatible non-critical regions
to modestly favor secondary-structure stabilityI/V/L → Q/E/K only at predicted exposed positions
to reduce aggregation tendency and improve solubility without disrupting membrane insertion
Why this matters
For me, the interesting part of this exercise is that protein engineering starts to look similar to architectural or material design:
- some regions behave like load-bearing structure
- some regions behave like interface surfaces
- some regions tolerate modification
- some regions must remain intact for the whole system to function
So the goal is not maximum change, but targeted intervention with minimum disruption.
References
Chamakura et al. Mutational analysis of the MS2 lysis protein L
https://pmc.ncbi.nlm.nih.gov/articles/PMC5775895/Goessens et al. A synthetic peptide corresponding to the C-terminal 25 residues of phage MS2 coded lysis protein dissipates the proton motive force in E. coli membrane vesicles
https://europepmc.org/article/pmc/pmc454404Mezhyrova et al. In vitro characterization of the phage lysis protein MS2-L
https://www.oaepublish.com/articles/mrr.2023.28
Mutation Strategy Map — MS2 L Protein
Preserve Core
Keep functional regions intact
(C-terminal + LS motif)
Stabilize Structure
Reduce flexibility
(G → A, S/T → A)
Control Aggregation
Increase polarity at surface
(I/V → Q/E)
Maintain Function
Preserve membrane interaction
Avoid over-editing
Minimize Intervention
Prefer small, local changes
(single mutations)
Design Logic
Stabilize edges
Protect the core
Adjust the surface
A design-oriented approach to protein mutation strategy focusing on minimal and targeted intervention.