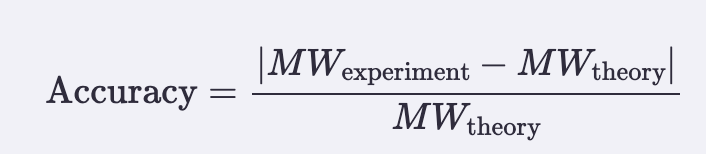About my project. Question 1 First, describe a biological engineering application or tool you want to develop and why.
I’d Like to Do I would like to develop an anthocyanin-based reporter system for Solanum aethiopicus. I would like it to use native genetic elements to fit in with my desire to do ethical biotechnology that fits in my own beliefs.
PART A Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming ~20–25% protein in meat: 500 g meat → 100–125 g protein With average amino acid mass = 100 Da ≈ 100 g/mol: that protein corresponds to 1.0–1.25 moles of amino acids, i.e. (6.0–7.5) × 10²³ amino acid molecules.
Part 1: Generate Binders with PepMLM Sequence Retrieval
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Mutated with A4V MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Binder Pseudo Perplexity Seq1 HHVPVVVLRHKX 16.326572 Seq2 WRYYAAVARWKE 13.897447 Seq3 HRYYPAAARWKX 8.531565 Control FLYRWLPSRRGG 20.63523127283615 Part 2: Evaluate Binders with AlphaFold3 Navigate to the AlphaFold Server: alphafoldserver.com For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried? In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder. Sequence 1 HHVPVVVLRHKX The sequence was submitted without the last X because it was flagged as an illegal character (it could stand for various amino acids). The peptide appears to be interacting with ipTM = 0.22 pTM = 0.85 pTM = 0.85 suggests the overall fold is very reliable, while ipTM = 0.22 says the predicted interaction/interface between chains is very poor. This means the protein’s global shape looks strong and likely meaningful, this is in line with it being a known and well illucidated human protein. It seems like a case where AlphaFold is confident about folding, but not about binding geometry.
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion DNA Polymerase - this is for amplifying the DNA by polymerisation. It is special because when used with its special buffer, it has up to 52× higher fidelity rate than ordinary Taq polymerase dNTPs- These are the building blocks that are used in the polymersiation by the DNA polymerase MgCl₂ - DNA polymerase enzyme needs magnesium ions to function properly (they are necessary cofactors for the thermostable enzyme) Optimized buffer - Phusion needs an optimized buffer because it uses a highly processive and high-fidelity DNA polymerase (originally a Pfu-like enzyme fused to a processivity domain) that is very sensitive to Mg²⁺, ionic strength, pH, and additives. Therefore these components must be balanced perfectly for the PCR not to fail in terms of processivity or fidelity. These items are added to the PCR: template DNA, primers, and molecular water. What are some factors that determine primer annealing temperature during PCR? Here’s the table simplified into bullet points:
Advantages of IANNs over Boolean genetic circuits Traditional genetic circuits (AND/OR/NOT) are great when the world can be cleanly thresholded into ON/OFF inputs and you only need discrete outputs. IANNs (genetic implementations of artificial neural networks) are useful when biology is messy—inputs are continuous, noisy, and correlated
Analog, not just digital
IANNs naturally support graded responses (continuous input → continuous output), not only Boolean truth tables. That lets you encode “how much” and “how confident,” not just “yes/no.”
General Homework Questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. -Cell-free can be used even when cold-chain conditions cannot be assured -Cell-free systems can be used without cloning and creating recombinant organisms Cell-free protein synthesis offers flexibility and a lot of control because you can directly tune reaction components (DNA amount, salts/cofactors, energy mix, chaperones, redox environment) and test many conditions rapidly without cell growth, regulation, or viability constraints. Cell-free expression is especially beneficial for rapid prototyping of constructs/circuits and also producing products especially if they are tocix to living expression systems. It can also enable on-demand expression (e.g., freeze-dried systems) and reactions requiring tightly controlled chemistry. This is great for making vaccines on demand on a mission to Mars for example. Describe the main components of a cell-free expression system and explain the role of each component. For General Systems Cell extract (lysate) or purified translation system — supplies the machinery that makes protein (ribosomes, tRNAs, translation factors, enzymes).
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. I intend to measure the presence of cyclotide activity using the GFP signal from a reporter construct. I also intend to measure the extent of membrane lysis caused by the presence of my cyclotides. Controls to make sure the system is working. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. A. eGFP reporter signal (cyclotide activity screen) What is measured
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Make a note on your HTGAA webpages including:
what you contributed to the community bioart project I change 3 pixels during the lecture, I wish I got a screen shot!
what you liked about the project, and I like the real-time collaboration on something artistic
Subsections of Homework
Week 1 HW: Principles and Practices
About my project.
Question 1 First, describe a biological engineering application or tool you want to develop and why.
I’d Like to Do
I would like to develop an anthocyanin-based reporter system for Solanum aethiopicus.
I would like it to use native genetic elements to fit in with my desire to do ethical biotechnology that fits in my own beliefs.
Development of a Cisgenic Anthocyanin-Based Visible Reporter System in Solanum aethiopicum
Overview
Reporter systems are important for studying biotechnology and understanding how life works. In resource-strained contexts, these kinds of experiments are hard to do due to high costs and equipment needs. They traditionally use elements like fluorescent or luminescent proteins from jellyfish and fireflies. The additional problem on an ethical level is that they rely on species-mixing which is contrary to the beliefs of some religions.
In this project, the aim is to develop and demonstrate the efficacy of a reporter system that does not rely on species mixing. To do this, the project will use strictly cisgenic, same species elements to build the reporter system, using Solanum aethiopicum as a novel model organism.
Question 2 Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
I would like its usage to be straight forward as a gene-edited technology that does not outcompete its wild counterparts. It contributes towards an ethical future because it allows people with limited resources to safely carry out analyses in Solanum aethiopicum to understand gene function and maybe design new varieties with better traits that match their needs. All this can be done without mixing species which matters to people who don’t want to do this because of their spiritual and personal beliefs.
Question 3 Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Purpose: What is done now and what changes are you proposing?
Now, gene-edited technologies such as vaccines are not treated as “genetically modified”, they are regulated by the same organisation that regulates medicines in Zambia. I think this is not a good idea. Although my technology fits this description, I don’t want to normalise and naturalise these technologies. They are still technologies that should be traced as such. option1I propose separate legislation that speaks directly to gene edited products like mine where no new DNA is introduced from another organism. Currently these products are treated as though they are natural, that does not sit well with me. I believe there should be traceability and vigilance even though it is perfectly safe when it is deployed to the public.
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
The buy-in of indigenous researchers. They need to opt to use Solanum aethiopicus and my reporter system to do routine plant biotechnology investigations especially about traits that are applicable to their context such as drought tolerance or pathogen resistance. Option2The authorities (the Ministry) could make it a recommended model organism for the nation’s researchers to use.
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
The gene editing could cause not unintended consequences. Maybe the anthocyanin expression may not work to a degree where phenotypes are easily visible. Option3The idea is that this technology makes it easy for researchers to see gene expression cheaply and easily. It does not work if you need high tech equipment to detect the anthocyanin expression from the reporter system. So its cost must be kept low
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
People might not want to use a new model organisms, perhaps they want to stick to Arabidosis or Medicago for their research.
Question 4 Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
3
1
2
• By helping respond
NA
NA
NA
Foster Lab Safety
• By preventing incident
1
NA
NA
• By helping respond
NA
NA
NA
Protect the environment
• By preventing incidents
1
2
NA
• By helping respond
NA
NA
NA
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
1
• Feasibility?
2
3
1
• Not impede research
3
2
1
• Promote constructive applications
3
2
1
Question 5 Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
I would opt for keeping the cost of the technology low through some kind of subsidy framework and I would also look at the involvement of the Ministry in encouraging uptake of the model organism and its specially developed reporter systems
Answers to Questions
Why read, write & measure polymers? To understand and exploit natural and designed properties like chirality and reactivity
How many basepairs do we need to synthesize? from less than 960 bp because that is the average bacterial protein size. There are plenty that are much smaller such as heat shock proteins and cyclotides in plants
Why use a custom panel from twist? Their panels show exceptional performance across low variant allele frequencies, that is one reason.
Week 2 HW: DNA Read Write and Edit
Part 1
Part 2 Gel Art
Part 3
3.1. Choose your protein.
I’ve chosen Brick1 protein from Arabidopsis thaliana.
This protein could be overexpressed in yeast cells and then harvested by lysing the cells. I’m not sure about cell-free methods that could be used.
3.5. [Optional] How does it work in nature/biological systems?
A single gene encodes proteins as shown below. Here is the AtBRICK1 mRNA, the highlighted part is the actual part that will encode the protein. The rest are untranslated regions which carry regulatory elements amongst other things.
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming ~20–25% protein in meat:
500 g meat → 100–125 g protein
With average amino acid mass = 100 Da ≈ 100 g/mol: that protein corresponds to 1.0–1.25 moles of amino acids, i.e. (6.0–7.5) × 10²³ amino acid molecules.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Beef goes through the alimentary canal and is broken down to its smallest components before nutrients are absorbed into the person. The DNA is broken down in this process and does not enter the nuclei of the eater for them to take on new traits through the food they eat.
Why are there only 20 natural amino acids?
There are a combinination of reasons for this, including
evolutionary history
chemical functionality
evolution of genetic code
a. Evolutionary Selection
Early life likely used a smaller set of amino acids that were easily formed through prebiotic chemistry. Experiments such as the Miller Urey experiment showed that several amino acids (e.g., glycine, alanine, and aspartate) can form under early Earth conditions. Over time, evolution gradually expanded the set until it stabilized around 20 amino acids, which provided sufficient chemical diversity for proteins to perform many biological functions.
Once the genetic code became fixed in early life, it became difficult for evolution to add many new amino acids because doing so would require major changes to tRNA molecules, aminoacyl-tRNA synthetases, and codon assignments.
b. Chemical Diversity Is Already Sufficient
The 20 amino acids provide a wide range of chemical properties, allowing proteins to perform diverse roles:
Property
Example Amino Acids
Function
Nonpolar / hydrophobic
Valine, leucine
Protein core stability
Polar
Serine, threonine
Hydrogen bonding
Charged
Lysine, glutamate
Enzyme catalysis, ionic interactions
Aromatic
Phenylalanine, tryptophan
Stacking interactions, light absorption
Special structure
Proline, glycine
Structural flexibility
This set already covers most chemical behaviors needed for enzyme catalysis, structural stability, and molecular recognition.
c. Genetic Code Constraints
The genetic code contains 64 codons:
61 codons encode amino acids
3 codons are stop signals
These codons map to the 20 amino acids, meaning multiple codons often code for the same amino acid (degeneracy). Expanding the number of amino acids significantly would require reorganizing this coding system, which evolution tends to avoid because it could disrupt many proteins at once.
d. Efficiency and Error Minimization
The standard 20 amino acids create a system that is robust to mutations. Many single-base changes in DNA produce amino acids with similar chemical properties, reducing harmful effects on proteins. This suggests the genetic code evolved to minimize translation errors.
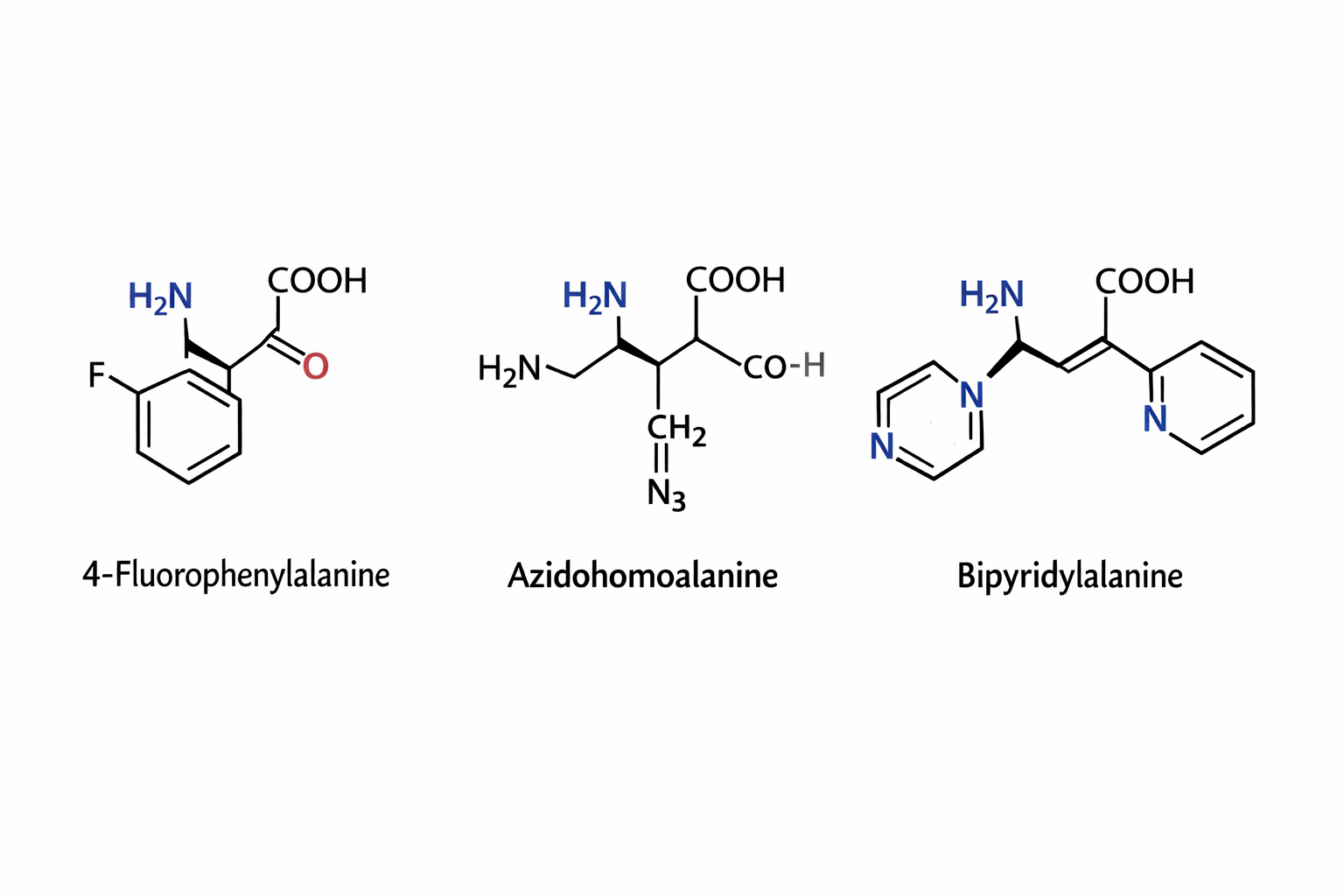
Can you make other non-natural amino acids? Go on. Design some new amino acids.
The three synthetic amino acids shown 4-fluorophenylalanine, azidohomoalanine, and bipyridylalanine are examples of noncanonical amino acids designed to extend the chemical capabilities of proteins beyond the twenty naturally occurring amino acids. By modifying the side chains while retaining the basic amino acid backbone, researchers can introduce new chemical properties into proteins for research, biotechnology, and biomedical applications.
4-Fluorophenylalanine is a modified analog of phenylalanine in which a fluorine atom replaces a hydrogen on the aromatic ring. The presence of fluorine alters the electronic properties and hydrophobicity of the side chain while maintaining a similar size to natural phenylalanine. This makes it useful in protein folding studies and structural biology, where it can act as a probe in spectroscopic techniques such as ¹⁹F NMR. Because fluorine is rarely found in natural proteins, its signal can be easily detected, allowing researchers to monitor conformational changes and interactions within proteins.
Azidohomoalanine contains an azide (-N₃) functional group, which is highly useful in bioorthogonal chemistry. This amino acid can be incorporated into newly synthesized proteins in place of methionine, allowing researchers to attach fluorescent dyes, affinity tags, or other molecules using “click chemistry” reactions. As a result, azidohomoalanine is widely used for protein labeling, tracking protein synthesis, imaging cellular processes, and studying protein turnover in living cells.
Bipyridylalanine includes a bipyridine moiety, a structure well known for its ability to bind metal ions such as iron, copper, or zinc. When incorporated into proteins, it can create artificial metal binding sites, enabling the design of engineered metalloenzymes, biosensors, or catalytic proteins. This amino acid can also be used to study metal protein interactions and to construct proteins with new catalytic or electronic properties.
Overall, synthetic amino acids like these expand the chemical diversity available to proteins. They allow scientists to probe protein structure, control biochemical reactions, introduce novel catalytic functions, and develop advanced tools for imaging and biotechnology, thereby playing an important role in modern protein engineering and synthetic biology.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids can form in nature if the right chemical conditions exist. Amino acids have been detected in space rock and gases. This shows that they are a part of the universe’s natural building blocks, which can become more complex through further chemical reactions.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A standard α-helix made from D–amino acids will be left-handed because the chirality flips the preferred backbone geometry; therefore -L amino acids favour right-handed α-helices, while D amino acids favour the mirror-image left-handed form.
Can you discover additional helices in proteins?
You can find additional helices if new conditions are used that improve resolution or biological context. The dynamics of the protein change depending on the conditions that were used to crystalise it and which is used to ellucidate the protein structure.
Why are most molecular helices right-handed?
Most biological molecules are right-handed because Earth biology is built almost entirely from L-amino acids, and the stereochemistry of L-amino acids makes the right-handed α-helix the lowest-energy, most stable geometry.
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
Why do many amyloid diseases form β-sheets?
Can you use amyloid β-sheets as materials?
Amyloid diseases often form β-sheets because misfolded proteins can stack into a highly stable “cross-β” structure that hides exposed hydrophobic/backbone surfaces and then seeds more proteins to misfold the same way. These fibrils are hard for cells to get rid of, so they accumulate and disrupt cellular function. This may lead to disease.
Yes, amyloid β-sheets can be used as materials because amyloid fibrils are strong, stiff, and chemically resistant nanofibers. They can be engineered into hydrogels, scaffolds, coatings, or templates for assembling other materials.
Design a β-sheet motif that forms a well-ordered structure.
Part B My Protein
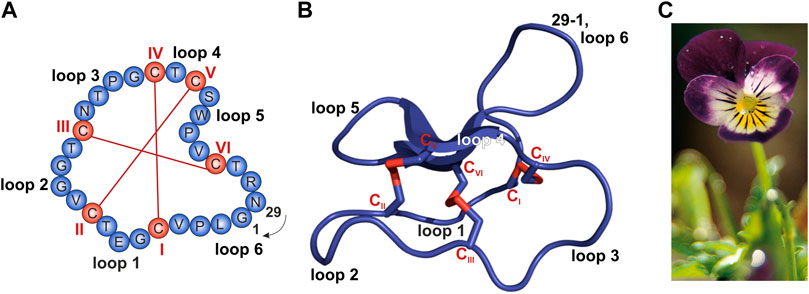
I chose cycloviolacin cyclotide protein from Viola odorata
Sequence:
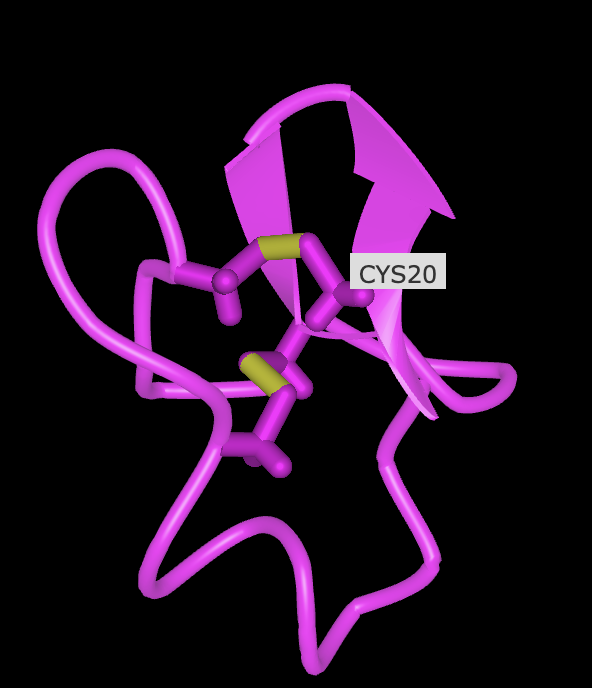
CAESCVYIPCTVTALLGCSCSNRVCYNGIP
Sequence and structure of the prototypic cyclotide kalata B1 and photograph of Viola tricolor. (A,B) The cyclized peptide kalata B1 (C) Viola tricolor
From
Conzelmann C, Muratspahić E, Tomašević N, Münch J and Gruber CW (2022) In vitro Inhibition of HIV-1 by Cyclotide-Enriched Extracts of Viola tricolor. Front. Pharmacol. 13:888961. doi: 10.3389/fphar.2022.888961
Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
-Paste your amino acid sequence here
my_sequence = “CAESCVYIPCTVTALLGCSCSNRVCYNGIP”
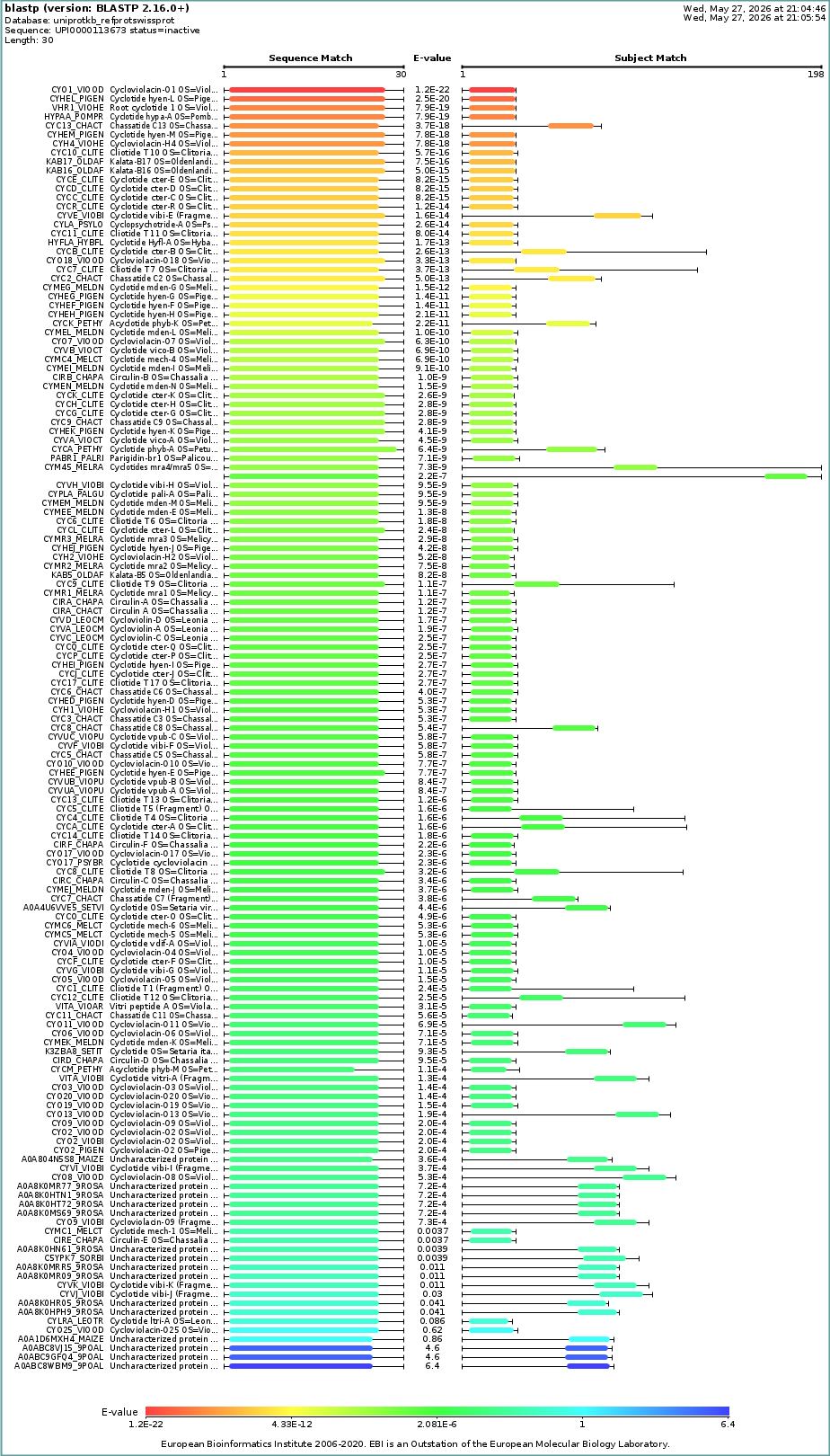
Sequence Length: 30 Amino Acid Frequencies: C: 6 S: 3 V: 3 A: 2 Y: 2 I: 2 P: 2 T: 2 L: 2 G: 2 N: 2 E: 1 R: 1
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
- Does your protein belong to any protein family?
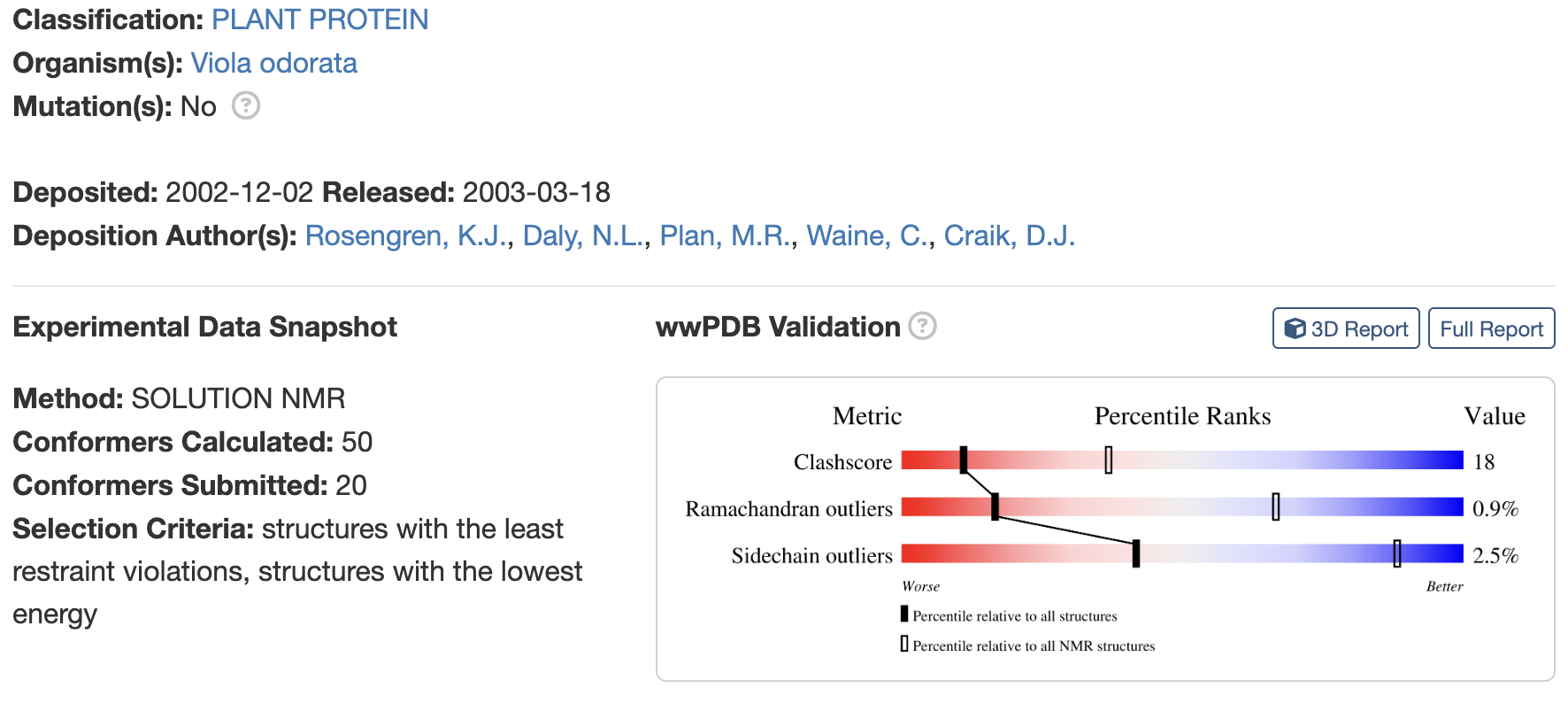
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Are there any other molecules in the solved structure apart from protein?
no
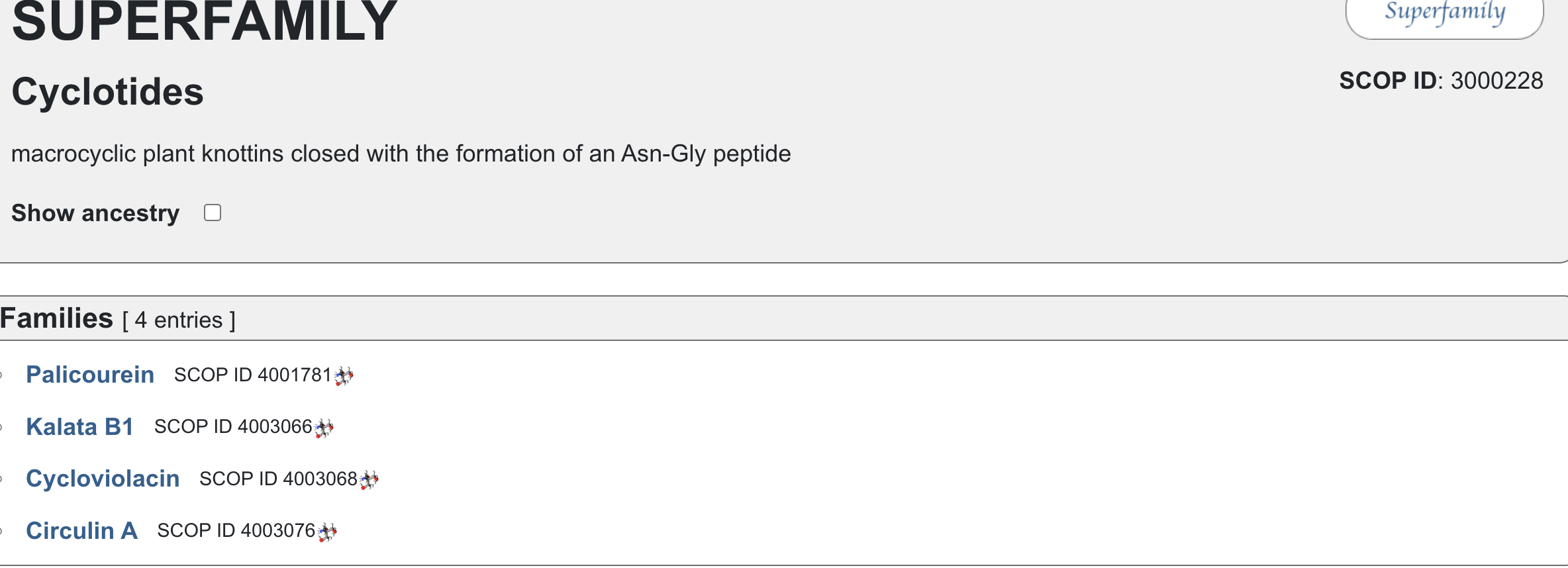
Does your protein belong to any [structure classification family]
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
see below
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
No, it has binding domains though
R
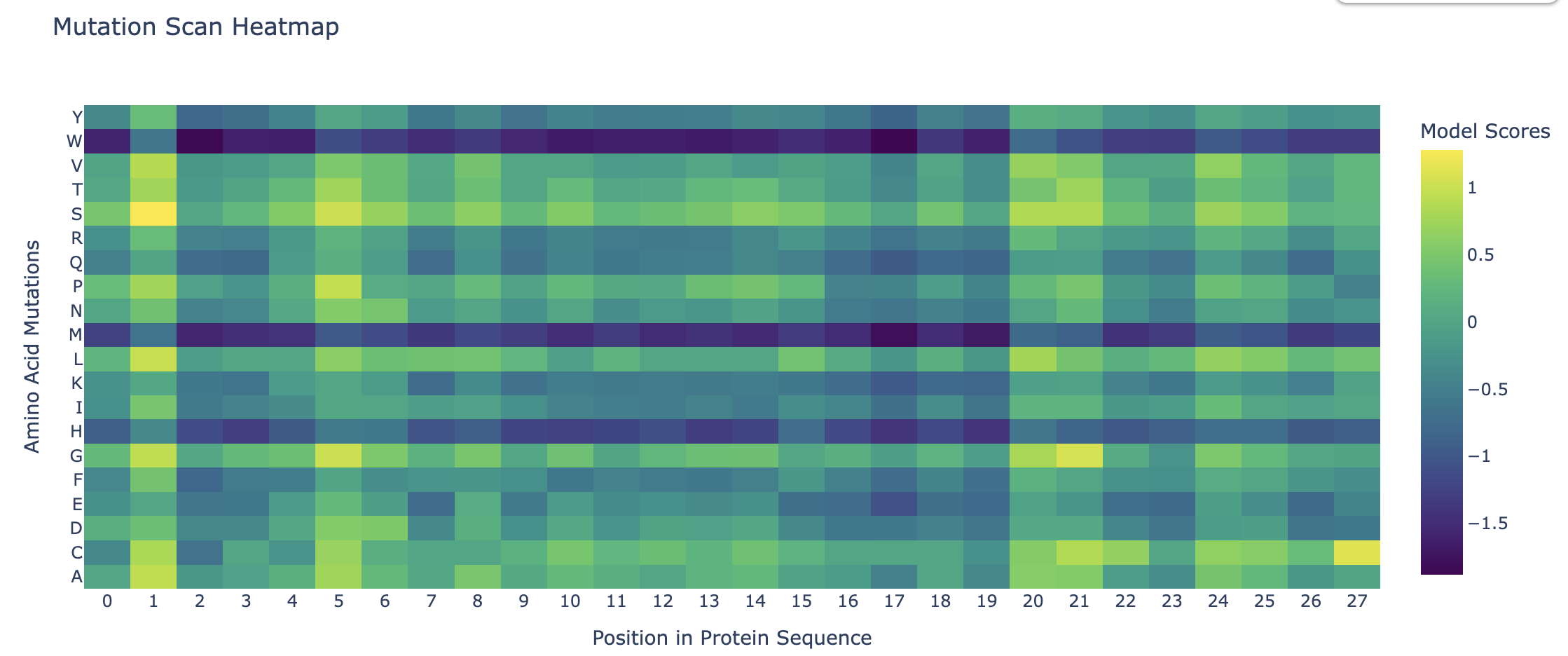
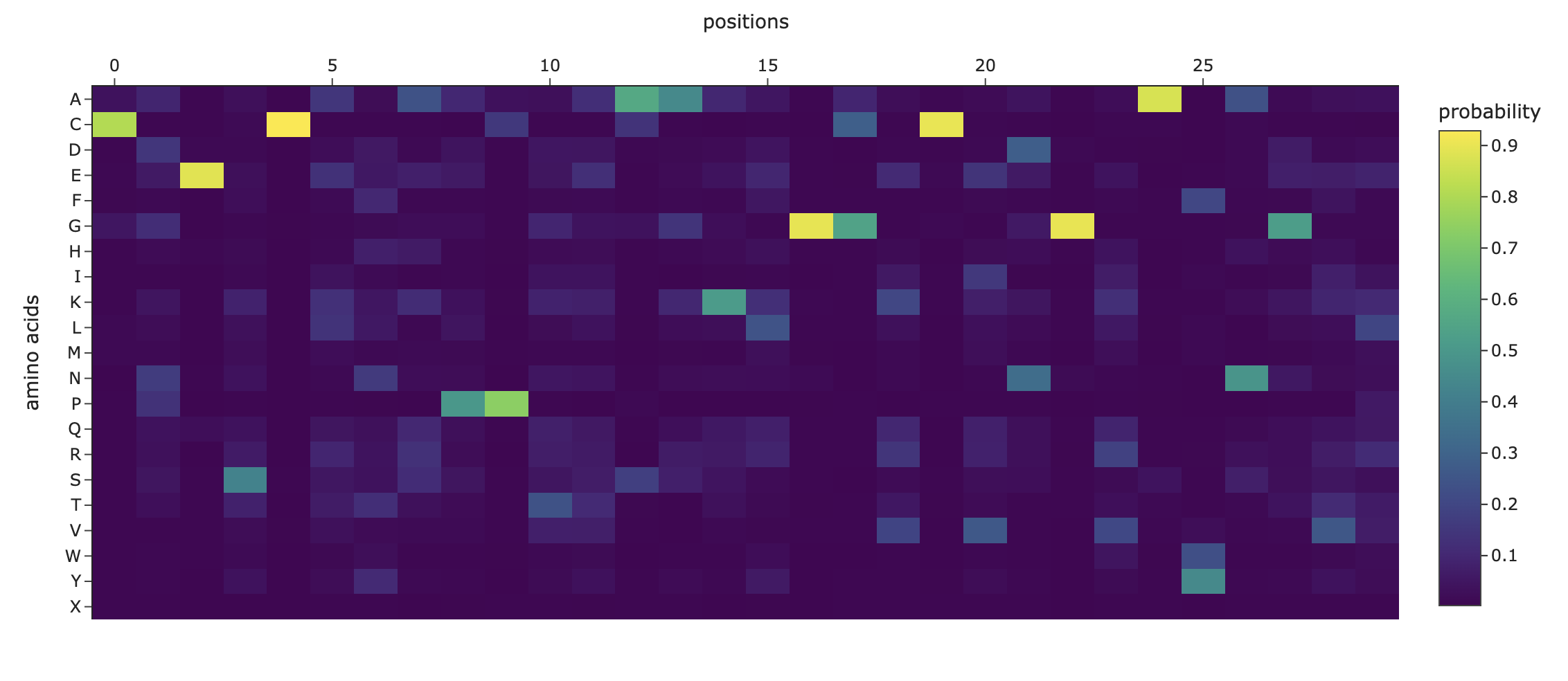
Deep Mutational Scans
Darker columns may indicate low likelihood of mutations at the residue, probably because it is structurally or functionally important.
Mutated with A4V
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
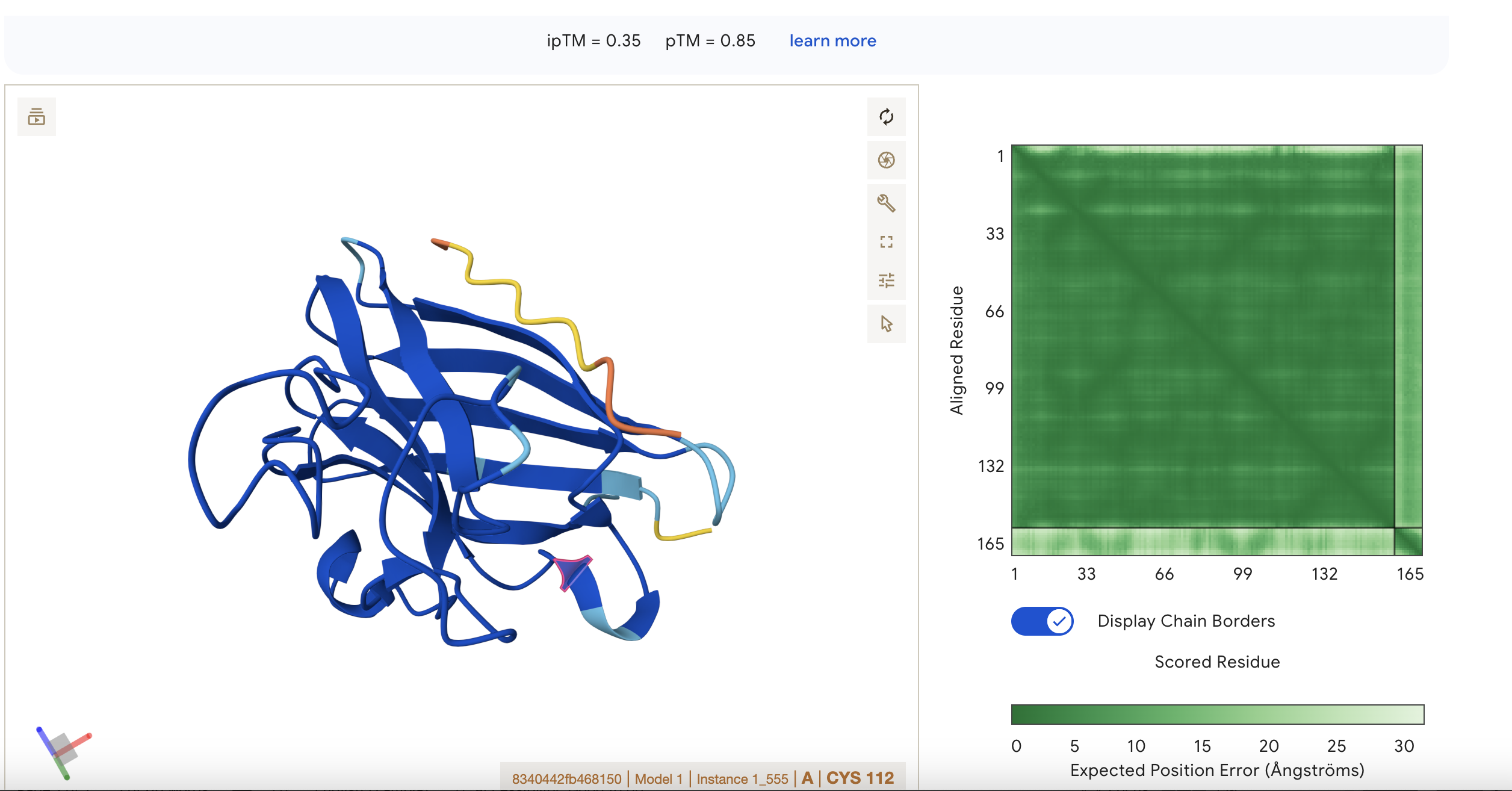
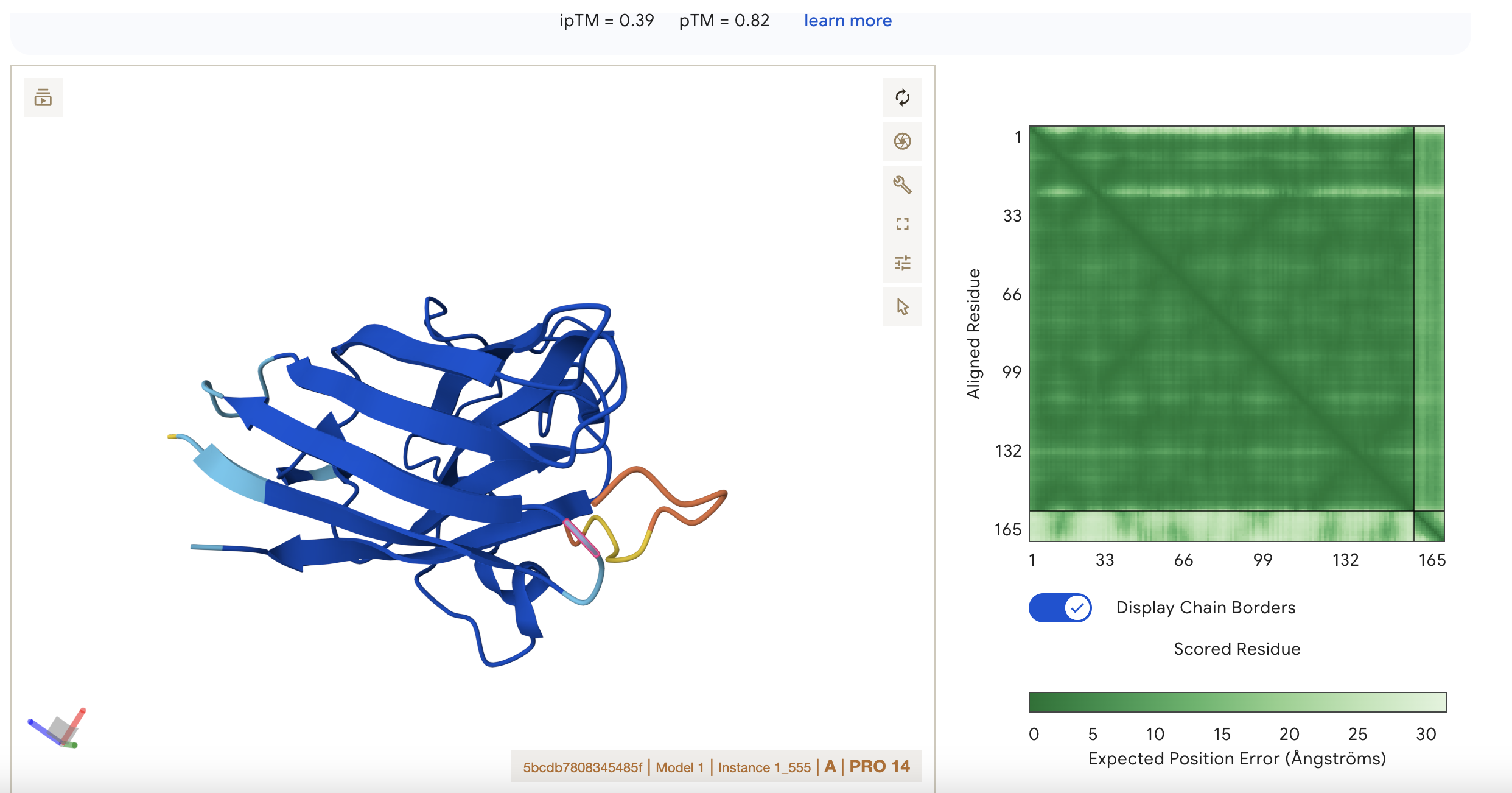
Sequence 1 HHVPVVVLRHKX
The sequence was submitted without the last X because it was flagged as an illegal character (it could stand for various amino acids).
The peptide appears to be interacting with
ipTM = 0.22 pTM = 0.85
pTM = 0.85 suggests the overall fold is very reliable, while ipTM = 0.22 says the predicted interaction/interface between chains is very poor.
This means the protein’s global shape looks strong and likely meaningful, this is in line with it being a known and well illucidated human protein. It seems like a case where AlphaFold is confident about folding, but not about binding geometry.
Localisation: Near beta barrel
Engagement with β-barrel region: not exact but close
Approach to the dimer interface: not exact but close
Appear surface-bound: Yes
Appear partially buried: No
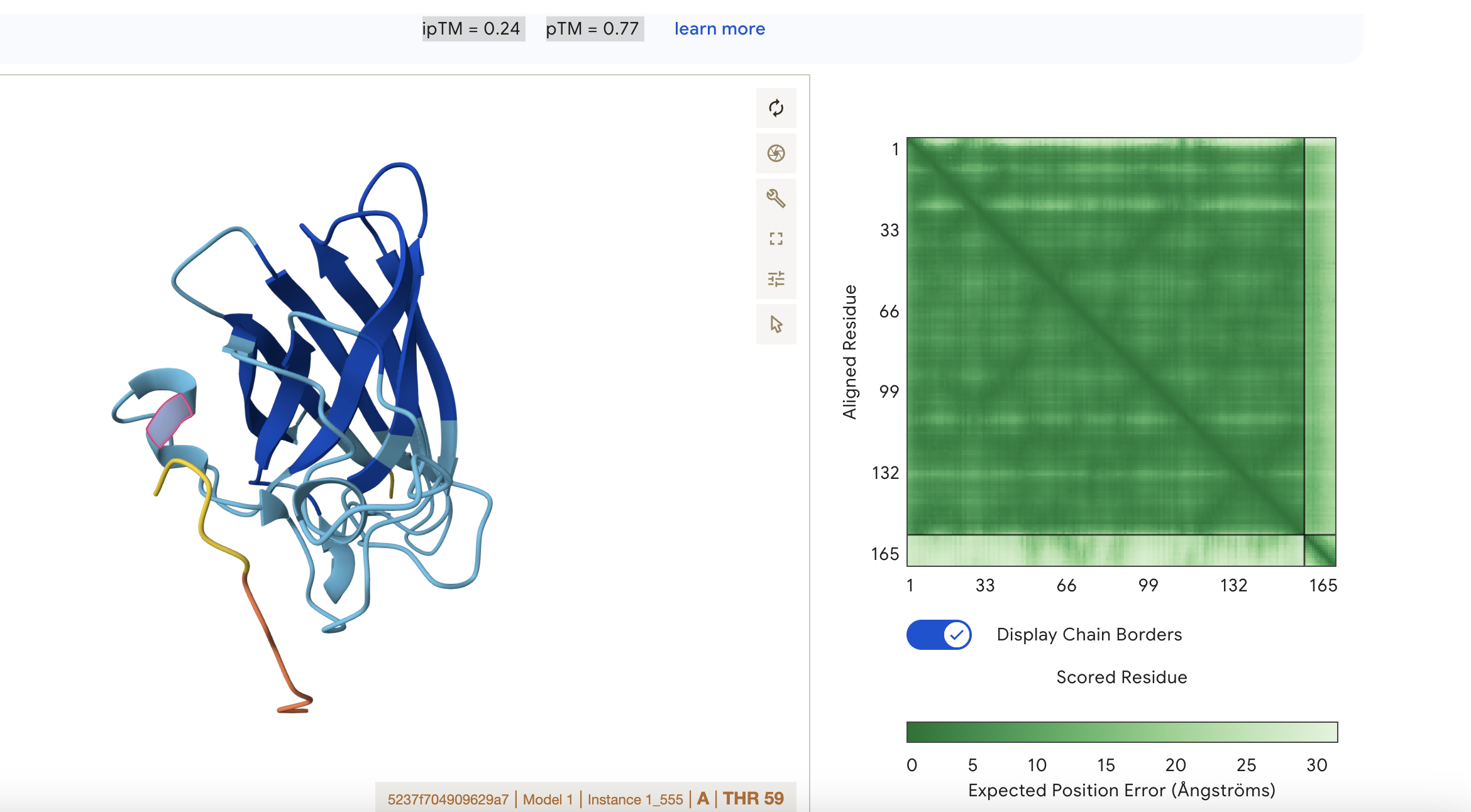
Sequence 2 WRYYAAVARWKE
ipTM = 0.24 pTM = 0.77
This means that the model is confident about the fold of the chain itself, but not about how chains interact.
pTM = 0.77: the overall protein fold looks reasonably reliable; this is in a fairly good range for the global shape of the modeled structure
ipTM = 0.24: the predicted interface between subunits is very poor, so AlphaFold is not confident that the chains are positioned correctly relative to each other.
Localisation: Near alpha helix
Engagement with β-barrel region: None
Approach to the dimer interface: None
Appear surface-bound: Yes
Appear partially buried: No
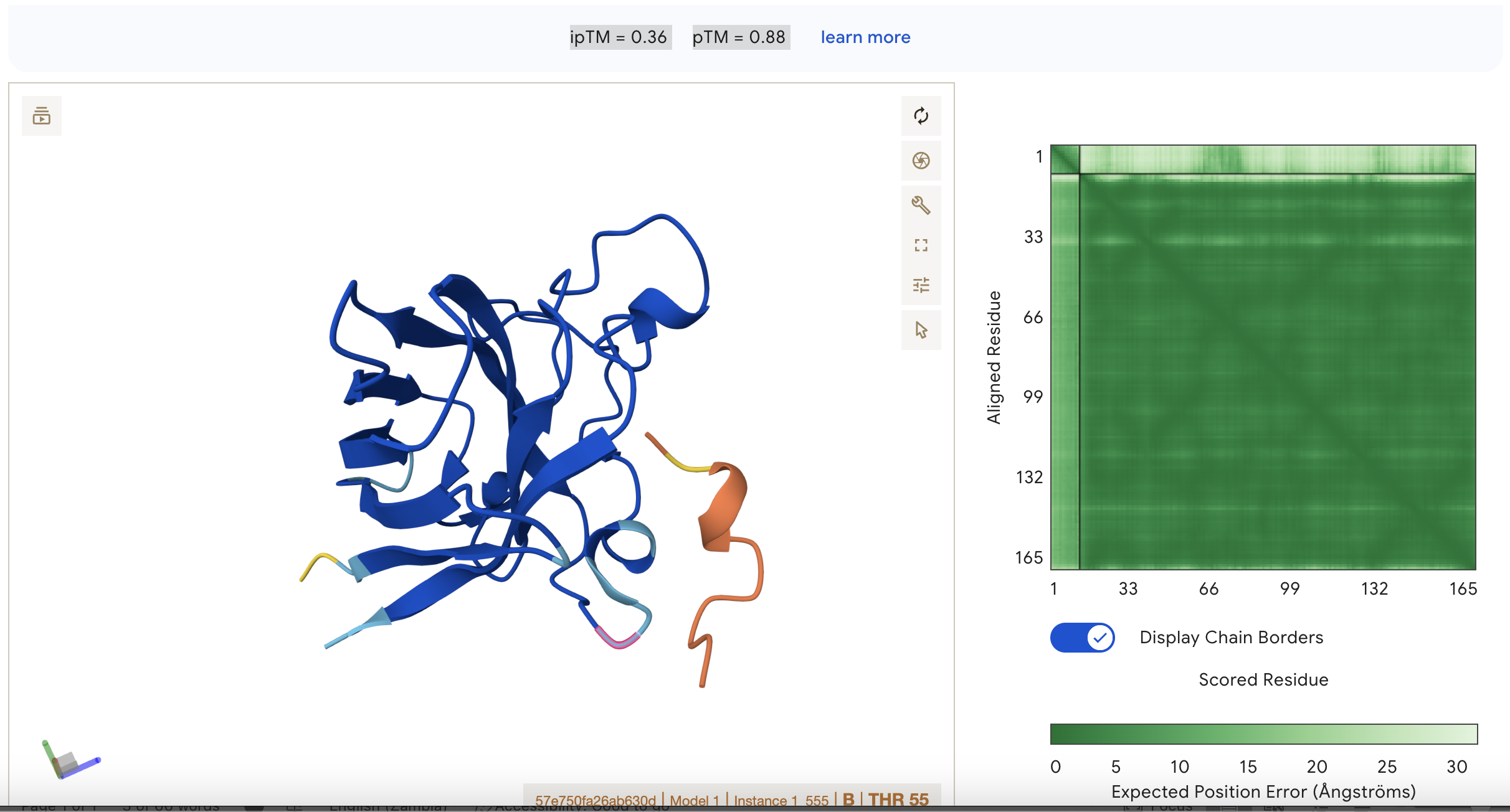
Sequence 3 HRYYPAAARWKX
ipTM = 0.36
pTM = 0.88
This suggests that the model is fairly confident about the structure but the binding interaction is still under a threshold of 0.5
This represents the second best binding in the data set however.
Localisation: Side of dimer interface
Engagement with β-barrel region: partial
Approach to the dimer interface: on the side
Appear surface-bound:yes
Appear partially buried: no
Sequence 4 FLYRWLPSRRGG
ipTM = 0.39 pTM = 0.8
pTM 0.8 suggests the model is fairly confident about the global arrangement of the structure as a whole.
ipTM 0.39 is low, so AlphaFold is not confident about the relative positioning of the interacting subunits. It is the highest score however so this represents the best binding possibility from this set of results.
There is low confidence of interaction between the short peptide and the protein.
Localisation: Side of dimer interface
Engagement with β-barrel region: partial
Approach to the dimer interface: on the side
Appear surface-bound: yes
Appear partially buried: no
Peptiverse Prediction
📊 Results
Input
Property
Prediction
Value
Unit
HHVPVVVLRHKX
💦 Hydrophobicity (GRAVY)
Non-hemolytic
1302.7
Probability
Input
Property
Prediction
Value
Unit
HHVPVVVLRHKX
💧 Solubility
Soluble
0.998
Probability
HHVPVVVLRHKX
🩸 Hemolysis
Non-hemolytic
0.022
Probability
HHVPVVVLRHKX
🔗 Binding Affinity
Weak binding
5.789
pKd/pKi
HHVPVVVLRHKX
📏 Length
12
aa
HHVPVVVLRHKX
⚖️ Molecular Weight
1302.7
Da
HHVPVVVLRHKX
⚡ Net Charge (pH 7)
2.02
HHVPVVVLRHKX
🎯 Isoelectric Point
11.00
pH
HHVPVVVLRHKX
💦 Hydrophobicity (GRAVY)
0.08
GRAVY
📊 Results
Input
Property
Prediction
Value
Unit
WRYYAAVARWKE
💦 Hydrophobicity (GRAVY)
Non-hemolytic
1598.8
Probability
Input
Property
Prediction
Value
Unit
WRYYAAVARWKE
💧 Solubility
Soluble
0.997
Probability
WRYYAAVARWKE
🩸 Hemolysis
Non-hemolytic
0.031
Probability
WRYYAAVARWKE
🔗 Binding Affinity
Weak binding
6.678
pKd/pKi
WRYYAAVARWKE
📏 Length
12
aa
WRYYAAVARWKE
⚖️ Molecular Weight
1598.8
Da
WRYYAAVARWKE
⚡ Net Charge (pH 7)
1.77
WRYYAAVARWKE
🎯 Isoelectric Point
9.70
pH
WRYYAAVARWKE
💦 Hydrophobicity (GRAVY)
-0.93
GRAVY
📊 Results
Input
Property
Prediction
Value
Unit
HRYYPAAARWKX
💦 Hydrophobicity (GRAVY)
Non-hemolytic
1400.8
Probability
Input
Property
Prediction
Value
Unit
HRYYPAAARWKX
💧 Solubility
Soluble
1.000
Probability
HRYYPAAARWKX
🩸 Hemolysis
Non-hemolytic
0.012
Probability
HRYYPAAARWKX
🔗 Binding Affinity
Weak binding
6.426
pKd/pKi
HRYYPAAARWKX
📏 Length
12
aa
HRYYPAAARWKX
⚖️ Molecular Weight
1400.8
Da
HRYYPAAARWKX
⚡ Net Charge (pH 7)
2.84
HRYYPAAARWKX
🎯 Isoelectric Point
10.28
pH
HRYYPAAARWKX
💦 Hydrophobicity (GRAVY)
-1.32
GRAVY
📊 Results
Input
Property
Prediction
Value
Unit
FLYRWLPSRRGG
💦 Hydrophobicity (GRAVY)
Non-hemolytic
1507.7
Probability
Input
Property
Prediction
Value
Unit
FLYRWLPSRRGG
💧 Solubility
Soluble
0.608
Probability
FLYRWLPSRRGG
🩸 Hemolysis
Non-hemolytic
0.047
Probability
FLYRWLPSRRGG
🔗 Binding Affinity
Weak binding
6.361
pKd/pKi
FLYRWLPSRRGG
📏 Length
12
aa
FLYRWLPSRRGG
⚖️ Molecular Weight
1507.7
Da
FLYRWLPSRRGG
⚡ Net Charge (pH 7)
2.76
FLYRWLPSRRGG
🎯 Isoelectric Point
11.71
pH
FLYRWLPSRRGG
💦 Hydrophobicity (GRAVY)
-0.71
GRAVY
Question: Do peptides with higher ipTM also show stronger predicted affinity?
The two peptides with the highest iPTM have higher binding affinity
Question: Are any strong binders predicted to be hemolytic or poorly soluble?
The control peptide FLYRWLPSRRGG is predicted to be poorly soluble
Question: Which peptide best balances predicted binding and therapeutic properties?
HRYYPAAARWKX
It has good binding affinity, good range isoelectric point, desirable net charge, smaller in molecular weight, and has the best solubility.
Choose one peptide you would advance and justify your decision briefly
I would advance with HRYYPAAARWKX because in the fist assay, it did not do so well but on closer inspection using the other tools it seems to perform better. When I looked at it using Alphafold, it interacted in the region most likely to disrupt dimer formation unlike the first peptide. Then in the therapeutic analysis, it also had better metrics than the other peptides on the most critical variables such as solubility. I would like the therapeutic to be soluble as possible to reduce toxicity and retention in the body’s long term fat deposits and for it to be easily excreted.
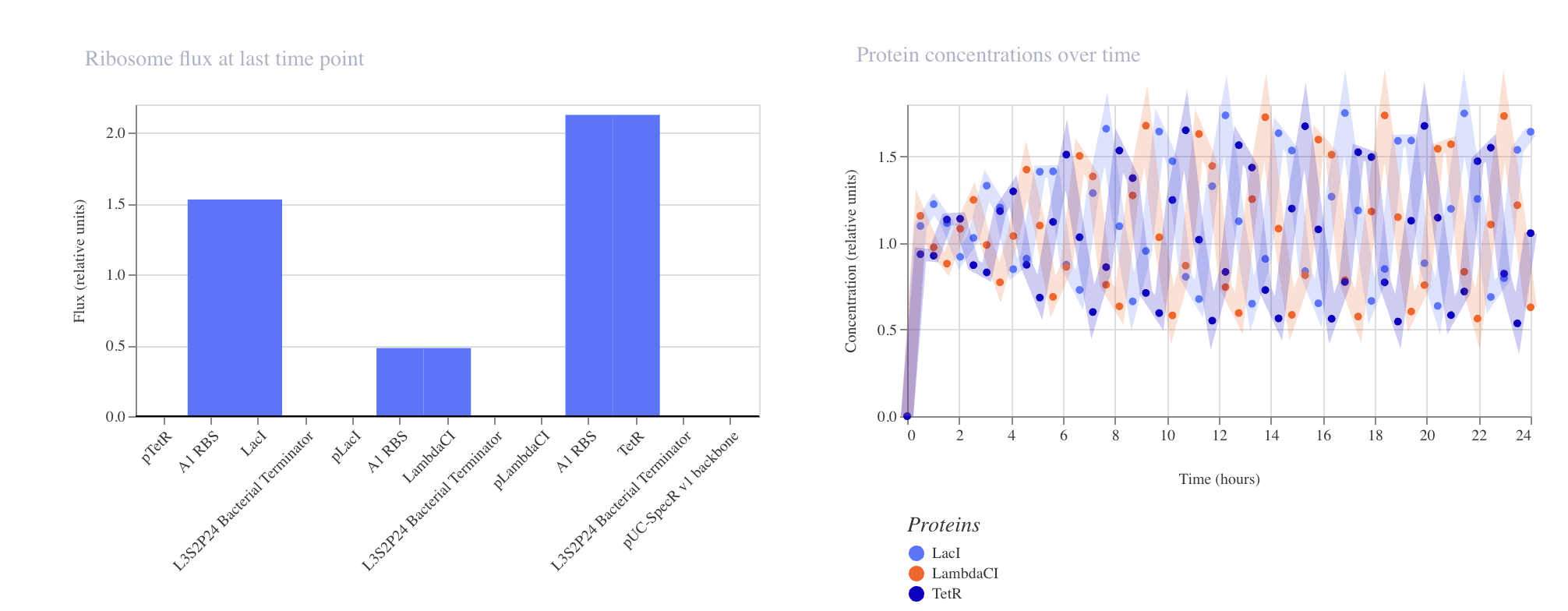
Week 6 HW: Genetic Circuit Part 1
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion DNA Polymerase - this is for amplifying the DNA by polymerisation. It is special because when used with its special buffer, it has up to 52× higher fidelity rate than ordinary Taq polymerase
dNTPs- These are the building blocks that are used in the polymersiation by the DNA polymerase
MgCl₂ - DNA polymerase enzyme needs magnesium ions to function properly (they are necessary cofactors for the thermostable enzyme)
Optimized buffer - Phusion needs an optimized buffer because it uses a highly processive and high-fidelity DNA polymerase (originally a Pfu-like enzyme fused to a processivity domain) that is very sensitive to Mg²⁺, ionic strength, pH, and additives. Therefore these components must be balanced perfectly for the PCR not to fail in terms of processivity or fidelity.
These items are added to the PCR: template DNA, primers, and molecular water.
What are some factors that determine primer annealing temperature during PCR?
Here’s the table simplified into bullet points:
Primer length: Longer primers have higher melting temperature (Tm), so it is necessary to use a higher annealing temperature for longer ones.
GC content: More G–C pairs (3 H-bonds each) increase Tm, so higher GC → higher annealing temperature.
Primer sequence (especially 3′ end): Stronger matches at the 3′ end increase effective Tm and allow higher annealing temperature.
Mg²⁺ concentration: Higher free Mg²⁺ stabilizes DNA duplexes, increasing Tm and annealing temperature; Mg²⁺ is also bound by dNTPs and DNA.
Monovalent cations (Na⁺, K⁺): Higher salt stabilizes DNA and increases Tm, but can compete with Mg²⁺ binding.
Buffer additives (DMSO, formamide, glycerol): These destabilize DNA and lower Tm (e.g., 10% DMSO lowers Tm by ~5.5–6°C), so annealing temperature must be reduced.
Polymerase and buffer system: Different polymerases and buffers (e.g., Phusion HF vs. GC) have different optimal annealing temperatures and salt/Mg²⁺ conditions.
Amplicon (product) Tm: The product’s Tm also affects optimal annealing temperature
Specificity vs. yield trade-off:
Lower annealing temperature may lead to more non-specific binding, primer dimers, lower yield of correct product.
Higher annealing temperature may lead to higher specificity but may reduce yield if too high.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Restriction digestion
PCR
What it does
Cuts existing DNA at specific restriction sites to release a linear fragment
Synthesises a new linear DNA fragment by amplifying a target region from a template
Flexibility
Limited to fragments bounded by existing (or engineered) sites
Very flexible: any region, any length (within polymerase limits), with custom ends
Sequence requirements
Needs restriction sites at desired boundaries; otherwise you must engineer them first
Only needs primer-binding sites; you can design primers anywhere
Flexibility
Limited to fragments bounded by existing (or engineered) sites
Very flexible: any region, any length (within polymerase limits), with custom ends
Fidelity
No new synthesis, so no new errors
Depends on polymerase; high-fidelity enzymes (e.g., Phusion) give very low error rates but ordinary Taq can introduce errors
Yield
Typically moderate, limited by starting DNA amount
Can generate high yield from very little template
Time
Usually up to 3 hrs for incubation + purification
upt to 3 hrs to run a full program of 33 cycles + purification. Involves more steps
Requires primer design, sometimes gradient PCR is needed to optimise primers for correct annealing temperature
Risk of unwanted changes
No sequence change (except if partial digestion or star activity. Buffer choice and incubation must be done carefully)
Risk of polymerase errors are there but these can be lowered with high-fidelity enzymes). Sometimes primer-dimer artifacts may be there.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To be “Gibson-ready,” digested fragments and PCR products must meet
Have correct overlap design,
Have correct ends, and
Have clean, intact DNA
1) Confirm overlaps are designed correctly
Gibson assembly needs homology overlaps between adjacent fragments.
Overlap length: typically 20–40 bp per junction (this may vary for GC-poor or repetitive sequences).
Orientation: overlaps must match the intended adjacency (5′ end of one fragment overlaps the 3′ end of the next).
Sequence content: overlaps that are extremely GC-rich must be avoided, also highly repetitive, or form strong hairpins/dimers.
Reading frame / features: if coding regions are to be fused, then checks must be made for:
frame continuity
start/stop codons
linkers/tags
RBS/promoter boundaries
2) Make sure the ends produced are compatible with Gibson
Gibson relies on 5′ exonuclease activity to create 3′ single-stranded overhangs from double-stranded DNA ends—so linear DNA is needed with the correct overlap sequences at the ends.
If inserts are PCR-ed
Overlaps are directly added in the 5′ tails of primers (the template-binding region is only part of the primer; the overlap tail becomes part of the product).
A high fidelity must be used
Leftover primers/dNTPs/polymerase must be removed using a cleanup kit.
If a vector is restriction-digested
The vector must be confirmed to be fully linearized by running a gel and if two enzymes were used, it is necessary to confirm they cut exactly where desired.
Critical for correct ends
If the PCR was done with a polymerase that leaves 3′ A-overhangs (like Taq), that’s not ideal for Gibson. A proofreading polymerase (Q5/Phusion) should be used or a reaction must be done to polish the ends.
For vectors, DpnI treatment can be used if the vector template was amplified by PCR (to remove methylated parental plasmid).
3) Clean Intact DNA and Wrong Concentrations
These issues commonly break Gibson:
Wrong concentration ratio (too much vector or too much insert). Typical starting point is 2–3× molar excess insert over vector for 2-piece assemblies.
Residual salts/ethanol/guanidine from cleanup columns, this inhibits enzymes.
Primer dimers / nonspecific bands, this causes parts to assemble into junk. If there are multiple bands, it is simpler to gel-purify the correct band for use.
Incomplete digestion, this gives uncut plasmid which increases background colonies. This is fixed by longer digest, fresh enzyme, gel-purified linear bands, or additon of a negative control (digested vector-only Gibson).
How does the plasmid DNA enter the E. coli cells during transformation?
Transformation can be done using different ways:
Chemical competence
Electro competence
Barrier:E. coli has a double memberance (an outer membrane and inner membrane leaflets). DNA normally can’t cross because it’s large and negatively charged.
Charge repulsion: DNA and the bacterial surface are both largely negative, so if there is no assitance, they repel .
Chemical competence—binding step: Divalent cations (e.g., Ca²⁺) shield/bridge charges, letting DNA stick to the cell surface.
Chemical competence—entry step: A brief heat shock creates a temporary membrane permeability change, helping surface-bound DNA move into the cell.
Electrocompetence—pore creation: A strong, brief electric pulse causes transient nanopores in the membrane(s) (dielectric breakdown).
Electrocompetence—DNA movement: The electric field helps drive DNA toward/through these pores (field-driven transport + diffusion).
Recovery: In both methods, membranes reseal, and cells need recovery time to repair stress and begin expressing traits like antibiotic resistance (if relevant).
Establishment: Successful transformation means DNA reaches the cytoplasm and avoid degradation—plasmids replicate; linear DNA is often degraded (unless recombined).
Describe another assembly method in detail (such as Golden Gate Assembly)
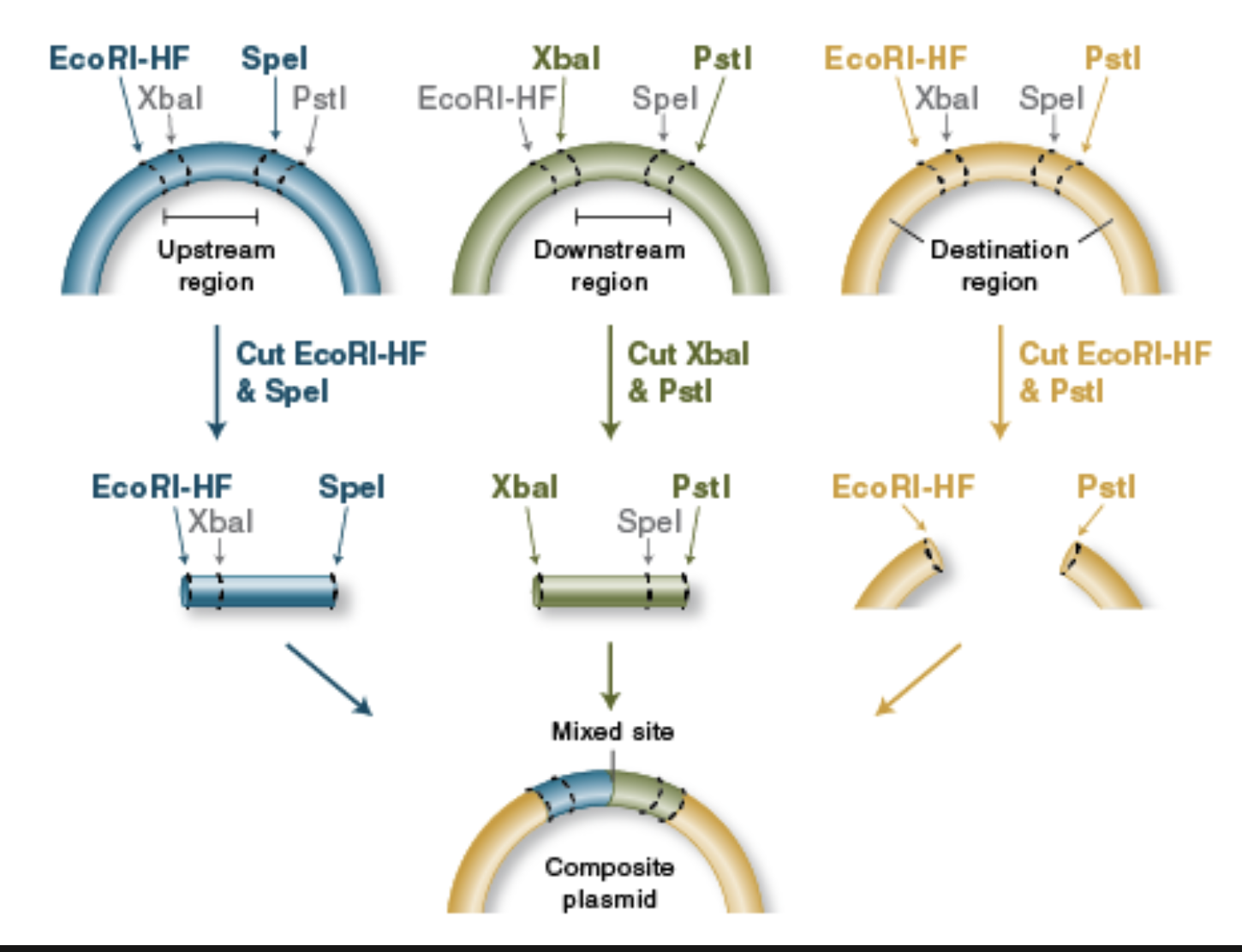
BioBrick Assembly
Standardized parts: BioBrick assembly uses DNA “parts” (promoters, RBSs, CDSs, terminators, etc.) that are flanked by a standard prefix and suffix containing specific restriction sites, so different labs can combine parts predictably.
Restriction enzymes define the join: Classic BioBrick (RFC10) uses four key restriction enzymes—EcoRI and XbaI in the prefix, SpeI and PstI in the suffix—so you can cut parts and vectors in a consistent way to prepare compatible ends.
Digest donor insert and recipient vector: To add a new part downstream of an existing part, you typically digest:
the insert (new part) with enzymes that expose the correct ends, and
the destination plasmid with the matching enzymes to open it at the insertion point.
Compatible “mixed” ends enable ligation: A key trick is that XbaI and SpeI generate compatible overhangs; that means a fragment cut with XbaI can ligate to one cut with SpeI, allowing ordered assembly of part A + part B.
Scar formation: When an XbaI end ligates to a SpeI end, the junction becomes a short “scar” sequence that is no longer recognized by either XbaI or SpeI. This “locks” the assembly and allows further rounds of building.
Idempotent design enables iterative building: After ligation, the combined part is still flanked by the standard prefix and suffix at the outer ends, so it can be treated like a new BioBrick part and assembled again with the same rules.
Verification: After transformation, you confirm correct assembly using colony PCR / diagnostic restriction digest and usually Sanger sequencing across junctions (especially important if coding sequences must stay in-frame, since the scar can affect protein fusions).
From New England Biolabs
Asimov Kernal
Week 7 HW: Genetic Circuit Part 2
Advantages of IANNs over Boolean genetic circuits
Traditional genetic circuits (AND/OR/NOT) are great when the world can be cleanly thresholded into ON/OFF inputs and you only need discrete outputs. IANNs (genetic implementations of artificial neural networks) are useful when biology is messy—inputs are continuous, noisy, and correlated
Analog, not just digital IANNs naturally support graded responses (continuous input → continuous output), not only Boolean truth tables. That lets you encode “how much” and “how confident,” not just “yes/no.”
Multi-input pattern classification Boolean circuits scale poorly as you add inputs (truth tables explode). IANNs can combine many weak signals into a robust classification—closer to how real cells integrate signals.
Tolerance to noise and variability Neural-style weighted sums + thresholds can be more forgiving than logic gates when promoter strengths drift, copy number varies, or environments fluctuate.
Decision boundaries beyond simple logic Boolean circuits implement crisp logic partitions. IANNs can implement complex nonlinear decision boundaries (e.g., “positive if this cocktail of biomarkers matches a pattern”), which is often what you want in diagnostics or environmental sensing.
Compact representations for complex behaviors For some tasks, an IANN can represent a complex mapping with fewer distinct regulatory parts than an equivalent explicit logic circuit that enumerates all cases.
Potential for tunability/adaptability In principle, weights (interaction strengths) can be tuned to adjust behavior without redesigning the whole circuit (though “learning in vivo” is still a hard frontier; most are trained/tuned ex vivo and then built).
A useful application: a living classifier for inflammatory disease states (multi-biomarker diagnostic)
Goal
Engineer a probiotic E. coli (or a gut commensal chassis) that detects an IBD-like inflammatory state in the gut and produces an output only when the overall biomarker pattern matches “inflammation,” not when any single marker spikes.
Inputs (example set)
Let the cell sense 5 gut-associated signals (each continuous, noisy, and not individually decisive):
x1: nitrate / reactive nitrogen species proxy (inflammation-associated)
x4: oxygen tension proxy (higher oxygen near inflamed mucosa)
x5: a host-derived inflammatory metabolite (or a quorum/metabolite proxy correlated with dysbiosis)
Each sensor produces a graded transcriptional signal proportional to concentration (not a binary “present/absent”).
Key point: the IANN outputs high only when the pattern matches, not when one input is high. That reduces false positives compared with an OR gate.
Why an IANN is better than Boolean here
A Boolean circuit would require you to pre-decide exact logic, but real biology doesn’t cleanly separate signals into ON/OFF. The IANN can implement a soft decision. i.e. multiple moderate signals can sum to a confident positive, and one spurious spike may not be enough.
Limitations an IANN might face (practical constraints)
Parts burden and cellular load More regulators, promoters, and wiring increases metabolic burden, slows growth, and can destabilize behavior.
Context dependence and drift Promoter strength changes with growth phase, media, host environment, plasmid copy number, and mutations—effectively changing “weights” over time.
Noise and signal coupling Sensors can cross-react; hidden nodes may inadvertently respond to unrelated metabolites, shifting the decision boundary.
Training/tuning is nontrivial Unlike silicon, you can’t easily backprop in cells. Most designs require careful calibration of transfer functions and iterative build-test cycles.
Dynamic timescales Many biological responses are slow (minutes–hours). If the goal needs fast decisions, performance may be limited.
Scaling limits (orthogonality bottleneck) Large IANNs need many orthogonal regulators/sensors that don’t interfere—still a major constraint in genetic engineering.
Safety and containment (for therapeutic outputs) If the output is a drug/effector, you need fail-safes (kill switches, dependency circuits, containment). These add complexity and can interact with the IANN.
Multilayer Perceptron
Fungal Materials and Synthetic Biology
Fungal materials like mycelium composites, mushroom-based leather, packaging, and insulation are increasingly used as sustainable alternatives to traditional materials. Mycelium composites serve as building insulation, bricks, packaging that replaces Styrofoam, and leather alternatives for fashion, all produced in just 7 days using agricultural waste like straw. These materials offer significant advantages: they are biodegradable, compostable, store CO₂, require minimal energy, are fire-resistant (burning cleanly to water and CO₂), and are ethical animal-free alternatives. However, they have notable disadvantages including lower structural strength (30 psi vs. 4000 psi for concrete), making them unsuitable for load-bearing structures, sensitivity to moisture and deformation in rain, degradation over time that limits long-distance shipping, and still-high costs at small scales requiring standardization for industry adoption.
Genetic engineering of fungi focuses on enhancing their capabilities as cell factories and material producers. Key goals include improving production of bioactive compounds like artemisinin, insulin, vitamins, and astaxanthin; tailoring mycelium materials with specific structural properties like higher compressive strength and moisture resistance; enhancing waste degradation enzymes for biofuel and bioremediation; implementing synthetic transcriptional regulation for programmable gene networks; and engineering novel metabolic pathways for high-value chemicals difficult to synthesize chemically. These engineering efforts leverage fungi’s natural ability to produce complex secondary metabolites and their established industrial use for enzymes and organic acids.
Synthetic biology in fungi offers distinct advantages over bacteria, particularly because fungi are eukaryotes with organelles enabling proper post-translational modifications like glycosylation and protein folding, which bacteria cannot perform adequately.
Fungi excel at efficiently secreting proteins (ideal for industrial enzymes), naturally produce complex secondary metabolites like antibiotics, and have industrial track records for producing vitamins and organic acids.
Filamentous fungi uniquely form mycelium networks for material applications that bacteria cannot replicate, and they are more robust to industrial stress conditions. While bacteria offer faster growth and simpler genetic manipulation, fungi are superior for eukaryotic protein production, complex natural products, and bio-based materials, making them ideal for cell factory applications and material science applications aligned with synthetic biology advances.
Week 9 HW: Cell Free Systems
General Homework Questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
-Cell-free can be used even when cold-chain conditions cannot be assured
-Cell-free systems can be used without cloning and creating recombinant organisms
Cell-free protein synthesis offers flexibility and a lot of control because you can directly tune reaction components (DNA amount, salts/cofactors, energy mix, chaperones, redox environment) and test many conditions rapidly without cell growth, regulation, or viability constraints.
Cell-free expression is especially beneficial for rapid prototyping of constructs/circuits and also producing products especially if they are tocix to living expression systems. It can also enable on-demand expression (e.g., freeze-dried systems) and reactions requiring tightly controlled chemistry. This is great for making vaccines on demand on a mission to Mars for example.
Describe the main components of a cell-free expression system and explain the role of each component.
For General Systems
Cell extract (lysate) or purified translation system — supplies the machinery that makes protein (ribosomes, tRNAs, translation factors, enzymes).
Genetic template (DNA or mRNA) — the instructions for what protein to make; DNA needs transcription first, mRNA can be translated directly.
Transcription components (if using DNA) — RNA polymerase + NTPs to make mRNA from DNA.
Translation substrates — amino acids (building blocks) and energy-carrying nucleotides (mainly ATP/GTP) to run translation.
Energy regeneration mix — keeps ATP/GTP levels up so the reaction lasts longer and yields more protein.
Salts/ions + buffer — sets the chemical environment (pH and ion balance) so enzymes and ribosomes work properly.
Stabilizers/processing helpers (optional) — reducing agents, chaperones, cofactors, membrane mimics, and nuclease/protease control to improve folding, function, and stability of the product.
For E coli
Component (E. coli CFPS)
What it is (typical examples)
Role in the system
E. coli S30 cell extract (lysate)
Crude extract containing ribosomes, tRNAs, translation factors, metabolic enzymes (often made from BL21-derived strains)
Core expression system that performs transcription/translation and supplies many supporting enzymes
DNA template
Plasmid DNA (most common) or linear PCR product; includes promoter + RBS + coding sequence + terminator
Blueprint for the protein; design of promoter/RBS strongly controls expression level
Enables functional expression of membrane proteins or cofactor-dependent enzymes
Why is energy provision regeneration critical in cell-free systems?
Translation is energy-intensive because charging tRNAs and moving the ribosome along mRNA consumes ATP/GTP at multiple steps. Transcription also draws on NTP pools, if using a DNA template, the system must spend NTPs to make mRNA. Without regeneration, the reaction stalls fast: ATP/GTP drop, ADP/AMP and inorganic phosphate build up, and the system loses driving force and efficiency. Therfore energy balance affects yield and fidelity: low energy can reduce protein yield and sometimes increase incomplete/aberrant products.
Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
One method to ensure continuous ATP supply could be to use an ATP regeneration module that continually converts ADP to ATP.
e.g.
phosphoenolpyruvate (PEP) + pyruvate kinase (PK)
Add an energy “fuel” molecule (PEP) and the enzyme pyruvate kinase.
PK transfers phosphate from PEP to ADP:
PEP + ADP → pyruvate + ATP
As the cell-free system consumes ATP (making ADP), PK converts that ADP back into ATP, keeping ATP levels high enough for sustained expression.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic vs eukaryotic cell-free expression (expressing GFP in each)
Feature
Prokaryotic Cell Free Systems
Eukaryotic Cell Free Systems
Source of machinery
Bacterial ribosomes + factors
Eukaryotic ribosomes + factors
Speed / yield
Typically fast and often high-yield for many proteins
Often slower and can be lower-yield, but depends on platform
Setup complexity
Generally simpler/cheaper
Often more expensive; more complex components
Template requirements
Works well with DNA; common to use T7 promoter + bacterial RBS
Often prefers capped mRNA or eukaryotic translation signals (this is platform-dependent)
Protein folding environment
Good for many soluble proteins but limited eukaryotic folding machinery
Better suited to some eukaryotic folding needs; can support more complex proteins
Post-translational modifications
Minimal
Can support some PTM-related processes depending on system (still limited vs living cells)
Typical best use
Rapid prototyping + high-throughput expression of simple proteins
Proteins needing a more eukaryote-like translation/folding context
Protein choice in each system: GFP (and why)
1) Prokaryotic system (E. coli cell-free) to produce GFP
GFP is a soluble, relatively robust protein that folds well in bacterial contexts.
Prokaryotic CFPS is ideal for fast, high-yield expression, so GFP gives a strong, quick fluorescence readout for verifying the system and optimizing conditions.
It’s a standard reporter for tuning DNA concentration, Mg²⁺/K⁺ balance, and energy regeneration because output is easy to measure.
Expected output
Rapid increase in fluorescence as GFP accumulates (often within hours, depending on system).
2) Eukaryotic system (wheat germ or rabbit reticulocyte CFPS) to produce GFP
GFP is still easy to detect, so it’s a simpler way to validate a eukaryotic CFPS setup.
Demonstrates eukaryotic translation control (e.g., dependence on mRNA features like cap/poly(A), or different 5′/3′ UTR effects—platform dependent) without the extra complication of difficult-to-fold proteins.
Using GFP in a eukaryotic system is a good baseline before moving to proteins that truly require eukaryotic translation/folding environments.
Expected output
Fluorescence increase that may have different kinetics and yield than E. coli CFPS, but still provides a clear functional readout.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
In an E. coli cell-free system, optimising expression of LacY (the lactose permease, an MFS transporter) primarily requires preventing aggregation of hydrophobic transmembrane segments and promoting correct insertion into a membrane-like environment during synthesis. When LacY is translated in a purely aqueous reaction, it commonly precipitates or adopts nonfunctional conformations because the nascent helices lack a suitable hydrophobic phase.
My experiment would include a membrane mimic from the start of the reaction to enable co-translational capture and insertion. I could include liposomes because they are often preferred when the experimental goal is functional reconstitution, since they allow LacY to be embedded in a bilayer context, although insertion efficiency and orientation may vary.
After selecting a membrane mimic that reduces aggregation, my next focus would be the quality of folding and insertion. Reaction conditions should be tuned to avoid overwhelming the folding and insertion capacity of the system. Excessive expression drive (for example, very high template concentration or very strong transcription) can cause LacY to accumulate faster than it can insert and fold, increasing aggregation and reducing the proportion of functional protein.
Energy regeneration is also critical because membrane protein synthesis may benefit from extended reaction durations. If ATP and GTP are depleted early, translation stalls before substantial correctly inserted protein accumulates. My effective energy regeneration strategy would help maintain nucleotide triphosphate levels and sustains translation long enough to improve overall production.
Successful optimisation would be assessed using multiple readouts rather than total yield alone. Useful metrics include the total amount of LacY produced, the fraction captured in the membrane mimic versus insoluble material, and most important of all, evidence of functional behaviour consistent with correctly folded LacY in a membrane environment.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Possible reasons:
Not enough regeneration of energy molecules - change my energy regeneration module
Folding of functional protein is not possible optimally- I would try other chaperones and reaction conditions
DNA template is not good- Maybe the promotors are not idea or other regulatory elements. I would test out other parts.
Synthetic Minimal Cells
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output? The synthetic minimal cell would function as a protected screening reactor that converts a biological “unknown” into a quantitative biosensor signal. The input is a plant-derived cyclotide/peptide extract (or fraction) added to the synthetic cell containing a LacI–sfGFP regulatory module and Tx/Tl machinery. The output is a measurable sfGFP signal (end-point fluorescence and/or time-course kinetics) that reports how the peptide perturbs the LacI–sfGFP module (e.g., changes in repression strength, expression rate, or signal stability). This output is used as a proxy for bioactivity and to stratify hits into mechanism-like response classes.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation? Yes, but not optimally. A bulk (non-encapsulated) Tx/Tl reaction can produce the GFP readout, but encapsulation improves practical performance by stabilizing the reaction microenvironment, buffering dilution and inhibitors from crude extracts, and enabling more consistent kinetics and comparability across samples. Encapsulation also supports portability and modular handling (e.g., dried “synthetic cells” that are rehydrated in the field), which is harder to maintain with open bulk reactions.
Could this function be realized by genetically modified natural cell? Not really for the intended screening goal, because cyclotides and related bioactive peptides can be toxic at the concentrations required for discovery and validation, which can collapse cell viability and confound interpretation. A living cell also introduces additional layers (stress responses, transport limits, metabolism, degradation, growth effects) that can mask direct bioactivity and reduce the clarity of the readout, especially when working with crude plant mixtures.
Describe the desired outcome of your synthetic cell operation. The desired outcome is a reliable, repeatable decision signal that identifies which African plant extracts/fractions contain cyclotide-like peptides with meaningful antiparasitic potential and prioritizes them into a short list of “hits.” Practically, success looks like: (i) clear, quantifiable changes in sfGFP output relative to controls, (ii) consistent response patterns across replicates and across preparation batches, and (iii) a set of top candidates that can be carried forward to structural integration (linking activity to known/predicted cyclotide scaffolds) and template definition for downstream peptide design and validation.
Design all components that would need to be part of your synthetic cell.
What would the membrane be made of? A lipid vesicle membrane is the most straightforward choice, because it mimics biology and is compatible with protein pores/channels if needed. A practical design would use phospholipid liposomes.
What would be encapsulated inside (enzymes, small molecules)? Inside the synthetic cell, the minimal set is everything required to run a LacI–sfGFP transcription/translation reaction reliably and report on perturbations caused by cyclotide-containing samples:
Tx/Tl machinery: ribosomes, tRNAs, translation factors (as part of an extract or purified mix)
Transcription components: RNA polymerase appropriate to the promoter used (e.g., T7 promoter)
Genetic program: DNA template encoding sfGFP under LacI control, plus the regulatory DNA elements
Regulator: LacI protein (preloaded) or DNA encoding LacI (so the system can establish repression internally)
Building blocks: amino acids, NTPs
Energy system: ATP/GTP supply plus an energy regeneration module
Buffering/ions: Mg2+, K+ and pH buffer to keep the reaction in its productive window
Stability helpers e.g. chaperones
Which organism would the Tx/Tl system come from? Is bacterial OK, or is mammalian needed? Bacterial is my ideal for this application, and an E. coli-based Tx/Tl system is the best fit for me in this cases. The readout is sfGFP expression controlled by LacI, which is a bacterial regulator and does not require mammalian transcriptional machinery or mammalian-only inducible systems.
How will the synthetic cell communicate with the environment? Communication can be designed in three tiers, depending on what must cross the membrane:
Pores/channels: the membrane can include a general or defined diffusion pore so small molecules and peptides can access the internal reaction without fully compromising compartment integrity.
Compartment-protective gating (best for crude extracts): If crude plant mixtures contain inhibitors that could crash the Tx/Tl chemistry, the membrane strategy can be tuned to admit the relevant molecular size range while excluding larger inhibitory components, effectively functioning as a filter. This makes encapsulation meaningfully better than bulk cell-free reactions.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Genes: lacI, sfGFP, T7 RNAP (T7 gene 1) or sigma-70 promoter with endogenous RNAP, plus one membrane pore gene (mscL, ompF for small-molecule access)
How will you measure the function of your system?
General GFP fluorescence, dose-dependent changes in GFP fluorescence, difference in performance between experiments with pores/channels and those without.
Applications of Freeze-Dried Cell Free Systems
One-sentence pitch A low-cost, single-use “kachasu safety strip” that flags dangerous adulterants sometimes added to boost “kick” (e.g., nitrogen fertilizer residues and related contaminants) using a simple, unmistakable color warning.
How it works (3–4+ sentences) A drop of spirit is applied to a paper strip (or a sealed sachet test) that wicks the sample into multiple reaction spots. Each spot contains a freeze-dried, cell-free colorimetric module tuned to a different high-risk adulterant class relevant to local practice, such as ammonium/urea-type nitrogen signals (fertilizer-associated), abnormal oxidants, and other broad “toxic adulterant” proxies, plus a control spot to confirm the test ran correctly. The outputs appear as a small set of traffic-light indicators (green = no flag, red = dangerous flag) designed for non-technical interpretation. The tool is framed as harm reduction: it does not certify safety, but it quickly identifies batches that are high-risk and should not be sold or consumed.
Societal challenge / market need Adulteration of informal spirits with non-food chemicals can cause poisoning, organ damage, and community outbreaks, and consumers often have no way to tell what has been added. Lab testing is expensive and inaccessible, while enforcement-only approaches often fail to reduce harm on the ground. A rapid, field-stable screening tool supports community health workers, vendors, and buyers with immediate risk information and can reduce preventable injury and death.
Addressing cell-free limitations (activation, stability, one-time use) Activation is triggered by the sample itself: the spirit rehydrates the freeze-dried reactions. Stability is handled through foil barrier packaging, desiccants, and stabilizing excipients so strips can be stored without cold chain in local conditions. One-time use is appropriate for batch screening; the design stays low-cost by using paper microfluidics and simple color outputs, and reliability is improved by including internal controls and conservative thresholds (flagging “danger likely” rather than trying to quantify exact concentrations).
Genes in Space
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Background
Long-duration spaceflight exposes astronauts to microgravity and radiation, which can alter gene expression, RNA processing, and protein synthesis. Neurodevelopmental and synaptic pathways are especially relevant because cognition, sensory processing, and behavioral performance are mission-critical. Fragile X biology (studied the process in plants before) is scientifically interesting because it links RNA structure, control, and neuronal function, offering a model for how stressors might shift RNA–protein regulation. Studying Fragile X–related translation in cell-free systems enables controlled, low-mass experiments that inform astronaut health risk, countermeasure design, and fundamental principles of gene regulation in space-relevant conditions.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Molecular/genetic target FMR1 5′UTR CGG-repeat RNA elements and FMRP-mediated translational regulation, measured using CGG-repeat reporter constructs in a eukaryotic cell-free transcription–translation system.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Microgravity-like transport changes and radiation-like stress can perturb RNA folding, RNA–protein interactions, and translation kinetics. The FMR1 CGG-repeat region is a structured RNA element whose repeat length influences translation and can promote abnormal translation behaviors. FMRP is an RNA-binding translational regulator central to neuronal function. Together, CGG-repeat RNA and FMRP provide a sensitive “stress test” for how space-relevant conditions alter translation control. A cell-free approach isolates these mechanisms from whole-cell confounders, enabling direct measurement of repeat-length–dependent translation shifts under space-like constraints.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Space-relevant stressors (altered mixing/transport and radiation-like oxidative/nucleic-acid stress) amplify CGG-repeat–dependent disruption of translation and RNA stability, and they alter the modulatory effect of FMRP on translation. The reasoning is that structured repeat RNAs can be sensitive to changes in biophysical environment (diffusion, crowding, redox balance) and to damage that affects RNA integrity or ribosome progression. If space-like stress worsens ribosome stalling or shifts translation dynamics, repeat-containing templates should show larger drops in protein output and/or altered kinetics compared to non-repeat controls. Goal: quantify how repeat length and FMRP presence interact with space-relevant stressors to change translation output, providing a mechanistic basis for neurorelevant risk assessment and countermeasure screening.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Test a panel of cell-free templates encoding sfGFP with matched 5′UTRs containing: no repeat (control), short CGG, premutation-range CGG, and long CGG. Run reactions under baseline conditions and under two perturbations: a microgravity-relevant transport condition (reduced convection/mixing) and a radiation-relevant stress proxy (oxidative/nucleic-acid stress). Include ± added purified FMRP (or an FMRP surrogate) to test regulation. Measurements: sfGFP fluorescence time-course (rate, onset, plateau), plus RNA integrity at endpoint (template stability). Controls: no-template and no-stress controls.
Week 10 HW: Imaging and Measurement
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
I intend to measure the presence of cyclotide activity using the GFP signal from a reporter construct. I also intend to measure the extent of membrane lysis caused by the presence of my cyclotides. Controls to make sure the system is working.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
A. eGFP reporter signal (cyclotide activity screen)
What is measured
Fluorescence intensity of eGFP as a function of time (e.g., every 2–5 minutes for 30–120 minutes) and the final endpoint value.
No DNA template (background fluorescence from reagents)
No cyclotide/extract (baseline expression)
IPTG positive control (confirms LacI system is responsive)
Sample-only control (extract + no reporter) to detect autofluorescence or quenching
“Vehicle” control (buffer used for extract prep)
How cyclotide “activity” is interpreted
B. Liposome membrane lysis (direct membrane disruption)
This is to confirm the activity of candidate cyclotide.
What is measured Quantifiable proxy for liposome leakage/lysis using dye or haemoglobin-leakage: fluorescence increase/decrease as dye is released or dequenched
Turbidity/optical density shift: absorbance change as vesicles break/aggregate (less specific than dye leakage)
How it will be measured (spectrophotometer/plate reader)
Fluorescence is read over time after adding cyclotide extract/fraction.
Calculate percent leakage:
0% leakage = buffer control
100% leakage = detergent/lysis control (e.g., a known surfactant that fully disrupts membranes)
What is reported
% leakage at fixed time (e.g., 10 min, 30 min)
Leakage kinetics (rate)
EC50-like comparison between extracts/fractions (if doing dilutions)
Critical controls
Buffer-only (no lysis baseline)
Positive lysis control (complete disruption reference)
Extract-only without dye liposomes (to check optical interference)
C. Hemolysis assay (biological relevance / toxicity proxy)
What is measured
Release of hemoglobin from red blood cells after exposure to cyclotide extracts.
How it will be measured
Spectrophotometric absorbance of supernatant after incubation and pelleting intact cells.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
I would apply DNA construct design and in silico editing to assemble the LacI–sfGFP plasmid in Benchling. I would also use basic bioproduction techniques, including bacterial transformation, plasmid preparation, and Sanger sequencing for verification. For functional testing, I would use cell‑free reactions (TXTL) to measure GFP expression directly from the plasmid. Optionally, I might also use basic data analysis / modeling (e.g., in a notebook) to quantify fold‑change and compare time courses between induced and uninduced reactions.
Waters Part I — Molecular Weight
Calculated MW from the eGFP sequence (with LE + His6 tag)
Using the provided sequence (includes “LEHHHHHH”), the theoretical intact mass is about 28.006 kDa (28,006.6 Da, average mass).
MW using the adjacent charge-state approach (from Figure 1)
Using adjacent peaks in the intact ESI spectrum (e.g., 933.804 and 903.715 m/z are adjacent charge states), the charge is 30+ for the 933.804 peak, giving an intact mass near 28.0 kDa. Using multiple adjacent pairs and averaging gives MW of 28,005.44 Da (28.005 kDa)**.
Accuracy (ppm error vs theory):
Charge state of the zoomed-in peak (~1473 m/z) in the intact spectrum
Yes. The isotope spacing in the zoom is consistent with ~19+
(\Delta m/z \approx 1/z)).
Waters Part 2 — Secondary and Tertiary Structure
Difference between native vs denatured conformations and how MS shows it
-Denatured proteins are unfolded/extended and typically pick up more charges
This shifts the envelope to lower m/z and also broader charge-state distributions.
-Native proteins remain compact and usually show fewer charges, giving a narrower distribution at higher m/z.
In Figure 2, the denatured spectrum shows many higher-charge peaks, while the native spectrum concentrates into fewer, lower-charge states at higher m/z.
Charge state of the native peak at 2800 m/z
Using the native spectrum mass (28 kDa), a peak near 2800 m/z corresponds to about 10+
This is consistent with the native envelope shown.
Waters Part III — Peptide Mapping (Primary Structure)
How many Lys (K) and Arg (R) are in eGFP?
K = 20, R = 6 (total cleavage residues = 26).
How many peptides from tryptic digestion?
Trypsin rules are cuts occur after K/R unless followed by P), this gives a total of 27 tryptic peptides.
How many chromatographic peaks between 0.5 and 6 min (>10% rel. abundance)?
From Figure 5a, there are 21 prominent peaks in the specified range (0.61, 0.79, 1.20, 1.43, 1.80, 1.85, 1.93, 2.17, 2.26, 2.54, 2.78, 3.27, 3.53, 3.59, 3.70, 4.30, 4.48, 4.64, 4.87, 5.06, 5.43).
Does peak count match predicted peptide count?
No. There are 27 predicted peptides for 21 chromatographic peaks . This can happen because some peptides are not abundant or elute together. Ionisation extent may also vary.
Identify m/z, charge z, and compute singly charged mass for the peptide in Figure 5b
Major peak: m/z 525.767
Isotope spacing is 0.5 Th and z = 2+
!(Equation)[Singlycharged.png]
Identify the peptide and calculate ppm error
Mass spectrum for the peptide is in Figure 5b.
% sequence confirmed by peptide mapping
The coverage map reports 88% identified.
Waters Part IV — Oligomers (KLH CDMS)
Using the reported subunit masses (7FU = 340 kDa; 8FU = 400 kDa), the expected oligomer masses are:
7FU decamer: 10 × 340 kDa = 3.40 MDa, consistent with the 3.4 MDa peak.
8FU didecamer (these can be interpreted as 2 decamers; 20-mer): 20 × 400 kDa = 8.0 MDa, consistent with the 8.33 MDa peak.
8FU 3-decamer (30-mer): 30 × 400 kDa = 12.0 MDa, consistent with the 12.67 MDa peak.
8FU 4-decamer (40-mer): 40 × 400 kDa = 16.0 MDa, expected near 16 MDa; this is not a strong labeled peak in the provided spectrum.
Observed intact LC–MS molecular weight (from Part I ): 28.005 kDa
Mass error: −29 ppm (observed slightly lower than theoretical)
Week 11 HW: Bioproduction and Cloudlabs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Make a note on your HTGAA webpages including:
what you contributed to the community bioart project
I change 3 pixels during the lecture, I wish I got a screen shot!
what you liked about the project, and
I like the real-time collaboration on something artistic
what about this collaborative art experiment could be made better for next year.
It could be introduced at the beginning when we do gel art
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
The lysate makes the whole process work: it contains the ribosomes and all the enzymes that do transcription and translation outside the cell. Using BL21 (DE3) Star matters because it’s built for high expression and mRNA stability, and it already brings T7 RNA polymerase so T7 templates get transcribed strongly and consistently.
The salts and buffers are there to recreate the intracellular conditions the machinery expects. Potassium glutamate sets the main ionic environment, HEPES-KOH keeps pH steady around 7.5, magnesium glutamate supplies Mg²⁺ which ribosomes absolutely depend on, and the monobasic/dibasic phosphate pair strengthens buffering and supports longer reactions.
The energy/nucleotide system is what keeps the reaction alive for hours rather than minutes. Ribose and glucose feed the extract’s metabolism so ATP and GTP can keep being regenerated, while AMP/CMP/GMP/UMP and guanine act as nucleotide building blocks that the lysate can recycle back into usable NTPs for transcription and energy use.
The amino acids are simply the raw materials for protein. The 17-amino-acid mix covers most of what translation needs, while tyrosine (kept soluble at high pH) and cysteine (often unstable/limiting) are supplied separately so they don’t become the bottleneck.
Nicotinamide supports cofactor/redox balance (it necessary to keep the extract’s metabolism steady so it can keep regenerating energy and running properly).
Nuclease-free water is important for the environment of the reactions: it sets final concentrations without introducing DNases/RNases that would otherwise finish off the templates.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour PEP–NTP mix is built for fast transcription and translation
The NTPs are ready-to-use NTPs and there is a high-power energy donor (PEP-based). There are other additives such a spermidine and cAMP that boost short-run performance. The 20-hour NMP–ribose–glucose mix is masde for slower steadier transcription and translation. Instead of adding NTPs directly, it feeds the lysate basic precursors (NMPs + ribose + glucose + guanine) so the extract’s enzymes can regenerate nucleotide triphosphates and energy over time, using fewer “boost” additives. A different buffer/ionic balance helps to stay stable for long incubations.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Since there are precursors in the lysate, guanine can be used by such enzymes and components to make GMP, which can undergo phosphorylation to become GDP and GTP as needed.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
The proteins share the same beta-barrel core so the baseline constraints are shared All six sequences have long conserved blocks and similar length through the core. That basically implies “same fold”: the classic fluorescent protein beta-barrel with the chromophore buried inside. Practical implication in cell-free: they all depend on the same broad requirements — decent folding conditions, enough time to fold/mature, and oxygen to finish the chromophore chemistry.
The biggest functional divergence is at the chromophore-forming motif. The early region in the alignment shows where the proteins diverge sharply:
sfGFP / mTurquoise2 are extremely close to each other and keep the classic GFP lineage context.
mKO2, Electra2, mScarlet-I, mRFP1 share a different conserved backbone which is consistent with the DsRed derived families.
sfGFP sfGFP folds efficiently and matures relatively fast, hence the name super folding GFP. Like all GFP-like proteins it still needs oxygen for chromophore maturation, so low oxygen can delay or limit fluorescence.
mRFP1 mRFP1 matures more slowly than many modern reds fluorescent proteins, so the readout can lag behind actual expression in short reactions. It’s also generally dimmer than newer red variants, so it can look as thought the yeild is low even when protein is present.
mKO2 mKO2 is a bright orange protein with fairly quick maturation, which is good for cell-free systems. Its fluorescence can be pH-sensitive can suffer from photobleaching under strong light.
mTurquoise2 mTurquoise2 is a bright cyan FP with strong signal, but cyan proteins can be more sensitive to photobleaching and background. If there isn’t enough oxygen, it may not express to the optimal degree.
mScarlet_I mScarlet-I is engineered to be a true monomer with fast maturation and high brightness, which usually makes it one of the most reliable reds in cell-free. The main constraint is still oxygen dependence for chromophore formation, so sealed/low-oxygen conditions can reduce signal.
Electra2 Electra2 is photoswitchable, so the readout depends strongly on the illumination.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximise fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Protein: mRFP1 Biophysical property to improve: slow maturation (and therefore a delayed/underreported red signal)
Reagent to adjust: increase the experimantal duration by strengthening the ribose-glucose-NMP energy regeneration module, and increase nicotinamide so more cofactor is able to support the duration.
Expected effect: keeping nucleotide pools and ATP/GTP regeneration stable for longer should maintain transcription/translation pressure over many hours, while improved cofactor/redox support should reduce premature stalling and give the mRFP1 chromophore more time and capacity to mature, producing a higher final red fluorescence at 36 hours.