Week 5 HW: Protein Design Part 2
Part 1: Generate Binders with PepMLM
Sequence Retrieval
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Mutated with A4V MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
| Binder | Pseudo Perplexity | |
| Seq1 | HHVPVVVLRHKX | 16.326572 |
| Seq2 | WRYYAAVARWKE | 13.897447 |
| Seq3 | HRYYPAAARWKX | 8.531565 |
| Control | FLYRWLPSRRGG | 20.63523127283615 |
Part 2: Evaluate Binders with AlphaFold3
- Navigate to the AlphaFold Server: alphafoldserver.com
- For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
- Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
- In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
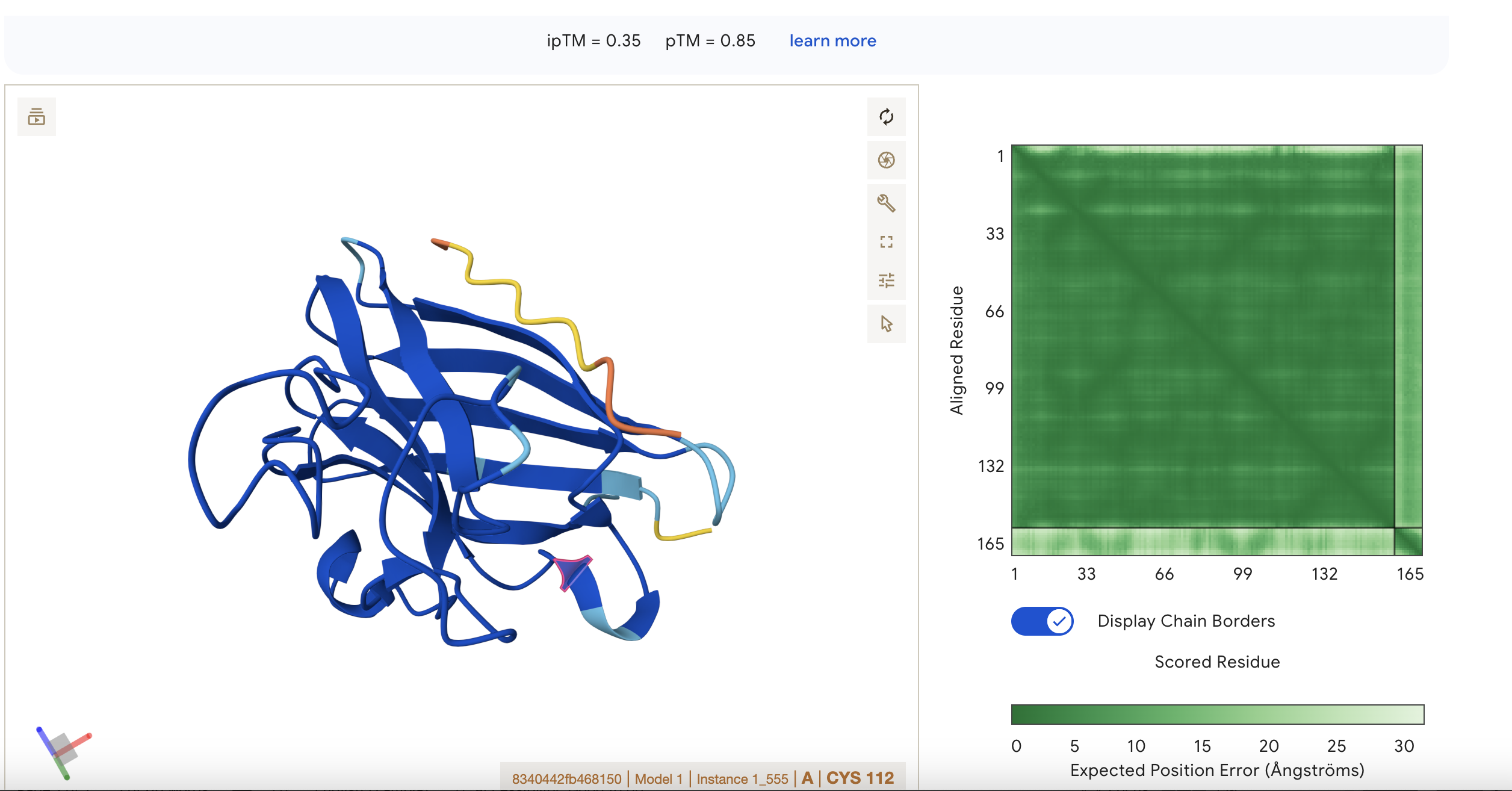
Sequence 1 HHVPVVVLRHKX The sequence was submitted without the last X because it was flagged as an illegal character (it could stand for various amino acids). The peptide appears to be interacting with ipTM = 0.22 pTM = 0.85 pTM = 0.85 suggests the overall fold is very reliable, while ipTM = 0.22 says the predicted interaction/interface between chains is very poor. This means the protein’s global shape looks strong and likely meaningful, this is in line with it being a known and well illucidated human protein. It seems like a case where AlphaFold is confident about folding, but not about binding geometry.
Localisation: Near beta barrel Engagement with β-barrel region: not exact but close Approach to the dimer interface: not exact but close Appear surface-bound: Yes Appear partially buried: No
Sequence 2 WRYYAAVARWKE
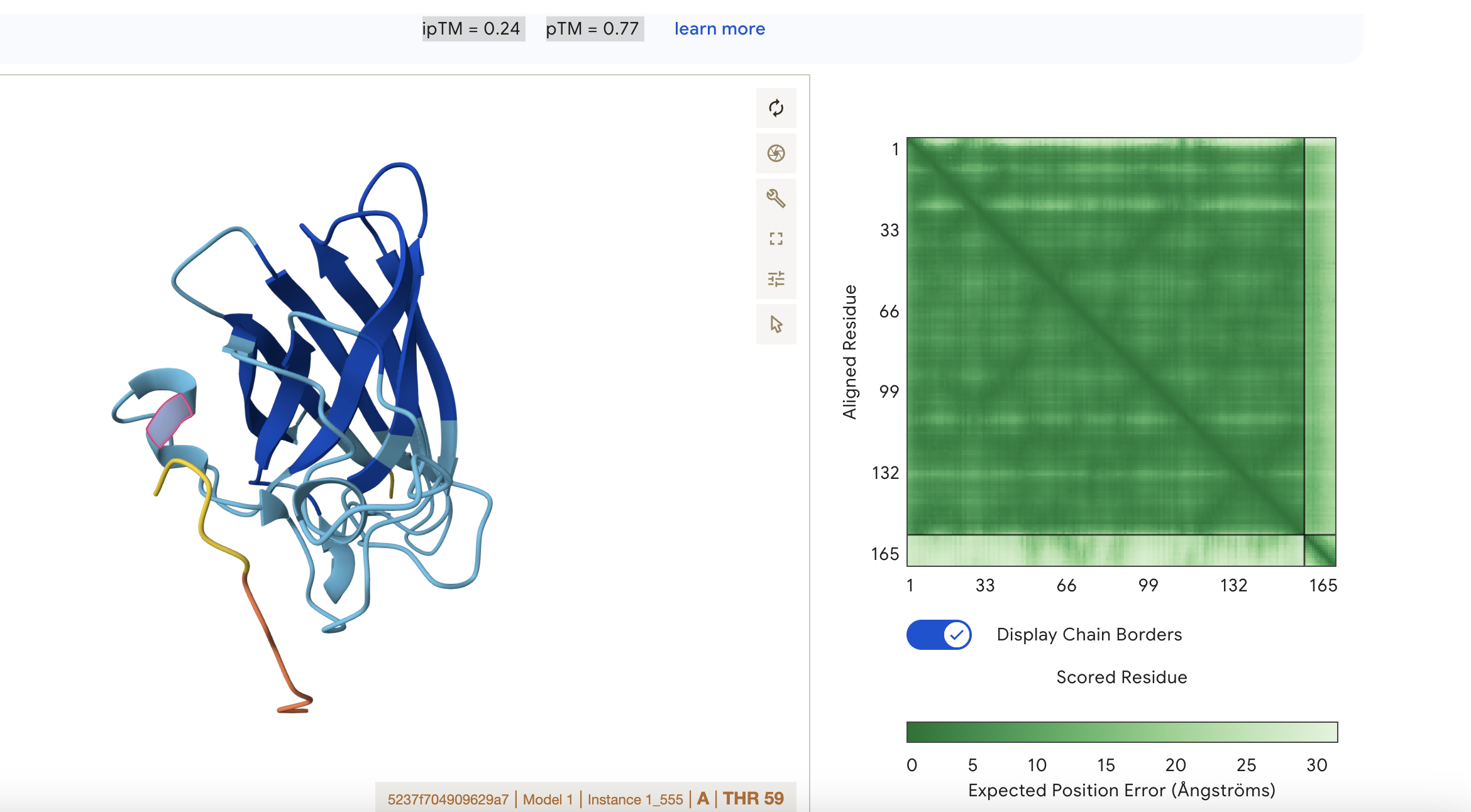
ipTM = 0.24 pTM = 0.77 This means that the model is confident about the fold of the chain itself, but not about how chains interact.
pTM = 0.77: the overall protein fold looks reasonably reliable; this is in a fairly good range for the global shape of the modeled structure
ipTM = 0.24: the predicted interface between subunits is very poor, so AlphaFold is not confident that the chains are positioned correctly relative to each other.
Localisation: Near alpha helix Engagement with β-barrel region: None Approach to the dimer interface: None Appear surface-bound: Yes Appear partially buried: No
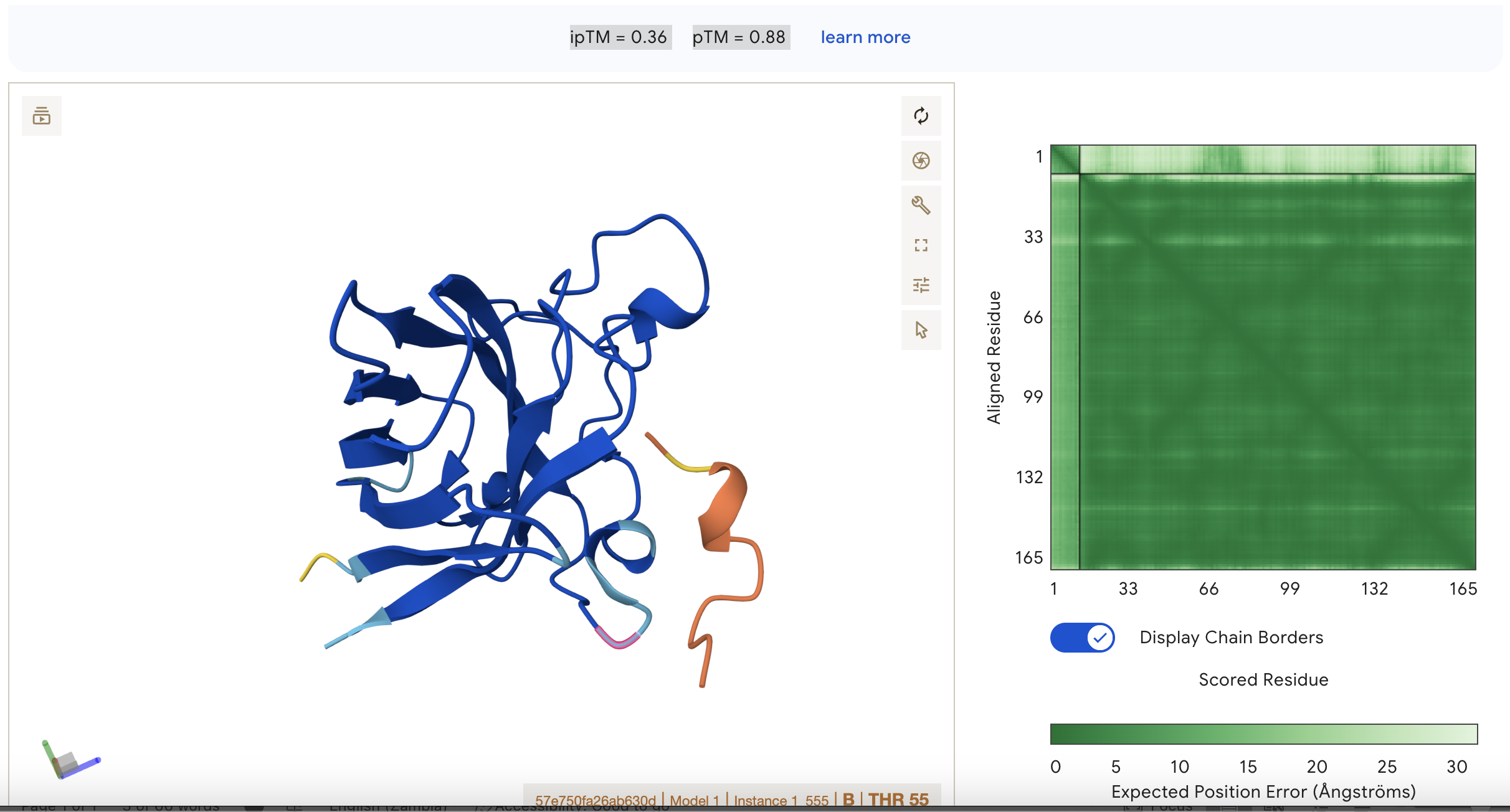
Sequence 3 HRYYPAAARWKX ipTM = 0.36
pTM = 0.88
This suggests that the model is fairly confident about the structure but the binding interaction is still under a threshold of 0.5 This represents the second best binding in the data set however. Localisation: Side of dimer interface Engagement with β-barrel region: partial Approach to the dimer interface: on the side Appear surface-bound:yes Appear partially buried: no
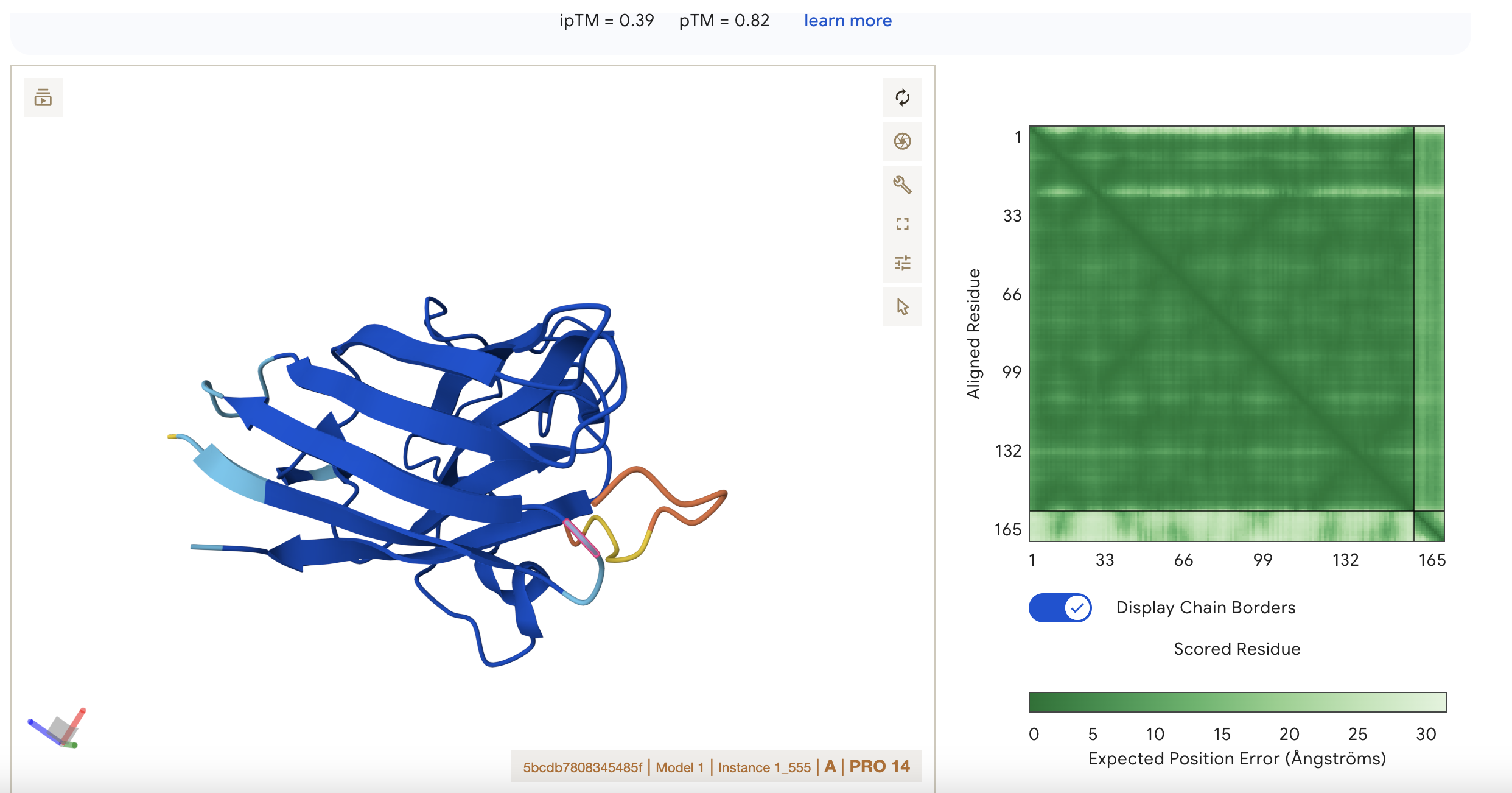
Sequence 4 FLYRWLPSRRGG ipTM = 0.39 pTM = 0.8
- pTM 0.8 suggests the model is fairly confident about the global arrangement of the structure as a whole.
- ipTM 0.39 is low, so AlphaFold is not confident about the relative positioning of the interacting subunits. It is the highest score however so this represents the best binding possibility from this set of results. There is low confidence of interaction between the short peptide and the protein.
Localisation: Side of dimer interface Engagement with β-barrel region: partial Approach to the dimer interface: on the side Appear surface-bound: yes Appear partially buried: no
Peptiverse Prediction
📊 Results
| Input | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| HHVPVVVLRHKX | 💦 Hydrophobicity (GRAVY) | Non-hemolytic | 1302.7 | Probability |
| Input | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| HHVPVVVLRHKX | 💧 Solubility | Soluble | 0.998 | Probability |
| HHVPVVVLRHKX | 🩸 Hemolysis | Non-hemolytic | 0.022 | Probability |
| HHVPVVVLRHKX | 🔗 Binding Affinity | Weak binding | 5.789 | pKd/pKi |
| HHVPVVVLRHKX | 📏 Length | 12 | aa | |
| HHVPVVVLRHKX | ⚖️ Molecular Weight | 1302.7 | Da | |
| HHVPVVVLRHKX | ⚡ Net Charge (pH 7) | 2.02 | ||
| HHVPVVVLRHKX | 🎯 Isoelectric Point | 11.00 | pH | |
| HHVPVVVLRHKX | 💦 Hydrophobicity (GRAVY) | 0.08 | GRAVY |
📊 Results
| Input | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| WRYYAAVARWKE | 💦 Hydrophobicity (GRAVY) | Non-hemolytic | 1598.8 | Probability |
| Input | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| WRYYAAVARWKE | 💧 Solubility | Soluble | 0.997 | Probability |
| WRYYAAVARWKE | 🩸 Hemolysis | Non-hemolytic | 0.031 | Probability |
| WRYYAAVARWKE | 🔗 Binding Affinity | Weak binding | 6.678 | pKd/pKi |
| WRYYAAVARWKE | 📏 Length | 12 | aa | |
| WRYYAAVARWKE | ⚖️ Molecular Weight | 1598.8 | Da | |
| WRYYAAVARWKE | ⚡ Net Charge (pH 7) | 1.77 | ||
| WRYYAAVARWKE | 🎯 Isoelectric Point | 9.70 | pH | |
| WRYYAAVARWKE | 💦 Hydrophobicity (GRAVY) | -0.93 | GRAVY |
📊 Results
| Input | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| HRYYPAAARWKX | 💦 Hydrophobicity (GRAVY) | Non-hemolytic | 1400.8 | Probability |
| Input | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| HRYYPAAARWKX | 💧 Solubility | Soluble | 1.000 | Probability |
| HRYYPAAARWKX | 🩸 Hemolysis | Non-hemolytic | 0.012 | Probability |
| HRYYPAAARWKX | 🔗 Binding Affinity | Weak binding | 6.426 | pKd/pKi |
| HRYYPAAARWKX | 📏 Length | 12 | aa | |
| HRYYPAAARWKX | ⚖️ Molecular Weight | 1400.8 | Da | |
| HRYYPAAARWKX | ⚡ Net Charge (pH 7) | 2.84 | ||
| HRYYPAAARWKX | 🎯 Isoelectric Point | 10.28 | pH | |
| HRYYPAAARWKX | 💦 Hydrophobicity (GRAVY) | -1.32 | GRAVY |
📊 Results
| Input | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| FLYRWLPSRRGG | 💦 Hydrophobicity (GRAVY) | Non-hemolytic | 1507.7 | Probability |
| Input | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| FLYRWLPSRRGG | 💧 Solubility | Soluble | 0.608 | Probability |
| FLYRWLPSRRGG | 🩸 Hemolysis | Non-hemolytic | 0.047 | Probability |
| FLYRWLPSRRGG | 🔗 Binding Affinity | Weak binding | 6.361 | pKd/pKi |
| FLYRWLPSRRGG | 📏 Length | 12 | aa | |
| FLYRWLPSRRGG | ⚖️ Molecular Weight | 1507.7 | Da | |
| FLYRWLPSRRGG | ⚡ Net Charge (pH 7) | 2.76 | ||
| FLYRWLPSRRGG | 🎯 Isoelectric Point | 11.71 | pH | |
| FLYRWLPSRRGG | 💦 Hydrophobicity (GRAVY) | -0.71 | GRAVY |
Question: Do peptides with higher ipTM also show stronger predicted affinity? The two peptides with the highest iPTM have higher binding affinity Question: Are any strong binders predicted to be hemolytic or poorly soluble? The control peptide FLYRWLPSRRGG is predicted to be poorly soluble Question: Which peptide best balances predicted binding and therapeutic properties? HRYYPAAARWKX It has good binding affinity, good range isoelectric point, desirable net charge, smaller in molecular weight, and has the best solubility. Choose one peptide you would advance and justify your decision briefly
I would advance with HRYYPAAARWKX because in the fist assay, it did not do so well but on closer inspection using the other tools it seems to perform better. When I looked at it using Alphafold, it interacted in the region most likely to disrupt dimer formation unlike the first peptide. Then in the therapeutic analysis, it also had better metrics than the other peptides on the most critical variables such as solubility. I would like the therapeutic to be soluble as possible to reduce toxicity and retention in the body’s long term fat deposits and for it to be easily excreted.