Week 1 HW: Principles and Practices

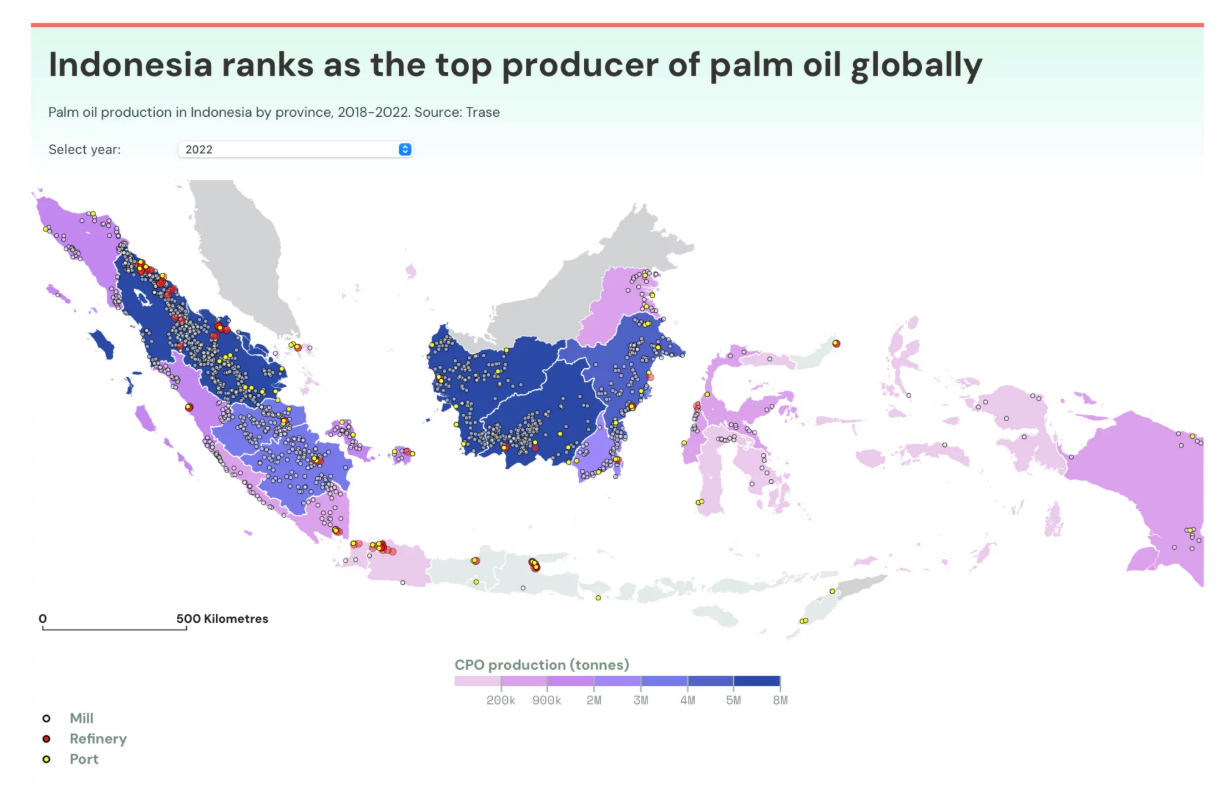
- Biological Engineering Application to Develop The application I want to develop is an innovation in yeast that produces linoleic acid and oleic acid. This innovation would enable the bioproduction of alternative oils besides palm oil. Palm oil plantations in Indonesia cover approximately 16 to 17.3 million hectares in 2024–2025, making Indonesia the largest producer of palm oil in the world (Badan Pusat Statistik). This large land footprint poses environmental and social challenges, and therefore developing microbial production of essential fatty acids could offer a sustainable alternative.

Governance/Policy Goals to Ensure an “Ethical” Future
Main Policy Goal: Ensure that this engineered yeast innovation contributes to an ethical future by preventing harm, safeguarding the environment, and fostering equitable social outcomes.
Sub-Goals:
Environmental Protection and Ecosystem Safety • Prevent unintended release of engineered yeast into natural ecosystems. • Establish environmental risk assessments before commercial deployment.
Health and Biosafety Safeguards • Standardize safety evaluations for laboratory handling and consumer exposure. • Protect laboratory workers and end-users from possible hazards.
Social Equity and Economic Justice • Avoid technologies that negatively impact smallholder farmers dependent on palm oil. • Create inclusive pathways for technology transfer and benefits sharing.
- Three Potential Governance “Actions”
For each proposed governance action below, I address Purpose, Design, Assumptions, and Risks of Failure & Success.
Option 1 — Environmental and Biosecurity Regulation
Purpose:
Currently, standards for environmental biosafety of engineered microbes vary widely across jurisdictions. I propose a regulatory system requiring formal environmental risk assessment before commercialization.
Design:
• Actors involved: national regulators (e.g., ministries of agriculture, food safety authorities), academic scientists, and industry stakeholders.
• Requirements: engineered yeast strains must undergo environmental impact assessment before approval for scaled fermentation.
Assumptions:
• There is adequate technical capacity within regulatory agencies to evaluate ecological risks.
• Stakeholders comply with registration and audit processes.
• Potential uncertainty exists in predicting long-term ecological interactions.
Risks of Failure & Success:
• Failure: Weak assessments may allow accidental releases that disrupt ecosystems.
• Success: A robust regulatory framework could become a model for safe biotech deployment, but may increase cost and bureaucracy.
Option 2 — Research Incentives and Inclusive Collaboration
Purpose:
To reduce socioeconomic resistance to innovation and ensure inclusion of diverse stakeholders, especially smallholder farmers traditionally linked to palm oil production.
Design:
• Actors: national research funding bodies, universities, small and medium enterprises (SMEs), and farmer cooperatives.
• Mechanism: financial incentives for collaborative research and shared benefit programs that involve farmers in providing feedstock or production services.
Assumptions:
• Farmers and local producers are willing to participate if clear economic benefits are provided.
• There may be uncertainty in designing fair incentive schemes and ensuring broad participation.
Risks of Failure & Success:
• Failure: Incentives may be insufficient to motivate meaningful participation.
• Success: Shared benefits strengthen local economies and ensure broader acceptance of the technology.
Option 3 — Product Safety Standards and Consumer Labeling
Purpose:
At present, there are no specific safety standards or labels for microbial-derived oils. I propose establishing product testing and labeling requirements to ensure transparency and consumer confidence.
Design:
• Actors: national standardization bodies, certification agencies, and manufacturers.
• Requirements: safety testing for human consumption and clear labeling indicating microbial origin and composition.
Assumptions:
• Consumers value transparency and will choose certified products.
• Market demand for alternative oils may grow, though adoption rates are uncertain.
Risks of Failure & Success:
• Failure: Regulation could become overly burdensome and slow market entry.
• Success: Improved public trust and market differentiation for safe, high-quality products.
| Does the option: | Option 1 | Option 2 | Option 3 |
|---|---|---|---|
| Enhance Biosecurity | |||
| • By preventing incidents | 1 | 2 | 2 |
| • By helping respond | 1 | 2 | 2 |
| Foster Lab Safety | |||
| • By preventing incident | 1 | 2 | 2 |
| • By helping respond | 1 | 2 | 2 |
| Protect the environment | |||
| • By preventing incidents | 1 | 2 | 2 |
| • By helping respond | 1 | 2 | 2 |
| Other considerations | |||
| • Minimizing costs and burdens to stakeholders | 3 | 1 | 2 |
| • Feasibility? | 1 | 2 | 2 |
| • Not impede research | 2 | 1 | 2 |
| • Promote constructive applications | 2 | 1 | 2 |
- Prioritized Governance Strategy and Recommendation
Based on the scoring and qualitative assessment, I would prioritize a combined governance approach centered on Option 1 (Environmental & Biosecurity Regulation) and Option 2 (Research Incentives and Inclusive Collaboration), while treating Option 3 (Product Safety and Labeling) as a secondary, downstream mechanism.
Primary Priority: Option 1 + Option 2 (Combined Approach)
This combination balances risk prevention, ethical responsibility, and innovation continuity, which is essential for deploying engineered yeast systems at scale.
Rationale for Prioritization
Why Option 1 is Foundational
Option 1 received the strongest scores for:
Preventing biosecurity incidents
Protecting ecosystems
Enhancing laboratory and environmental safety
For a living, self-replicating system such as engineered yeast, preventing harm must precede commercialization. Environmental containment and biosafety regulation are therefore non-negotiable. Without this foundation, even socially beneficial technologies risk public backlash, regulatory shutdown, or irreversible ecological harm.
However, Option 1 alone carries trade-offs:
Higher regulatory costs
Increased administrative burden
Potential slowing of early-stage research
This is why it should not stand alone.
Why Option 2 is Essential for Ethical Impact
Option 2 scored best for:
Minimizing economic burdens
Not impeding research
Promoting constructive and inclusive applications
‘Week 2 Lecture Prep’
A. Homework Questions from Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
DNA replication in living systems is carried out by DNA polymerase, an enzyme that selects complementary nucleotides according to Watson–Crick base pairing rules. The intrinsic error rate of a high-fidelity replicative polymerase in human cells (such as DNA polymerase δ or ε) after proofreading is approximately 10⁻⁷ errors per base incorporated, and when combined with post-replicative mismatch repair, the final mutation rate drops to roughly 10⁻⁹ to 10⁻¹⁰ errors per base per cell division.
To appreciate the scale of this fidelity, we can compare it mathematically to the size of the human genome. The haploid human genome contains approximately 3 × 10⁹ base pairs. If the effective error rate after proofreading and repair is ~10⁻¹⁰ per base per replication, then the expected number of replication errors per cell division is:
(3×10^9 bases) × (10^−10 errors/base) = 0.3 errors per genomeThis means that, on average, fewer than one mutation occurs per genome per cell division. Without proofreading (10⁻⁷ error rate), the expected number would be:
(3×10^9) × (10^−7) = 300 errors per replicationThus, proofreading and mismatch repair reduce errors roughly 1,000-fold, preventing hundreds of potentially deleterious mutations each cell cycle. Biology resolves the apparent discrepancy between the immense genome size and nonzero polymerase error rate through layered error control systems: nucleotide selectivity (base pairing energetics), 3′→5′ exonuclease proofreading, mismatch repair, and cell-cycle checkpoints. Together, these mechanisms push the effective fidelity to a level compatible with organismal survival over billions of cell divisions.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The second question concerns coding capacity. The genetic code uses four nucleotides (A, T, G, C) arranged in triplets (codons). The total number of possible codons is: 4^3 =64
Of these, 61 encode amino acids and 3 are stop codons. Because 20 amino acids are encoded by 61 sense codons, the genetic code is degenerate (redundant). Most amino acids are encoded by more than one codon.
Consider an average human protein of approximately 400 amino acids. If we assume (for simplicity) that each amino acid can be encoded on average by 3 synonymous codons (since 61 codons / 20 amino acids ≈ 3), then the number of possible DNA sequences that could encode the same protein is approximately:
3^400
Using logarithms to estimate magnitude:
log10(3^400)=400log10(3)≈400×0.477=190.8
So:
3^400 ≈ 10^191
This is an astronomically large number of possible DNA sequences that could encode a single 400-amino-acid protein. Even if we refine the estimate using the actual codon degeneracy distribution, the number remains on the order of 10¹⁸⁰–10¹⁹⁰ possibilities.
However, in practice, not all these sequences function equally well. Several biological constraints drastically reduce the set of viable coding sequences:
Codon bias and tRNA abundance Organisms prefer certain synonymous codons because corresponding tRNAs are more abundant. Using rare codons can slow translation, cause ribosome stalling, or reduce protein yield.
mRNA secondary structure Synonymous substitutions alter GC content and folding. Strong hairpins near the 5′ end can inhibit ribosome binding and translation initiation.
Translational kinetics and co-translational folding The speed at which ribosomes move influences how nascent polypeptides fold. Codon choice affects elongation rate and therefore final protein structure.
Regulatory motifs within coding regions Coding DNA may contain splice enhancers, microRNA binding sites, RNA-binding protein motifs, or nucleosome positioning signals. Changing synonymous codons can disrupt these.
GC content and genomic stability Extremely high or low GC content affects DNA stability, methylation, and chromatin organization.
Error robustness Some codon choices minimize the impact of point mutations (conservative amino acid substitutions), improving evolutionary robustness.
Thus, although mathematics suggests ~10¹⁹⁰ theoretical sequences for a typical human protein, functional biology constrains that space to a much smaller subset compatible with transcriptional efficiency, translational fidelity, folding kinetics, regulatory integration, and evolutionary stability.
B. Homework Questions from Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Why can’t you make a 2000bp gene via direct oligo synthesis?
The most commonly used method for oligonucleotide synthesis today is solid-phase phosphoramidite chemistry, first developed in its modern form in the late 1970s and early 1980s. In this approach, nucleotides are added stepwise to a growing DNA chain that is anchored to a solid support, typically controlled pore glass (CPG). Each synthesis cycle consists of four chemical steps: detritylation (removal of the 5′ protecting group), coupling of an activated nucleoside phosphoramidite, capping of unreacted chains, and oxidation to stabilize the phosphodiester linkage. The critical feature of this method is its high coupling efficiency—typically around 99–99.5% per step—which allows automated synthesizers to produce oligos reliably up to about 150–200 nucleotides in length. Despite the emergence of enzymatic DNA synthesis platforms, phosphoramidite solid-phase chemistry remains the industrial standard because of its speed, scalability, and compatibility with chemical modifications. The difficulty in synthesizing oligonucleotides longer than approximately 200 nucleotides arises from compounding yield loss at each synthetic cycle. Even very small inefficiencies accumulate exponentially. If we assume a coupling efficiency of 99% (0.99 per cycle), the overall full-length yield for an oligo of length n is approximately:
Yield=(0.99)^n
For a 200-nt oligo:
(0.99)200≈0.13
This means only about 13% of synthesized molecules are full-length, while the rest are truncated products. If coupling efficiency is slightly better, say 99.5%:
(0.995)200≈0.37
Even then, only ~37% of molecules are complete. Beyond yield loss, side reactions accumulate, including depurination (especially of adenine), incomplete detritylation, and chemical damage during repeated acid exposure. These chemical stresses increase with each cycle, leading to structural defects and decreasing purity. Purifying full-length products from a complex mixture of near-length fragments becomes increasingly inefficient as length increases.
For a 2000 base pair gene, direct chemical synthesis becomes essentially impractical. Using the same yield calculation with 99% efficiency:
(0.99)^2000≈1.8×10^−9
This corresponds to less than one full-length molecule per billion synthesized chains. Even at 99.5% efficiency:
(0.995)^2000≈4.5×10^−5
Only 0.0045% of molecules would be full-length. In addition to catastrophic yield loss, cumulative chemical damage and sequence errors would make the product highly heterogeneous. The probability of incorporating at least one incorrect base also increases with length, further reducing functional product yield.
Therefore, long genes (e.g., 2000 bp) are not synthesized directly as single oligos. Instead, they are constructed by assembling multiple shorter oligos (typically 60–200 nt) using enzymatic methods such as PCR assembly, Gibson assembly, or ligase cycling reaction. This hybrid approach leverages the high precision of short chemical synthesis while using enzymatic replication machinery to produce long, accurate DNA molecules. In summary, solid-phase phosphoramidite chemistry remains the dominant method for oligo synthesis, but exponential yield decay and cumulative chemical damage impose a practical upper limit of ~200 nucleotides. For kilobase-scale genes, biology’s enzymatic polymerases are far more efficient and accurate than purely chemical stepwise synthesis, which is why modern gene synthesis relies on assembly rather than direct extension.
C. Homework Question from George Church
- [Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
In animals, the “essential amino acids” are those that cannot be synthesized de novo in sufficient quantities and therefore must be obtained from the diet. Across essentially all animals, the ten essential amino acids are:
Histidine (H), Isoleucine (I), Leucine (L), Lysine (K), Methionine (M), Phenylalanine (F), Threonine (T), Tryptophan (W), Valine (V), and Arginine (R) (Arginine is considered essential in many animals, especially during growth and development.)
These ten are indispensable because animals lack complete biosynthetic pathways for producing their carbon skeletons from central metabolic intermediates. In contrast, plants and many microorganisms retain full anabolic pathways for synthesizing these amino acids from precursors such as oxaloacetate, pyruvate, phosphoenolpyruvate, and ribose-5-phosphate. Thus, “essential” reflects a metabolic limitation, not a chemical impossibility.
The “Lysine Contingency,” as discussed in synthetic biology (notably in George Church’s work), refers to engineering organisms so that their survival depends on an externally supplied amino acid—classically lysine—often by reassigning lysine codons or making essential genes lysine-dependent. This creates a biological containment strategy: the organism cannot survive without an environmental lysine supply.
Knowing that lysine is one of the ten universally essential amino acids reframes this strategy in two important ways.
First, from a metabolic standpoint, animals (including humans) already depend entirely on dietary lysine. That means lysine availability is environmentally controllable at the ecosystem level. If an engineered organism were made lysine-dependent beyond natural capacity (for example, by deleting lysine biosynthesis genes or recoding lysine codons), its survival would be tightly constrained outside controlled conditions. The contingency exploits a natural metabolic vulnerability.
Second, from a systems perspective, lysine is deeply integrated into protein structure and function. It is:
Positively charged at physiological pH
Critical in DNA-binding proteins
Frequently used in active sites
A major target of post-translational modifications (acetylation, ubiquitination)
Because lysine is common and functionally versatile, replacing or reassigning it at the codon level imposes strong selective pressure. If lysine codons are recoded (for example, reassigned to a non-standard amino acid), the organism becomes dependent not just on lysine availability, but on a synthetic amino acid supply chain. This significantly increases biocontainment robustness.
However, the universality of lysine as essential in animals also highlights a limitation: since lysine is abundant in many natural environments (soil microbes, decaying biomass, gut ecosystems), absolute environmental exclusion is unrealistic. Thus, the contingency works best when combined with genetic code reassignment, not merely metabolic auxotrophy.
- [Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions? An effective AA:AA code would be based on interaction classes, which can be expressed as combinations of physicochemical properties:
Electrostatic Complementarity Positive ↔ Negative (Lys/Arg ↔ Asp/Glu)
Hydrophobic Packing Nonpolar ↔ Nonpolar (Leu/Ile/Val/Phe clustering via entropy-driven burial)
Hydrogen Bonding Pairs Donor ↔ Acceptor (Ser/Thr/Tyr/Asn/Gln/His)
Aromatic Stacking / Cation–π Phe/Tyr/Trp ↔ aromatic or Lys/Arg
Covalent Crosslinking Cys ↔ Cys (disulfide bonds)
Thus, instead of a 4-letter base alphabet, the amino acid “alphabet” effectively reduces to interaction categories. If we classify amino acids into simplified groups:
Positive (+): K, R, H
Negative (−): D, E
Polar (P): S, T, N, Q, Y
Hydrophobic (H): A, V, L, I, M, F, W
Special (S): C, G, P
Then AA:AA interactions can be approximated as a matrix of favorable energy pairings, not a discrete triplet code.
[(Advanced students)] Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or devise one of your own: https://arpa-h.gov/explore-funding/programs/boss https://www.darpa.mil/research/programs/smart-rbc https://www.darpa.mil/research/programs/go
I propose a response to the BioStabilization Systems (BoSS) program centered on engineering a programmable anhydrobiotic preservation platform for biologics and living cell therapies. The core hypothesis is that long-term room-temperature stability can be achieved by combining three natural stress-tolerance strategies—intracellular vitrification, protein phase stabilization, and membrane glass transition control—into a unified, manufacturable preservation system compatible with existing bioprocess pipelines. Nature already provides proof-of-concept. Tardigrades, brine shrimp cysts, and certain plant seeds survive near-complete desiccation by replacing intracellular water with protective disaccharides (e.g., trehalose), intrinsically disordered proteins (IDPs) such as LEA proteins, and by forming glassy cytoplasmic matrices that immobilize biomolecules. The preservation mechanism is fundamentally thermodynamic: degradation reactions scale with molecular mobility, which itself depends on temperature relative to the glass transition temperature (Tg). Reaction rate can be approximated by Arrhenius behavior: k=Ae^−Ea/RT Lowering temperature reduces reaction rate, which is why cold chains work. However, increasing Tg above storage temperature similarly suppresses mobility and therefore degradation. If a preserved biologic is embedded in a vitrified matrix with Tg ≈ 60–80°C, then at 25°C the system operates far below Tg, placing it in a glassy regime with dramatically reduced diffusion-driven damage. The objective is therefore not to “cool” the system, but to chemically immobilize it. The proposed platform, termed VitraCell, has three integrated components. First, transient intracellular loading of trehalose using reversible membrane poration or transporter engineering to replace bulk water before drying. Second, co-expression or exogenous addition of synthetic LEA-mimetic peptides that form reversible protective networks around proteins and membranes. Third, encapsulation within a thin-film biodegradable polymer matrix engineered to raise Tg and buffer humidity fluctuations. Unlike traditional lyophilization, this approach aims to produce a controlled intracellular glass state without ice crystal formation or osmotic collapse. A simple quantitative framing illustrates feasibility. Suppose a biologic protein degrades at 5% per month at 4°C due to residual hydrolysis. If molecular mobility is reduced 100-fold by vitrification (a conservative estimate from glassy-state kinetics literature), then the effective degradation rate constant k becomes k/100. Shelf life, inversely proportional to k, extends 100-fold. A 1-month stability profile becomes ~8 years under comparable chemical constraints. While real systems are more complex, this order-of-magnitude reasoning demonstrates why mobility suppression can rival refrigeration. For cell therapies, viability rather than mere molecular stability is the key challenge. The project will focus on reversible metabolic arrest through controlled desiccation combined with membrane phase stabilization. Membrane leakage probability increases sharply when lipid bilayers cross their phase transition temperature (Tm). By incorporating cholesterol-mimetic stabilizers and trehalose-mediated headgroup spacing, we shift effective Tm and preserve bilayer integrity during drying and rehydration. Viability targets for Phase I would be ≥50% recovery after 30 days at 25°C, benchmarked against cryopreserved controls. Manufacturing compatibility is central. All excipients must be GRAS or rapidly translatable; drying processes must integrate with GMP workflows; reconstitution must occur in under 10 minutes with sterile diluent. Phase II would involve stress-testing under temperature cycling (4–45°C), humidity variation, and mechanical shock to simulate real-world distribution failures currently catastrophic for cold-chain products.
The broader impact is systemic. If even 50% of biologics could be stabilized without −80°C storage, logistics costs—currently estimated in the tens of billions annually—would fall dramatically. Stockpiling for pandemic response becomes feasible without cryogenic infrastructure. Rural and low-resource settings gain access to therapies currently restricted to tertiary centers. In essence, this proposal reframes the cold chain problem as a materials thermodynamics problem rather than a refrigeration problem. By engineering a reversible intracellular glass state inspired by extremophiles and implemented with scalable biomaterials, BoSS can replace temperature control with molecular immobilization—achieving room-temperature biologics not by fighting entropy with freezers, but by restructuring the energetic landscape of biological matter itself.