Engineered Biosensors For the Detection of Illegal Mining Pollutants Week One’s Principles and Practice class taught us the foundations of ethics, safety, and governance using biotechnology. While pondering ideas for the bioengineered tool or application, I was inspired by the battle against the ongoing menace of small-scale illegal mining in Ghana propularly known as “Galamsey”.
Homework Part 0: Basics of Gel Electrophoresis I have watched all the lecture slides and reciatation videos.
Part 1: Benchling & In-silico Gel Art I created a benchling account and imported the Lambda DNA
Python Script for Opentrons Artwork This has been the most interesting and somewhat challenging assignment so far. I chose make an artistic design based on the adrinkra symbols. The adinkra symbols are a set of visual symbols from Ghana, created by the Akan people to represent philosophical concepts, historical events, and social proverbs.
Part A. Conceptual Questions Question 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Answer
A dalton is a unit of mass used to express the mass of atoms, molecules, and other subatomic particles.
Part A: SOD1 Binder Peptide Design A peptide binder is a short, engineered protein fragment usually <50 amino acids that binds to specific targets. It functions as a powerful, cost-effective, and stable alternative to larger antibiotics or small-molecule drugs. A peptide binder is used to modulate, degrade, or inhibit disease-related proteins, especially those that are deemed undruggable due to the absence of clear binding pockets.
Assignment: DNA Assembly Question 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Answer
A PCR master mix is a pre-formulated, ready-to-use solution containing all the components required for PCR, except the DNA template and primers. It usually includes Taq DNA polymerase, deoxynucleotide triphosphates (dNTPs), magnesium ions, and an optimized reaction buffer at precise concentrations to ensure efficient and reproducible DNA amplification.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) Question 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Answer
An IANN is a nonlinear computer model designed to mimic the structure and function of biological neurons in the brain. It usually has multiple inputs and outputs, where neurons process weighted inputs to generate output signals.
General Homework Questions Question 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Answer
Cell-free protein synthesis is a biotechnology technique for producing proteins in a test tube using biological machinery extracted from a cell.
Homework: Final Project For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. Answer
One of the aspects I will measure is the expression levels and enzymatic activity of the four colorimetric reporters in response to known concentrations of their respective target metals in a cell-free system. The colorimetric reporters are LacZ (Blue, for lead detection), Crtl (Orange, for arsenic detection), BpsA(Purple, for mercury detection) ad MelA(Brown, for cadmium detection)
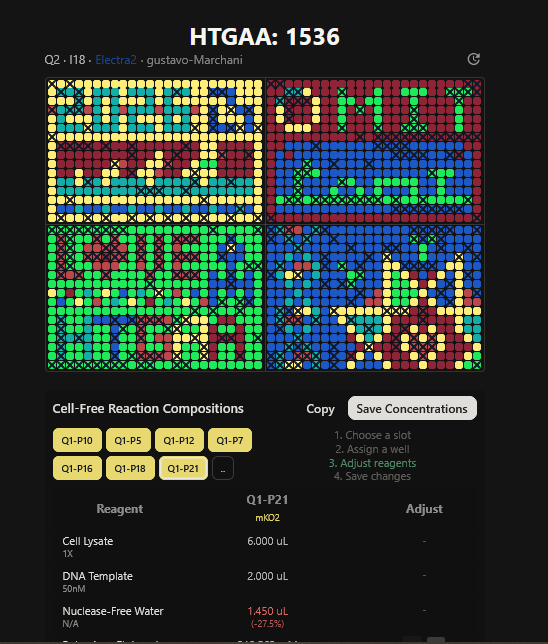
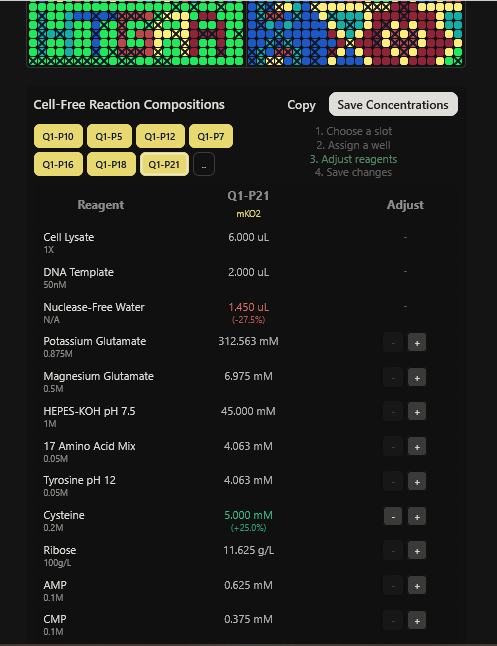
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork I contributed to the HTGAA global artwork canvas, which drew inspiriration from the reddit r/Place experiment. I helped in filling the yellow space pixels in the blue box at the lower left corner of the artwork, and also helped in some of the other designs. In all, I contributed 10 pixels to the global artwork canvas and was ranked 66th among the top contributors. I enjoyed working with everyone to create beautiful designs from the chaos of pixels 😹.
The Lecture this week focused on genome engineering with Jef Boeke, George Church, and John Glass delivering wonderful lectures. Additionally, the recitation was centered on CRISPR, with TA Ice leading the discussion.
I also worked on my final project.
This week’s lecture was led by Dr.Renee Wegrzyn. She shed light on her time at ARPA-H, Transfyr.ai, her new biotechnology AI start-up, and the background work that goes into scientific breakthroughs and failures. It was very insightful; she drew parallels on how most innovations are adopted in other industries before scientists have the chance to use them.
TA Ice leads this week’s recitation, focused on Golden Gage Assembly. Additionally, TA Suvin took us through the requirements for final projects.
This week’s lecture featured a coast-to-coast interaction between SynBioBeta in San Jose and MIT in Boston, with Christina Agapakis and Michel Chen leading the session. It focused on protein design and synthesis as well as rapid prototyping. The interaction also featured pixel art collaboration between HTGAA 2026 and SynBioBeta.
There was no recitation this week. I worked on my final project and had mock presentation sessions with my node.
Subsections of Homework
Week 1 HW: Principles and Practices
Engineered Biosensors For the Detection of Illegal Mining Pollutants
Week One’s Principles and Practice class taught us the foundations of ethics, safety, and governance using biotechnology. While pondering ideas for the bioengineered tool or application, I was inspired by the battle against the ongoing menace of small-scale illegal mining in Ghana propularly known as “Galamsey”.
Illegal mining is the extraction of minerals, metals, or other resources without proper authorization, permits, or compliance with national laws or regulations. It leads to the destruction of forests, leading to the loss of biodiversity, land degradation, and water pollution of rivers and groundwater with pollutants such as mercury, cyanide, and arsenic. Water pollution from galamsey activities is causing chronic diseases as pollutants seep into the water supply undetected.
I wish to explore the development of a microbial testing kit that uses genetically engineered non-pathogenic microbes to detect metal pollutants such as mercury, cyanide, and arsenic associated with small-scale mining activities in Ghana. The bioengineered microbe should be housed in a sealed, single-use microfluidic cartridge that will generate a visible signal when pollutant concentrations exceed defined thresholds. This approach will be a low-cost, rapid, and field-deployable environmental monitoring tool that can support public health by preventing the use of contaminated water supply and aid remediation efforts by facilitating the tracking of pollutants without the direct release of bioengineered organisms in the environment.
Escherichia coli will serve as an ideal engineered biosensor for detecting mining pollutants because it can be genetically engineered to couple potent specific sensing elements with standardized reporter outputs. It has native regulatory systems responsive to mercury, cyanide, and arsenic that can be integrated with plug-and-play genetic circuits that convert toxin recognition into a visible or measurable signal, such as fluorescence or luminescence. Engineered E. coli biosensors have been successfully demonstrated for mercury using mer-regulated promoters, for arsenic using ars operon regulators, and for cyanide through redox- and respiration-linked sensing systems, highlighting their sensitivity, specificity, and applicability for environmental monitoring in contaminated water systems. Making it a practical platform for environmental monitoring in mining-impacted regions.
Governance & Policy Goals for Ethical Usage
I chose these policy goals due to the project being a contained diagnostic synthetic biology tool, not a system meant for environmental release. As such, the primary ethical risks center on contaiment, missue and social impact.
Policy Goals
Biological Containment and Preventing Harm.
Prevent environmental release
Prevent horizontal gene transfer
Ensure post-use inactivation
Responsible Use and Misuse Prevention
Restrict access to live biological material
limit modification and replication
Ensure appropriate interpretation of results
Environmental and Social Protection
Avoid stigmatization or punitive misuse of data
Support remediation and public health responses
Protect vulnerable communities
Accessibility and Constructive Innovation
Maintain affordability
Avoid impeding legitimate research
Encourage local adoption and trust
Governance Actions
Option 1. Build-In Dual Contatiment
Purpose
Currently, biosensors are often regulated based on organism release risk. This option shift goverance upsteam by embedding safety directly into design.
Biotechnology companies to facilitate manufacturing.
Biosafety regulators such as the Environmental Protection Agency (EPA) and the National Biosafety Authority (NBA) for approval standards.
Funders: biosafety enforcement through grants and investments.
Assumptions
Containment systems remain reliable across conditions
Kill switches remain evolutionarily stable.
Risks of Failure & Success
Failure: Manufacturing defects or improper disposal
Success risk: over-reliance on technical fixes leading to reduced oversight
Option 2. Device-Level Regulatory Certification
Purpose
More governance from organism-based oversight to diagnostic-device style regulation, similar to water quality strips or pregnancy kits.
Design
Certification based on performance, containment, disposal, and shelf life.
Independent validation studies
Periodic recertification.
Actors
National environmental agencies: defining acceptable detection thresholds
Biosafety authorities: monitor post approval compliance and certify containment, inactivation, and disposal protocols.
standards organizations: develop testing, labelling, and performance standards.
Independent academic validators: conduct third-party performance and safety evaluations to provide credibility and transparency.
Assumptions
Regulators have the capacity to evaluate synthetic biology devices.
Certification increases public trust.
Risks of Failure & Success
Failure: slow approval processes.
Success risk: compliance cost excludes small innovators.
Option 3: Controlled Distribution and Stewardship
Purpose
Prevents misuse while ensuring ethical use.
Design
Distribution through approved institutions such as the EPA, NGOs and Universities.
Basic user training.
Standardize results reporting templates.
No access to cell or DNA.
Actors
Local environmental agencies: distribute kits to approved users, aggregate and interpret monitoring data.
NGOs and community organizations: act as community intermediaries, train users, and support ethical use.
Universities and extension services: provide technical training and oversight, update protocols as science evolves, and support data quality and analysis.
Local governments: coordinate response actions, ensure data is used for public health, not punishment, and set rules on who can deploy kits.
Assumptions
Training reduces misuse
Institutions act in the community’s interest.
Risk of Failure and Success
Failure: informal redistribution
Success risk: limited access in remote areas
Governance Scoring
Scores
1 = Most Effective
2 = Moderatly Effective
3 = Minimally Effective
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
1
2
Foster Lab Safety
• By preventing incident
1
1
2
Protect the environment
• By preventing incidents
1
1
2
Other considerations
• Preventing misuse
1
1
1
• Minimizing costs and burdens to stakeholders
2
2
3
• Community Protection
2
2
1
• Feasibility?
1
2
2
• Not impede research
1
2
2
• Promote constructive applications
1
1
2
Prioritization and Recommendation
I would prioritize a combined strategy of Option 1 (Dual containment) as a non-negotiable baseline, Option 2 (Device-level certification) for clarity and trust, and Option 3 (Controlled distribution) selectively in high-risk or sensitive contexts. This layered approach balances technical safety, regulatory clarity, and social responsibility. The primary trade-offs are increased development cost and reduced flexibility, but this is justified by a substantial reduction in ecological, ethical, and reputational risk.
Assignment Week 2 Lecture Prep
Homework Questions from Professor Jacobson
Question 1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Question 2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Answers
Question 1.
The error rate of DNA polymerase is approximately 1 mistake for every 106 added during DNA replication. The intrinsic 3’ to 5’ exonucleolytic proofreading activity of DNA polymerase removes the mismatch bases and lowers the replication error rate to about 108 nucleotides. When this is combined with the post-replication mismatch repair mechanisms, the overall error rate is reduced to better than 1 in 109 nucleotides. The human genome consists of approximately 3.1 to 3.2 billion base pairs (3×10^10) due to the combined accuracy of DNA polymerase, 3’ to 5’ exonucleolytic proofreading activity, and post-replication repair. The error rate compared to the human genome is less than one mistake per genome per cell division cycle.
Biology deals with the discrepancy through a multilayered correction system that consists of polymerase accuracy, 3’ to 5’ exonucleolytic proofreading activity, post-replicational mismatch, and redundancy in DNA sequences, which prevent the massive number of errors that would occur otherwise.
Question 2.
The genetic code consists of 4 nucleotide bases that code for 20 amino acids. mRNA reads nucleotides in triplets called codons, resulting in 64 possible codon combinations. The average human protein is composed of approximately 300 to 500 amino acids, and most amino acids are encoded by two to six different codons. There is a huge number of possible DNA sequences for any given protein approximatly X450 combinations, where X is the average number of codons per amino acid.
In practice, however not all codon combinations are equally effective code for expression due to codon usage bias. Most cells do not have equal amounts of tRNAs for every codon and prefer optimal codons, which enhance translation efficiency and protein production. Some other reasons are
The use of suboptimal codons slows tranlastion leading to protein misfolding and
Homework Questions from Dr. LeProus
Question 1. What’s the most commonly used method for oligo synthesis currently?
Question 2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
Question 3. Why can’t you make a 2000bp gene via direct oligo synthesis?
Answers
Question 1.
The most commonly used method for oligo synthesis currently is solid-phase phosphoramidite chemistry. It was developed by Caruthurs in 1981 and has become the industry standard because it allows for easy automation, rapid, and cost-effective production of custom oligonucleotides of 150-200 nucleotides in length.
Question 2.
It is difficult to make oligonucleotides longer than 200 nucleotides via direct chemical synthesis due to the cumulative effect of inefficiencies such as depurination, loss of yield, and accumulation of truncated sequences in each coupling step. By the 200 nucleotide, the fraction of full-length correct oligonucleotides becomes very low while truncated sequences and error-containing sequences increase, making further synthesis and purification increasingly difficult.
Question 3.
You cannot make a 2000bp gene via direct oligo synthesis because it is not feasible due to the cumulative effect of increasing low yields and errors in phosphoramidite chemistry as the chain length of nucleotide increases. Even at 200 nucleotide purification is difficult, much less at 2000 nucleosides, where the high number of truncated sequences and low yields would make the purification process impractical and the error rate unacceptably high.
Homework Questions from George Church
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
Question 1. [Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Question 2. [Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions?
Question 3. [(Advanced students)] Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or devise one of your own:
An amino acid is an organic molecule that consists of a basic amino group (-NH2), an acidic carboxyl group (-COOH), and an organic R group that is unique to each amino acid. They are organic compounds that serve as the fundamental building blocks of proteins, which are essential for repairing tissue, building muscle, and driving nearly all cellular functions.
Essential amino acids are amino acids that cannot be synthesized from scratch by organisms fast enough in sufficient quantities to supply their demands and must therefore be obtained from their diets. Essential amino acids are crucial for protein synthesis, tissue repair, and immune function. The 10 essential amino acids are Arginine, Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, and Valine. Nine of the essential amino acids are essential in humans, with the exception of Arginine, which is generally only essential in infants and many non-human species, particularly in strict carnivores such as felines, reptiles, avian, and some fish.
The lysine Contingency was a genetically engineered fail-safe performed by Dr. Henry Wu in Jurassic Park. The fail-safe was meant to knock out the ability of dinosaurs to produce the essential amino acid Lysine, forcing them to rely on synthetic supplements from the park’s staff. To ensure that, in the event of a dinosaur breakout, it would not survive long enough to damage global ecosystems.
Based on my understanding of essential amino acids, using Lysine as a bioengineered fail-safe was not the right choice. Lysine is an integral part of the metabolic process; it is needed for collagen formation, calcium formation, and energy production, and might seem to be a good target for a failsafe mechanism. However, all known animals lack the ability to synthesize lysine in adequate amounts but derive it from their food sources, primarily plants. Thus, Dr. Wu and the other scientists at InGen (International Genetics Technologies) essentially broke a feature in the dinosaur genome that was already broken in nature, assuming dinosaurs could actually produce lysine in adequate amounts in the first place. In nature, herbivores obtain lysine from feeding on plants, and carnivores obtain it by feeding on other animals. The Lysine contingency essentially forced the dinosaurs into the food web; as such, any dinosaur that escaped the park could survive by just consuming their normal diet in the natural environment, which ironically is lysine-rich in nature.
Reference
Bechor, O., Smulski, D.R., Van Dyk, T.K., LaRossa, R.A. and Belkin, S., 2002. Recombinant microorganisms as environmental biosensors: pollutants detection by Escherichia coli bearing fabA′:: lux fusions. Journal of Biotechnology, 94(1), pp.125-132.
Beese, L.S., Derbyshire, V. and Steitz, T.A., 1993. Structure of DNA polymerase I Klenow fragment bound to duplex DNA. Science, 260(5106), pp.352-355.
Benserhir, Y., Salaün, A.C., Geneste, F., Pichon, L. and Jolivet-Gougeon, A., 2022. Recent Developments for the Detection of Escherichia Coli Biosensors Based on Nano-Objects—A Review. IEEE Sensors Journal, 22(10), pp.9177-9188.
Bilal, M. and Iqbal, H.M., 2019. Microbial-derived biosensors for monitoring environmental contaminants: Recent advances and future outlook. Process Safety and Environmental Protection, 124, pp.8-17.
Dieudonné, A., Prévéral, S. and Pignol, D., 2020. A sensitive magnetic arsenite-specific biosensor hosted in magnetotactic bacteria. Applied and Environmental Microbiology, 86(14), pp.e00803-20.
Cai, S., Shen, Y., Zou, Y., Sun, P., Wei, W., Zhao, J. and Zhang, C., 2018. Engineering highly sensitive whole-cell mercury biosensors based on positive feedback loops from quorum-sensing systems. Analyst, 143(3), pp.630-634.
Hou, Y. and Wu, G., 2018. Nutritionally essential amino acids. Advances in Nutrition, 9(6), pp.849-851.
I tried to create a design in Benchling, after many trials and errors, I managed to make a pattern by using double and triple digests of restriction enzymes.
I think it looks like a Y.
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in the recitation (NCBI, UniProt, Google), obtain the protein sequence for the protein you chose.
Answer
I picked the BMP1 protein (Bone morphogenetic protein 1). It is a secreted metalloprotease encoded by the BMP1 gene in humans. It belongs to the astacin M12A family of proteases and plays a central role in extracellular matrix assembly by cleaving precursor proteins into the mature functional forms. Growing up, I never had a bone fracture or dislocation, but my brother had a fracture in his left hand, which made me curious about the proteins and genes that drive bone formation. (https://www.uniprot.org/uniprotkb/P13497/entry)
The protein sequence for BMP1 protein on Uniprot is an isoform that has been chosen as the canonical sequence.
The sequence is as follows:
MPGVARLPLLLGLLLLPRPGRPLDLADYTYDLAEEDDSEPLNYKDPCKAAAFLGDIALDEEDLRAFQVQQAVDLRRHTARKSSIKAAVPGNTSTPSCQSTNGQPQRGACGRWRGRSRSRRAATSRPERVWPDGVIPFVIGGNFTGSQRAVFRQAMRHWEKHTCVTFLERTDEDSYIVFTYRPCGCCSYVGRRGGGPQAISIGKNCDKFGIVVHELGHVVGFWHEHTRPDRDRHVSIVRENIQPGQEYNFLKMEPQEVESLGETYDFDSIMHYARNTFSRGIFLDTIVPKYEVNGVKPPIGQRTRLSKGDIAQARKLYKCPACGETLQDSTGNFSSPEYPNGYSAHMHCVWRISVTPGEKIILNFTSLDLYRSRLCWYDYVEVRDGFWRKAPLRGRFCGSKLPEPIVSTDSRLWVEFRSSSNWVGKGFFAVYEAICGGDVKKDYGHIQSPNYPDDYRPSKVCIWRIQVSEGFHVGLTFQSFEIERHDSCAYDYLEVRDGHSESSTLIGRYCGYEKPDDIKSTSSRLWLKFVSDGSINKAGFAVNFFKEVDECSRPNRGGCEQRCLNTLGSYKCSCDPGYELAPDKRRCEAACGGFLTKLNGSITSPGWPKEYPPNKNCIWQLVAPTQYRISLQFDFFETEGNDVCKYDFVEVRSGLTADSKLHGKFCGSEKPEVITSQYNNMRVEFKSDNTVSKKGFKAHFFSDKDECSKDNGGCQQDCVNTFGSYECQCRSGFVLHDNKHDCKEAGCDHKVTSTSGTITSPNWPDKYPSKKECTWAISSTPGHRVKLTFMEMDIESQPECAYDHLEVFDGRDAKAPVLGRFCGSKKPEPVLATGSRMFLRFYSDNSVQRKGFQASHATECGGQVRADVKTKDLYSHAQFGDNNYPGGVDCEWVIVAEEGYGVELVFQTFEVEEETDCGYDYMELFDGYDSTAPRLGRYCGSGPPEEVYSAGDSVLVKFHSDDTITKKGFHLRYTSTKFQDTLHSRK
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize Google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
[Example from Codon Optimization Tool | Twist Bioscience while avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI]
Answer
Codons need to be optimized for use due to the codon usage bias in the heterologous host organisms. The codon usage bias is due to variations in tRNA abundance in different organisms, which directly impacts translation speed and accuracy. When a gene from one organism is expressed in another, such as a human gene in bacteria, the mismatch in codon preference can cause ribosomes to stall at rare codons, leading to reduced protein yield, truncated proteins, or misfolding. Thus, codons are optimized to ensure the efficient expression of proteins in heterologous host organisms.
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Answer
In a cell-dependent system, the Bone morphogenetic protein 1 can be produced using recombinant plasmid cloning technology. This would work by inserting the DNA sequence coding for the BMP1 protein into a plasmid such as E.coli. The DNA sequence should be optimised for the chosen plasmid. The plasmid should have a promoter, start and stop codons, regulator sequences, and a terminator. The plasmid is then introduced into bacteria via transformation. Inside the bacteria, RNA polymerase will bind to the promoter and transcribe the DNA coding region in RNA. Which then binds to the ribosome and tRNA reads the codons and assembles amino acids and peptide chains fold into the BMP1 protein.
Part 4: Prepare a Twist DNA Synthesis Order
4.1 Create a twist account
I created a twist account
I chose to build an insert sequence for the luciferase. The luciferase gene encodes an enzyme that catalyzes a bioluminescent reaction, producing light in the presence of its substrate (luciferin), ATP, and oxygen. It is widely used as a reporter gene to study gene expression.
I first started the build by using the T7 promoter and added the Shine-Dalgarno sequence as the ribosome binding site.
I added a start codon
I imported the luciferase gene from NCBI and tried to copy out the coding sequence for luciferase. I used Benchling to optimize the coding sequence for insertion into E.coli plasmid.
I then inserted the optimized luciferase coding sequence into the build, added a 6x his tag,a stop codon, and a T7 terminator
I uploaded the sequence into Twist and chose the pTwist Amp High Copy vector. I then downloaded the construct as GenBack and imported it into Benchling.
(i) What DNA would you want to sequence (e.g., read) and why?
Answer
I would like to sequence the whole genome and transcriptome of rice varieties grown in Ghana. I would focus on genes involved in nitrogen use efficiency, drought tolerance, and yield stability.
Coming from an agricultural biotechnology background, sequencing rice DNA for nitrogen use efficiency will improve food security by reducing fertilizer dependency while maintaining yield levels. Additionally, nitrogen metabolism is tightly linked to drought stress and carbon metabolism, thus sequencing can reveal alleles that enhance resilience under variable rainfall.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Answer
I would use a hybrid sequencing strategy combining third-generation PacBio HiFi sequencing (developed by Pacific Biosciences) and second-generation Illumina sequencing (developed by Illumina) because each technology addresses different challenges of plant genomics. The genome of Oryza sativa contains many repetitive elements and duplicated gene families, which are difficult to assemble accurately using short reads alone. PacBio HiFi produces long, highly accurate reads that can span repetitive regions and resolve structural variants such as insertions, deletions, and gene duplications. This is especially important when studying nitrogen use efficiency genes, which may exist in multiple similar copies or be influenced by regulatory structural variation. Long-read sequencing therefore enables the creation of a high-quality de novo assembly of locally adapted rice varieties, ensuring that important region-specific alleles are not missed.
However, PacBio alone is not sufficient for large-scale comparative or expression studies. Illumina sequencing provides extremely high depth at a lower cost per base, making it ideal for population-level SNP discovery, genome polishing, and RNA sequencing. Since nitrogen use efficiency is strongly influenced by gene regulation, Illumina RNA-seq would allow precise quantification of gene expression under different nitrogen treatments. Combining long-read structural resolution with high-depth short-read accuracy ensures reliable variant detection, strong transcriptomic analysis, and cost efficiency. Together, this hybrid approach provides the comprehensive genomic insight needed to improve nitrogen use efficiency, enhance sustainable fertilizer management, and support precision breeding strategies in rice.
For PacBio HiFi sequencing, the input would be high-molecular-weight genomic DNA (15-25 kb fragments).
Library Preparation:
Extract intact genomic DNA.
Size selection.
Ligate hairpin adapters to create circular SMRTbell templates.
Polymerase binding (no amplification required).
Sequencing & Base Calling:
DNA polymerase is immobilized in Zero-Mode Waveguides (ZMWs).
Fluorescently labeled nucleotides are incorporated.
Each base emits a distinct fluorescence signal.
Circular consensus sequencing (multiple passes) improves accuracy to >99.9% (Wenger et al., 2019).
Output:
Long high-fidelity reads (10–25 kb)
FASTQ files with quality scores
Ideal for resolving repetitive plant genome regions
Long-read sequencing is especially important because plant genomes are repeat-rich and structurally complex (Michael & VanBuren, 2020).
For Illumina Sequencing, the input would be fragmented DNA of 300-500 bp or cDNA for RNA sequencing.
Library Preparation:
DNA fragmentation
End repair and adapter ligation
PCR amplification
Cluster generation via bridge amplification
Sequencing & Base Calling:
-Sequencing-by-synthesis
Reversible terminator nucleotides incorporated one at a time
Fluorescent imaging determines base identity
High per-base accuracy (>99%)
Output:
Millions to billions of short reads (100–150 bp), which are ideal for gene expression quantification and polishing assemblies
5.2 DNA Write
I would design and synthesize a synthetic nitrogen-sensing genetic circuit that could be introduced into rice to improve nitrogen uptake and fertilizer responsiveness.
Instead of simply overexpressing a transporter gene (which can cause metabolic imbalance), I would engineer a smart, feedback-controlled genetic circuit that activates nitrogen uptake genes only under low-nitrogen conditions. The genetic circuit would consist of:
A low-nitrogen inducible promoter.
A synthetic transcriptional activator module.
A nitrogen transporter gene.
A fluorescent reporter for monitoring.
A terminator sequence.
The core gene would be NRT2.1 (high-affinity nitrate transporter, which is involved in nitrate uptake under nitrogen-limited conditions.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Answer
To synthesize the nitrogen-responsive genetic circuit containing the NRT2.1 module, I would use phosphoramidite-based solid-phase DNA synthesis combined with enzymatic assembly methods such as Gibson Assembly.
In this approach, short DNA oligonucleotides are chemically synthesized base-by-base through iterative cycles of deprotection, nucleotide coupling, capping, and oxidation. Because individual oligos are typically limited to ~200 bp, overlapping fragments would then be assembled into the full-length construct using enzymatic assembly in a single reaction using Gibson assembly.
This method is precise, scalable, and well-suited for modular plant genetic circuit design. However, limitations include length constraints requiring multi-fragment assembly, potential synthesis errors that accumulate with longer sequences, and challenges with high-GC or repetitive regions. Therefore, the final construct would require sequence verification to ensure accuracy before plant transformation.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
Beyond plants, I would be very interested in editing the genome of Aedes aegypti, the mosquito species that transmits diseases such as dengue, Zika, and yellow fever. The goal would be to reduce the transmission of these viruses through gene drives or other targeted genome editing strategies. Specifically, I would target genes involved in fertility or pathogen susceptibility, such as those encoding reproductive proteins or viral receptor proteins in the mosquito midgut. For example, disrupting a key fertility gene could reduce mosquito population density, while modifying viral receptor genes could make mosquitoes resistant to virus infection, breaking the disease transmission cycle.
My rationale for editing Aedes aegypti is both public health and environmental impact. Vector-borne diseases affect millions worldwide, especially in tropical regions, and current control methods (insecticides, habitat elimination) are often insufficient, costly, or ecologically harmful. Gene editing offers a precise, sustainable solution that can complement traditional control strategies.
(ii) What technology or technologies would you use to perform these DNA edits and why?
For this, I would use CRISPR-Cas9-based gene drives, which allow a targeted gene to be copied preferentially to offspring, ensuring rapid spread of the desired trait through wild populations.
CRISPR-Cas9, which is currently the most precise and widely used genome editing technology for insects and other organisms. CRISPR-Cas9 enables targeted modifications of DNA by creating double-strand breaks at specific genomic locations, which are then repaired by the cell’s own repair machinery, allowing for insertions, deletions, or gene replacement. This technology is ideal for engineering traits such as reduced fertility or virus resistance in mosquitoes.
However, careful containment, ecological risk assessment, and ethical considerations would be critical because of the potential for irreversible effects in wild populations.
How CRISPR-Cas9 Edits DNA
Targeting – A single-guide RNA is designed to complement a specific DNA sequence in the mosquito genome, adjacent to a protospacer adjacent motif.
Cutting – Cas9 endonuclease binds the sgRNA and introduces a double-strand break at the targeted site.
Repair – The mosquito cell repairs the break via:
Non-Homologous End Joining → introduces small insertions/deletions (indels), which can disrupt gene function.
Homology-Directed Repair → if a DNA template is provided, precise sequence changes can be introduced, e.g., inserting a virus-resistance allele.
Preparation and Inputs
Before editing, careful design and preparation are required:
Design steps
Identify the target genes critical for fertility or viral susceptibility.
Design sgRNAs that minimize off-target effects using computational tools.
Design donor DNA templates if precise sequence insertion is needed.
Inputs
sgRNA – synthesized guide RNA targeting the mosquito gene.
Cas9 protein or Cas9-expressing plasmid/mRNA.
Donor DNA template.
Embryos or cultured mosquito cells – Aedes aegypti embryos are typically microinjected with these components.
The delivery will be a microinjection of CRISPR components into fertilized mosquito eggs, which is standard for germline editing, ensuring heritable changes.
CRISPR-Cas9 has some limitations, which are editing efficiency can be low because not all injected embryos survive, and only a fraction carries the intended mutation.
The precision of the edits is also variable: when DNA breaks are repaired through non-homologous end joining, unpredictable insertions or deletions can occur, and homology-directed repair is often inefficient in embryos. Off-target effects are another concern, as the guide RNA may bind unintended genomic sites, causing unwanted mutations.
Additionally, while CRISPR-based gene drives can spread edited traits through populations, they require careful ecological risk assessment to avoid unintended consequences, and scaling up edits for population-level interventions demands extensive breeding and monitoring. Despite these challenges, CRISPR remains the most practical and validated method for achieving heritable and targeted genetic modifications in mosquitoes.
References
Michael, T.P. & VanBuren, R., 2020. Building near-complete plant genomes. Current Opinion in Plant Biology, 54, pp.26–33.
Thomsen, H.C. et al., 2014. Glutamine synthetase: role in nitrogen metabolism and crop productivity. Frontiers in Plant Science, 5, p.465.
The sequence of sequencers: The history of sequencing DNA
https://pmc.ncbi.nlm.nih.gov/articles/PMC4727787/
Wang, W. et al., 2018. Genetic variation in ARE1 mediates grain yield by modulating nitrogen utilization in rice. Nature Communications, 9, p.735.
Wenger, A.M. et al., 2019. Accurate circular consensus long-read sequencing improves variant detection and genome assembly. Nature Biotechnology, 37, pp.1155–1162.
Xu, G. et al., 2012. Plant nitrogen assimilation and use efficiency. Annual Review of Plant Biology, 63, pp.153–182.
I also used Chat-gtp to guide me in the steps, and design procedures used in read , write and edit DNA questions.
Week 3 HW: Lab Automation
Python Script for Opentrons Artwork
This has been the most interesting and somewhat challenging assignment so far.
I chose make an artistic design based on the adrinkra symbols. The adinkra symbols are a set of visual symbols from Ghana, created by the Akan people to represent philosophical concepts, historical events, and social proverbs.
I picked the Nsaa symbol, which is a type of woven cloth renowned for its quality. It is the symbol of excellence, genuineness, and authenticity.
I used the GUI at opentrons-art.rcdonovan.com.to generate an artistic design for the Nsaa symbol.
I then used the design coordinates from the Opentrons Automation Art Interface to write the code in Google Colab.
Writing the code to ensure that the colors were dispensed at the correct coordinates and there was no cross-contamination of pipette tips was very tricky. I wrote my first attempt and used the Gemini 2.5 flash in Google Colab to optimize and debug errors in the code.
When writing the code, I noticed that the color wells available were red, green, and orange, so I used green and red for the design.
This table is the documentation of how I used Google Genimi 2.5 flash in Google Colab to help debug my code.
Error
Prompt
Fix
General execution error
Please explain this error: “Sorry, I ran into an error, could you try again?”
The issue was caused by an indentation error in the for loops iterating over the ‘Green’ and ‘Red’ coordinates. The indentation was corrected and metadata fields were updated with placeholder values.
AttributeError: ‘Location’ object has no attribute ‘moves’
Please explain this error
The Location object does not have a moves method. The correct Opentrons API method is move. Replacing moves with move resolved the issue.
Pipette dispense error (no liquid)
Please explain this error
The pipette attempted to dispense without aspirating first. The fix was to add aspiration steps before dispensing both ‘Green’ and ‘Red’ solutions.
Tip not dropped error
The robot is reporting that the tip was not dropped
The protocol likely stopped earlier due to incorrect aspiration logic. The aspiration volumes were revised to explicitly match dispense volumes, allowing the protocol to complete and drop the tip properly.
KeyError: Labware well names not found
Please explain this error
The labware 'opentrons_96_aluminumblock_generic_pcr_strip_200ul' does not use well names like ‘A1’, ‘B1’, or ‘C1’. It was replaced with 'corning_96_wellplate_360ul_flat', which supports standard 96-well naming.
Cross-contamination error
Please explain this error
The same pipette tip was used for both green and red solutions. The fix was to add a drop_tip() step after finishing with green and pick up a new tip before handling red.
Visualization color missing
Coordinate (9.9, -16.5) for green doesn’t have a color showing in the visualization. What is the fix?
There was a typo in the Green coordinate list. (9.9, 16.5) was listed instead of (9.9, -16.5). Correcting the coordinate fixed the visualization.
Question 1: Revolutionizing sample preparation: a novel autonomous microfluidic platform for serial dilution
The paper I chose is titled “Revolutionizing sample preparation: a novel autonomous microfluidic platform for serial dilution” by Dries Vloemans et al. The paper presented a novel, standalone, and fully automated microfluidic platform for the stepwise preparation of serial dilutions without the need for any active elements.
Dilution is a standard fluid operation that is widely employed in the sample preparation of many biochemical assays. It serves multiple essential functions, such as sample mixing with certain reagents at specific dilution ratios, reducing sample matrix effects, and bringing target analytes within the linear assay detection range, among many others.
Traditionally, dilution relies either on manual pipetting, which is labor-intensive and prone to human error, or automated laboratory liquid handling systems, which are bulky, expensive, and unsuitable for point-of-care use. The goal of the authors was to develop a passive, self-contained microfluidic platform that could execute serial dilution in a controlled, programmable, and reproducible manner.
The key findings of the paper include demonstrating that the proposed automated microfluidic platform can perform precise and reproducible serial dilutions without pumps or active control systems. The hydrophobic burst valves reliably metered out defined liquid volumes, enabling accurate dilution ratios such as 2X, 5X, and 10X. It also showed that effective mixing could be achieved through the incorporation of sequential expansion chambers. Which were geometrically optimized to promote passive mixing as fluids pass through them, eliminating the need for mechanical agitation. Additionally, it demonstrated the platform’s compatibility with relevant biological fluids like blood and the integration of a capillary-driven SIMPLE pumping mechanism to allow the device to operate in a fully self-powered manner and complete dilution sequences within short time frames after user activation.
Fig. 1 a) Conceptual design of the dilution module illustrating the 3 microfluidic units that are used for plug metering, merging and mixing, and the positions of the different valving elements (single-coated (sc) and double-coated (dc) HBVs, and hydrophobic barrier (HB)). b) Configuration and working principle of the different valves with their respective theoretical burst pressure profiles. The sc HBV contains a hydrophobic coating at the bottom channel wall, while the dc HBV is treated hydrophobically at both the top and bottom walls, resulting in varying burst pressures. The HB comprises a hydrophobic-treated filter paper, which allows air passage but forms a physical barrier for the liquid, hence, inducing a very high burst pressure. c) Conceptual exploded view of the integrated microfluidic device for autonomous multistep serial dilution, illustrating the top ‘dilution’ and bottom ‘pumping’ layer. The top dilution layer comprises 3 serially coupled dilution modules (5× DF), connected with a connection hole to the bottom pumping layer, holding the prefilled working liquid and wedge-shaped filter paper (Whatman grade 598) of the SIMPLE pump unit.
Fig. 2 a) Snapshots of the different liquid manipulations within a dilution module (DF = 5×) illustrating the working principle. (i and ii) The coordinated burst action of HBVs with different burst strengths is used to first isolate a precisely metered sample liquid (2 μL, blue), after which the excess is removed to the storage channel. (iii and iv) The metered sample liquid is next merged with a prefilled diluent (8 μL, yellow), after which (v and vi) the combined plug is sent through a sequence of expansion chambers in which it is mixed into a homogeneous solution. b) Detailed schematics of plug merging, and working principle of the microfluidic air bridge (top). Illustration of failed downstream plug manipulation when no blocking channel is used due to air intake via the microfluidic air bridge (bottom). c) Close-up of the expansion chambers, illustrating the three ongoing principles that are used within the mixing process: increase of diffusion interface, parabolic flow profile, and lateral plug distribution. Dashed and full arrows indicate air and liquid flow, respectively.
Question 2: What I intend to do with automation tools for my final project
Using my first idea, which involves developing a biosensor kit for the detection of illegal mining pollutants. The automation tools used would be a combination of Python-based liquid handling, 3D-printed assay holders, and could-based design tool like Google Nebula.
Here is a rough idea of the automation tools I might end up using:
Using Opentrons OT-2 to dispense growth media and mix microbial cultures with chemical regents.
Using PLateLoc to seal the plate.
Using XPeel to remove the seal after incubation.
Measuring fluorescence and color intensity using PHERAstar plate reader.
Using Ginkgo Nebula to design synthetic genetic circuits for microbial biosensors and simulate sensor response behaviors.
Final Project Ideas
I have submitted my final project ideas in the slide deck that was provided for committed listeners
Vloemans, D., Pieters, A., Dal Dosso, F. and Lammertyn, J., 2024. Revolutionizing sample preparation: a novel autonomous microfluidic platform for serial dilution. Lab on a Chip, 24(10), pp.2791-2801.
Week 4 HW: Protein Design - Part I
Part A. Conceptual Questions
Question 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Answer
A dalton is a unit of mass used to express the mass of atoms, molecules, and other subatomic particles.
The average percentage of protein in meat is about 20-30% of its total weight.
Using 30% as the average amount of protein in meat.
Mass of protein = 500 × 30% = 150g
Moles of amino acids = mass/molar mass = 150/100 = 1.5mol
Calculating the number of amino acid molecules in the meat:
Therefore, there are 9.0× 1023 amino acid molecules in 500g of meat.
Question 2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Answer
This is due to digestion breaking down food such as meat and fish into basic molecules like amino acids, fatty acids, and sugars. The DNA present in Food is therefore broken down and rendered nonfunctional; as such, it is not directly incorporated into our genetic structure.
Question 3. Why are there only 20 natural amino acids?
Answer
There are only 20 natural amino acids due to evolutionary optimization and biochemical efficiency. The 20 amino acids offer a balanced range of properties such as polarity, charge, and hydrophobicity that allow proteins to fold properly and perform diverse biological functions. The limit on the amino acids due to the degeneracy of the genetic code protects against mutations, ensures accurate protein synthesis, and metabolic efficiency.
Question 5 Where did amino acids come from before enzymes that make them, and before life started
Answer
Amino acids were most likely formed before enzymes and life began through abiotic chemical processes, fuelled by the exposure of simple molecules such as methane, ammonia, hydrogen, water, and carbon dioxide to energy sources.
This was demonstrated by Stanley Miller and Harold Urey’s 1953 experiment, which showed that when gases are subjected to electrical sparks, amino acids such as glycine, aspartic acid, and alanine are formed. Additionally, amino acids have been detected in the Murchison meteorite, which suggests that amino acid precursors were formed through photochemical and catalytic reactions.
Question 6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Answer
An α-helix is a secondary protein structure in which the polypeptide chain is twisted into a coil. a-Helices are crucial for protein stability and function. There are 3.6 amino acids per turn of the helix, with each turn having a height of 0.54nm.
D and L configurations describe the stereochemistry of amino acids based on the position of the amino group in a Fischer projection.
L-amino acids have the amino group (NH2) on the left, while D-amino acids have it on the right. This makes them mirror images of each other.
An α-helix made using D-amino acids instead of the naturally occurring L-amino acids is expected to form a left-handed α-helix. This is due to the chirality of amino acids. In natural biological systems, proteins are made almost entirely of L-amino acids, which form right-handed α-helices. Since D-amino acids are mirror images of L-amino acids, when D-amino acids form an α-helix, they produce a mirror image structure of the typical right-handed helix. Thereby producing a left-handed α-helix.
Question 7. Can you discover additional helices in proteins?
Answer
It is possible to discover additional helices in proteins using a combination of experimental and computational methods. In certain environmental conditions, proteins adopt other helical conformations. Some examples of these are: 310 helix, which has 3.0 residues per turn and hydrogen bonds between residues i and i+3. Additionally, π Helix with 4.4 residues per turn and hydrogen bonding patterns of i to i+5.
Several experimental and computational methods are used to discover helices in proteins. One key technique is X-ray crystallography, which enables the determination of the three-dimensional coordinates of atoms in proteins. This technique reveals backbone angles and hydrogen-bonding patterns. Researchers then analyse the revealed structure to determine if the chains form a known helix or an uncharacterized one.
Additionally, computational tools, such as the Protein Data Bank, store thousands of protein structures, allowing scientists to use algorithms to scan for recurring backbone structures that may not match any known helices. If consistent and stable patterns are observed repeatedly across unrelated proteins, these patterns may suggest the existence of a new type of helix. Molecular dynamics simulations can also be used to predict whether alternative backbone conformations are stable, and researchers can design proteins based on the simulations and test whether they fold into new helices.
Question 8. Why are most molecular helices right-handed?
Answer
Most molecular helicases are right-handed because it is the most thermodynamically stable and energetically favorable conformation. This is due to the chirality, where right-handed folding maximizes hydrogen bonding and minimizes steric hindrance, making them more stable than left-handed helices.
Question 9 Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
Answer
A β-sheet is a fundamental form of a secondary protein structure consisting of β-strands that are linked laterally by hydrogen bonds, forming a zig-zag and twisted pleated sheet configuration.
β-sheets aggregate due to their inherent structural tendency to form intermolecular hydrogen bonds and bury hydrophobic residues, which results in the creation of amyloid fibrils.
The driving force of beta sheet aggregation is is the combination of thermodynamic stabilization through dehydration and the maximization of weak interactions in environments with high concentrations of misfolded protein intermediates.
Question 10 Why do many amyloid diseases form β-sheets?
Can you use amyloid β-sheets as materials?
Answer
Amyloid diseases are a group of rare diseases caused by the abnormal misfolding and aggregation of proteins into insoluble fibrils known as amyloids. These fibrils accumulate in tissue and organs, disrupting normal function and potentially leading to organ failure. Some amyloid diseases are Alzheimer’s disease and parkinson’s disease.
Amyloid diseases form β-sheets due to misfolded proteins caused by mutations, stress, or aging, which expose β-strand regions that can form hydrogen bonds with strands from other proteins. The exposed β-strands then form exceptionally stable and low-energy β-structures that run perpendicular to the fibril axis, forming insoluble, self-propagating fibrils via hydrophobic interactions.
Amyloid sheets can be used as materials due to amyloid fibrils being extremely strong, resistant to heat and chemical degradation, and self-assembling in nature. Not all amyloids are pathological; organisms such as E.coli and yeast naturally produce functional amyloids for structural and biological roles. As such, short peptides that deliberately form controlled β-sheets fibrils can be used to produce materials.
Part B: Protein Analysis and Visualization
Question 1
I chose the aequorin protein. While thinking of what protein to select for this section of the assignment, I decided to pick an interesting protein found in a sea organism and stumbled upon the Crystal jelly (*Aequorea victoria), a bioluminescent hydrozoan jellyfish.
Aequorin is a photoprotein that emits blue light in direct response to binding to calcium ions (Ca 2+), making it a calcium-sensitive photoprotein.
Honestly, I first chose aequorin because it is a bioluminescent protein and I love glowing stuff :). However, after reading more on it, I discovered it has played an important role in paving the way for the discovery and application of green fluorescent protein (GFP) and transformed the observation of molecular proteins inside cells. Groundbreaking work on awquorin and its related fluorescent proteins contributed to Osamu Shimomura, Martin Chalfie, and Roger Y. Tsien winning the Nobel Prize in Chemistry in 2008.
I selected the Aequorin-1 protein with an annotation score of 4/5 from Uniprot.
The length of the Aequorin-1 is 196 amino acids, and the most frequent amino acid is D (Aspartic acid), which appears 18 times throughout the sequence.
I run a blast for the homologs of the Aequorin-1 protein using Uniprot
I found 250 homologs of the Aequorin-1 protein
Aequorin-1 belongs to the EF-hand calcium-binding protein superfamily, which is a group of eukaryotic proteins that play a central role in cellular signaling, regulation, and homeostasis.
Question 3
The structure of the Aequorin-1 protein was deposited on 2004-03-05 and released on 2004-12-28 by Deng, L., Markova, S.V., Vysotski, E.S., Liu, Z.J., Lee, J., Rose, J., Wang, B.C. X-ray Diffraction was used to determine the 3d strcuture of the Aequorin protein.
The protein has a good quality structure and a resolution of 1.70 Å.
Using SCOP, I found out that Aequorin-1 belongs to the calmodulin-like structural family.
It has calcium ions (Ca2+) that form part of its structure
Question 4
I used PyMol to visualize the Aequorin-1 protein.
Aequorin-1 protein as a Cartoon
Aequorin-1 protein as a Ribbon
Aequorin-1 protein as a Ball and Stick
I colored the cartoon structure of Aequorin-1, highlighting the helices in red, the beta sheets in yellow, and the loops in green. Based on the image generated, Aequorin-1 has a significant amount of alpha helices and no beta sheets.
I tried to use a code to count the number of helices and sheets
The helices atom count was 1076 atoms, and the atom sheet count was 44 atoms
I highlighted the hydrophobic elements in orange and the hydrophilic elements in cyan.
Based on the color distribution, there are more hydrophilic elements than hydrophobic elements.
I visualized the surface of the protein, and it didn’t have any holes in it.
Part C. Using ML-Based Protein Design Tools
Question 1 - Deep Mutational Scans
Just like in part B, I chose the Aequorin-1 protein. I then inserted the mutation scan in Google Colab and ran it. I used the relative mode when I ran the mutation scan for the protein sequence.
This was the Mutation Scan Heatmap of the Aequorin-1 protein.
I tried to read the mutation scan heatmap and asked Gemini for a guide on how to read the map.
Based on the information it gave me, the X-axis showed the positions in the protein sequence, and the Y-axis represented the 20 standard amino acids that the wild-type residue at a given position
Additionally, a high positive score (e.g., a bright yellow cell) indicates that the language model predicts the mutated amino acid is significantly more likely to occur at that position than the original wild-type amino acid. Conversely, a highly negative score (e.g., a dark purple cell) suggests the mutation is much less likely.
Using this information to read the map and pick a bright yellow residue at X = 84, Y = S, and Z = 1.941 (mutation score).
E (Glutamic acid) is at position 84 of the Aequorin-1 protein sequence, with a score of 1.941, which is a high positive score, and the cell could be bright red. This means it is highly likely that E (Glutamic acid) will mutate into S (Serine) at position 84 of the Aequorin-1 protein sequence.
Question 2 - Latent Space Analysis
I used the provided sequence dataset to embed proteins in reduced dimensionality.
I placed the Aequorin-1 protein in the resulting map and coloured it red, while making all the other proteins blue.
The Aequorin-1 was located at the following 3D t-SNE coordinates:
TSNE1: -3.6272, TSNE2: 0.3001, TSNE3: -6.5145
The proteins that were close to the Aequorin-1 protein in the Latent space analysis were:
When I tried to determine the relation between them, I discovered they all possess the EF-hand calcium-binding motif except for Resp1.
C2. Protein Folding
I used ESMFOLD to fold the Aequorin-1 protein
When I compared it to the structure of the Aequorin-1 protein in RCSB, I noticed there were differences in both structures
I think the differences could be due to conformation variability or accuracy limitations. I verified that I correctly copied and inputted the correct Aequorin-1 protein sequence, so the discrepancy in the structure is not due to wrong protein sequence inputs. Additionally, the structure displayed by ESMFold did have a plddt value of 80.295, which means there is a high likelihood that the general backbone structure is accurate.
For mutant_1, I made some random changes in a small portion of the protein sequence, and for mutant_2, I made random changes to a larger part of the protein sequence.
There was not a drastic difference in the folding structures of Aequorin-1 protein in ESMFold and the Aequorin-1 protein Mutant_1, as well as the Aequorin-1 protein Mutant_2. Based on this, I can conclude that the protein is quite resilient to mutations.
C3. Protein Generation
I used the PDB file generated by ESMfold to inverse-fold the protein using ProteinMPNN.
The Amino Acid Probabilities heatmap generated after the inverse folding with ProteinMPNN predicts the likelihood of each amino acid occurring at every position along my input protein backbone structure.
By comparing the original Aequorin-1 protein to the new protein sequence generated by the inverse folding using ProteinMPNN, I noticed there was a significant difference between the two sequences. The sequence recovery rate was 41.33% this means that almost 60% of the protein residues were changed by ProteinMPNN. These changes might be due to ProteinMPNN’s design process, optimizing the sequence to best fit the provided 3D backbone structure. This means that some of the changes in the sequence were due to certain amino acids being substituted for others that are energetically or structurally more favorable in that specific local environment. I also noticed that both sequences maintained the same length of 196 residues despite the changes.
I entered the new protein sequence from ProteinMPNN into ESMFold, and the displayed structure resembled the one generated when I used ESMFold for the Aequorin-1 protein.
The documentation of how I used Gemini 2.5 flash in Google Colab.
Prompt
Error
Fix / Explanation
the protein structure that was shown after using EMSFOLD was different from the structure in RCSB. What could be the reason for the difference in structure
None
Explained differences between predicted (ESMFold) and experimental (RCSB) structures, including prediction vs experimental methods, accuracy limitations, missing ligands, conformational flexibility, and environmental conditions.
is there a way to tell if a protein is resilient to mutations based on its structure? Using emsfold
None
Clarified that ESMFold predicts structure but does not directly measure mutation resilience. Suggested using mutation scanning, stability prediction tools, and structural analysis for assessing resilience.
how can you interpret the pLDDT score
None
Explained that pLDDT ranges from 0–100 and indicates model confidence per residue. Provided interpretation ranges (very high, high, low, very low confidence) and how to use them to evaluate the reliability of predicted regions.
how do I get the backbone of your chosen PDB to run Inverse folding with ProteinMPNN
None
Explained that the pdb variable must be updated to the correct ESMFold-generated PDB file path before running ProteinMPNN. Provided instructions to modify the setup cell.
what does your fix do
None
Explained that the fix updated the pdb variable so ProteinMPNN uses the correct ESMFold-generated structure for inverse folding.
ok run your fix
FileNotFoundError: [Errno 2] No such file or directory: ’test_85b81/ptm0.805_r3_default.pdb.pdb'
Error caused by double .pdb extension being appended. Identified that get_pdb function incorrectly added .pdb to a filename that already had it.
(Implicit – after execution error)
None (after fix)
Modified the get_pdb function to properly handle local PDB paths without appending an extra .pdb extension. Ready for re-execution.
(Implicit – after successful execution)
None
Confirmed successful ProteinMPNN setup execution and instructed user to run the sequence generation cell.
explain the results of the Amino acid possibilities in the mutation heat scan after the inverse folding with proteinMPNN
None
Explained amino acid probability heatmap: axes (positions vs amino acids), color intensity representing probability, and interpretation for residue preference, conservation, and sequence diversity.
Part D. Group Brainstorm on Bacteriophage Engineering
Three students from the William and Mary Node and I formed a group for this part of the assignment.
We chose the goal of increasing the thermodynamic and structural stability of the Lysis protein while preserving its native fold and lytic function.
Is a limited understanding of the structural biology of lysis protein as a beginner to phage engineering
Increasing stability may not lead to improved function of the protein.
Here is a picture of the schematic of the pipeline
Reference
Greenwald, J., & Riek, R. (2010). Biology of amyloid: structure, function, and regulation. Structure, 18(10), 1244-1260.
Glyakina, A. V., Likhachev, I. V., Balabaev, N. K., & Galzitskaya, O. V. (2014). Right‐and left‐handed three‐helix proteins. II. Similarity and differences in mechanical unfolding of proteins. Proteins: Structure, Function, and Bioinformatics, 82(1), 90-102.
JPT Peptide Technologies. (n.d.). What are L- and D- amino acids? JPT Peptide Technologies. Retrieved February 28, 2026, from https://www.jpt.com/blog/l-d-amino-acids/
Makin, O. S., Atkins, E., Sikorski, P., Johansson, J., & Serpell, L. C. (2005). Molecular basis for amyloid fibril formation and stability. Proceedings of the National Academy of Sciences of the United States of America, 102(2), 315–320. https://doi.org/10.1073/pnas.0406847102
Niu, Z., Gui, X., Feng, S., & Reif, B. (2024). Aggregation Mechanisms and Molecular Structures of Amyloid‐β in Alzheimer’s Disease. Chemistry–A European Journal, 30(48), e202400277.
Sinnige T. (2022). Molecular mechanisms of amyloid formation in living systems. Chemical science, 13(24), 7080–7097. https://doi.org/10.1039/d2sc01278b
Week 5 HW: Protein Design - Part II
Part A: SOD1 Binder Peptide Design
A peptide binder is a short, engineered protein fragment usually <50 amino acids that binds to specific targets. It functions as a powerful, cost-effective, and stable alternative to larger antibiotics or small-molecule drugs. A peptide binder is used to modulate, degrade, or inhibit disease-related proteins, especially those that are deemed undruggable due to the absence of clear binding pockets.
My task this week is to design peptide binders for the SOD1 mutant.
Part 1: Generate Binders with PepMLM.
Question 1.
I retrieved the human SOD1 sequnce from Uniprot(P00441):
The A4V mutation is a mutation in the SOD1 gene that causes a rapidly progressive and aggressive form of familial amyotrophic lateral sclerosis(ALS). The mutation involves the substitution of A(alanine) for V (valine) at the 4th codon of the SOD1 sequence.
A (Alanine) is viewed as the 4th codon despite being in the 5th position in
the protein sequence, due to sequences being read/counted after the start codon M (Methionine). Which is often cleaved after translation.
Question 2 & 3.
I generated four peptides, each of length 12 amino acids and with a k value of 3, conditioned on the SOD1 mutant sequence using PepMLM.
The K value determines the number of the most probable tokens(amino acids) considered at each step when generating the peptide sequence. A low K value leads to the model almost always picking the most probable amino acids, resulting in peptides that are highly coherent but lack diversity, while a high K value leads to the model having a wider range of choices by including more amino acids in the selection pool. Which could lead to novel peptide sequences that might explore less common but potentially effective binding sites. However, there is also a higher risk of generating less optimal binding peptides.
Index
Binder
Pseudo Perplexity
1
WHYPVVALRLKK
19.111508
2
WRYPVVAAAWWE
13.402168
3
WRSPAVAVELGK
9.439937
4
WRYPAVGVALKK
10.208561
Question 4
I added the known SOD1-binding peptide “FLYRWLPSRRGG” to the list of generated amino acids. I used Gemini to write a code that would generate a pseudo perplexity score for “FLYRWLPSRRGG” based on the SOD1 Mutant sequence.
Index
Binder
Pseudo Perplexity
1
WHYPVVALRLKK
19.111508
2
WRYPVVAAAWWE
13.402168
3
WRSPAVAVELGK
9.439937
4
WRYPAVGVALKK
10.208561
5
FLYRWLPSRRGG
20.635231
Question 5
The pseudo perplexity score measures how expected or natural a peptide sequence looks to the PepMLM model when interacting with a protein. Lower scores usually indicate better potential binders, while higher scores indicate a potentially poor binder.
Based on their pseudo perplexity scores, I indicate PepMLM’s confidence in the binders.
High confidence indicates a potentially better binder.
Low coincidence indicates a potentially poor binder.
I used the alphafold server to evaluate the protein-peptide complex of the SOD1 mutant and the binding peptides.
WHYPVVALRLKK peptide
It has an ipmTM score of 0.37, and the peptide appears to localize near the β-barrel. It does not appear to be buried in the protein’s structure.
WRYPVVAAAWWE peptide
It has an ipmTM score of 0.22, and the peptide appears to localize near the dimer interface. It also does not appear to be buried in the protein’s structure
WRSPAVAVELGK peptide
It has an ipTM score of 0.24, and the peptide appears to localize near the β-barrel. It does not appear to be buried in the protein’s structure but is loosely associated with the surface of the protein.
WRYPAVGVALKK peptide
It has an ipTM score of 0.25, and the peptide appears to localize near the dimer interface. It also appears to be loosely associated with the surface of the protein.
FLYRWLPSRRGG peptide
It has an ipTM score of 0.35, and the peptide appears to localise near the dimer interface. It also appears to be loosely associated with the surface of the protein
The ipTM values indicate how confident AlphaFold is in the interaction between different protein chains in a complex.
The pTM scores also indicate the confidence in the overall shape of the whole complex.
The ipTM scores for the peptides were low. This means AlphaFold was not confident in the interactions between the peptides and the SOD1 A4 mutant.
I ranked the peptides from the highest score (Highest confidence level) to the lowest score (Lowest confidence level):
Peptide WHYPVVALRLKK had the highest ipTM score and was the only peptide generated by PepMLM that surpassed the score of the known SOD1 mutant binder FLYRWLPSRRGG.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
I used the Peptiverse to evaluate the generated peptides based on predicted binding affinity, solubility, hemolysis probability, net charge, and molecular weight.
Peptide WHYPVVALRLKK
It is soluble, non-hemolytic, has weak binding affinity, a net positive charge of 2.84, and a molecular weight of 1509.8 Daltons.
Peptide WRYPVVAAAWWE
It is soluble, non-hemolytic, has medium binding affinity, a net negative charge of -0.23, and a molecular weight of 1533.7 Daltons.
Peptide WRSPAVAVELGK
It is soluble, non-hemolytic, has weak binding affinity, a net positive charge of 0.76, and a molecular weight of 1312.5 Daltons.
Peptide WRYPAVGVALKK
It is soluble, non-hemolytic, has weak binding affinity, a net positive charge of 2.76, and a molecular weight of 1387.7 Daltons.
Peptide FLYRWLPSRRGG
It is soluble, non-hemolytic, has a weak binding affinity, a net positive charge of 2.76, and a molecular weight of 1507.7 Daltons.
I noticed all the peptides had solubility of 1 and were non-hemolytic. Their hemolytic probability ranges from 0.019 to 0.098.
When comparing ipTM scores with binding affinity, I observed that there was not a directly proportional relationship between the two. Some peptides with higher ipTM scores did not demonstrate stronger predicted binding affinity. For instance, peptide WRYPAVGVALKK had the highest ipTM score among the tested peptides but did not have the highest binding affinity. However, peptide WRYPVVAAAWWE, which had the lowest ipTM score, exhibited the highest binding affinity, with a medium binding affinity of 7.819.
Based on their properties, I would advance peptide WRYPAVGVALKK. This is because it has a good balance of properties as compared to the other peptides.
It has the lowest hemolytic probability (0.019), the second highest binding affinity (6.456), a good net charge of 2.76, and a low molecular weight (1387.7 Da), making it the ideal choice. Unlike peptide WRYPVVAAAWWE, which has the highest binding affinity but a net negative charge(-0.23) that would likely reduce its membrane penetration.
Part 4: Generate Optimized Peptides with moPPIt
I made a copy of the momPPIt Colab and inserted the SOD1 AV4 mutant sequence. I set the binder length to 12 amino acids and the number of samples to 4.
I selected hemolysis, solubility, specificity, and motif binding as the objectives. I select 3-6 as the motif binding regions to let the peptides bind to residues near the AV4 position.
MoPPIt generated the following peptide sequences
After comparing the peptides generated by moPPIt and my PepMLM peptides. I noticed the peptides generated by moPPIt had higher motif-binding scores and specificity. However, they had lower solubility scores and higher hemolytic scores, meaning they might be more prone to damage or rupture red blood cells.
I would evaluate the peptides generated by moPPIt before advancing them for clinical studies by first using computational tools such as GROMACS and alphafold to further validate their structure, target binding strength, stability, physicochemical properties, and potential toxicity.
After further validation, I would then select peptides that demonstrate high binding strength, stability, and no toxicity to advance to the next phase, which would involve animal studies to determine their therapeutic effects in living systems. Finally, I would ensure all safety and quality standards set by the regulatory agency are met before advancing the peptide for clinical(Human) trials.
Part B: BRD4 Drug Discovery Platform Tutorial
Part C: Final Project: L-Protein Mutants
The objective for this section of the assignment is to improve the stability and auto-folding of the lysis protein of an MS2-phage. Which might be the key to understanding how phages can potentially solve antibiotic resistance.
L-Protein Engineering | Option 1: Mutagenesis
I formed a group with a couple of students from the William and Mary Node. We decided to tackle Option 1: Mutagenesis of the L-Protein.
We run the Colab notebook to generate likely mutation positions in the L-protein. We used the generated Mutation heat map to identify possible positive mutations.
We also used the BLAST results for the L-protein provided in the Google Drive and performed sequence alignment using Clustal Omega to determine the conserved regions of the L-Protein to guide the mutant selection process.
Clustal Omega Sequence alignment results for the L-protein:
clustalo-I20260309-215356-0951-56325226-p2m
Conserved residues of the L-protein
The table below shows the meaning of the level of conservation of the L-protein. We avoided selecting mutants that occurred at residues of high conservation to avoid negatively affecting the function of the L-protein.
Symbol
Meaning
Conservation Level
*
Fully conserved residue (all sequences identical at that position)
⭐⭐⭐ Highest
:
Strongly conserved substitution (similar chemical properties)
⭐⭐ High
.
Weakly conserved substitution (some similarity)
⭐ Moderate
(space)
No conservation; residues differ significantly
❌ Low
We each came up with 5 mutants; 2 of my mutants had mutations in the soluble region of the L-protein, and the rest had mutations in the transmembrane region.
I selected these mutants based on the LLR (Log Likelihood Ratio) Score from the ESM Model Colab notebook.
The LLR scores quantify how much more or less likely a mutated amino acid is at a specific position compared to the wild-type (original) amino acid, according to the protein language model. A positive LLR score suggests that the mutated amino acid is more likely to appear at that position than the wild-type amino acid, implying the mutation might be beneficial or stabilizing. However, a negative LLR score indicates that the mutated amino acid is less likely, suggesting the mutation might be detrimental or destabilizing to the protein’s function or structure.
LLR Scores for My Selected Mutants :
Soluble Region
Position 5: F -> Q ( LLR Score = 1.79524445533752)
Position 17: N -> R ( LLR Score = 1.32365107536315)
Transmembrane region
Position: 40 V -> L ( LLR Score = 1.79524445533752)
Position 50 K -> L (LLR Score = 2.56146419048309)
Position 65 R -> L ( LLR Score = 1.0260357856750488)
I chose these mutations because they have a high positive LLR score and are outside the highly conserved region of the L-protein genome; as such, they are more likely to have a positive effect on L-protein stabilization and autofolding.
We used AF2_Multimer to generate a Multimeric Assembly for each of our mutants.
Multimeric assembly refers to the process by which individual protein subunits, known as monomers, associate through non-covalent interactions to form larger, functional complexes.
Here is a multimeric assembly for my five mutants:
Based on the pLDDT, PTM, and ipTM scores, all the models showed poor scores, meaning Alpha Fold did not have confidence in their structure fold accuracy and prtein complex interfaces. However, model 2 stood out with the highest confidence in terms of fold accuracy, protein complex interfaces, and residue positioning.
Week 6 HW: Genetic Circuit - Part I
Assignment: DNA Assembly
Question 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Answer
A PCR master mix is a pre-formulated, ready-to-use solution containing all the components required for PCR, except the DNA template and primers. It usually includes Taq DNA polymerase, deoxynucleotide triphosphates (dNTPs), magnesium ions, and an optimized reaction buffer at precise concentrations to ensure efficient and reproducible DNA amplification.
The Phusion High-fidelity PCR Master Mix is a ready-to-use 2X mastermix designed for high-speed, high-fidelity PCR applications. It was developed by Finnzymes Oy and manufactured and controlled by New England Biolabs (NEB) under Thermo Fisher Scientific’s Band.
The components in the Phusion High-Fidelity PCR Master Mixcomponents are:
Phusion High-Fidelity DNA Polymerase:
It is a specialized thermostable enzyme engineered for high speed, accuracy, and processivity. It is responsible for synthesizing new DNA strands during PCR. It also possesses 5’ to 3’ polymerase activity and 3’ to 5’ exonuclease proofreading activity, which reduces error rates.
2X Phusion HF or GC Buffer:
It maintains the optimal chemical environment for enzyme activity by stabilizing polymerase and DNA, correcting pH and the ionic strength needed for effective amplification. GC buffer is used when working with templates with high GC content, as it aids the amplification of GC-rich templates that are difficult to denature.
dNTPs (Deoxynucleotide triphosphates)
They are the building blocks of DNA synthesis, the mix contains dATP, dTTP, dCTP, and dGTP. They are incorporated by DNA polymerase during PCR to extend the new DNA strand complementary to the template.
MgCL2 (Magnesium Ions)
It acts as a necessary cofactor for DNA polymerase enzyme activity.
DMSO (Dimethyl sulfoxide)
It is used to improve the amplification of difficult GC-rich templates by reducing secondary structures. It is not always included in the master mix.-
Question 2. What are some factors that determine primer annealing temperature during PCR?
Answer
Annealing temperature in PCR is the temperature at which primers hybridize/bind to their complementary sequences on the single-stranded DNA template during a PCR cycle. Typically, annealing temperature ranges from 50 to 65 degrees celcuis. Annealing temperature is very important because it determines the specificity and efficiency of amplification.
Annealing temperature is directly determined by factors such as primer length and sequence composition. Primers with longer lengths form more hydrogen bonds with the template DNA, increasing the stability of the primer-template complex, and thus require a higher annealing temperature to ensure binding. However, shorter primers bind less strongly and require lower annealing temperatures.
Additionally, primers that contain a higher proportion of guanine(G) and cytosine(C) form stronger bonds with the DNA template due to GC base pairs forming three hydrogen bonds, which require a higher annealing temperature when compared with the weaker two hydrogen bonds formed by adenine(A) and thymine(T).
Primer melting temperature, which is the temperature at which 50% of a DNA primer is separated from the primer-template duplex. Also plays a role in determining annealing temperature, as annealing temperature is usually 3-5 degrees Celsius below the melting temperature.
Question 3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other
Answer
A restriction enzyme digest is a molecular biology technique that uses restriction enzymes to cut DNA at specific recognition sequences known as restriction sites. However, the polymerase chain reaction (PCR) is a laboratory technique used to amplify specific segments of DNA, to generate millions to billions of copies of a target DNA sequence from a very small initial sample. They are both molecular biology techniques used to produce linear DNA fragments, but have different protocols, mechanisms, and ideal applications.
In terms of protocol, restriction enzyme digests involve incubating DNA with one or more restriction enzymes in a suitable buffer at 37 degrees celcuis for 1 to 2 hours, resulting in the cleavage of the DNA at defined sites to produce linear fragments with either blunt or sticky ends. However, PCR takes place in a Thermocycler and involves repeated cycles of denaturation, primer annealing, and extension, which exponentially amplify the target region of a DNA using primers, nucleotides, buffer, DNA template, and thermostable DNA polymerase.
PCR and restriction enzyme digests also differ in the mechanism by which DNA fragments are generated. PCR creates DNA fragments by synthesizing new DNA strands based on primer-defined boundaries, allowing the precise control of the start and end of the amplified fragment. While in restriction enzyme digests, DNA fragments are created by cutting existing DNA molecules at naturally occurring sites, with the size of a fragment depending on the location of the restriction site in the DNA sequence.
It is preferable to use PCR when your goal is to amplify and generate a specific DNA fragment from unknown or low-abundance DNA. It is useful when working with degraded or low-quality DNA needed for downstream applications. However, restriction enzyme digests are best for analyzing or modifying DNA based on sequence-specific cleavage sites. It is used when verifying plasmid constructs, cloning DNA, DNA fingerprinting, and linearizing circular DNA for gel electrophoresis.
Question 4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Answer
Gibson Assembly is a molecular cloning technique that enables the seamless joining of multiple DNA fragments in a single, isothermal reaction without the need for restriction enzymes.
To ensure that DNA sequences that have been digested and PCR-ed will be appropriate for Gibson cloning, you would have to clean the DNA using a purification protocol. This will remove buffer components that inhibit the activity of Gibson enzymes while concentrating the DNA fragments so they can assemble more efficiently.
Additionally, the DNA sequences must have overlapping regions, which would have been introduced when designing the PCR primers. The primers would both require homologous sequences at their 5′ends. The overlap on the forward primer should be complementary to the backbone insertion site, and the reverse primer should also contain a complementary overlap to the forward primer. These overlaps will ensure the 5’ exonuclease creates single-stranded overhangs and complementary overlaps anneal together during Gibson assembly.
Question 5. How does the plasmid DNA enter the E. coli cells during transformation?
Answer
Plasmid DNA is usually introduced into E. coli cells during transformation via either heat shock or electroporation.
Transformation via a heat shock involves using a sudden change in temperature to temporarily create pores in the cell walls of E. coli, which enables the plasmid DNA to enter the E.coli cells. The change in temperature is achieved by briefly heating the E.coli cells to 42 degrees celcusi for 30 to 60 seconds. Before heat shock, E. coli cells are treated with salts such as calcium chloride to neutralize the negative charges on the cell wall and membrane. The calcium ions reduce the electrostatic repulsion between DNA and the bacterial cell membrane. However, electroporation utilizes a short high voltage electric pulse to temporarily create pores in the cell wall and membrane of E.coli to allow the plasmid DNA to enter the E.coli cells. Electroporation has a higher transformation efficiency and works effectively with large plasmid DNA.
After transformation via either electroporation or heat shock, the E.coli cells are given a recovery period and incubated in nutrient-rich media so they can repair the pores in their membranes before they are cultured on agar containing antibiotics for selection.
Question 6. Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
Answer
Golden Gate assembly is a molecular cloning method that enables the seamless joining of multiple DNA fragments into a single construct in a single reaction using Type IIS restriction enzymes and T4 DNA ligase. It uses Type IIS restriction enzymes such as Bsal, BsmBI, and BbsI to cleave DNA at positions outside the restriction sites to create unique custom non-palindromic overhangs.
The overhangs created are designed to be unique and complementary, ensuring the DNA fragments ligate together in a specific order. Since the recognition sites are removed during Golden Gate assembly, the final construct is scarless and has no extra sequences left behind, making the method highly efficient.
I tried to model the Golden Gate assembly in Benchling. I created a new project in Benchling called “Golden Gate Assembly”.
I used Aequorea victoria green fluorescent protein (GFP) mRNA complete cds, which I obtained from NCBI as the insert, and the pYTK095 plasmid from Addgene (https://www.addgene.org/65202/), which encodes AmpR-ColE1 as the backbone.
I used Benchling’s Molecular Biology tools to simulate the Golden Gate Assembly. Finally, I validated the construct, confirming the proper insertion of the green fluorescent protein.
Assignment: Asimov Kernel
Question 1 & 2
I created a new repository named “William & Mary_Nana Agyei” and created a new notebook titled “Nana Agyei’s HW 6 Entry" to document my use of Asimov Kernel.
Question 3
I explored the Comparing promoters, Repressilator, and J23117 Promoter constructs in the Bacterial Demos Repository to familiarize myself with the parts of the various constructs and how they work. The UI of Asimov Kernel is pretty easy to use and visually appealing, making the exploration and tests easy and interesting.
Question 4
I noted down all the parts used to create the Repressilator in the Bacterial Demos repository and created a blank construct to recreate the Repressilator, which I named “Repressilator recreation.” I used the search function to find all the parts I had noted down.
I ran into a problem while searching for the pUC-SpecR v1 backbone. I could only find the pUC-SpecR v2 backbone, so I just copied the sequence for the pUC-SpecR v1 backbone from the Repressilator device and pasted it in the appropriate position of my construct.
After recreating the Repressilator, I tried to color the different components of the circuits
I run a simulation on my recreation of the Repressilator, and it seemed to work as expected
Question 5
I tried to build my own constructs using the parts in the Characterized Bacterials Part repository.
Construct 1. Luciferase gene expression
I create a genetic circuit construct to control the expression of the Luciferase gene. I based it on the luciferase gene construct I built in Benchling during week 2.
Based on the results for construct 1 below, the circuit is working properly, with each component turning on as expected.
Construct 2. Inducible Arabinose GFP reporter
I created a genetic circuit construct that would express green fluorescent protein (GFP) when arabinose is present.
Based on the results for construct 2, the genetic circuit is working properly. All its components turn on, and GFP is produced, with its concentration increasing over time before stabilizing at a steady level.
**Construct 3. Constitutive expression of the GFP gene
I created a genetic circuit construct that would constantly express green fluorescent protein (GFP) without any external input needed.
Based on the results for construct 3, the genetic circuit is working properly. All its components turn on, and GFP is produced.
Starčič Erjavec, M. (2020). Annealing Temperature of 55°C and Specificity of Primer Binding in PCR Reactions. In Synthetic Biology - New Interdisciplinary Science. IntechOpen. https://doi.org/10.5772/intechopen.85164
Week 7 HW: Genetic Circuits Part-II
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
Question 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Answer
An IANN is a nonlinear computer model designed to mimic the structure and function of biological neurons in the brain. It usually has multiple inputs and outputs, where neurons process weighted inputs to generate output signals.
Unlike traditional circuits that use signals to hit on and off thresholds, IANNs operate on analog computation and signals, which is more efficient for biological systems and allows them to process continuous chemical concentrations with much higher precision without losing information at binary thresholds. Additionally, IANNs are more robust to noise and biological variability because their distributed architecture prevents the failure of a single component from crashing the system. IANNs can also be trained to classify and adapt to nonlinear biological data through pattern recognition and learning, unlike traditional circuits, which are hard-coded for specific logic.
Question 2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Answer
A useful application for an IANN would be in an environmental biosensor for detecting and classifying the levels of heavy metal pollutants in water.
The IANN would take intracellular signals generated in response to the presence of different heavy metal pollutants, such as mercury, lead, cyanide, and arsenic, at varying concentrations. Each signal would be weighted and processed through a network of regulatory elements that would allow the cell to integrate all the signals simultaneously rather than responding to each pollutant independently.
The output would be graded expressions of different reporter proteins, where a particular color corresponds to a specific pollutant. The presence of multiple pollutants would also correspond to a specific colour. For example, green for mercury, blue for lead, and orange for mercury and lead being present at the same time. This would enable the biosensor to distinguish complex environmental conditions, rather than simply indicating the presence or absence, as in a traditional genetic circuit.
Some limitations I might face include difficulty in engineering the precise weights and stable interactions between components in a variable and noisy cellular environment. Additionally, the IANN would need to be trained on high-quality data for the unique water chemistry of the specific mining regions to prevent inaccurate readings.
Question 3. Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Answer
Below is a diagram of an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2. I drew it using Microsoft Word. I designed with a low-pass circuit in mind.
X1, X2 and B are the inputs. X1 is DNA encoding RNase A (a Csy4-like endoribonuclease), X2 is DNA encoding RNase B (a second endoribonuclease), and B (the Bais) is DNA encoding the fluorescent protein (FP) whose mRNA will be regulated. The TX are transcription nodes, they represent RNA polymerase reading the DNA from the inputs and producing mRNA molecules.
The red TL node on X1 represents translation. Its output RNase A is inhibitory and will cleave and silence other downstream mRNA. The black TL node on the X2 branch represents translation of RNase B mRNA into RNase B protein.
The circle with the blue outline is the core computational node of Layer 1. At this node, RNase A (−, red) arrives from the top and delivers an inhibitory signal. RNase A cleaves the RNase B mRNA, reducing how much RNase B protein gets made. This is the negative weight in the perceptron analogy.
X2 mRNA (+, black) arrives from the bottom and delivers a positive/permissive signal. It is the substrate being regulated. This is the positive weight.
The circle with the orange outline is the core computational node of layer 2. At this node, Modulated RNase B (−) arrives from Layer 1 at the top and delivers an inhibitory signal. That cleaves the fluorescent protein mRNA, suppressing translation. B mRNA (+) arrives from the bottom. This is the fluorescent protein mRNA produced by transcription of B, and it is the substrate being regulated in Layer 2.
The final black TL node represents the ribosome translating the surviving fluorescent protein mRNA into fluorescent protein. The blue circle represents the fluorescent protein Y. It is the final output of the entire multilayer perceptron. Y will be express when inputs of X1, X2 are lower than the input of B.
Assignment Part 2: Fungal Materials
Question 1. What are some examples of existing fungal materials, and what are they used for? What are their advantages and disadvantages over their traditional counterparts?
Answer
Fungal materials are sustainable, bio-based materials grown from mycelium.
Some examples of materials and their uses are:
Mycelium leather, like Mylo by Bolt Threads, is grown from fungal mats that are harvested and treated to create a soft and flexible material that mimics the texture and tensile strength of animal leather. It is used to produce fashion products such as bags, shoes, and jackets.
Mycelium Building blocks and insulation made from mycelium-bound biocomposites formed by growing fungi on cellulose-rich bases. They are used as acoustic tiles, insulation panels, and brick to construct structures.
Funagl paper and filtration membranes made from processing fungal filaments into a paper-like sheet using *Trametes versicolor, which are hydrophobic and fire-resistant. They are used in research for water filtration membranes that can attract heavy metals due to their chitin content.
Fungal materials, unlike their traditional counterparts such as plastics, forms, and animal/synthetic leather, offer several advantages. Biomaterials are highly sustainable due to their biodegradable nature and production from agricultural waste. By upcycling agricultural waste such as sawdust and rice husks, we can create valuable products that significantly reduce the carbon footprint. Additionally, they can be grown into specific shapes and do not require high-heat polymerization like plastics, which reduces manufacturing waste and energy consumption. In the case of mycelium leather, fungal materials can provide an ethical alternative to animal-derived products.
However, aside from their numerous benefits, fungal materials also have notable disadvantages. They generally exhibit lower mechanical strength and durability compared to their traditional counterparts. They are also sensitive to moisture and environmental conditions if not properly treated. Additionally, their growth-based manufacturing process is slower than traditional materials methods, which poses a challenge for scalability and widespread adoption. This also currently makes them more expensive due to the limited production scale.
Question 2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Answer
I would like to engineer fungi to act as a cooperative network that redistributes nutrients to improve soil structure and protect plants from disease. Mycorrhizal fungi species naturally form underground hyphal networks that connect plant roots and facilitate nutrient exchange. They can be engineered to sense deficiencies and transport key elements like nitrogen and phosphorus to areas where they are needed. They could be designed to secrete compounds that improve soil aggregation, water retention, and overall soil health, while producing antimicrobial molecules to suppress plant pathogens.
This idea would help create more sustainable agriculture by reducing reliance on chemical fertilizers and pesticides. By improving nutrient distribution, enhancing soil structure, and protecting plants from disease. Which would increase crop productivity while maintaining long-term soil health and minimizing environmental damage.
Performing synthetic biology in fungi as opposed to bacteria has certain advantages, such as the ability of fungi to secrete large quantities of proteins directly into their environment, which simplifies the protein purification process and makes fungi highly efficient cell factories. Additionally, fungi are eukaryotes and possess complex intracellular machinery, such as the Golgi apparatus and the endoplasmic reticulum, which enables them to perform critical post-translational modifications that are essential for the folding and stabilization of human-like proteins. This makes fungi very valuable for producing pharmaceuticals and enzymes.
Assignment Part 3: First DNA Twist Order
Answer
I have reviewed the Individual Final Project documentation guidelines.
I have also submitted the Google form with my Draft Aim, Final project summary,
HTGAA industry council selections, and shared a link to a Benchling folder with my DNA designs.
I designed a DNA insert sequence in my Benchling folder.
I downloaded the insert sequence as a FASTA file and inserted it into the pTwist Amp High Copy conal vector on the Twist Biosciences site. Visualized the plasmid and downloaded the plasmid sequence in GeneBank format and imported it into Benchling.
Week 9 HW: Cell-Free Systems
General Homework Questions
Question 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Answer
Cell-free protein synthesis is a biotechnology technique for producing proteins in a test tube using biological machinery extracted from a cell.
In terms of flexibility, cell-free protein synthesis is an open reaction environment. This allows for the direct addition, removal, and modification of any reaction component at any time. Additionally, unlike in living cells, there are no constraints related to cell viability, enabling the rapid prototyping of genetic constructs, testing different pH and temperature conditions, and the expression of proteins that would be toxic or lethal to living cells. In terms of control over experimental variables, in cell-free protein synthesis. Each component of the reaction, such as ATP concentration, pH, and amino acid ratios, can be independently tuned with exact precision, enabling more reproducible and targeted biological investigations.
Cell-free expression is more beneficial when producing proteins that would be lethal to host cells, incorporating non-natural amino acids, and for on-demand protein production, such as in portable biosensors. Additionally, it speeds up the testing of genetic constructs and designs by eliminating the waiting period for cloning and cell growth.
Question 2. Describe the main components of a cell-free expression system and explain the role of each component.
Answer
The main components of a cell-free expression system are:
Cell extract (Lysate):
It is the core of the system and provides the biological machinery needed to convert genetic information into protein. It is derived from organisms and contains ribosomes to carry out protein synthesis, tRNAs to deliver amino acids to the ribosomes, and enzymes to support transcription, translation, and energy metabolism.
DNA or mRNA templates:
They encode the protein of interest and serve as the blueprint for protein synthesis.
Energy Source and regeneration system:
It supplies the energy required for transcription and translation, as well as regenerates Adenosine Triphosphate (ATP) and Guanosine Triphosphate (GTP) from Adenosine Diphosphate (ADP) and Guanosine Diphosphate (GDP). To maintain high-efficiency protein production for hours. It includes molecules like ATP, GTP, and energy substrates such as Phosphoenolpyruvate and creatine phosphate.
Amino acids:
They are the building blocks used to assemble proteins by the ribosome.
Cofactors and Salts:
They maintain optimal conditions for enzyme activity, ribosome stability, and overall reaction efficiency. They usally inculde magnesium ions (Mg2) and potassium ions (K+).
Question 3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Answer
Energy provision regeneration is important in cell-free systems because transcription and translation, which drive protein synthesis, are energy-intensive processes that quickly deplete ATP present in the system. Therefore, efficient energy provision regeneration is required to prevent premature reaction termination and low protein yields.
An effective method to ensure continuous ATP supply is to use a phosphoenolpyruvate (PEP) based energy regeneration system. In the method, PEP acts as a high-energy phosphate donor in the presence of pyruvate kinase and transfers a phosphate group to ADP to regenerate ATP, providing the system with energy.
Question 4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Answer
Prokaryotic cell-free expression systems offer low cost, high and faster yield of proteins that are easy to optimize. However, they do not possess the cellular machinery for complex post-translational modifications such as advanced protein folding and glycosylation. On the other hand, eukaryotic cell-free expression systems closely mimic the environment of higher organisms and support the folding of complex proteins and membrane protein insertion, but are more expensive and slower compared to prokaryotic systems.
For a prokaryotic cell-free system, I would choose the green fluorescent protein (GFP) because it is a simple protein and does not require complex post-translational modifications such as glycosylation to function.
For a eukaryotic cell-free expression system, I would choose the Human Erythropoietin (EPO), which is a glycoprotein responsible for the production of red blood cells in the bone marrow. It requires glycosylation to function properly and is ideal for a eukaryotic cell-free expression system.
Question 5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Answer
A membrane protein is a protein molecule that is attached to or interacts with the membrane of a cell or organelle. It usually acts as a transporter, receptor, or enzyme that enables cells to communicate, obtain nutrients, and manage energy.
To optimize the expression of a membrane protein in a cell-free experiment, I would first consider an appropriate system to obtain the cell-free lysate. I would use a cell lysate from E. coli; it is cheap, has high yield, and would enable rapid screening by serving as a high-throughput testing platform. I would later use a eukaryotic cell lysate if the membrane protein requires post-translational modifications for its functionality. I would then design a DNA construct with a strong promoter for the detection of the protein.
I would add detergents, liposomes, or nanodiscs to the reaction to provide a membrane-like environment to support proper protein folding and co-translational insertion. The cell-free reaction would also include molecular chaperones and energy regeneration systems to enhance folding and sustain protein synthesis.
I would then perform parallel reactions with variables such as lipid composition, detergent type, DNA concentration, and temperature varied. I would evaluate the expression levels, solubility, and functionality of the membrane protein, selecting the best-performing conditions and refining them to maximize yield while ensuring proper folding and function of the membrane protein.
A challenge I would face during the experiment is protein aggregation due to hydrophobic transmembrane regions clamping together in aqueous environments. I would overcome this by including detergents, liposomes, or nanodiscs to provide a stable lipid environment. Another challenge is improper folding due to a lack of a full cellular machinery for correct protein conformation. I would address this challenge by adding a molecular chaperone and lowering the reaction temperature to promote proper protein folding. A final challenge would be low yield, which is common among membrane proteins due to their instability and translational difficulty. I would address the issue of low yield by adjusting ionic conditions, DNA concentrations, and maintaining efficient energy systems to ensure continuous protein synthesis.
Question 6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Answer
In a cell-free system the low yield of a target protein may be due to the following reasons:
Inefficient transcription or translation from the DNA template, this may be due to issues with the DNA construct, such as a weak promoter or poor ribosome binding site. Additionally, suboptimal codon usage or degraded DNA templates could also significantly impact transcription and translation.
Troubleshooting strategy:
Redesign the DNA construct with a strong promoter and an optimized ribosome binding site. Additionally, mRNA can be used directly instead of DNA to bypass the limitations of transcription.
Energy Depletion due to an insufficient energy regeneration system or insufficient ATP or GTP, which halts the reaction.
Troubleshooting strategy:
The energy regeneration system should be optimized fr the reaction or supplemented with additional ATP, GTP, and cofactors.
Protein misfolding or aggregation may occur if the protein is too large, complex, or membrane-associated, which leads to a low functional yield.
Troubleshooting strategy:
Lowering the temperature of the reaction can improve folding, and the inclusion of molecular chaperones, detergents, liposomes, or nanopores can also aid in improving folding in membrane proteins.
Homework Question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Question 1. Pick a function and describe it.
a. What would your synthetic cell do? What is the input, and what is the output?
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
c. Could this function be realized by a genetically modified natural cell?
d. Describe the desired outcome of your synthetic cell operation.
Question 2. Design all components that would need to be part of your synthetic cell.
a. What would the membrane be made of?
b. What would you encapsulate inside? Enzymes, small molecules.
c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (Hint: for example, if you want to use small molecule modulated promoters, like Tet-ON, you need mammalian)
d. How will your synthetic cell communicate with the environment? (Hint: are substrates permeable? or do you need to express the membrane channel?)
Question 3. Experimental details
a. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel,” pick the actual gene.)
b. How will you measure the function of your system?
Answer
Question 1.
a.
I would design a synthetic microbial cell that detects heavy metal contamination(lead, arsenic, mercury, and cadmium) in water samples from galamsey-affected communities. The input is river water possibly containing dissolved heavy metal ions at varying concentrations, and the output will be distinct colorimetric signals for each pollutant: blue for lead, orange for arsenic, purple for mercury, and brown for cadmium. The color will be produced inside the vesicle and visible through the membrane, making it readable by the naked eye.
b.
Yes, it can be realized without encapsulation in a cell-free Tx/TL system by directly exposing the reaction mixture to the water sample to be tested. This will allow the metal ions to interact with the sensing proteins to trigger reporter expression.
c.
Yes, its function can be realised by a genetically modified natural cell, such as a modified E. coli strain. The modified E. coli strain would express PbrR, ArsR, MerR, and CadC coupled to a colorimetric reporter.
d.
The desired outcome is that the synthetic cell produces a visible, distinct colorimetric signal in the presence of lead, arsenic, mercury, and cadmium, indicating which metals are present in the river sample.
Question 2.
a.
The synthetic cell will have a membrane composed of a phospholipid bilayer that is permeable to small ions but can retain DNA, ribosomes, and proteins inside. It could have a possible lipid composition of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPG), and Cholesterol. POPC would provide bilayer stability and fluidity across a wide temperature range, while POPE would add mechanical stability and reduce the membrane’s permeability to unwanted solutes. POPG will add a negative surface charge to stabilize the vesicle and improve its compatibility with the encapsulated E. coli Tx/Tl machinery. Cholesterol will modulate the fluidity of the membrane and reduce passive permeability to ions to ensure the entry of metal ions is controlled through expressed membrane channels rather than passive diffusion.
b.
The synthetic cell would encapsulate:
Genetic material for two plasmids that contain PbrR-LacZ + ArsR-CrtI circuits and MerR-BpsA + CadC-MelA circuits for heavy metal detection.
Tx/Tl machinery from E. coli.
A phosphocreatine and creatine kinase energy regeneration system to sustain the synthesis of the reporter proteins.
Heavy metal-sensing proteins: PbrR protein (lead sensor), ArsR protein (arsenic sensor), MerR protein (mercury sensor), CadC protein (Cadmium sensor).
Reporter substrates for each reporter protein.
Buffer and salts.
c.
The Tx/ Tl system will come from the E. coli BL21 (DE3) bacterial system. This is because the four metal-sensing proteins are of bacterial origin and function optimally in a bacterial biochemical environment. Additionally, the T7 promoter driving the circuits is compatible with the E. coli Tx/Tl machinery.
d.
The synthetic cell will communicate with the environment by incorporating specific membrane ion transporters for each target metal. To overcome the impermeability of the lipid bilayer to charged metal ions, which would prevent them from reaching the encapsulated sensing proteins. The possible transporters are the lead transporter protein (PbrT), the Glycerol uptake facilitator protein (GlpF) for arsenite entry, the mercuric transporter protein (MerT), and the CadA-associated transporter.
I will measure the function of the system through the appearance of a distinct color inside or outside the vesicles of the synthetic cells in correspondence to the detection of a heavy metal pollutant. Additionally, I can use an inductively coupled plasma mass spectrometry (ICP-MS) to measure both internal and external metal ion concentration after incubation to confirm the uptake and accumulation of metal ions.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
Answer
A band-aid or wound dressing with embedded freeze-dried cell-free circuits that would sample wound exudate, to detect the molecular signatures of specific bacterial pathogens, and in response produce and release precise antimicrobial peptides needed to counter the pathogens and prevent the infection of the wound.
How will the idea work, in more detail? Write 3-4 sentences or more.
Answer
The band-aid or dressing would have three layers. The innermost layer will be in contact with the wound and wick fluid outward through capillary action, in a similar way to how flow test strips work. The middle layer will be the core and contain freeze-dried cell-free microzones that are each programmed to recognize a different bacterial pathogen by detecting its unique RNA signature. When wound fluid rehydrates a zone and a matching pathogen RNA is present, the circuit switches on and produces both the specific antimicrobial peptide that kills that exact bacteria, and a visible color unique simultaneously. The outermost layer will be a breathable waterproof backing that retains moisture to keep the chemistry running. The colour will be displayed on the surface of the outermost layer for clinicians to read.
What societal challenge or market need will this address?
Answer
This idea would address the rise of antimicrobial resistance by enabling immediate pathogen-specific treatment through the dressing/ band-aid, which will eliminate the need for broad-spectrum antibiotics and delays associated with traditional diagnostic procedures. Additionally, it would also improve the outcomes for chronic wound patients by enabling continuous monitoring and targeted therapy to prevent infections and lower healthcare costs.
How do you envision addressing the limitations of cell-free reactions (e.g., activation with water, stability, one-time use)?
Answer
For this idea, activation with water would not be a limitation but rather a function of the design since wound fluid is the activation trigger of the band-aid/ dressing. The ability of the band-aid/ dressing would be ensured by freeze-drying and protective encapsulations that would preserve its sensitive components until use. The one-time use of cell-free systems would not be a limitation here but align with standard wound care practices, with each dressing delivering a fresh, effective dose.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Question 1. Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Answer
Astronauts are exposed to ionizing radiation such as cosmic rays and solar particle events during space exploration. Their exposure to these doses of radiation, which are almost 100 times higher than what reaches the Earth’s surface, does not imbue them with superpowers but causes cumulative DNA double-stranded breaks. This increases cancer risk and threatens the mission integrity of long-term space exploration missions. Currently, during space missions, radiation damage is monitored through passive dosimetry bags that measure only physical damage. I would like to propose the development of a rapid, low-resource cell-free tool to measure biological damage due to radiation exposure in real time.
Question 2. Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Answer
The target molecule would be phosphorylated histone H2AX (γH2AX). It is an early molecular marker for DNA double-stranded breaks and replication stress. It is detectable in blood, urine, and saliva. I think collecting saliva samples will be the best way to measure γH2AX.
Question 3. Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Answer
When ionizing radiation causes a double-stranded break in DNA, the cells trigger an emergency alarm and rapidly tag the nearby H2AX protein by attaching a phosphate group to it and convert it into γH2AX. γH2AX acts like a flare planted directly at the damage site with more DNA breaks, meaning more flares. Therefore, measuring can tell us exactly how much radiation damage an astronaut’s cells have suffered.
Question 4. Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Answer
My hypothesis involves using Biobits as a cell-free expression system to encode for an anti-γH2AX nanobody fused to a split fluorescent reporter to detect relevant concentrations of γH2AX in astronauts’ saliva samples to provide a quantitative, real-time index of cumulative DNA damage during spaceflights.
The reasoning behind this is that current diometry tells mission controllers how much radiation has been absorbed by the spacecraft. However, it cannot tell how much DNA damage a particular astronaut has sustained in a week versus last week. As identical doses of radiation may produce differing biological outcomes in individual astronauts due to their varying DNA repair capacity. A personalized repeated biological readout that would allow for dynamic mission decisions, such as flagging crew members with unexpectedly high DNA damage accumulation for reduced exposure duties, and aid in building a longitudinal biological dataset to set evidence-based radiation exposure limits for possible future deep space exploration missions.
Question 5. Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Answer
I would collect saliva from the astronauts before, during, and after high radiation events, and saliva collected from before going into space will serve as the control. The saliva will be added to rehydrated Biobits reactions encoding an anti-γH2AX nanobody split green fluorescent protein fusion. The γH2AX in the samples will bridge split GFP halves, reconstituting fluorescence proportional to γH2AX concentration. The intensity of the fluorescence measured will be used to plot a γH2AX concentration curve over the mission duration for each crew member.
Homework Part B: Individual Final Project
Question 1. I have added my final project slide to the committed listener ONE FINAL PROJECT IDEA deck.
Question 2. I have submitted the final project selection form.
Question 3. I have also begun planning how to write my final project documentation based on the HTGAA project guidelines.
Question 4. I have begun preparing my first DNA order.
Reference
Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 4th edition. New York: Garland Science; 2002. Membrane Proteins. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26878/
Calhoun, K. A., & Swartz, J. R. (2007). Energy systems for ATP regeneration in cell-free protein synthesis reactions. Methods in molecular biology (Clifton, N.J.), 375, 3–17. https://doi.org/10.1007/978-1-59745-388-2_1
Gregorio, N.E., Levine, M.Z. and Oza, J.P., 2019. A user’s guide to cell-free protein synthesis. Methods and protocols, 2(1), p.24.
Mah, LJ., El-Osta, A. & Karagiannis, T. γH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24, 679–686 (2010). https://doi.org/10.1038/leu.2010.6
Osaki, T. and Takeuchi, S., 2017. Artificial cell membrane systems for biosensing applications. Analytical chemistry, 89(1), pp.216-231.
Sachse, R., Dondapati, S.K., Fenz, S.F., Schmidt, T. and Kubick, S., 2014. Membrane protein synthesis in cell-free systems: From bio-mimetic systems to bio-membranes. FEBS letters, 588(17), pp.2774-2781.
Sharma, B., Moghimianavval, H., Hwang, S. W., & Liu, A. P. (2021). Synthetic Cell as a Platform for Understanding Membrane-Membrane Interactions. Membranes, 11(12), 912. https://doi.org/10.3390/membranes11120912
Yue, K., Zhu, Y., & Kai, L. (2019). Cell-Free Protein Synthesis: Chassis toward the Minimal Cell. Cells, 8(4), 315. https://doi.org/10.3390/cells8040315
Week 10 HW: Advanced Imaging and Measurement Technology
Homework: Final Project
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Answer
One of the aspects I will measure is the expression levels and enzymatic activity of the four colorimetric reporters in response to known concentrations of their respective target metals in a cell-free system. The colorimetric reporters are LacZ (Blue, for lead detection), Crtl (Orange, for arsenic detection), BpsA(Purple, for mercury detection) ad MelA(Brown, for cadmium detection)
Please describe all of the elements you would like to measure, and furthermore, describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail
Answer
I would measure the expression levels and enzymatic activity of the four colorimetric reporters by performing a series of serial dilutions of each target metal with cell-free reactions containing the two dual plasmids I would design. In a 384-well plate format using the Echo525 acoustic liquid handler. The reporter output will be quantified by absorbance spectroscopy on the Spark Plate Reader to capture all four reporter absorbance peaks simultaneously.
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Question 1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
Answer
Using the Expasy online calculator, the predicted amino acid sequence of eGFP has a molecular weight of 28006.60 Daltons and a theoretical pl of 5.90.
Question 2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Answer
The two charge states I selected from Figure 1 are 933.7349 and 903.7148. 933.7349 represents m/zn and 903.7148 represents m/zn+1.
Determine z for each adjacent pair of peaks (n,n+1).
Answer
m/zn = 933.7349
m/zn+1 = 903.7148
z = m/zn+1 / m/zn - m/zn+1
putting the values into the formula
z = 903.7148 / (933.7349 - 903.7148)
z = 903.7148 / 30.0201
z = 30.103
Z is aproximalty = 30
Therefore, the charge state at n is 30 and the charge state at n+1 is 31
Determine the MW of the protein using the relationship between m/zn, MW, and z.
Answer
m/zn = zn × H / zn
MW = (m/zn)
MW = (m/zn - H )× z
MW = (933.7349 - 1.00728) × 30
MW = 27,981.8286
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
Answer
Homework: Waters Part II — Secondary/Tertiary structure
We will analyze eGFP in its native, folded state and compare it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS-only analysis (no liquid chromatography, also known as “direct infusion” experiments) on the Waters Xevo G3-QToF MS.
Question 1. Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Answer
A native protein conformation refers to the properly folded, functional, and most thermodynamically stable three-dimensional shape of a protein under physiological conditions. However, a denatured protein conformation refers to the unfolded, disordered, inactive state of a protein. When a protein unfolds, it loses its secondary, tertiary, and quaternary structures, which make up its specific 3D shape, while its primary amino acid sequence remains intact. This exposes previously buried residues and increases its surface area.
A mass spectrometer can determine the differences in protein conformation by measuring the number of charges a protein acquires during ionization. In a native conformation, the protein remains compact and has fewer exposed sites for protonation, resulting in lower charge states, a narrow charge distribution, and sharp peaks at higher m/z ratios. While in a denatured conformation, the unfolded protein has exposed sites for protonation, resulting in higher charge states, a broader charge distribution, and peaks at lower m/z ratios.
In Figure 2, the denatured eGFP shows several closely spaced peaks reflecting high charge states of the unfolded protein. The native eGFP shows a few narrow, sharp peaks reflecting low charge states of the compact protein.
Question 2. Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell?
Answer
You cannot discern the charge state at 2800 m/z just by zooming in, but you can calculate it by using the difference in isotope spacing of the charge peaks at ~2800 m/z and dividing 1 by the answer.
Spacing = 2799.6365 - 2799.4199 = 0.2166
z = 1/ spacing = 1 / 0.2166
z = 4.616
The charge state of the peak at ~ 2800 is 4.
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
Question 1. How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
There were 19 monoisotopic peptides larger 500 daltons generated from tryptic digestion of eGFP.
Question 3. Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Answer
Based on Figure 5a, there are 25 peaks between 0.5 and 6 minutes in the eGFP peptide map.
Question 4. Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
Answer
The number of peaks and peptides is not identical. There are more peaks in the chromatogram.
Question 5. Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide (|M+H|+) based on its m/z, and z.
Answer
The most abundant peak of Figure 5b is 252.76712, and the second most abundant peak is 526.25918.
using the zoomed-in insert, the isotope peaks are:
monoisotopic 525.76712 m/z
+1 isotope 526.25918 m/z
+2 isotope 526.76845 m/z
+3 isotope 527.26098 m/z
finding the spacing between consecutive isotope peaks:
Therefore, the mass of the singly charged form of the peptide is 1050.527 Daltons.
Question 6. Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is the mass accuracy of measurement? Please calculate the error in ppm.
Answer
Accuracy = | MWexperiment - MWtheory| / MWtheory
Using 1050.5214 | 115-123 | FEGDTLVNR as the therotical molecular weight and |M+H|+ = 1050.527 Daltons as the experimantal molecular weight.
Question 7. What is the percentage of the sequence that is confirmed by peptide mapping?
Answer
The percentage of the sequence confirmed by peptide mapping is 88%.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
Answer
Using Table 1 and Figure 7, the masses of the oligomeric states are :
7FU Decamer is 3.40 MDa
8FU Didecamer is 8.33 MDa
8FU 3-Decamer is 12.67 MDa
8FU 4-Decame is 20 MDa
Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Answer
Our node did not visit Waters Immerse Lab, so I am just filling the table with the data from my previous calculations for eGFP.
Theoretical
Observed
PPM Mass Error
Molecular weight (kDa)
28006.60
27981.8286
-884.48
Week 11 HW: Bioproduction and Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I contributed to the HTGAA global artwork canvas, which drew inspiriration from the reddit r/Place experiment. I helped in filling the yellow space pixels in the blue box at the lower left corner of the artwork, and also helped in some of the other designs. In all, I contributed 10 pixels to the global artwork canvas and was ranked 66th among the top contributors. I enjoyed working with everyone to create beautiful designs from the chaos of pixels 😹.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Question 1. Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
Answer
E.coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA polymerase): Provides the core transcription and translation machinery, such as ribosomes, tRNAs, and enzymes, to the cell-free system. The included T7 RNA polymerase will drive high transcription efficiency from T7 promoters.
Salts / Buffer
Potassium Glutamate: stabilizes the activity of ribosomes and enzymes for efficient translation.
HEPES-KOH pH 7.5: It is a buffer that maintains the cell-free system at a physiologically optimal pH for enzymatic activity.
Magnesium Glutamate: supplies Mg2+ ions, which are essential cofactors for ribosome structure, ATP utilization, and transcription efficiency to the cell-free system.
Potassium phosphate monobasic: It supplies inorganic phosphate, which is essential for regenerating ATP from ADP during the energy-consuming processes of transcription and translation
Potassium phosphate dibasic: It forms a phosphate buffer with potassium phosphate monobasic to contribute phosphate groups, which are necessary for energy metabolism and nucleotide balance.
Energy / Nucleotide System
Ribose: Serves as a precursor for nucleotide regeneration and supports sustained transcription.
Glucose: Serves as an energy substrate and fuels the regeneration of ATP through glycolytic enzymes present in the cell-free system.
AMP: Adenosine Monophosphate serves as the fundamental building block for AP regeneration, which is used to power transcription and translation reactions.
CMP: Cytidine Monophosphate acts as the precursor for CTP, which is an essential nucleotide triphosphate for RNA synthesis during transcription in cell-free systems.
GMP: Guanosine Monophosphate acts as the precursor for GTP, which is essential for RNA synthesis and acts as the immediate energy source for protein synthesis.
UMP: Uridine Monophosphate serves as the precursor for UTP, which is essential for RNA polymerization and used in the synthesis of activated sugars like UDP-glucose.
Guanine: Functions as the purine base substrate in the salvage pathway to replenish GDP/GTP pools and provide essential energy for translation and protein synthesis.
Translation Mix (Amino Acids)
T7 Amino Acid Mix: Supplies most of the amino acids required for protein synthesis.
Tyrosine: Serves as a building block for protein synthesis and a functional marker for biochemical activity.
Cysteine: Acts as a reducing agent and enhances protein synthesis by maintaining a reduced environment, protecting thiol groups. It also acts as a precursor for glutathione synthesis.
Additives
Nicotinamide: acts as a cofactor precursor that supports redox balance and metabolic activity in cell lysate.
Backfill
Nuclease-free Water: is used to adjust the final volume of the reaction while preventing disruptions in transcription due to nucleic acid degradation.
Question 2. Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
Answer
The 1-hour PEP-NTP master mix supplies the cell-free reaction with immediate usable materials such as nucleotide triphosphates (NTPs) and phosphoenolpyruvate (PEP) to achieve really fast protein synthesis. However, it is not efficient due to its limited longevity of protein synthesis and high cost. On the other hand, the 20-hour NMP-Ribose-Glucose master mix supplies the cell-free reaction with raw materials such as nucleotides monophosphates (NMPs), ribose and glucose to build nuclotide triphosphates (NTPs) via metabolic regeneration. This results in a slower but more sustained and resource-efficient protein synthesis.
Question 3. Bonus question: How can transcription occur if GMP is not included but Guanine is?
Answer
Transcription can still occur when guanosine monophosphate (GMP) is not included in a cell-free system, but guanine is, due to the cell lysate containing active salvage pathway enzymes that convert the free guanine bases into functional guanosine triphosphate (GTP) that can be used for RNA synthesis.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Question 1. Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP
mRFP1
mKO2
mTurquoise2
mScarlet_I
Electra2
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
Answer
sfGFP: Superfold GFP has a very fast maturation time and enhanced folding efficiency. This allows it to fold correctly even under suboptimal conditions typical for a cell-free system. This results in early signal detection.
mRFP1: A red fluorescent protein has a low maturation time of around 20 to 50 minutes, which causes the production of fluorescence to lag behind protein synthesis. This leads to an underestimation of early protein yield levels.
mKO2: A basic orange fluorescent protein exhibits a moderate acid sensitivity and slow maturation. This causes the orange fluorescence to develop slowly and be reduced by a lightly acidic cell-free reaction system. It also exhibts a stong dependance on oxygen. In a cell free system where dissolved oxygen is consumed and not replenshid mKO2 chromophore maturation could be delayed or incomplete resulting in a reduction in the observed fluorescene relative to actual protein expression.
mTurquoise2: A cyan fluorescent protein has a very low pKA of 3.1, which makes it one of the most pH-stable fluorescent proteins available. Its robustness to acidic environments means any pH drift in a cell-free system will not compromise its readout.
mScarlet-I: Is an engineered red fluorescent protein that has been optimized for fast maturation compared to other red fluorescent proteins. This improves its real-time readout and gives it a high intrinsic brightness.
Electra2: Is an engineered blue fluorescent protein that was designed for improved brightness and folding. It is oxygen-dependent for chromophore maturation, which means fluorescence will not develop in anaerobic or poorly aerated cell-free systems.
Question 2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Answer
My hypothesis involves manipulating the ratio of nicotinamide to ribose to determine the net oxygen available for mK20 chromophore maturation and the level of fluoresecne produced. The goal I have in mind is to produce low, medium, and high fluorescence of mK02.
The rationale behind my hypothesis stems from mKO2 exhibiting a strong dependence on dissolved oxygen for chromophore maturation, making it sensitive to redox environments of cell-free reactions over extended incubation periods. Nicotinamide and ribose would serve as the primary levers governing the redox environment of the reaction. Ribose drives the metabolic flux of the reaction through the pentose phosphate pathway to regenerate NMP for sustained transcription and translation, but consumes dissolved oxygen as a byproduct. While nicotinamide serves as a precursor to NAD+ and governs the NAD+/NADH ratio of the cell-free system. When NAD+ is abundant, the NADH generated by metabolism is effectively re-oxidized, preserving dissolved oxygen for the maturation of the mK02 chromophore rather than allowing it to be consumed by competing metabolic reactions.
With that in mind, I believe pairing a high concentration of ribose to a low concentration of nicotinamide would create a hyperactive reducing environment that rapidly depletes dissolved oxygen, leaving mKO2 protein unable to complete the oxidation of its chromophore for maturation, producing a low fluorescence. Additionally, a balanced proportional increase in both ribose and nicotinamide would partially compensate for oxygen consumption by improving the cycling of NAD+ to support moderate chromophore maturation and produce medium fluorescence. Finally, a high concentration of nicotinamide paired with a low concentration of ribose would maximize NAD+ driven redox maintenance while limiting metabolic oxygen consumption, thereby preserving the oxidizing environment mK02 requires to produce a high fluorescence.
Question 3. The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
Answer
To compose my master mix to produce low, medium, and high fluorescence of the mK2O fluorescent protein, I used the cell-free optimization interface to adjust the composition of the cell-free reaction.
To produce a low fluorescence of the mK20 protein, I adjusted the Delta of nicotinamide to +0.500mM and a Delta % of 16%, while I adjusted the Delta of ribose to +5.875 g/L and a Delta % of 50.5%.
Potassium Glutamate 312.56 mM
HEPES-KOH pH 7.5 45.00 mM
Magnesium Glutamate 6.97 mM
Potassium phosphate dibasic 5.63 mM
Potassium phosphate monobasic 5.63 mM
17 Amino Acid Mix 4.06 mM
Tyrosine pH 12 4.06 mM
Cysteine 4.00 mM
Nicotinamide 3.63 mM
AMP 625.00 uM
CMP 375.00 uM
UMP 375.00 uM
Guanine 156.25 uM
Ribose 17.500 g/L
Glucose 1.250 g/L
Nuclease-Free Water 0.725 uL
To produce medium fluorescence of the mK20 protein, I increased the Delta of nicotinamide to +1.250 mM and a Delta % of 40.0%, while I adjusted the Delta of ribose to +4.750 g/L and a Delta % of 40.9%
Potassium Glutamate 312.56 mM
HEPES-KOH pH 7.5 45.00 mM
Magnesium Glutamate 6.97 mM
Potassium phosphate dibasic 5.63 mM
Potassium phosphate monobasic 5.63 mM
Nicotinamide 4.38 mM
17 Amino Acid Mix 4.06 mM
Tyrosine pH 12 4.06 mM
Cysteine 4.00 mM
AMP 625.00 uM
CMP 375.00 uM
UMP 375.00 uM
Guanine 156.25 uM
Ribose 16.375 g/L
Glucose 1.250 g/L
Nuclease-Free Water 0.800 uL
To produce a high fluorescence of the mK20 protein, I adjusted the Delta of nicotinamide to +2.500 mM and a Delta % of 80.00%, while I adjusted the Delta of ribose to +1.250g/L and a Delta % of 10.8%.
Potassium Glutamate 312.56 mM
HEPES-KOH pH 7.5 45.00 mM
Magnesium Glutamate 6.97 mM
Potassium phosphate dibasic 5.63 mM
Potassium phosphate monobasic 5.63 mM
Nicotinamide 5.63 mM
17 Amino Acid Mix 4.06 mM
Tyrosine pH 12 4.06 mM
Cysteine 4.00 mM
AMP 625.00 uM
CMP 375.00 uM
UMP 375.00 uM
Guanine 156.25 uM
Ribose 12.875 g/L
Glucose 1.250 g/L
Nuclease-Free Water 1.250 uL
Part 2
Following my hypothesis above, I only selected wells that housed the mKO2 fluorescent protein.
I first selected well Q1-P10, I then adjusted the level of nicotinamide to 3.625nM and ribose to 15.500g/L to produce a low fluorescence of the mK20 protein.
I then selected well Q1-P5 and adjusted the level of nicotinamide to 4.375 mM and ribose to 16.375g/L to produce medium fluorescence of the mK20 protein.
I selected well Q1-P12 and adjusted the level of nicotinamide to 5.625 mM and ribose to 12.875 g/L to produce High fluorescence of the mK20 protein.
I used wells Q1-P7, Q1-P16, and Q1-P18 as controls, with all the reagent volumes and concentrations left unchanged for those wells. They would enable me to determine whether the observed differences in fluorescence are truly due to the manipulation of ribose and nicotinamide by serving as reference points of what mKO2 fluorescence looks like under standard conditions.
Additionally, I selected Well Q1-P17 adjusted nicotinamide to 4.875 mM and ribose to 12.375 g/L to produce a medium-to-high fluorescence of the mK20 protein.
I selected Q1-P21 and adjusted the amount of nicotinamide to 5.375 mM and cysteine to 5.000 mM. I did this to test the effect of cysteine on the mK02 fluorescent protein. Cysteine is also a potential competing reductant that consumes dissolved oxygen and might suppress mKO2 chromophore oxidation while also being one of the building blocks for protein synthesis.
Question 4. The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
Answer
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Question 1. Use this simulation tool to create an interesting-looking cloud lab out of the Ginkgo Reconfigurable Automation Carts. This is just a minimal implementation so far, but I would love to see some fun designs!
Answer
I used the simulation tool to create a simple design of a cloud lab.
References
Kaida, A., & Miura, M. (2012). Differential dependence on oxygen tension during the maturation process between monomeric Kusabira Orange 2 and monomeric Azami Green expressed in HeLa cells. Biochemical and biophysical research communications, 421(4), 855–859. https://doi.org/10.1016/j.bbrc.2012.04.102
Week 12 HW: Building Genomes
The Lecture this week focused on genome engineering with Jef Boeke, George Church, and John Glass delivering wonderful lectures. Additionally, the recitation was centered on CRISPR, with TA Ice leading the discussion.
I also worked on my final project.
Week 13 HW: AI, SynBio and Scaling Health Innovation
This week’s lecture was led by Dr.Renee Wegrzyn. She shed light on her time at ARPA-H, Transfyr.ai, her new biotechnology AI start-up, and the background work that goes into scientific breakthroughs and failures.
It was very insightful; she drew parallels on how most innovations are adopted in other industries before scientists have the chance to use them.
TA Ice leads this week’s recitation, focused on Golden Gage Assembly. Additionally, TA Suvin took us through the requirements for final projects.
I also worked on my final project presentation.
Week 13 HW: Bio Design & Bio Fabrication
This week’s lecture featured a coast-to-coast interaction between SynBioBeta in San Jose and MIT in Boston, with Christina Agapakis and Michel Chen leading the session. It focused on protein design and synthesis as well as rapid prototyping. The interaction also featured pixel art collaboration between HTGAA 2026 and SynBioBeta.
There was no recitation this week. I worked on my final project and had mock presentation sessions with my node.




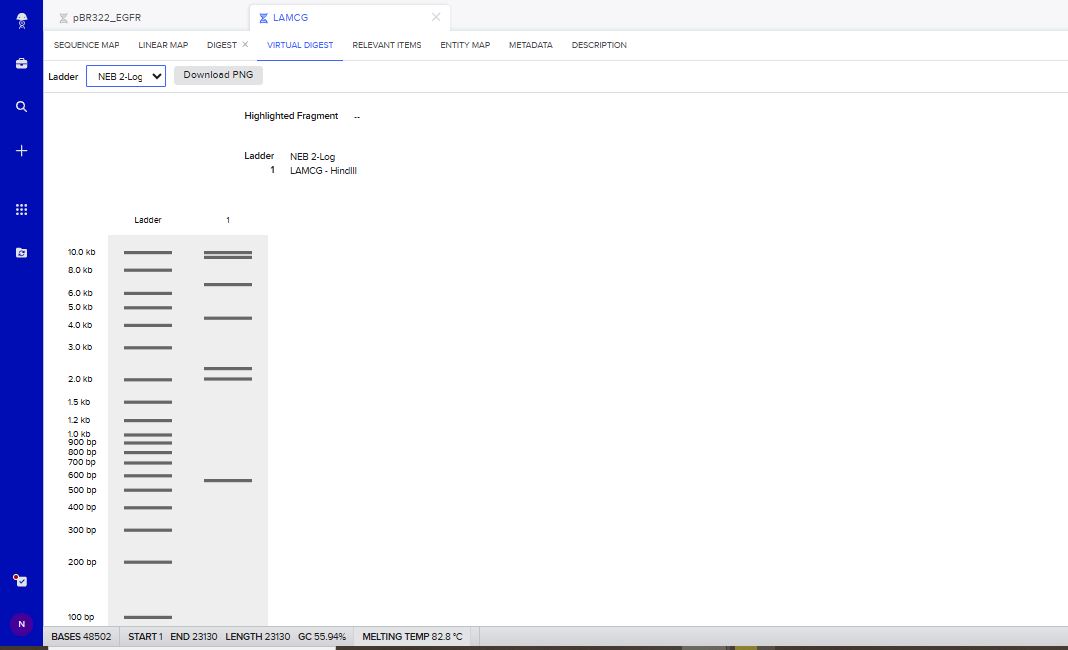
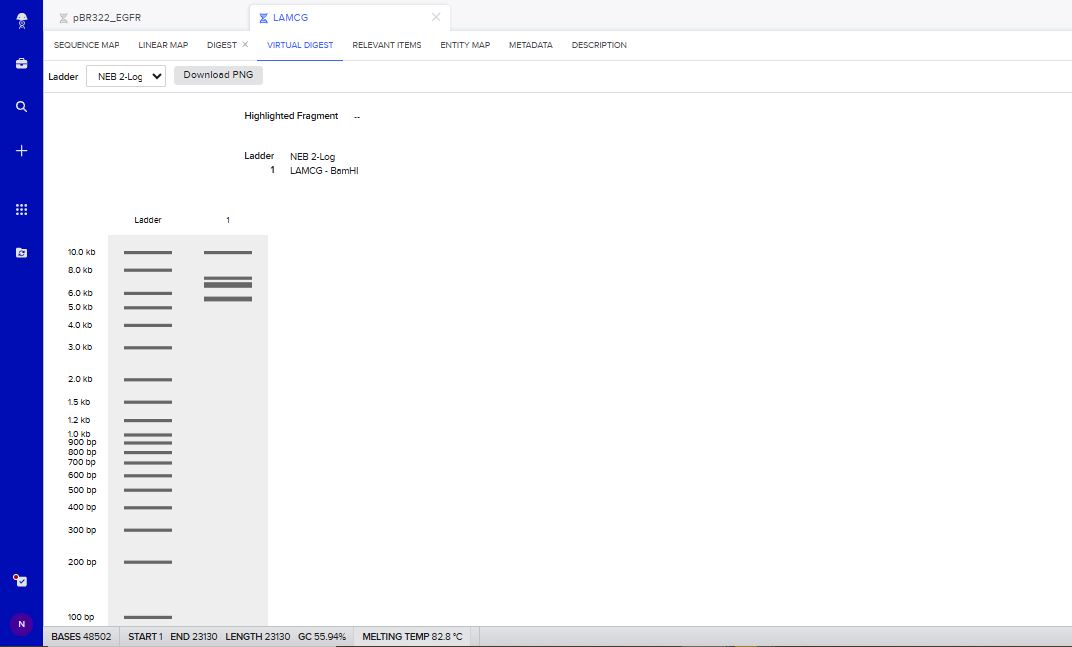
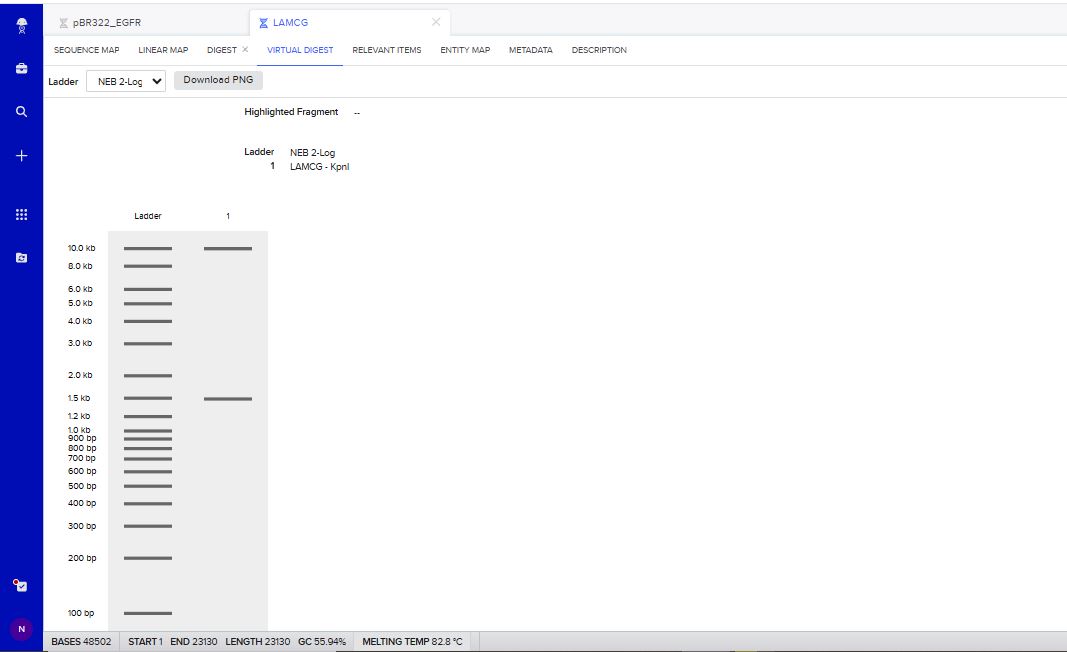
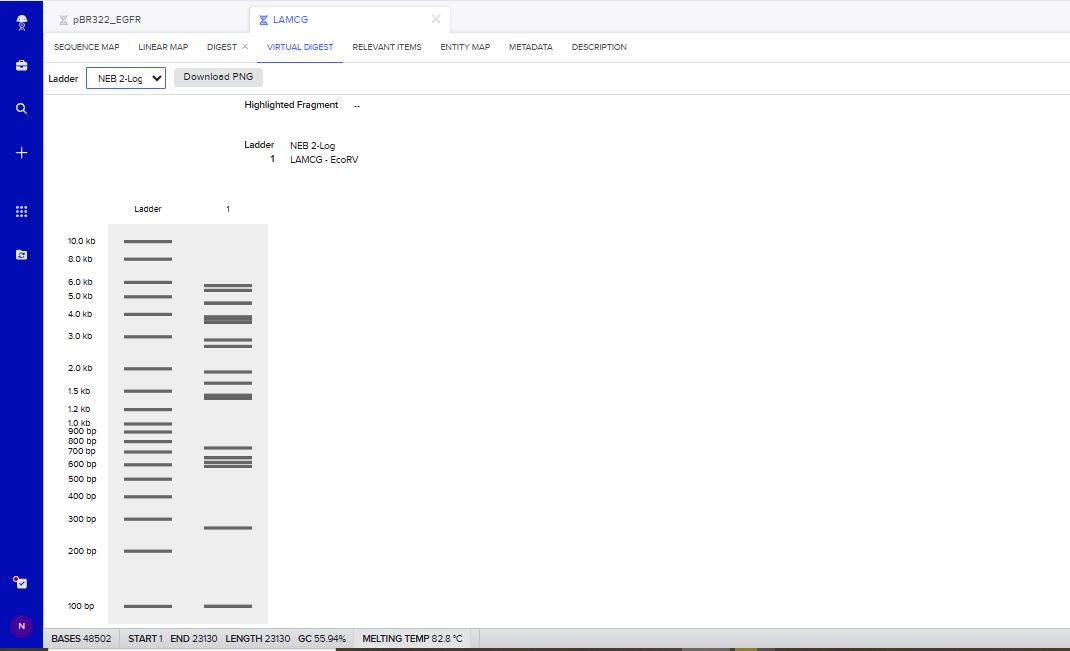
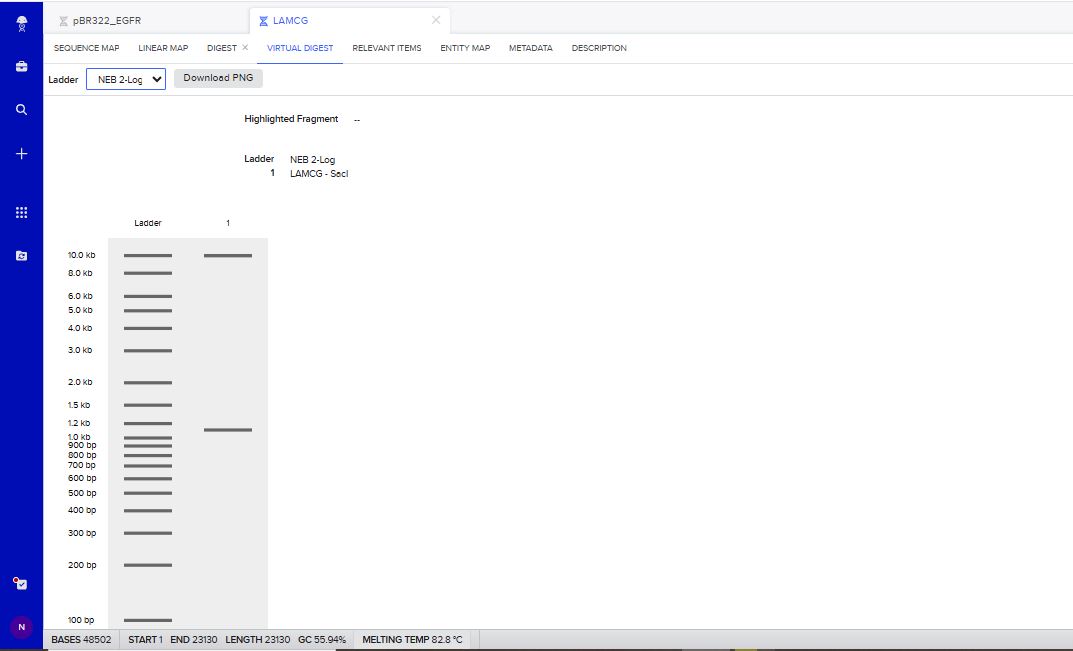
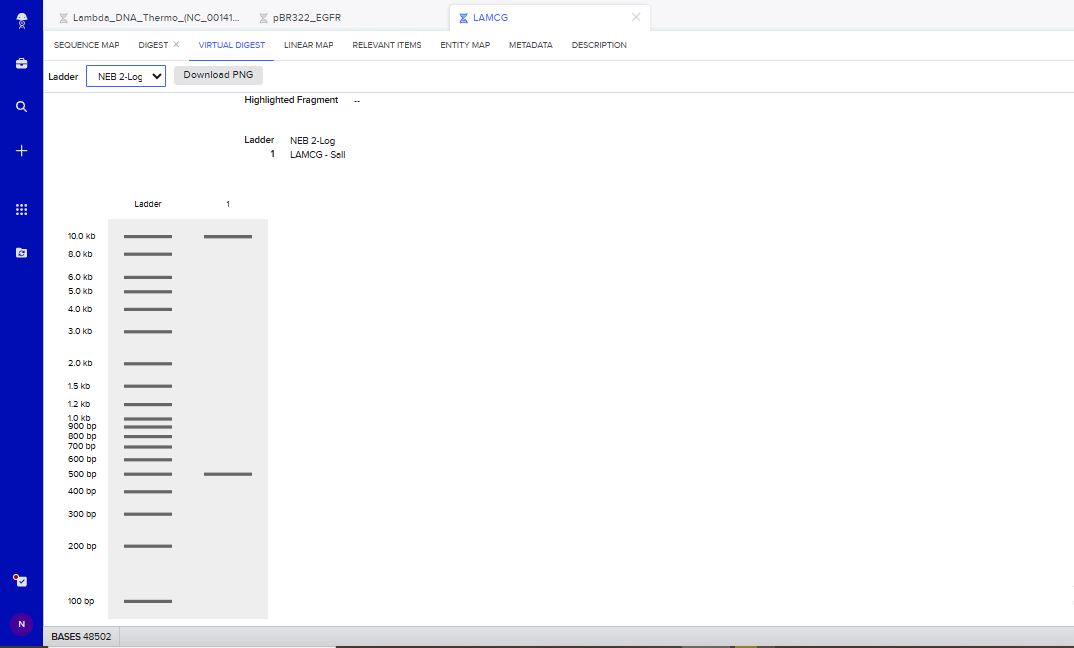
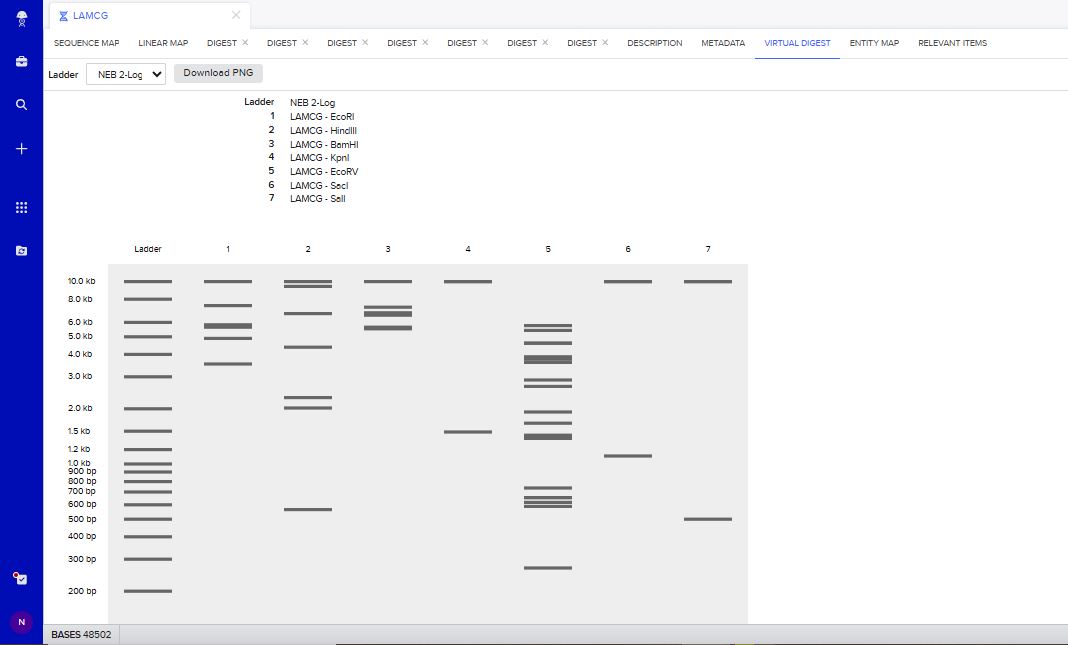
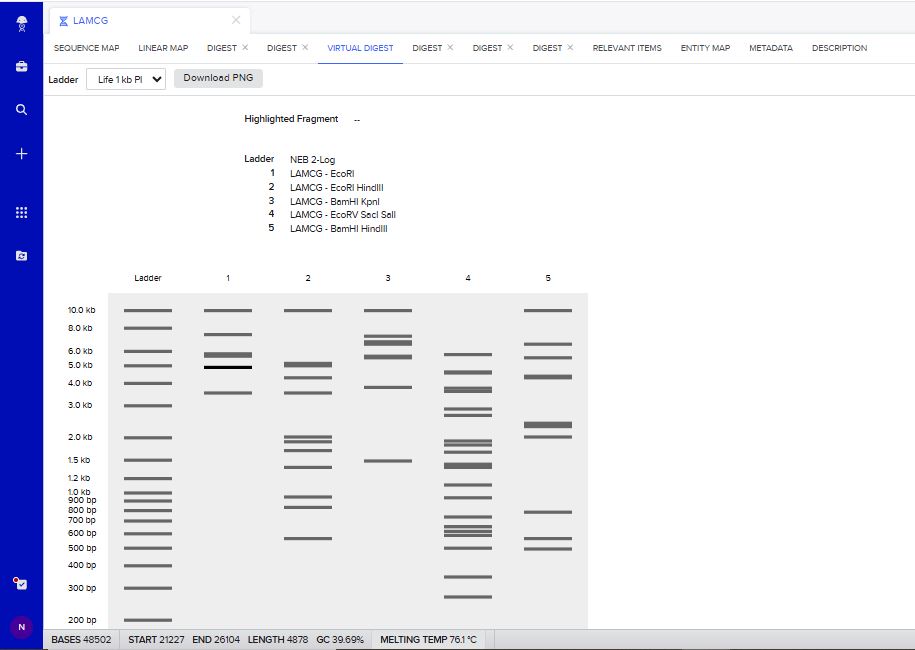
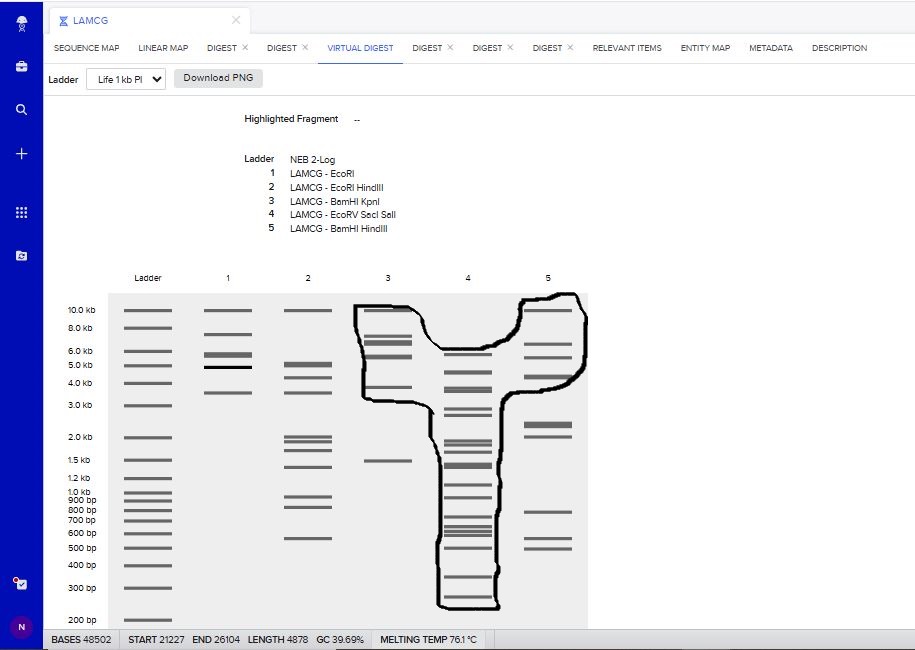
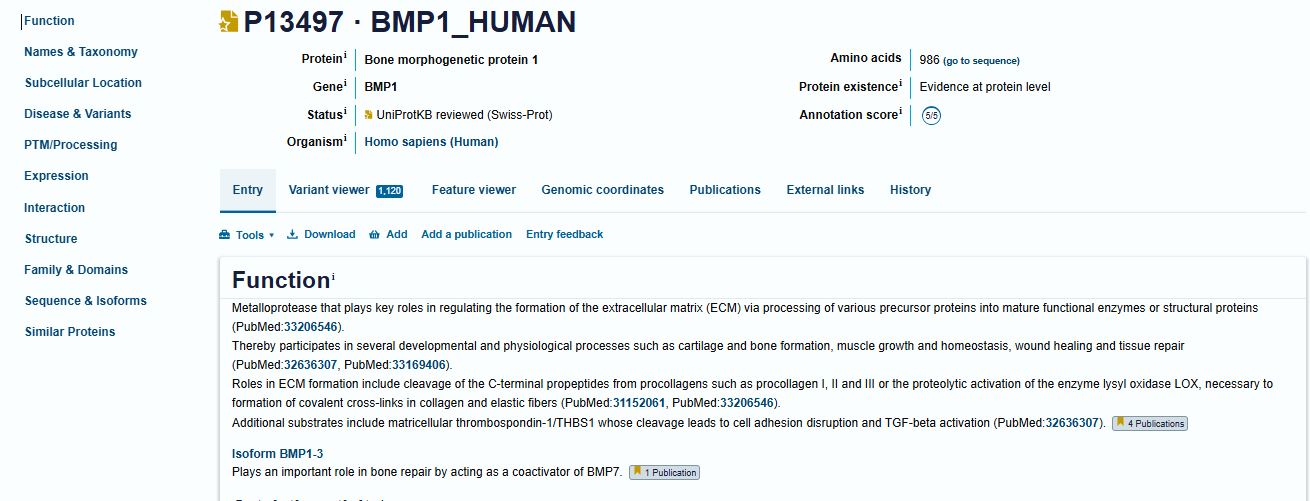
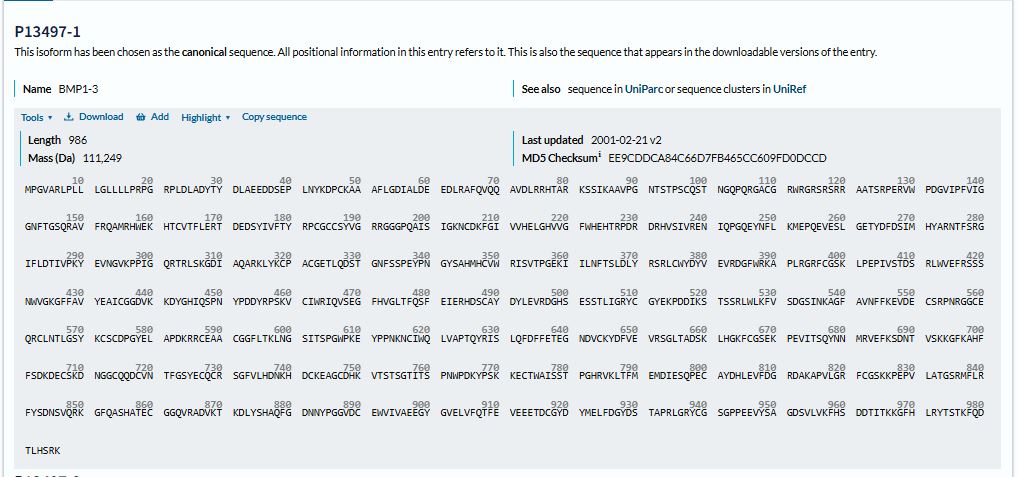
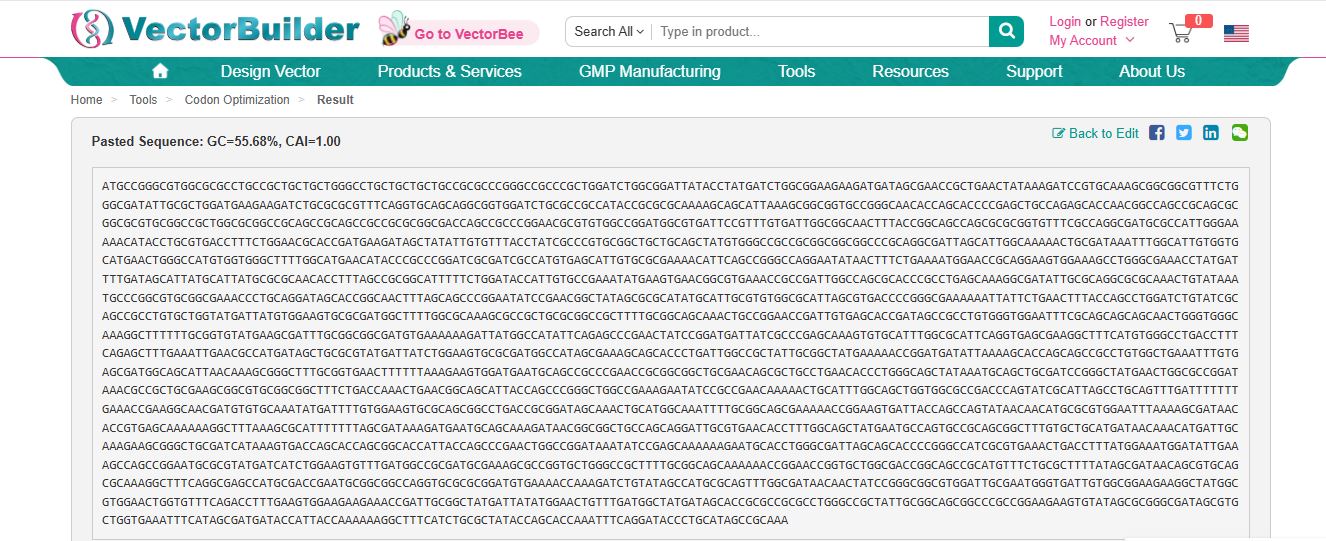
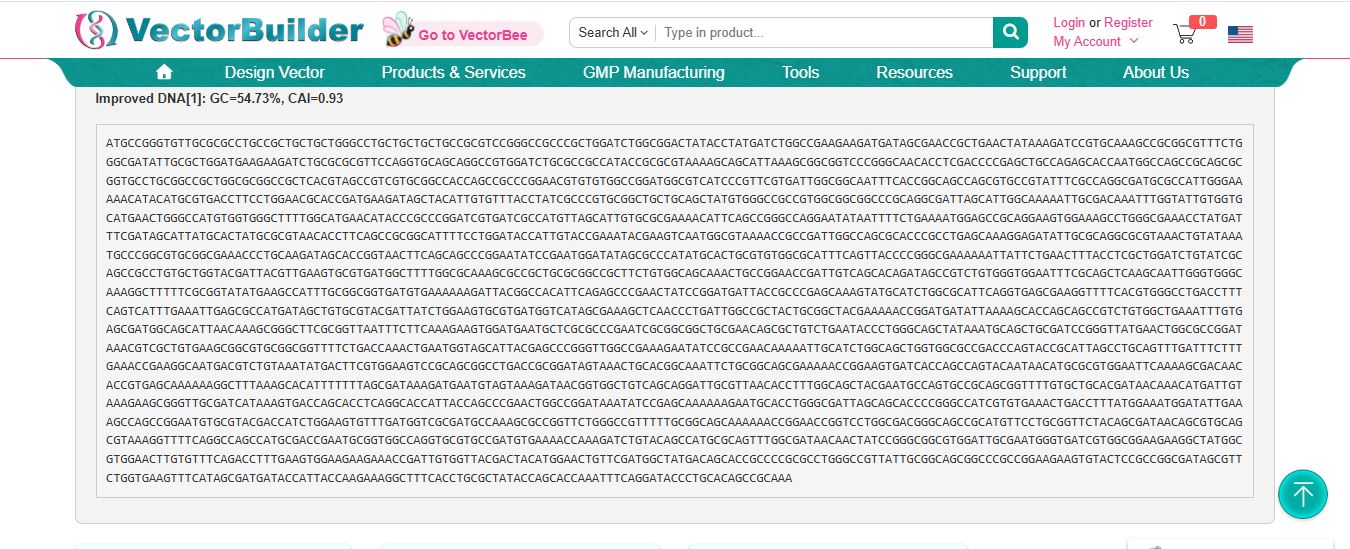
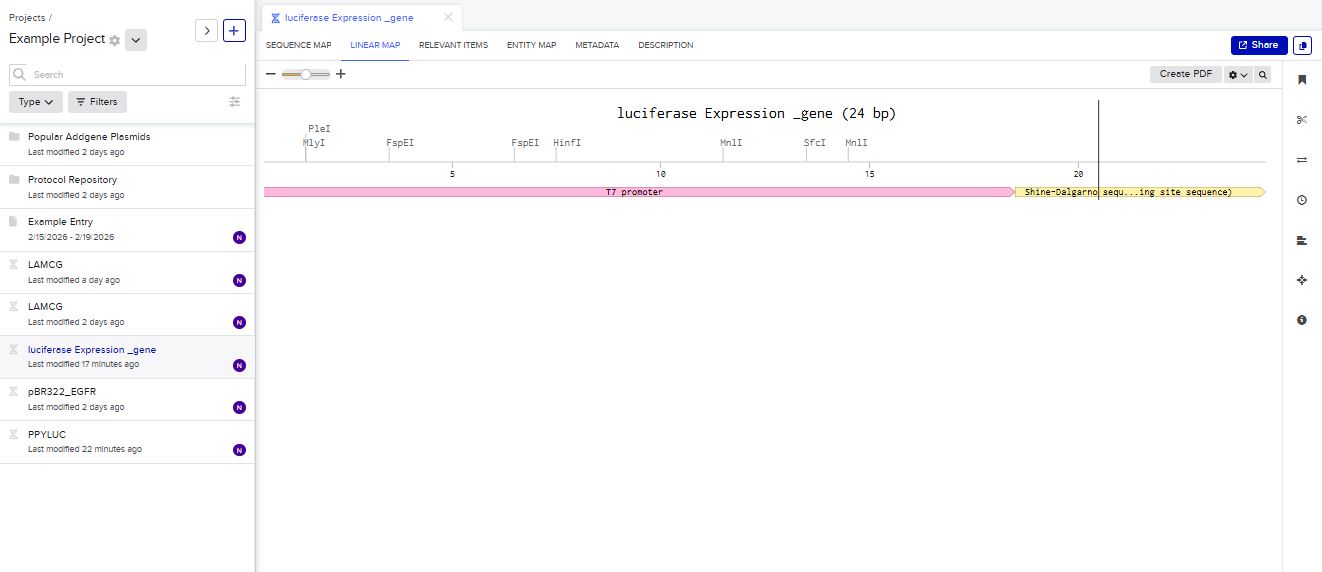
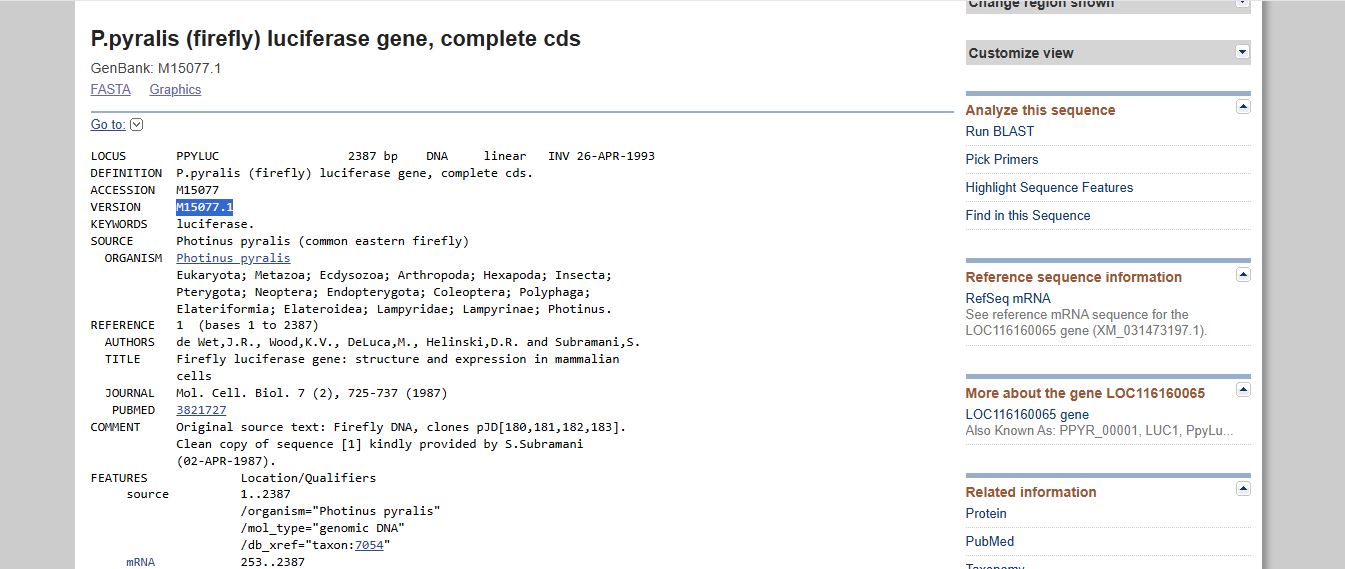
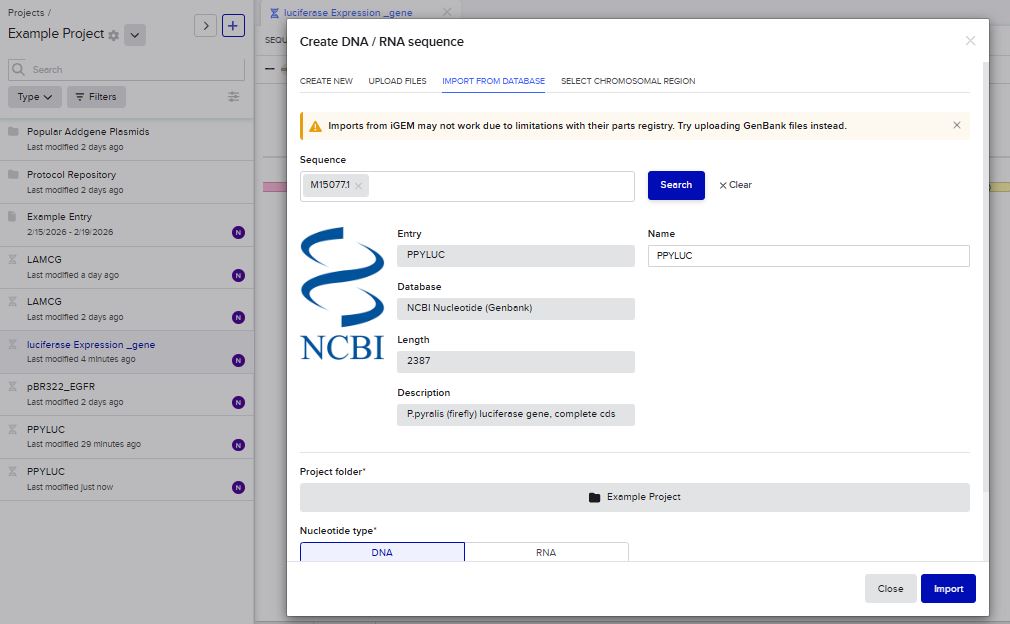
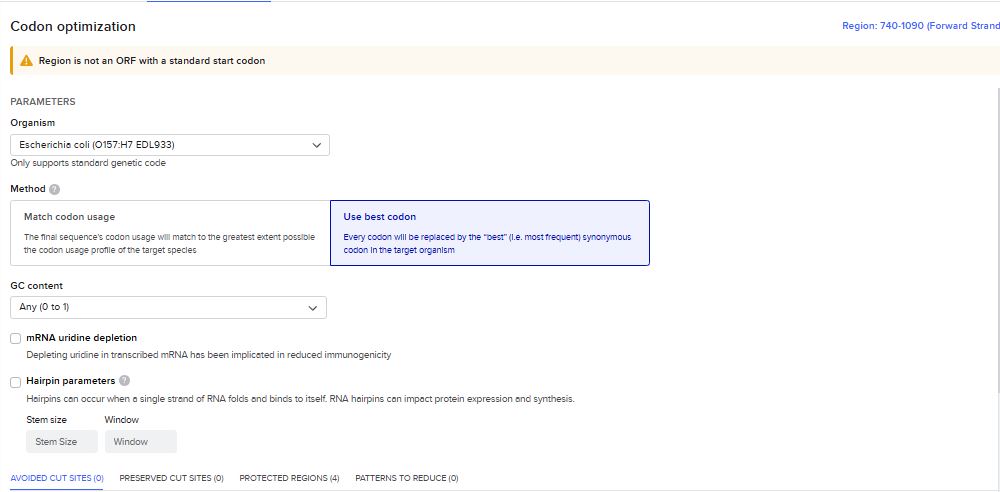
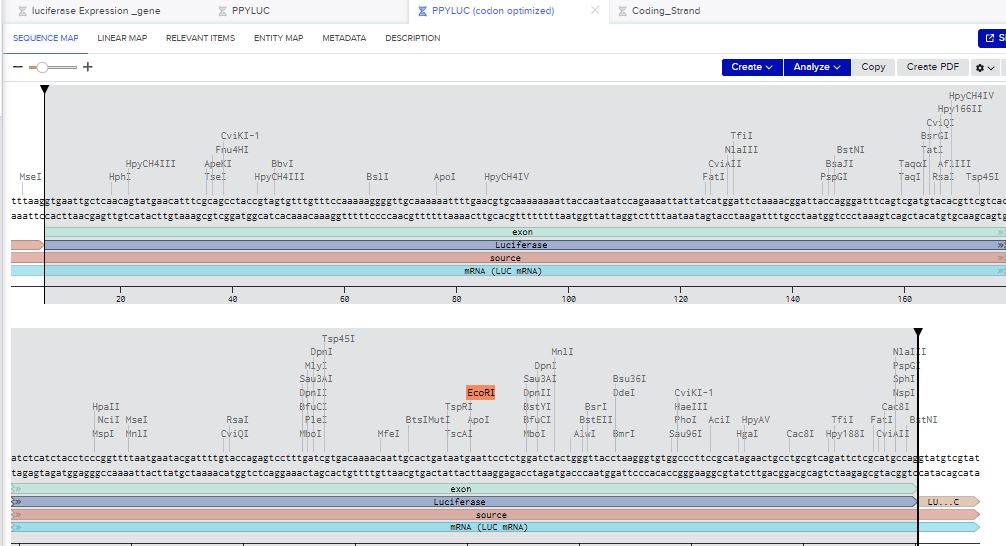
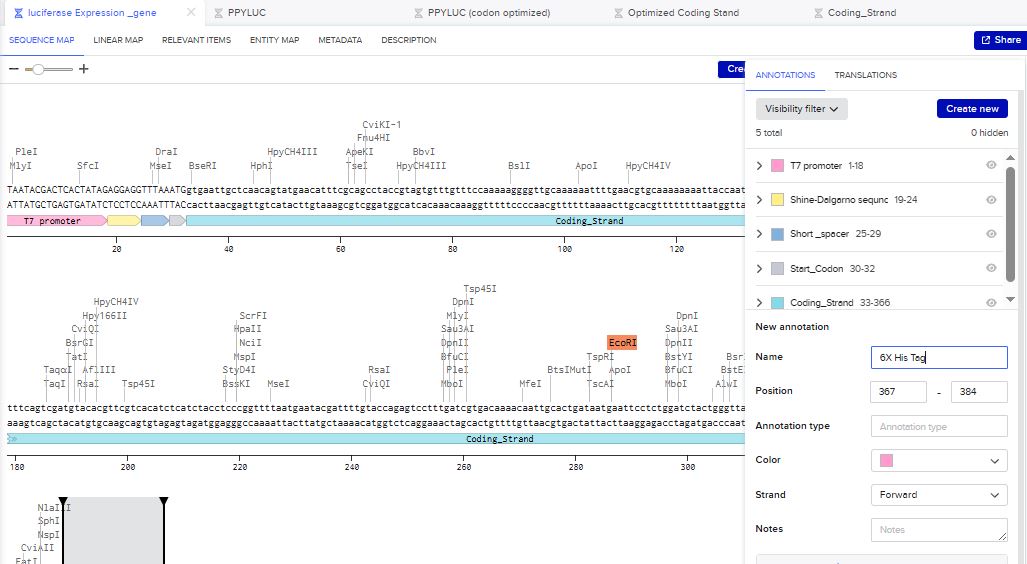
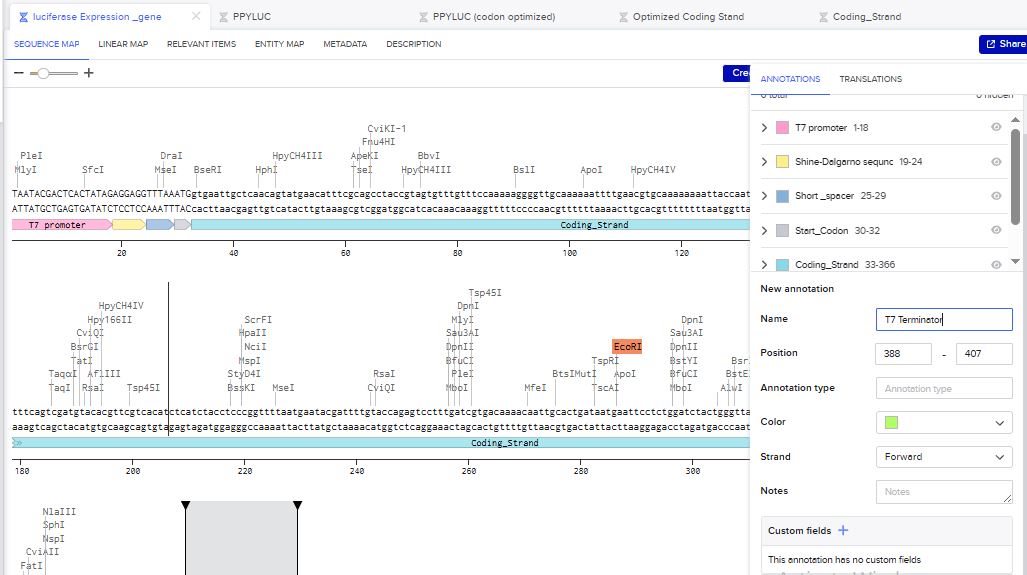
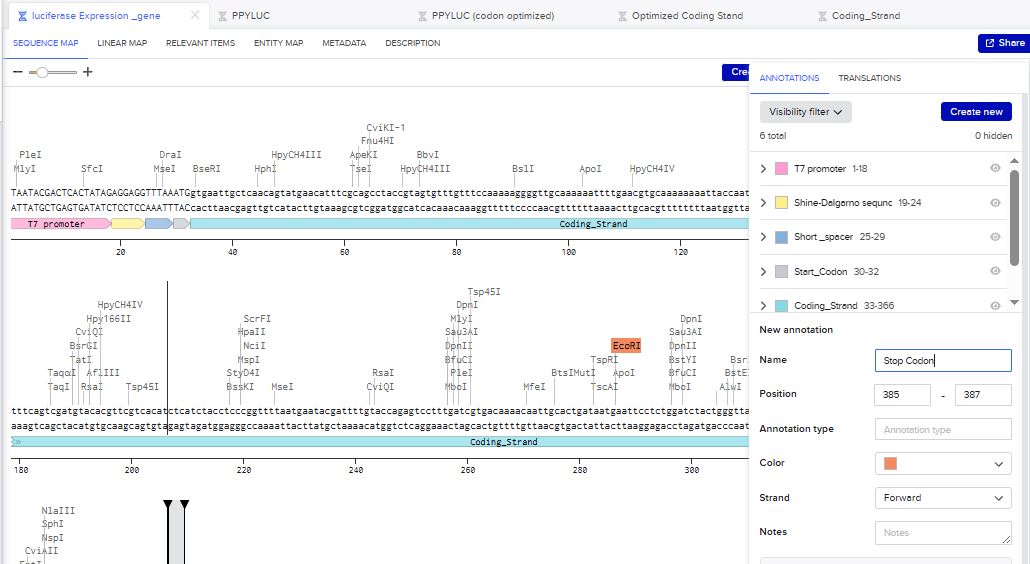
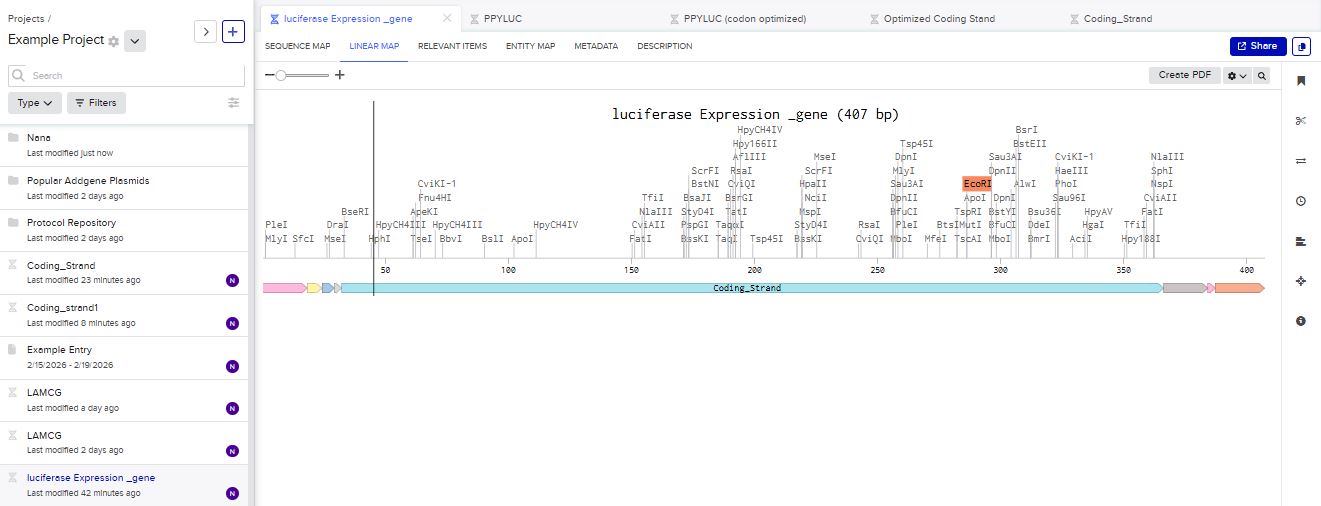
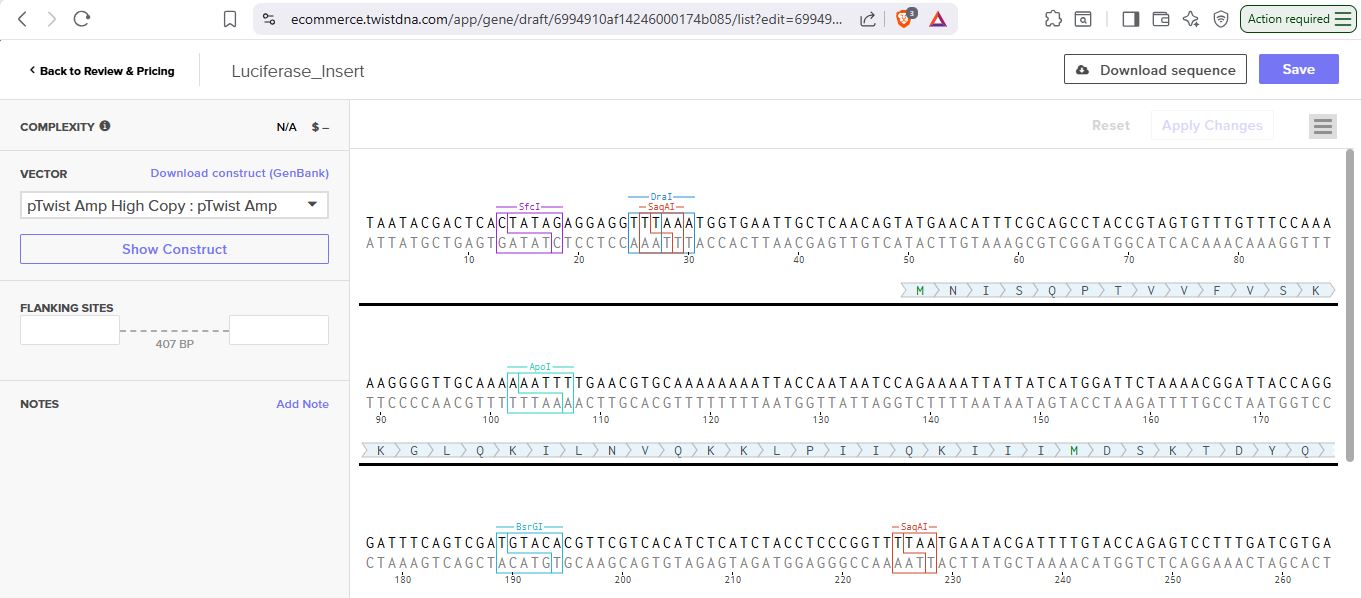
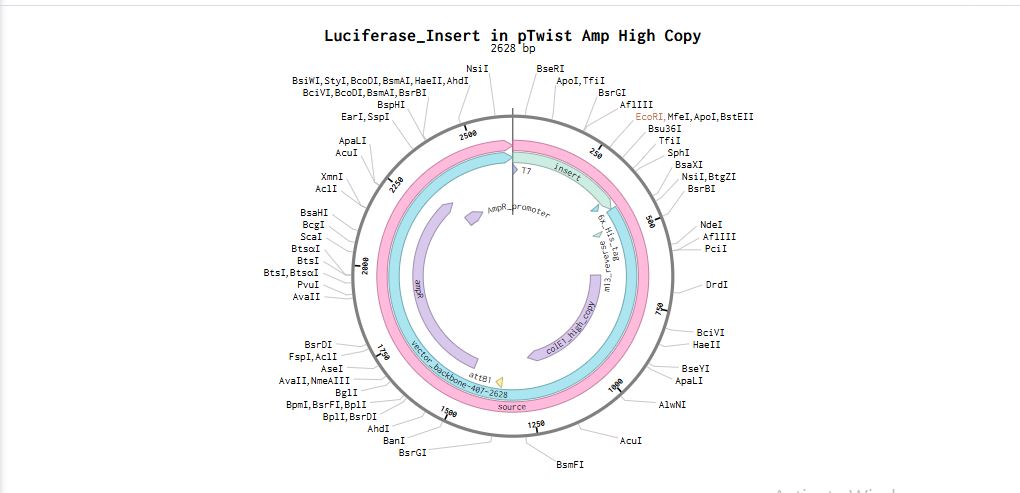


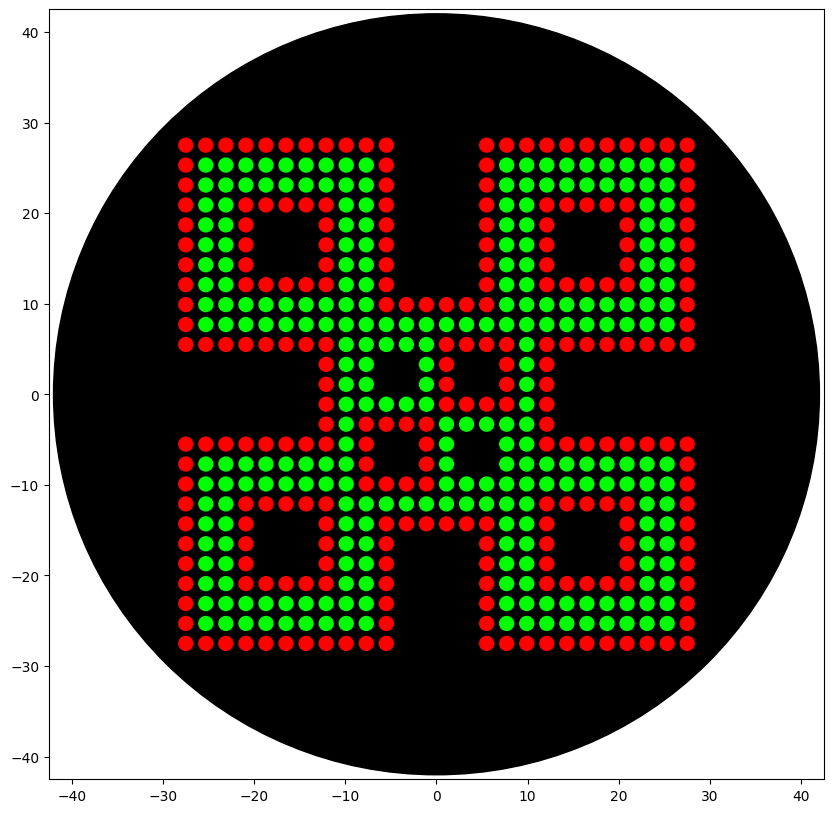
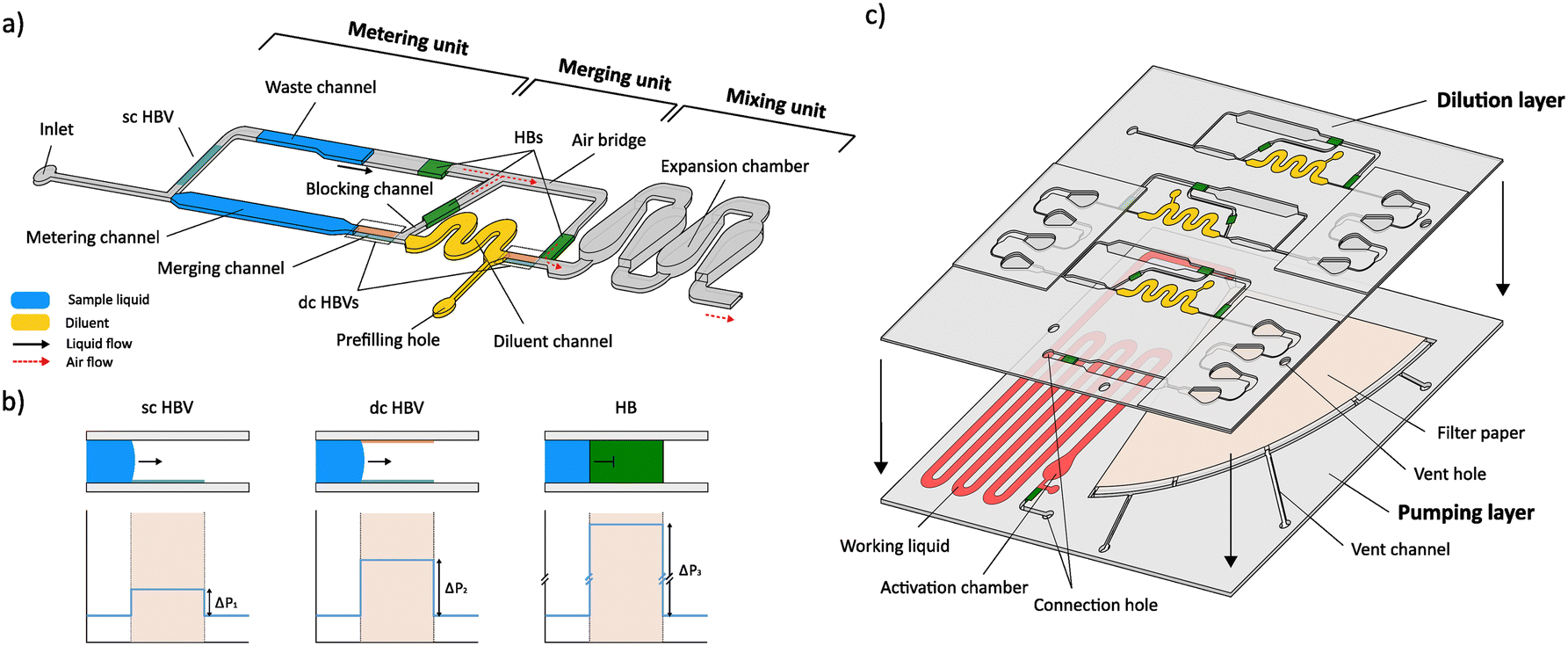
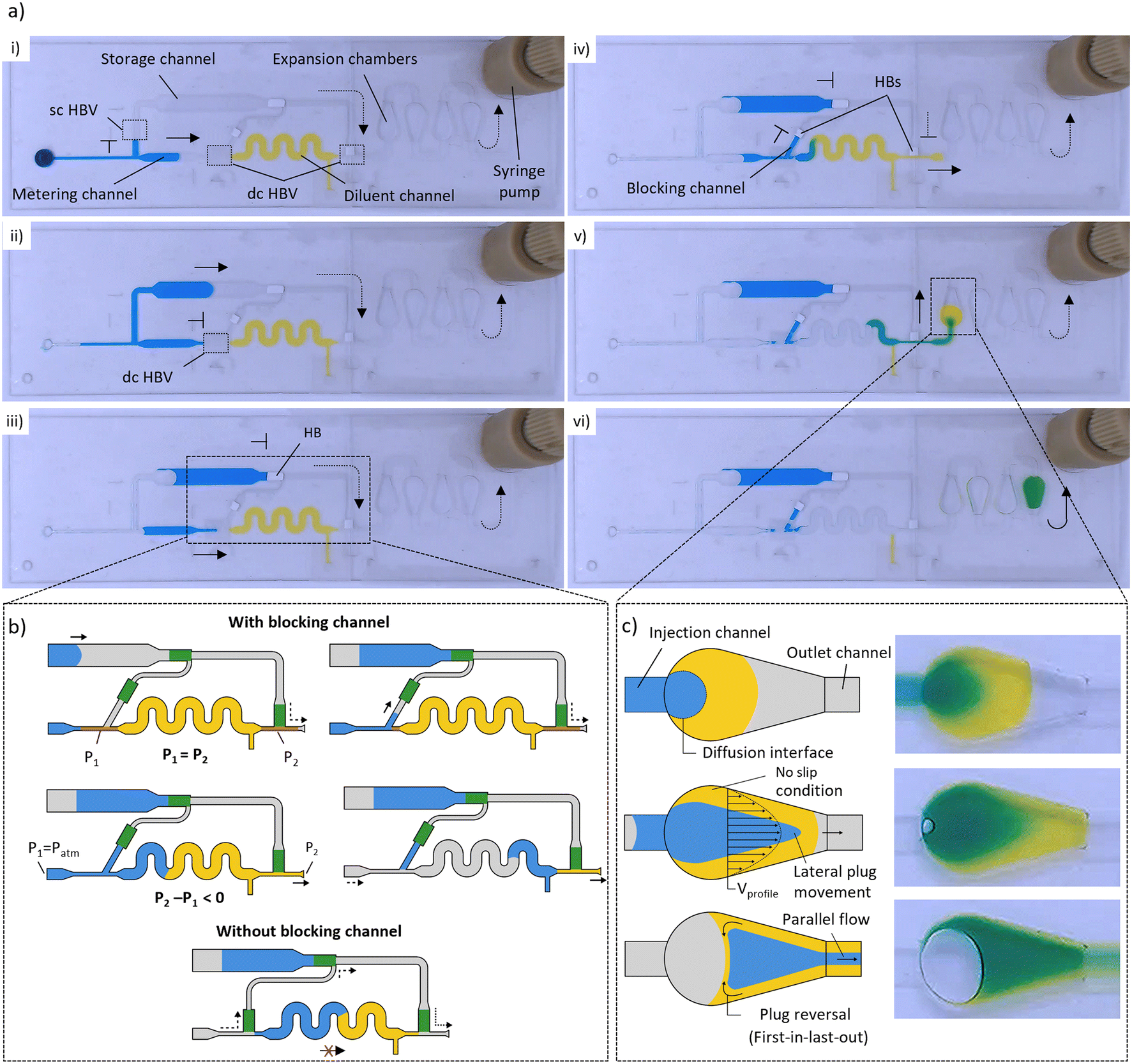


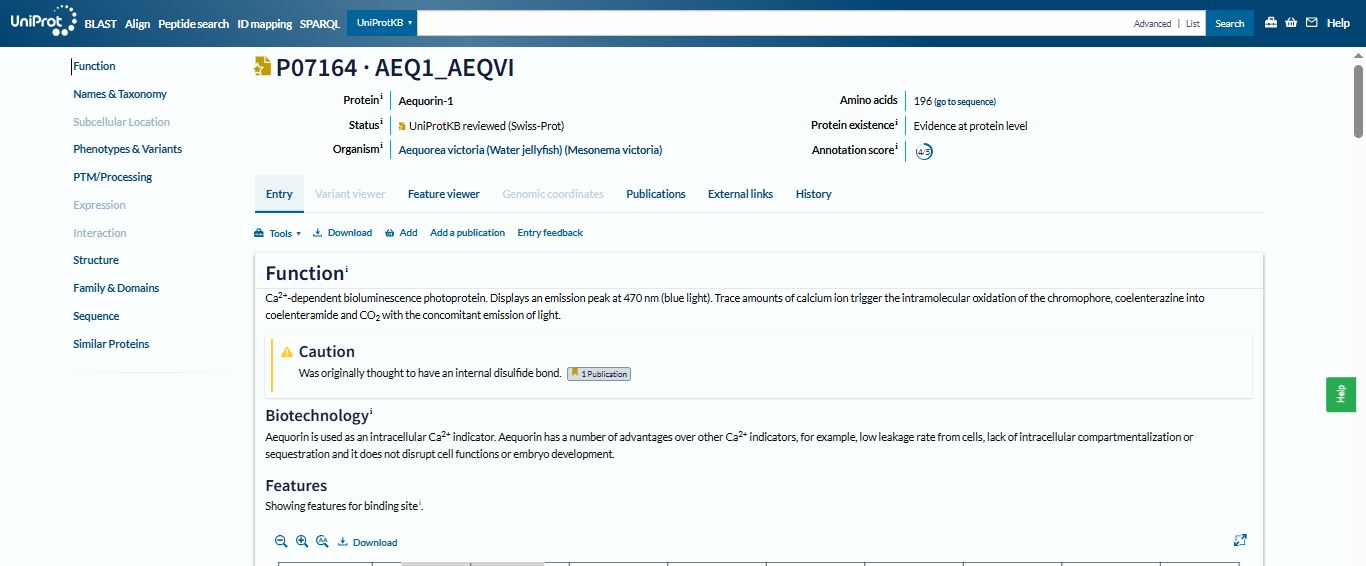
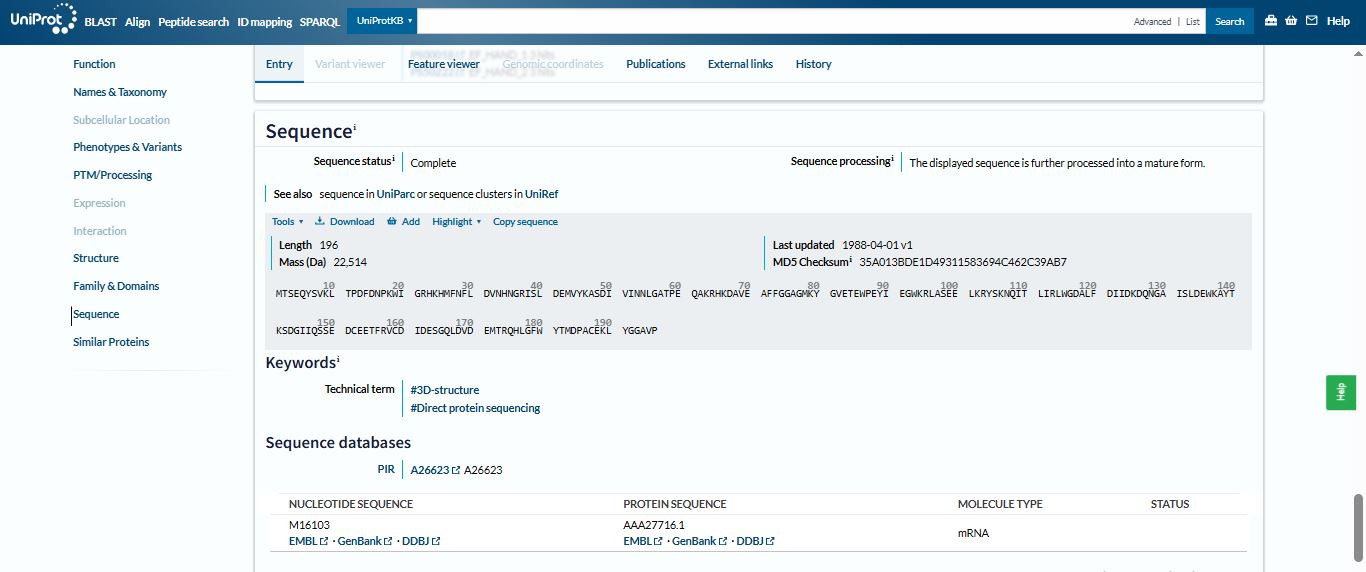
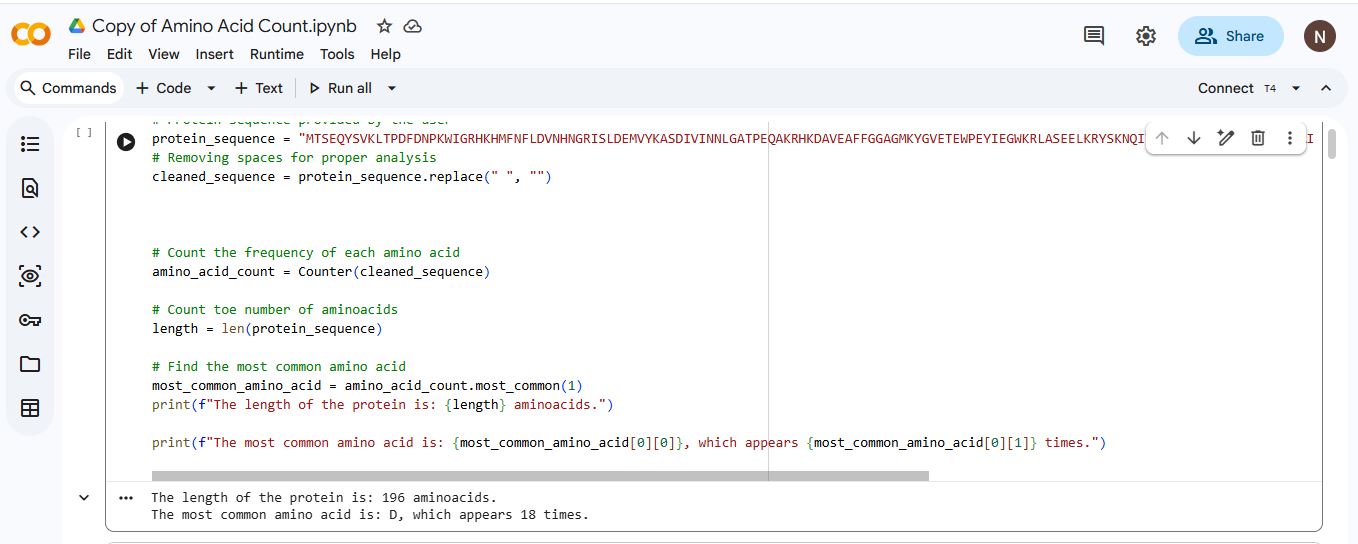
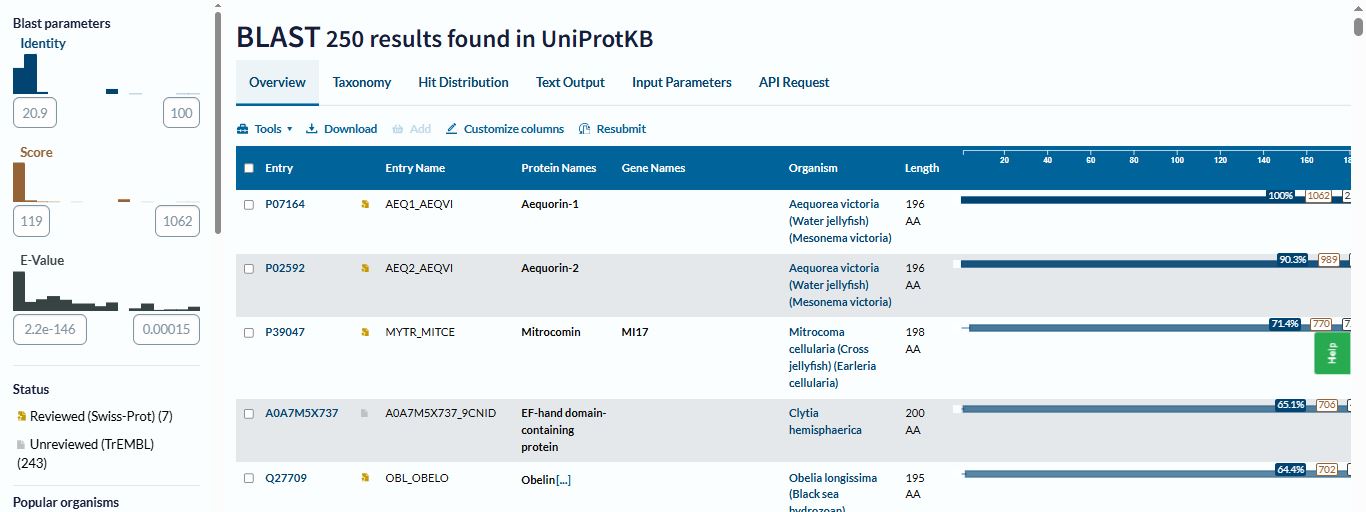
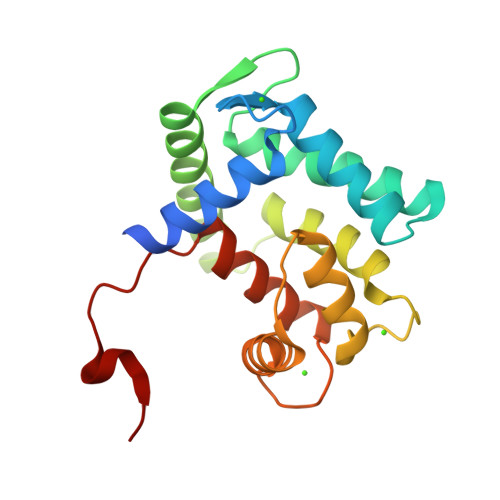
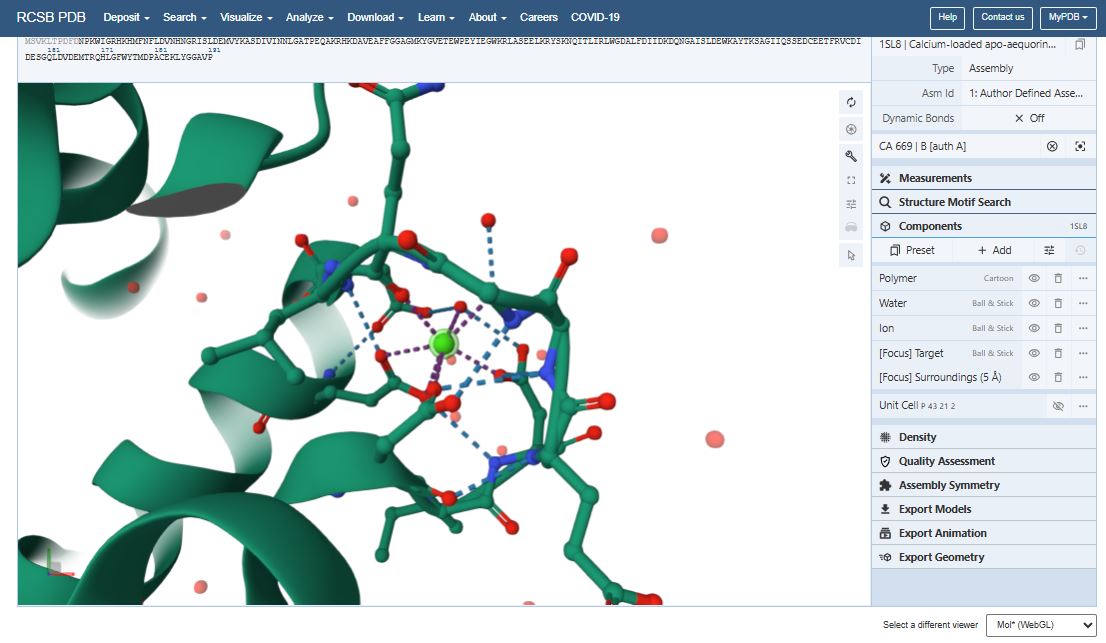
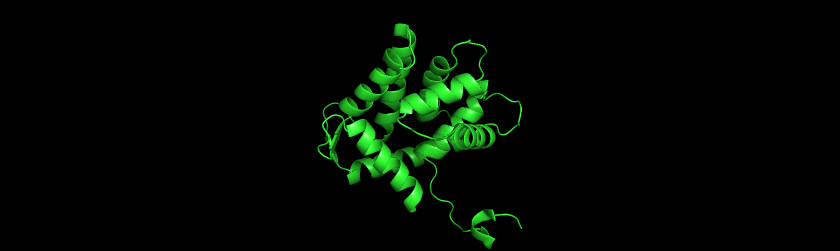

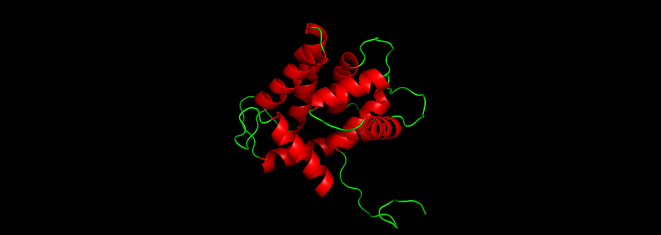
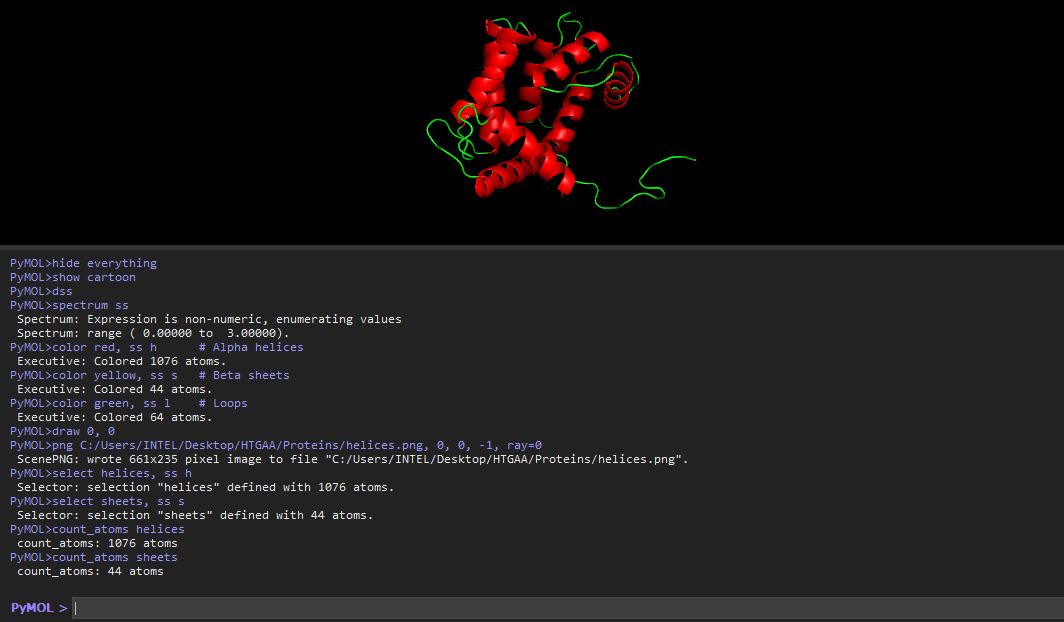
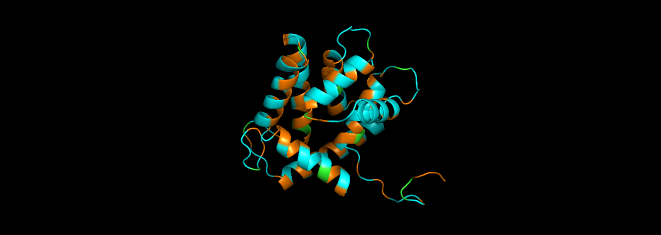
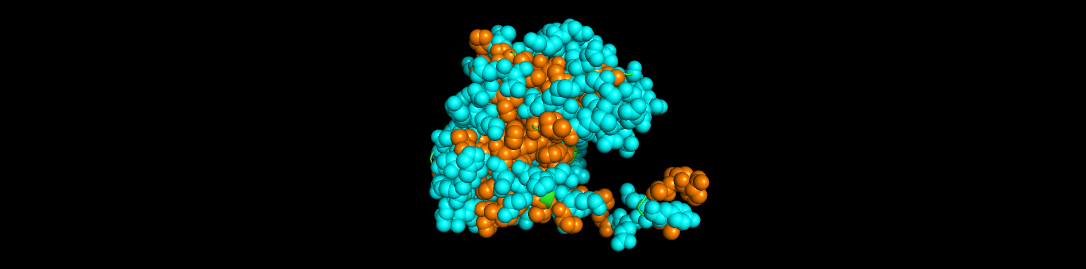
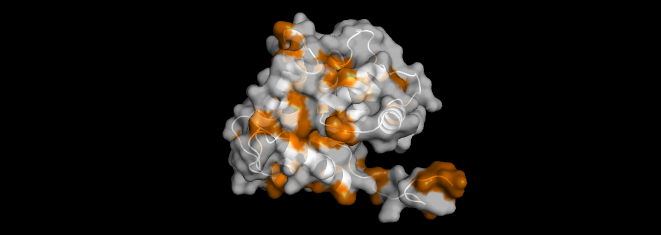
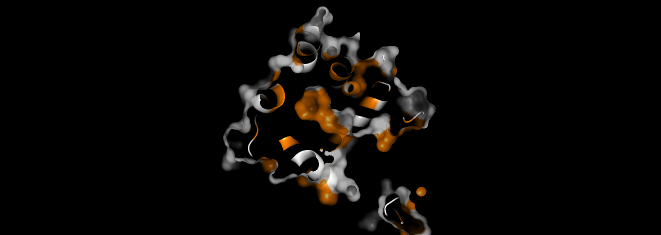
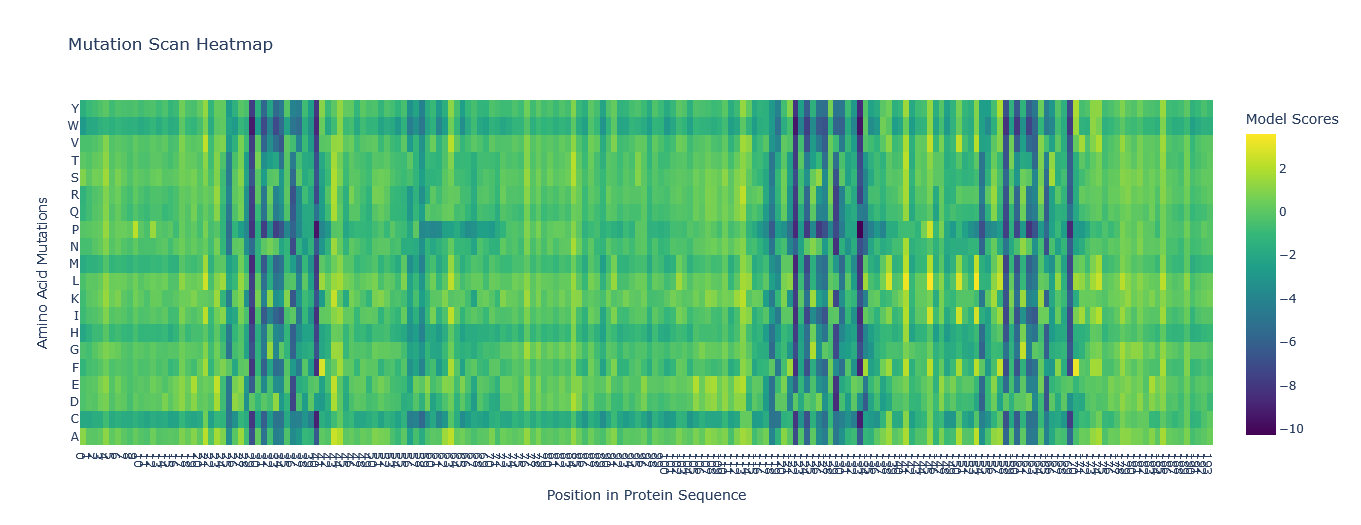
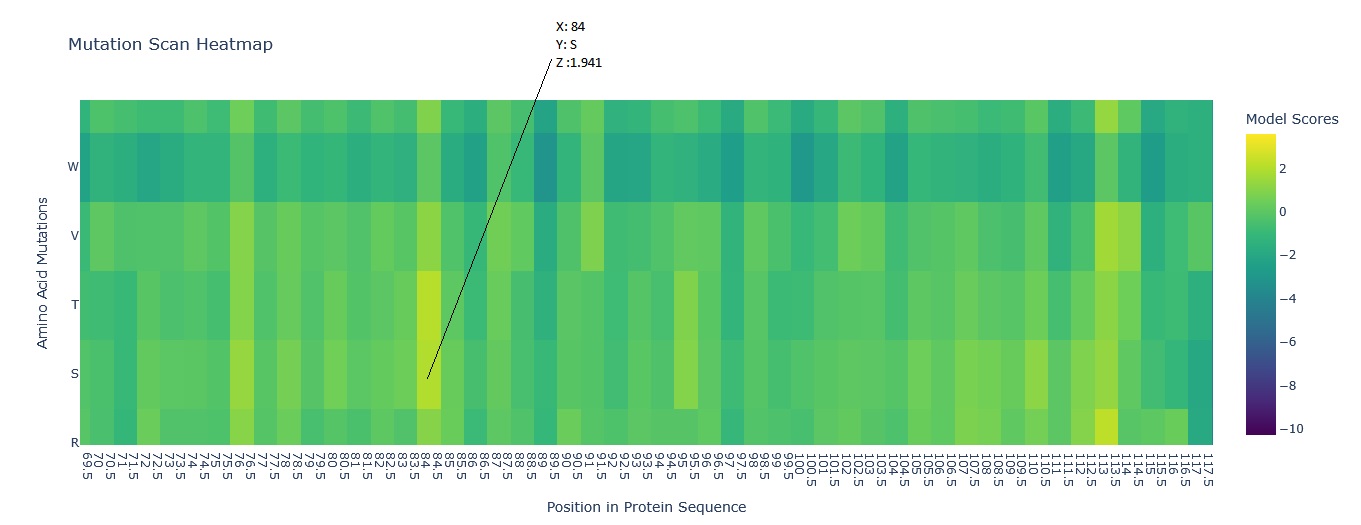
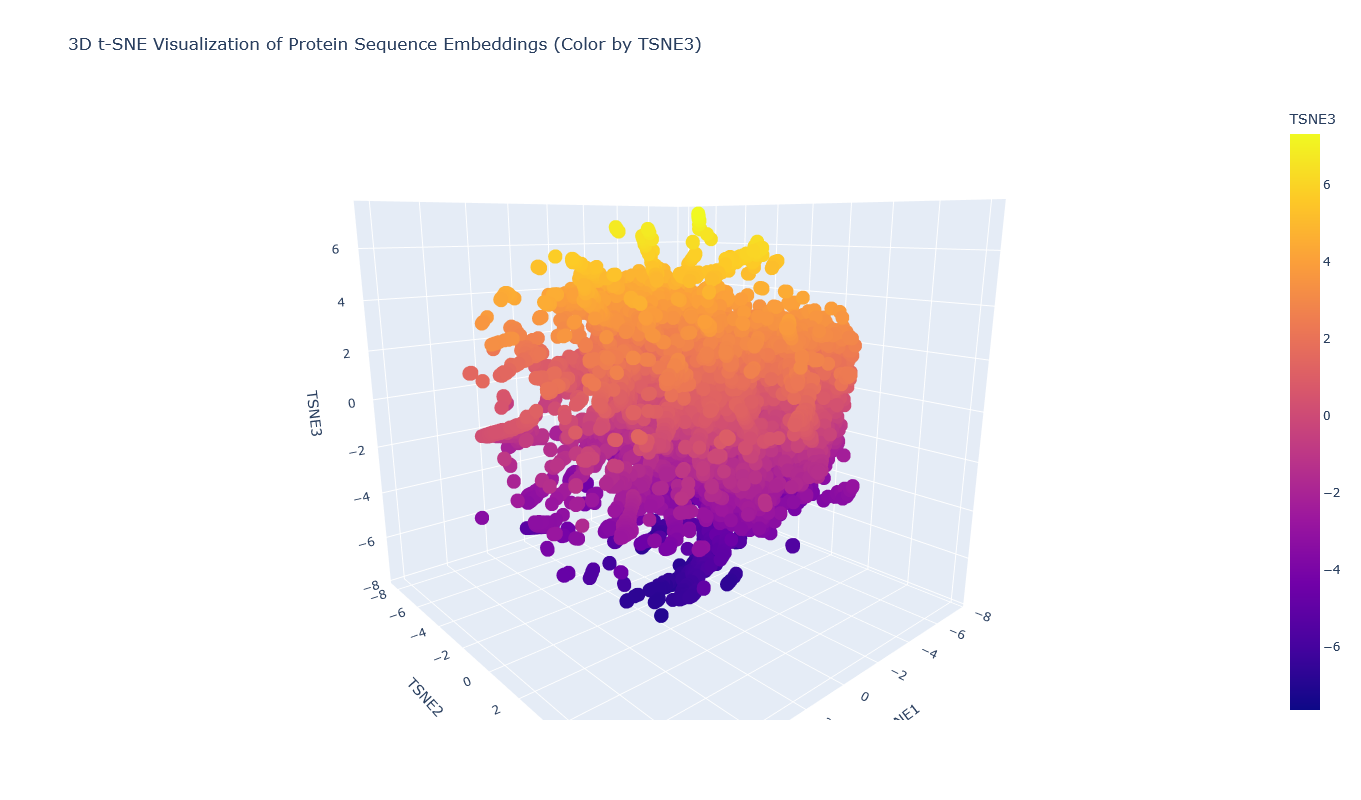
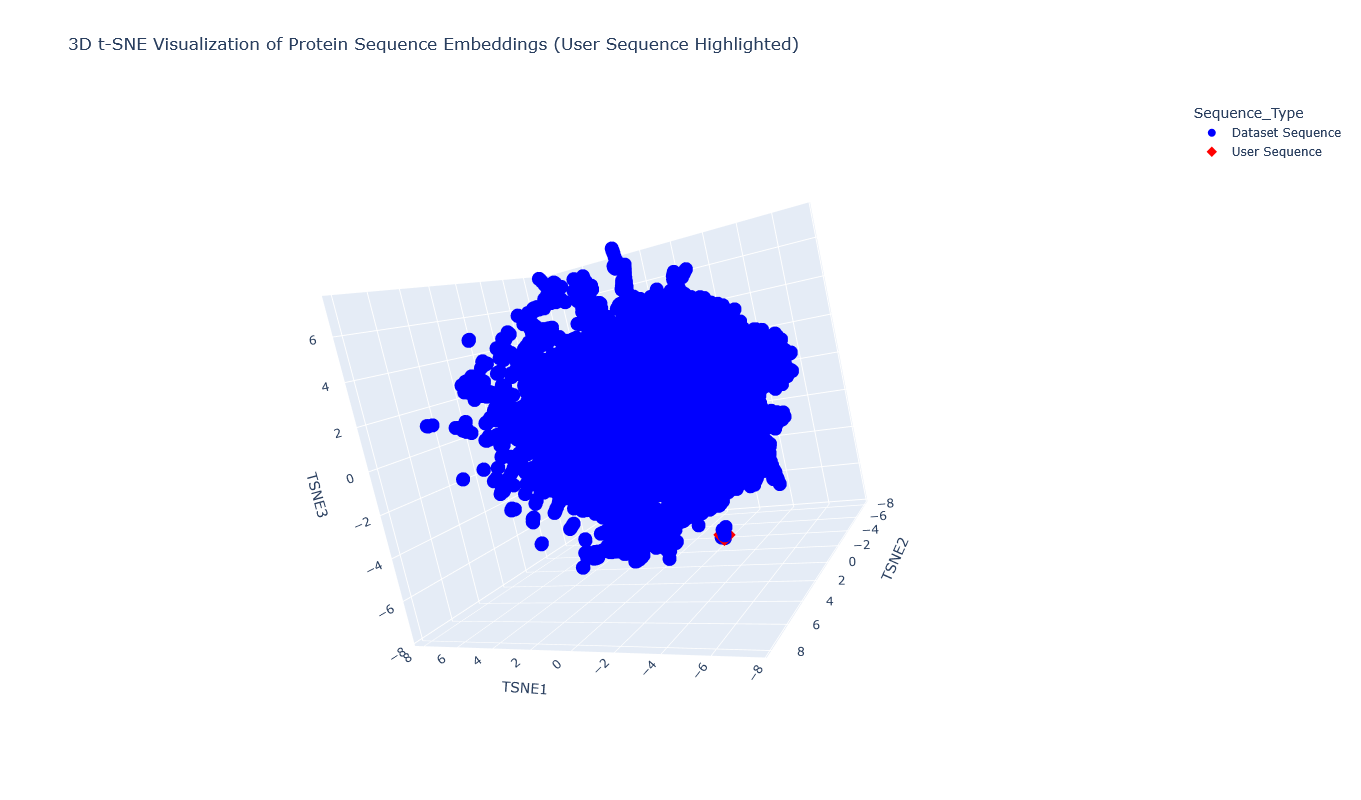
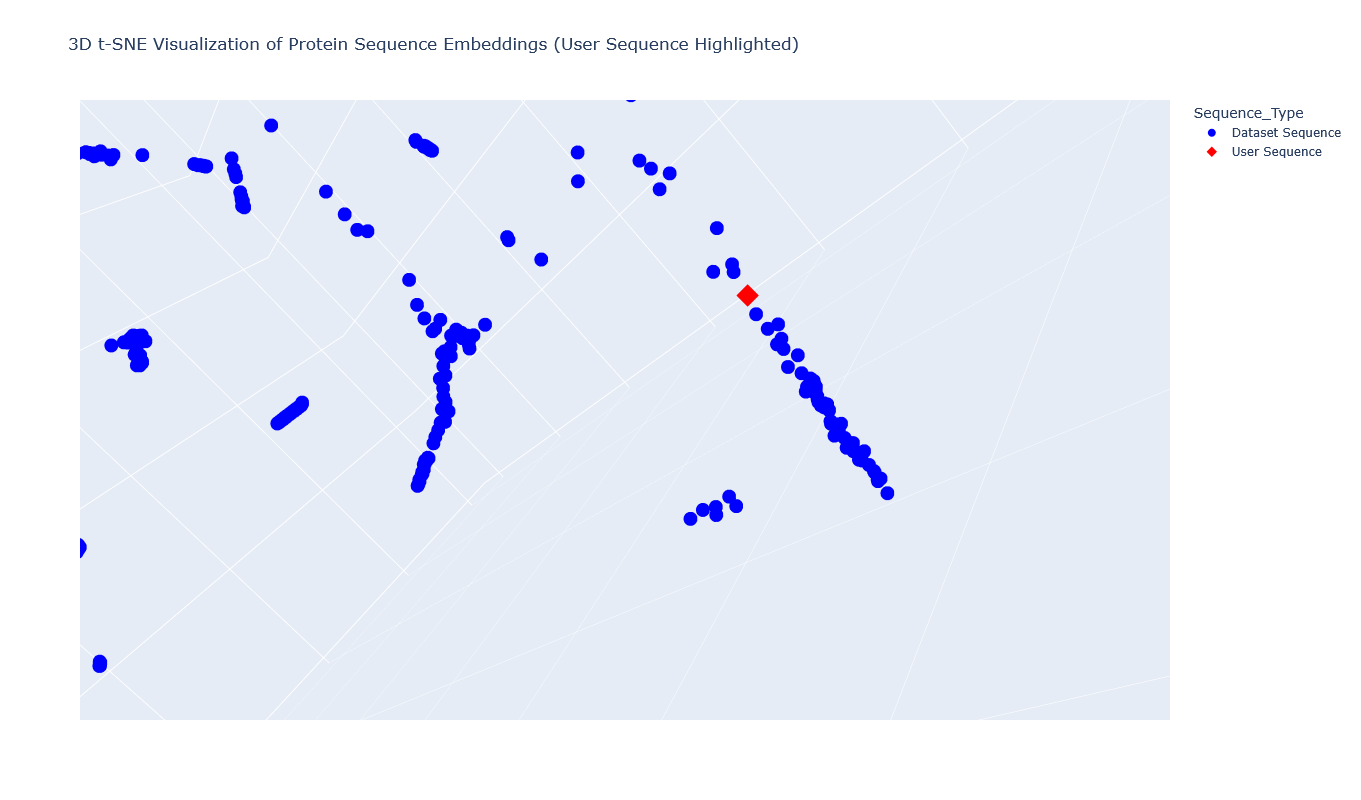

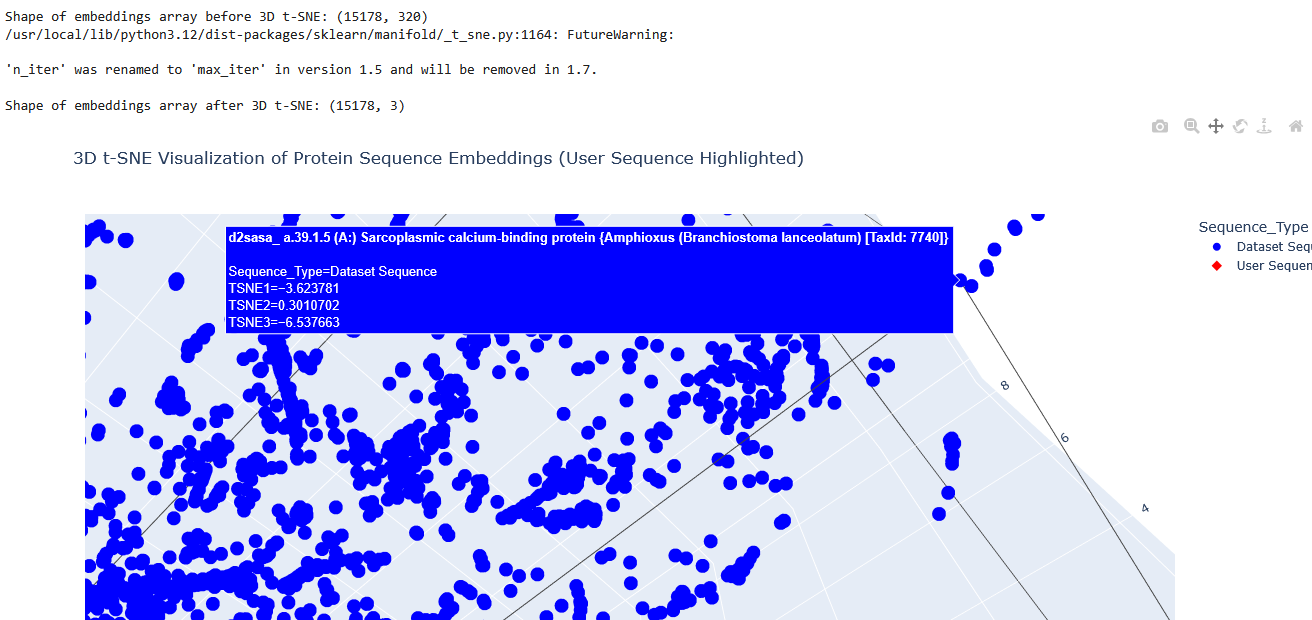
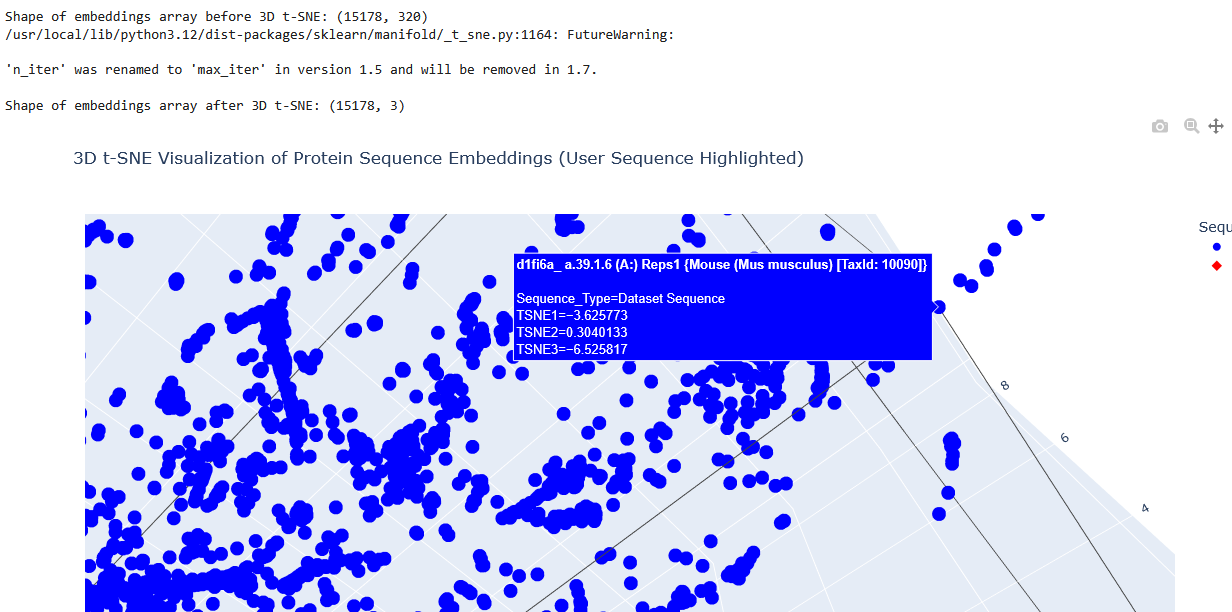
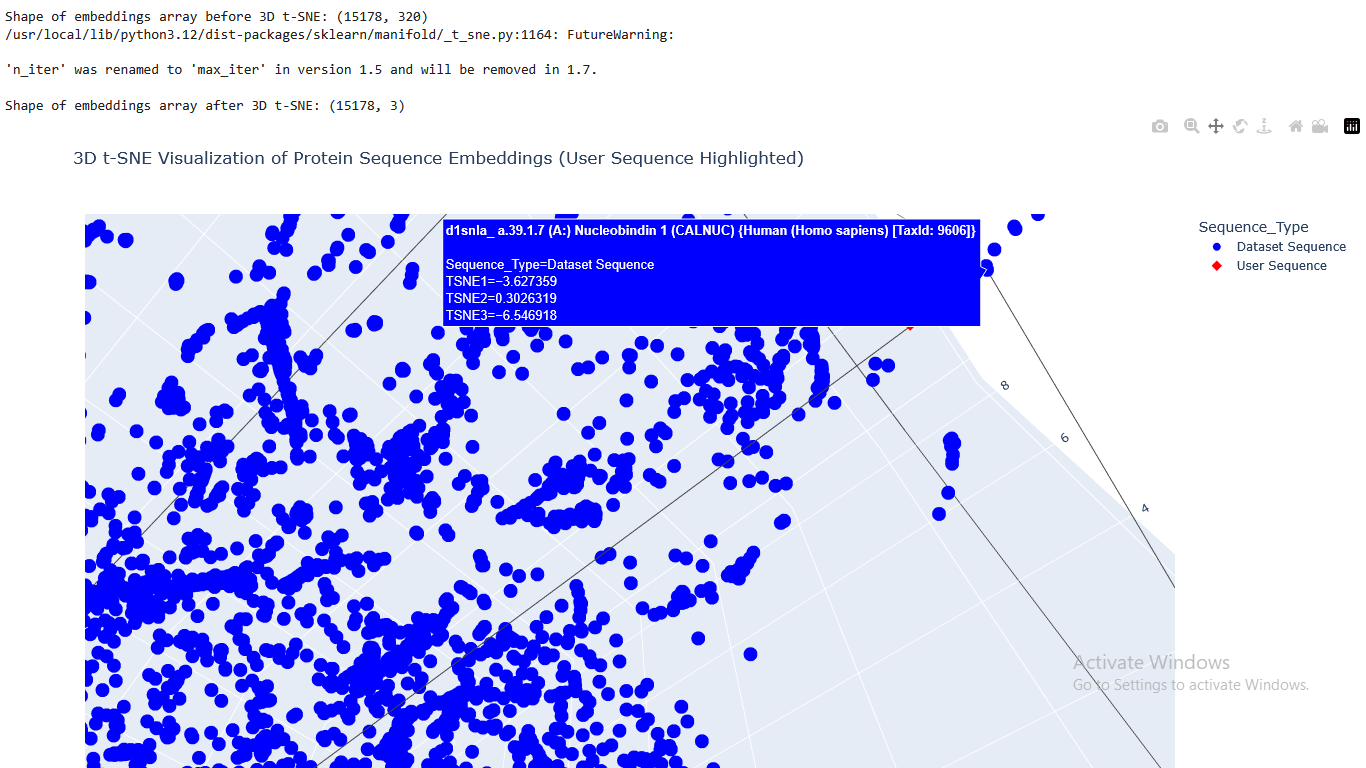
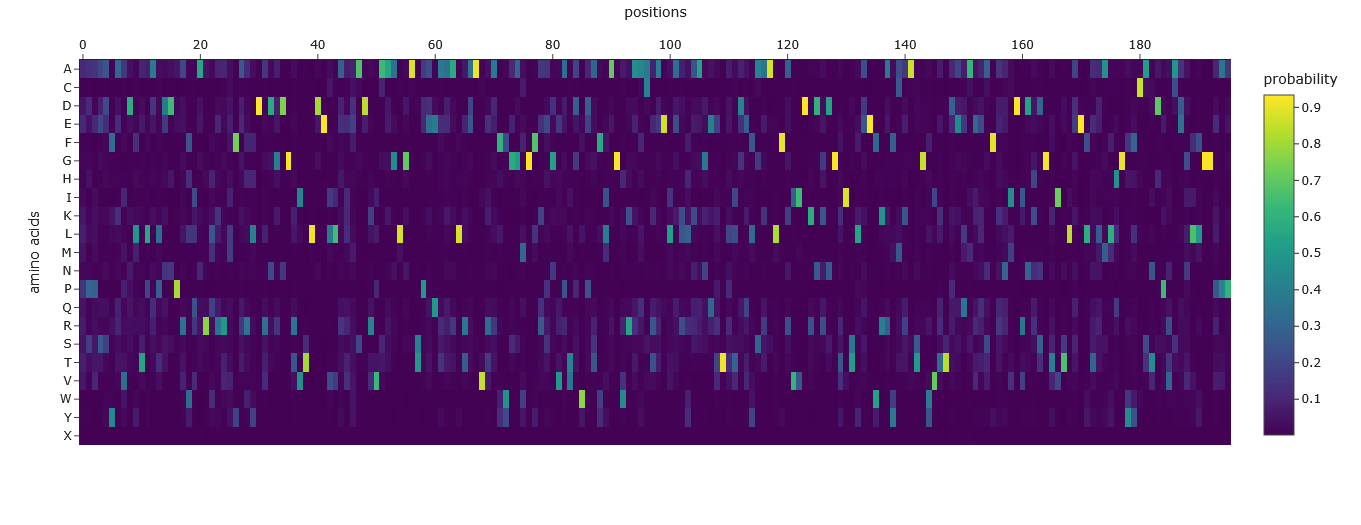
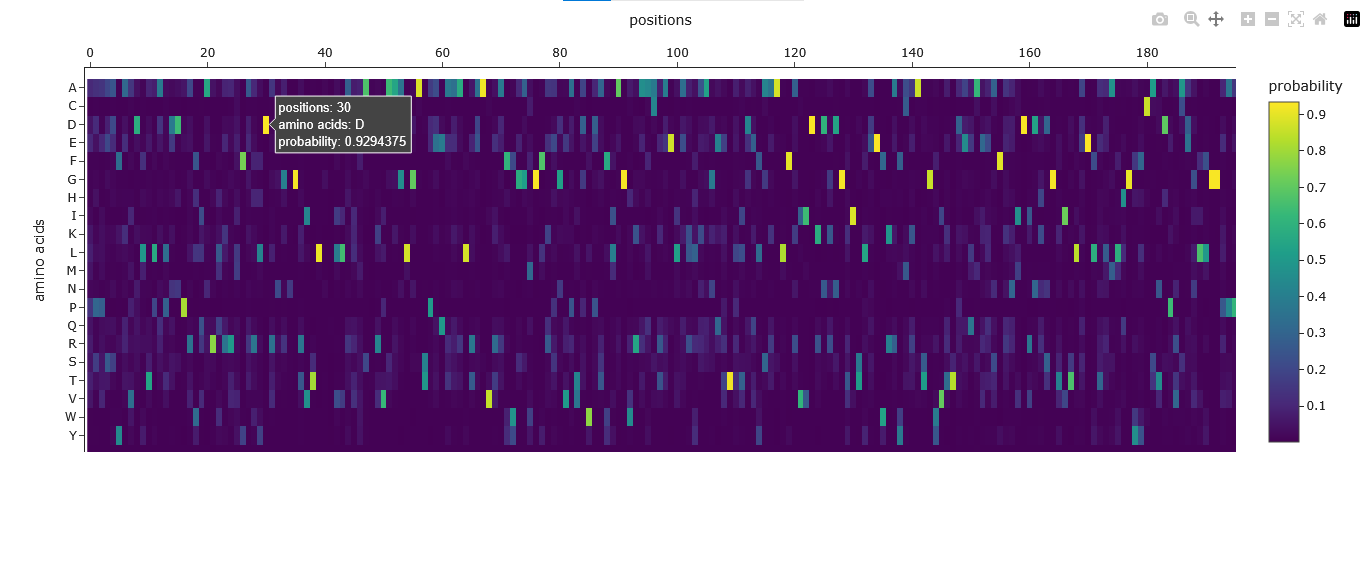
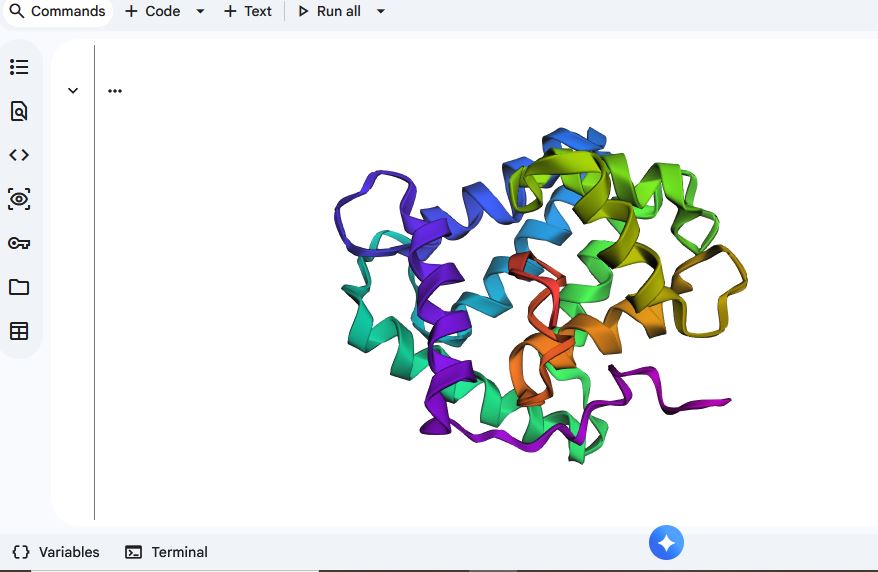

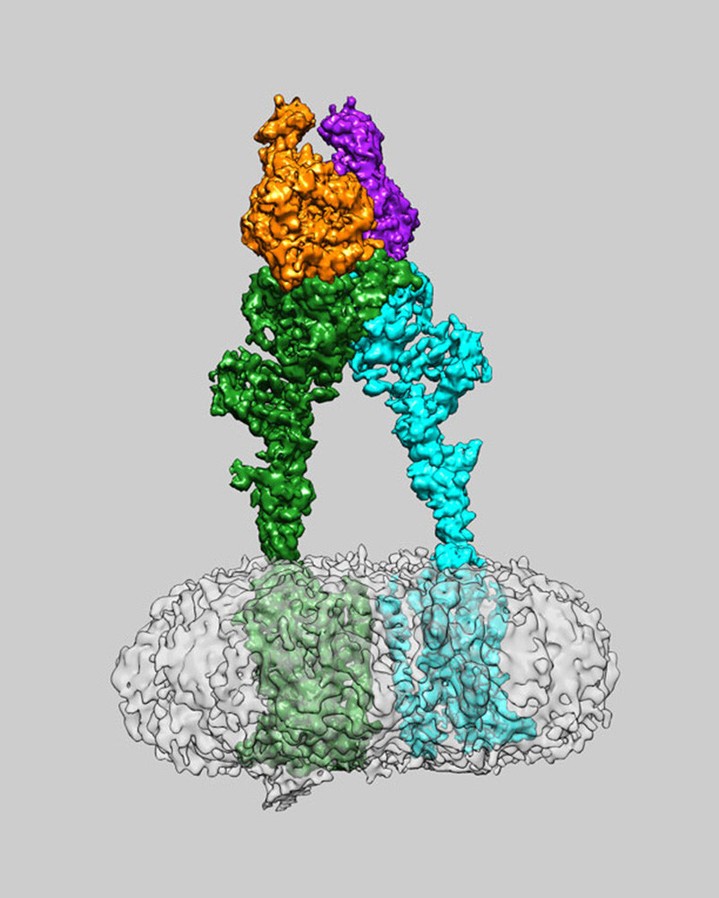
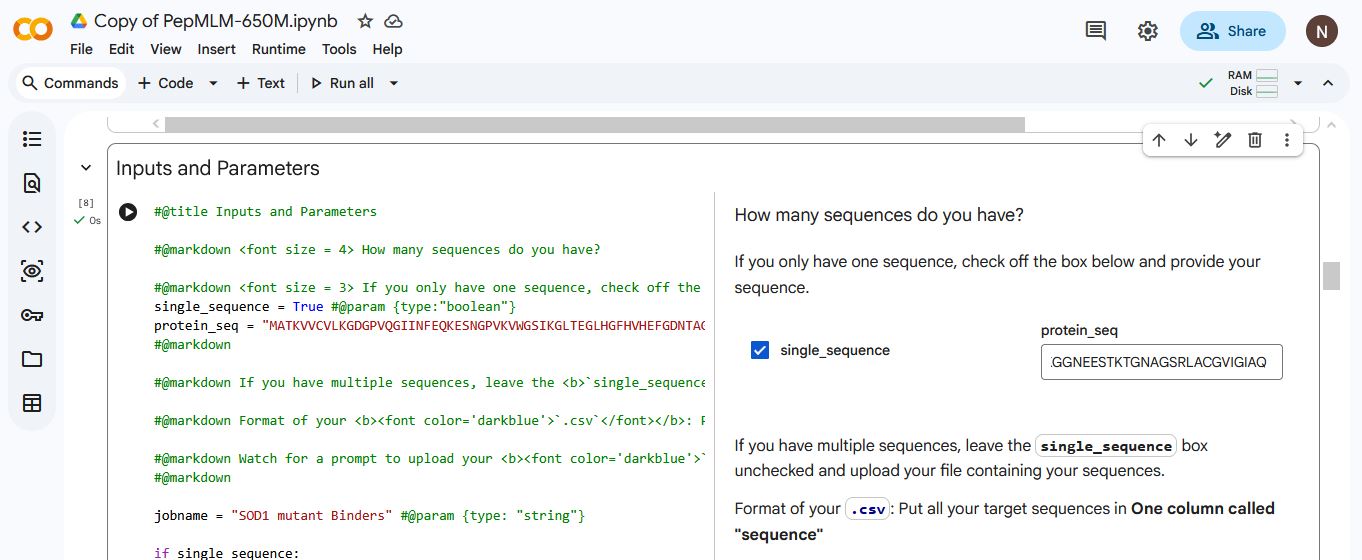
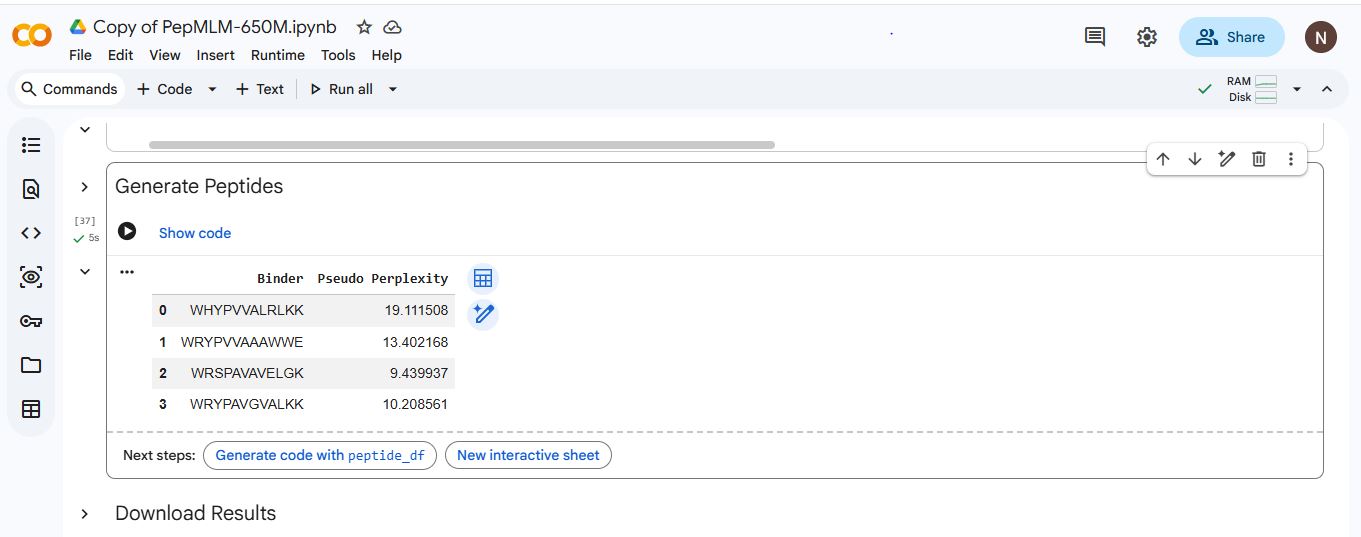
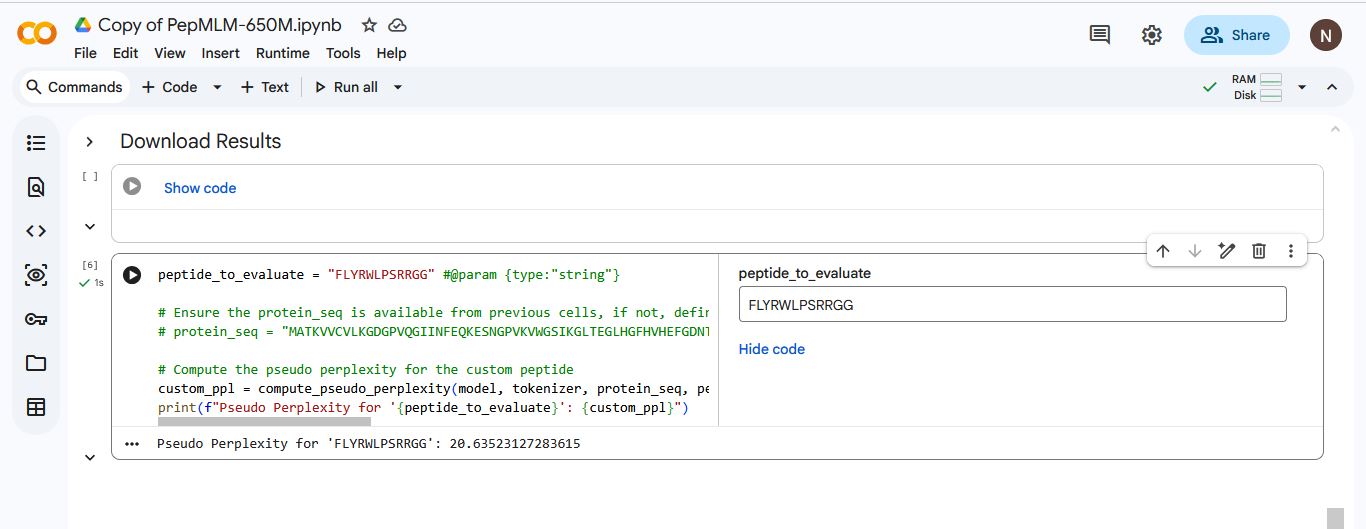
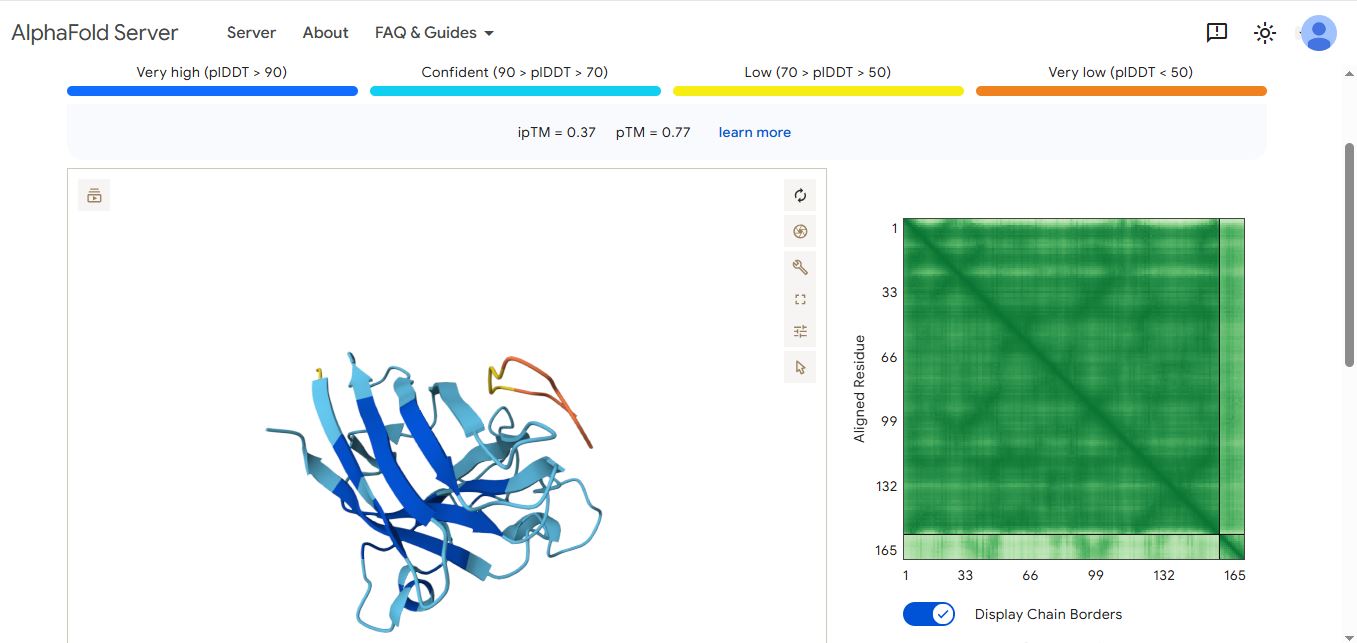
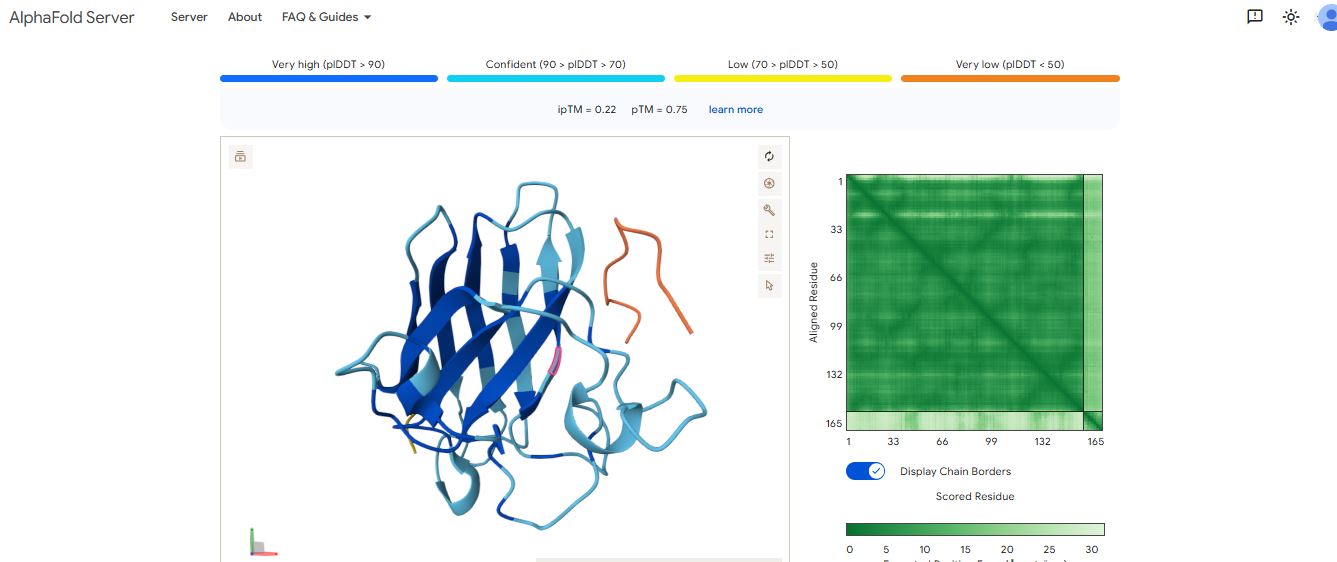
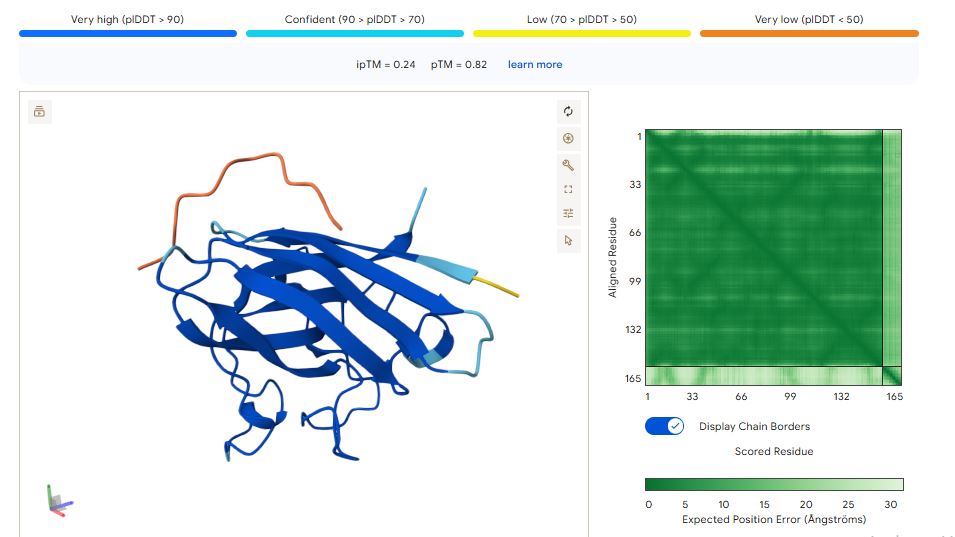
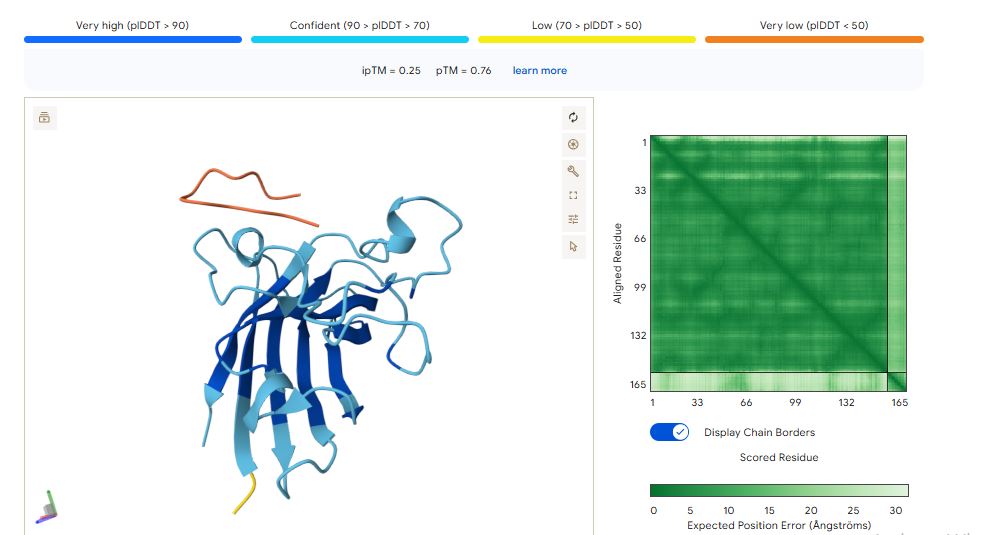
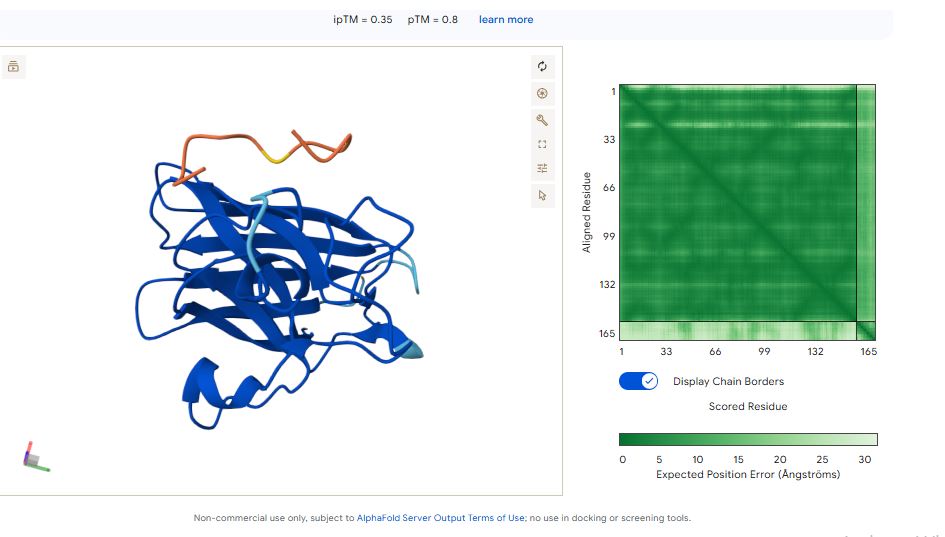
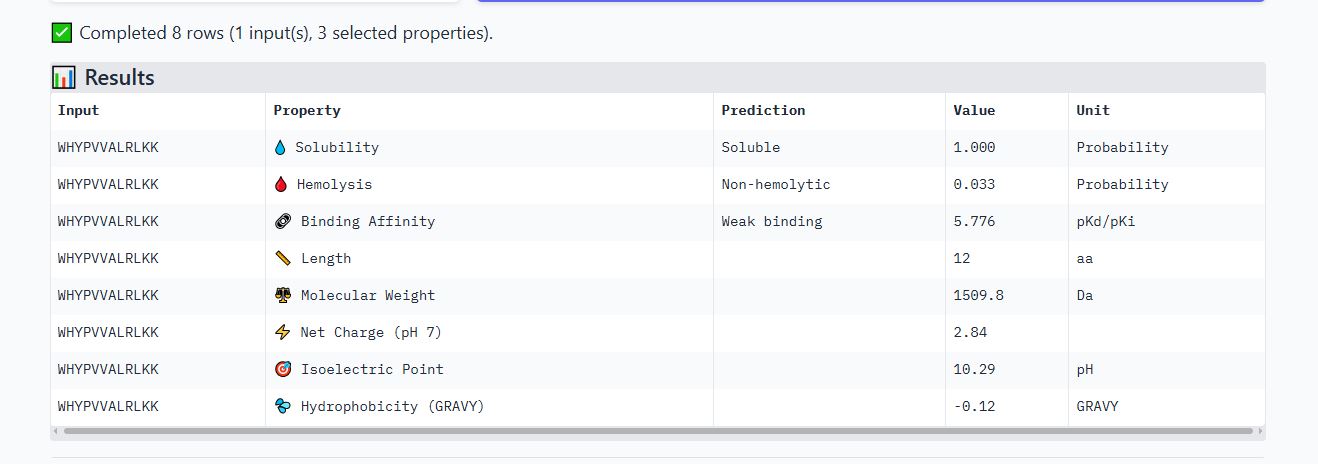
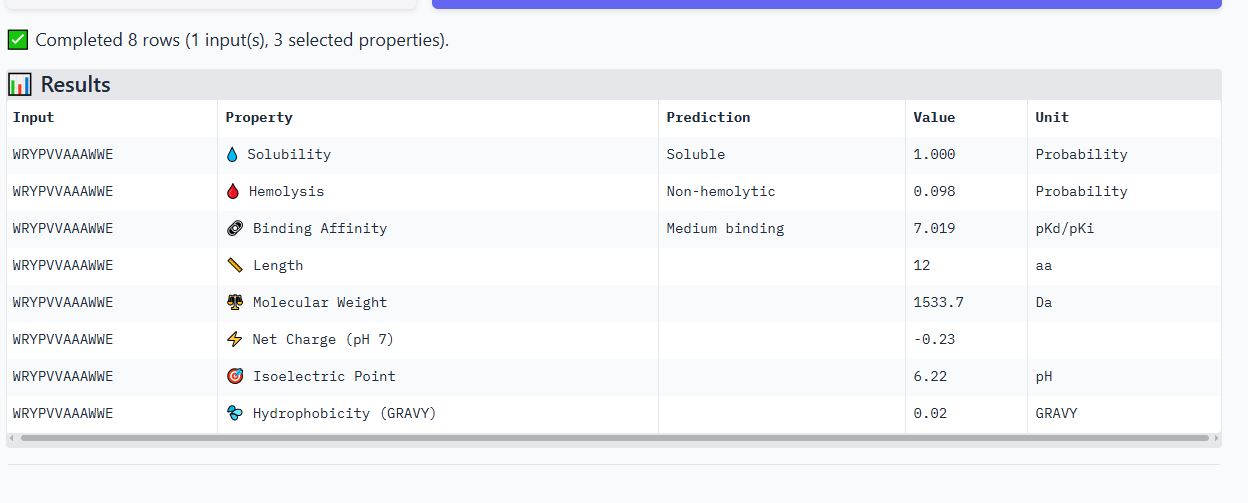
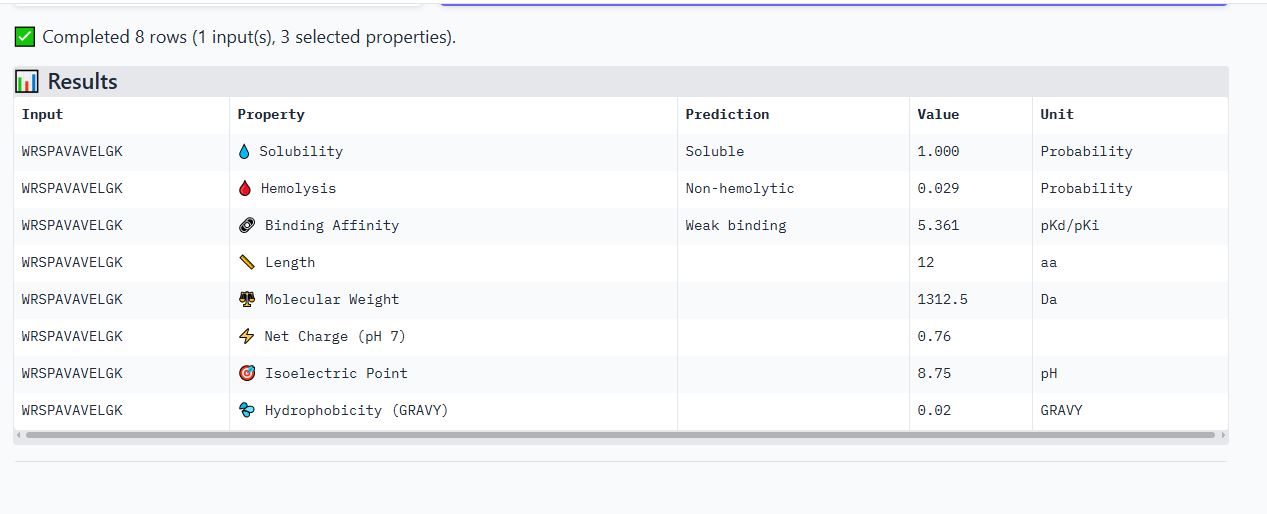
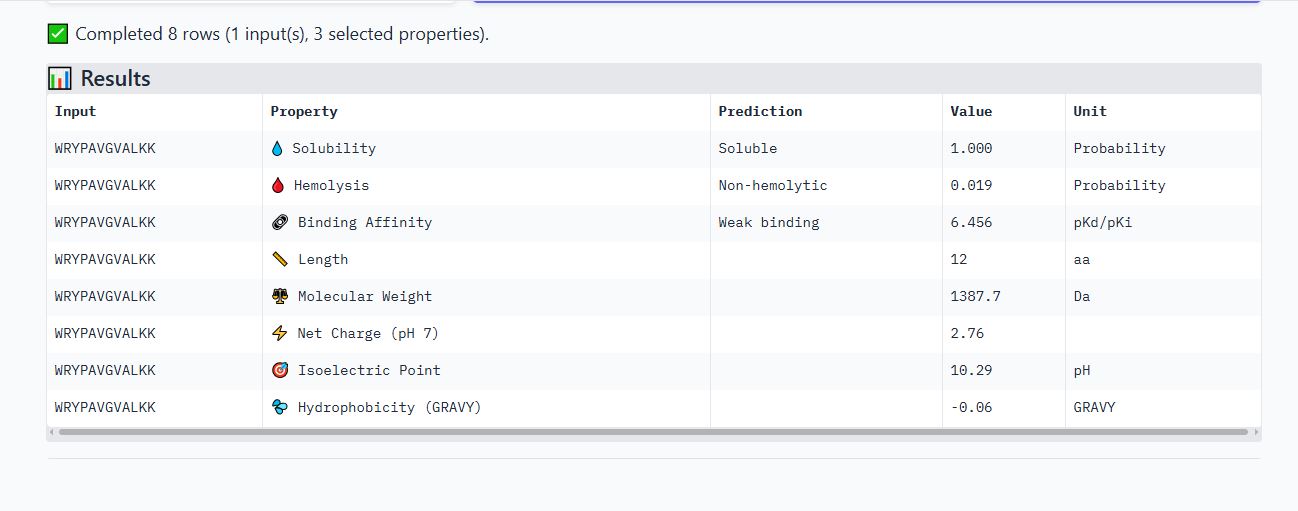
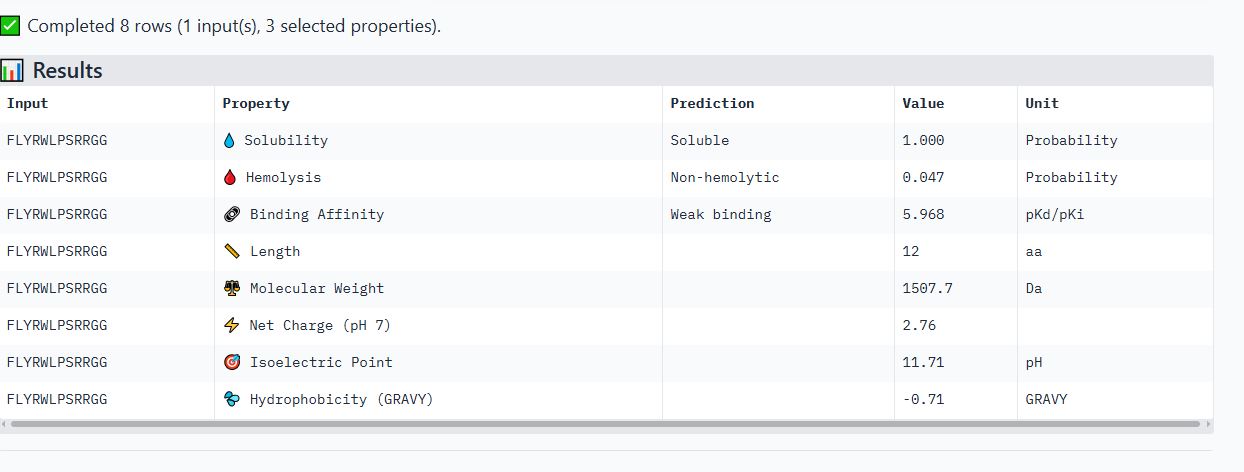
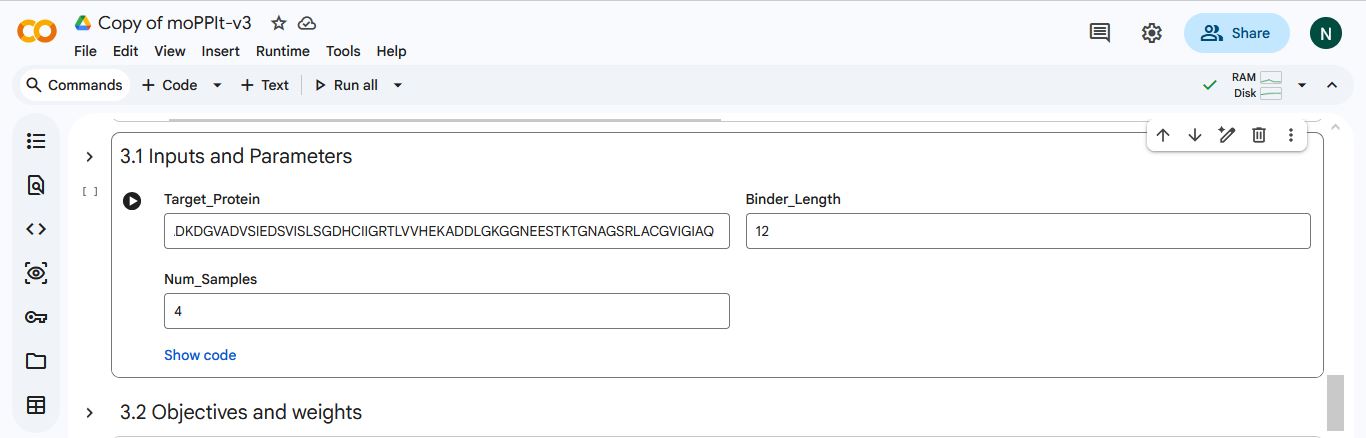
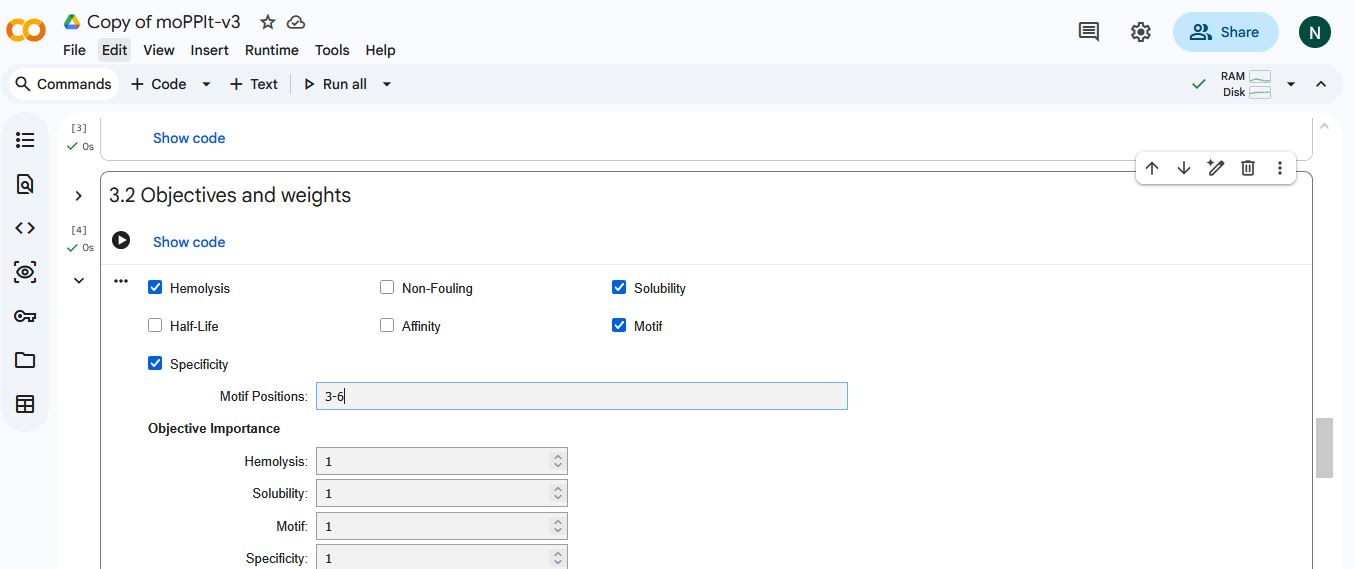
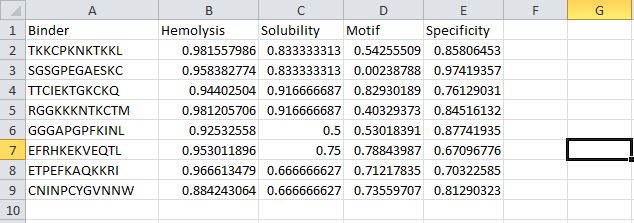




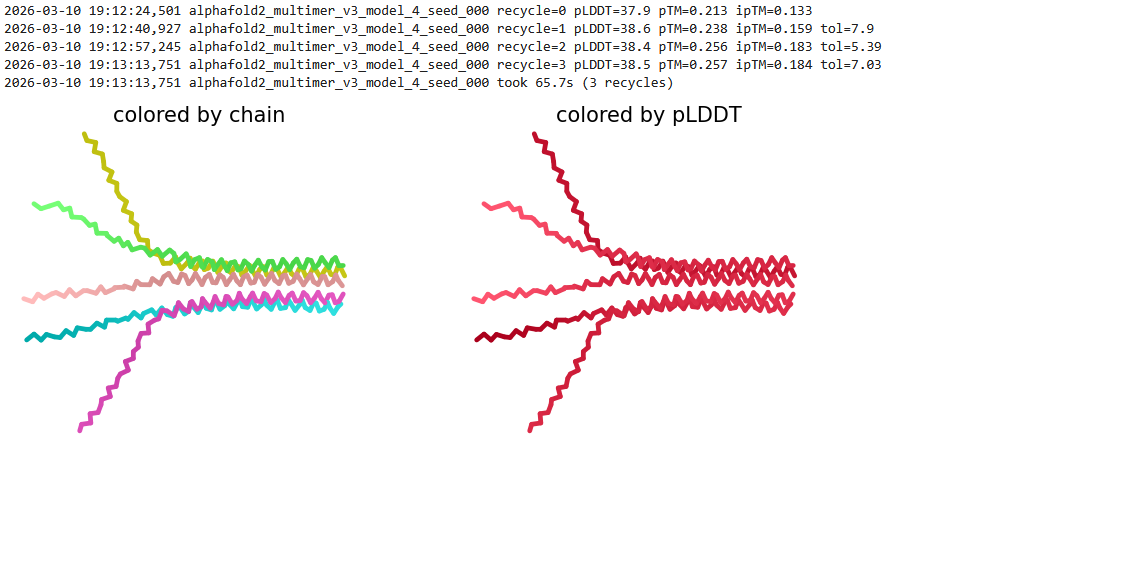
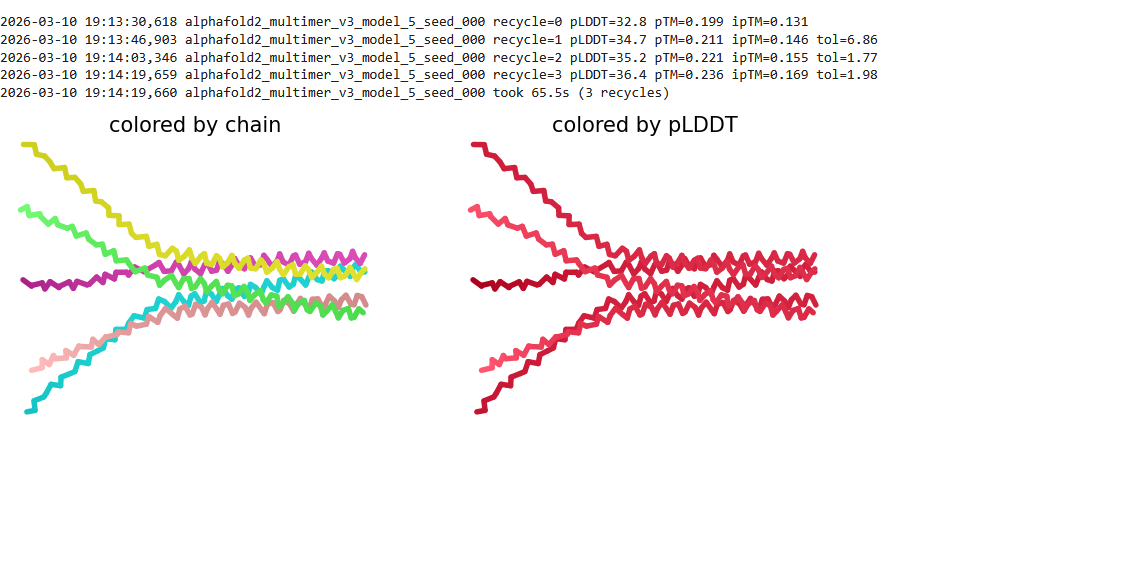
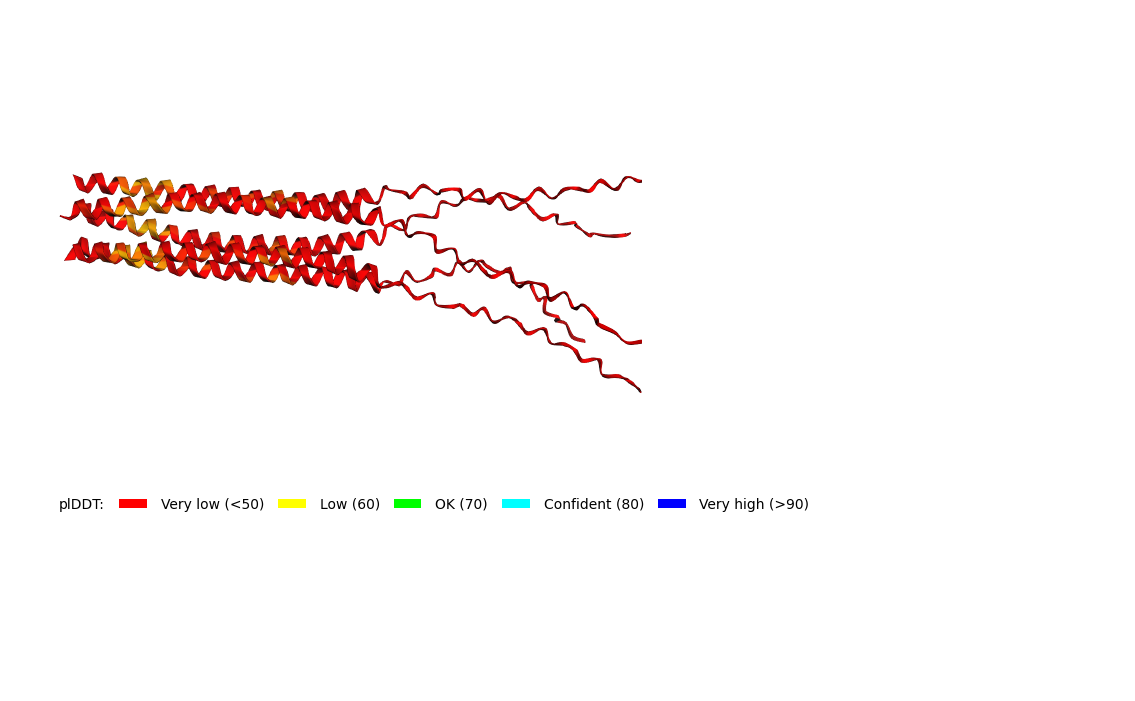
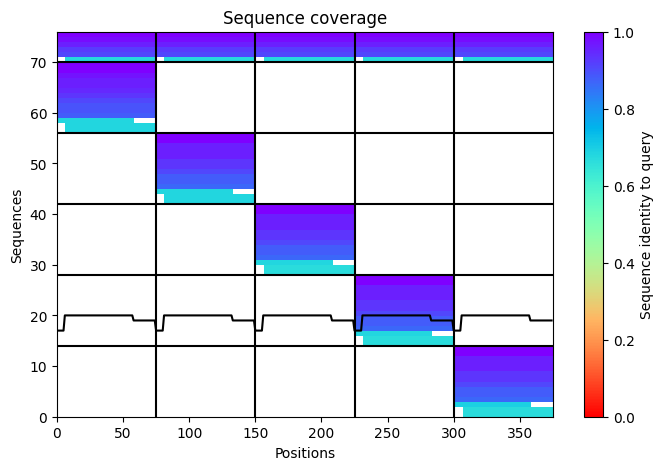
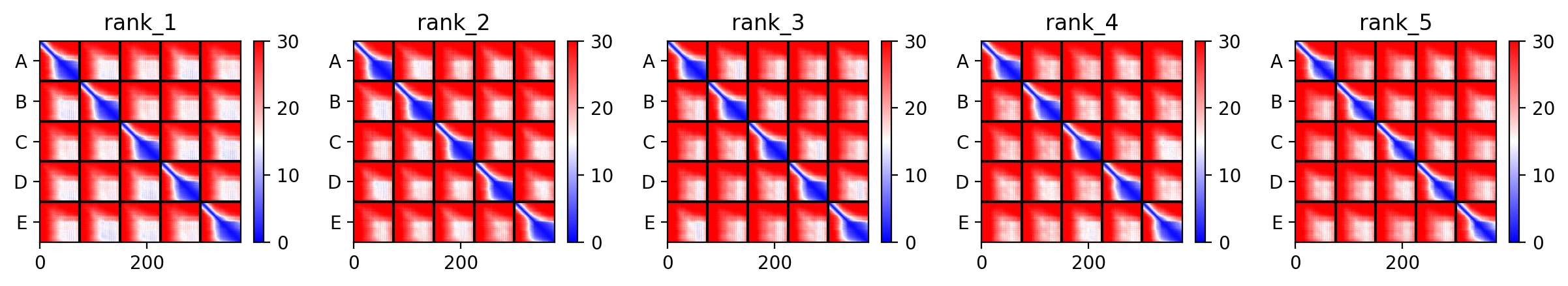
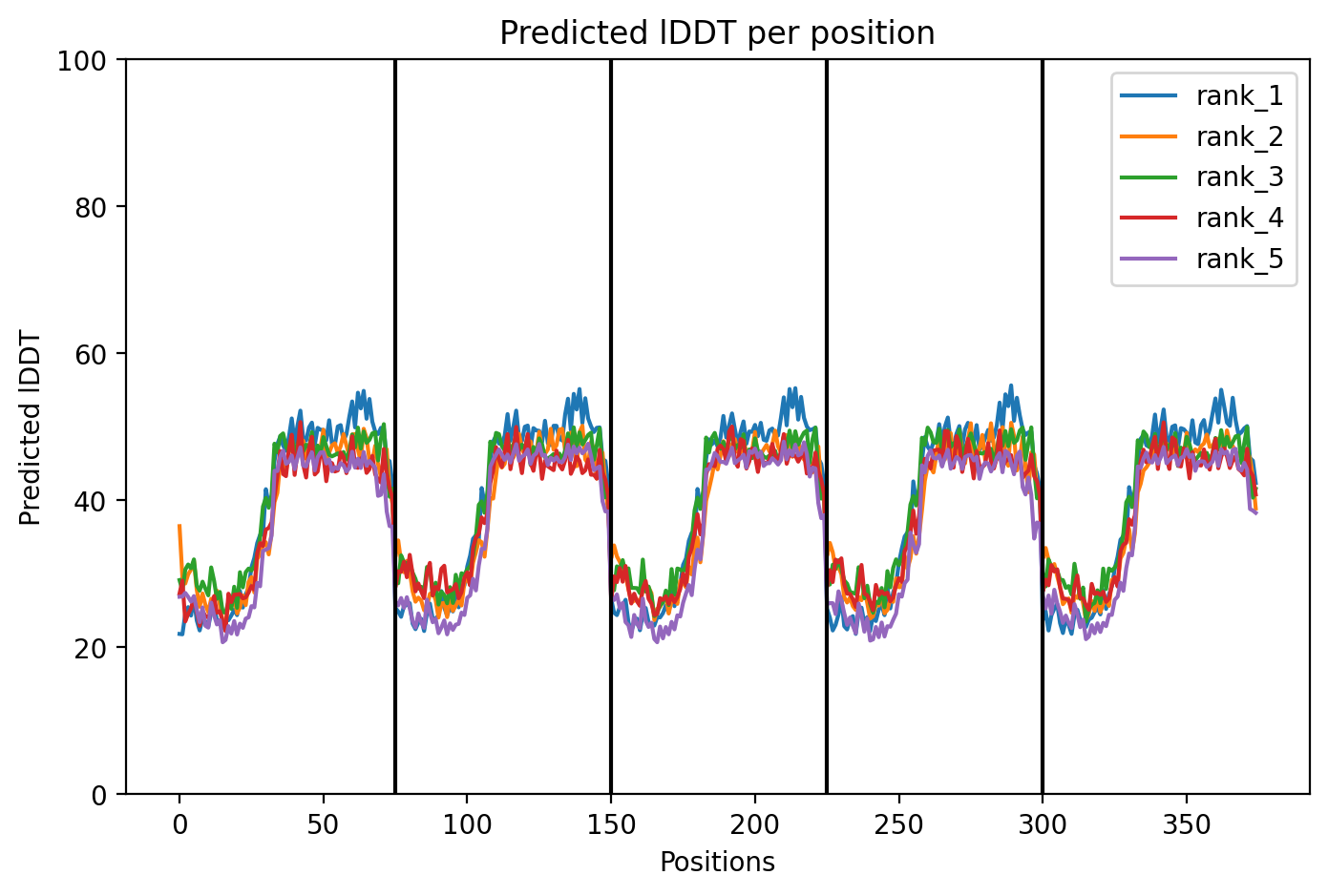
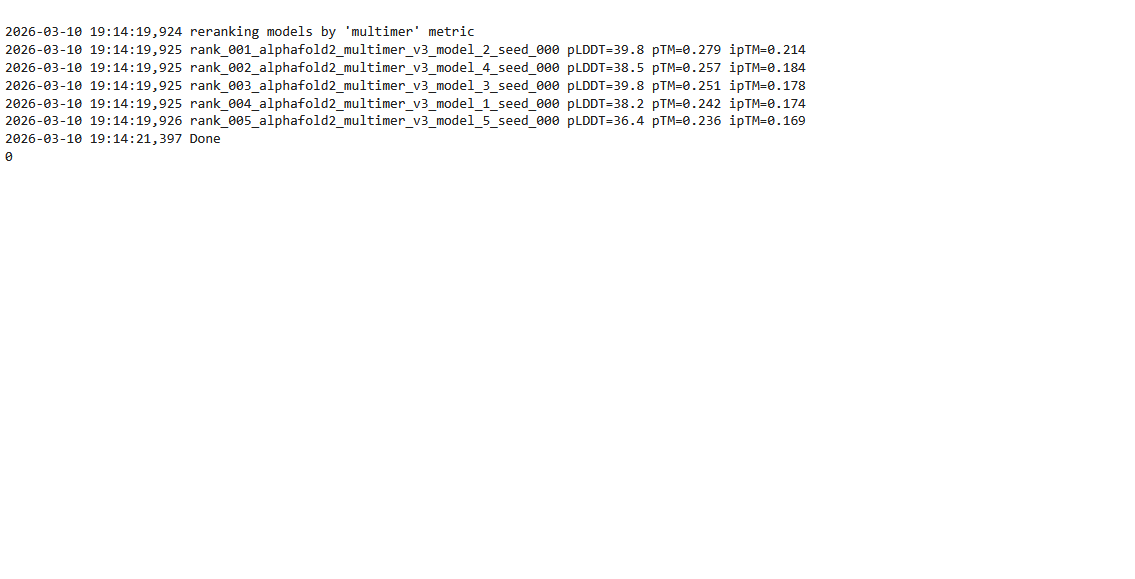
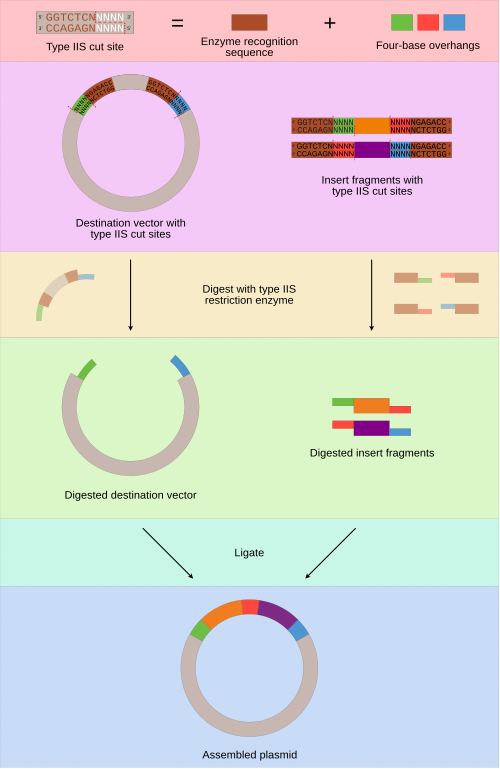
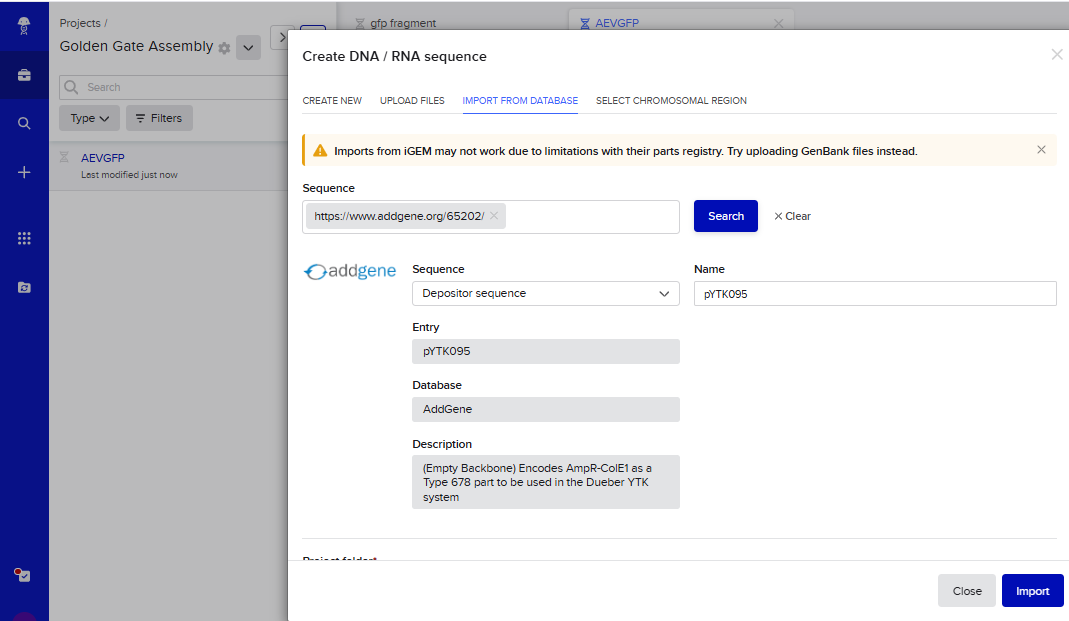
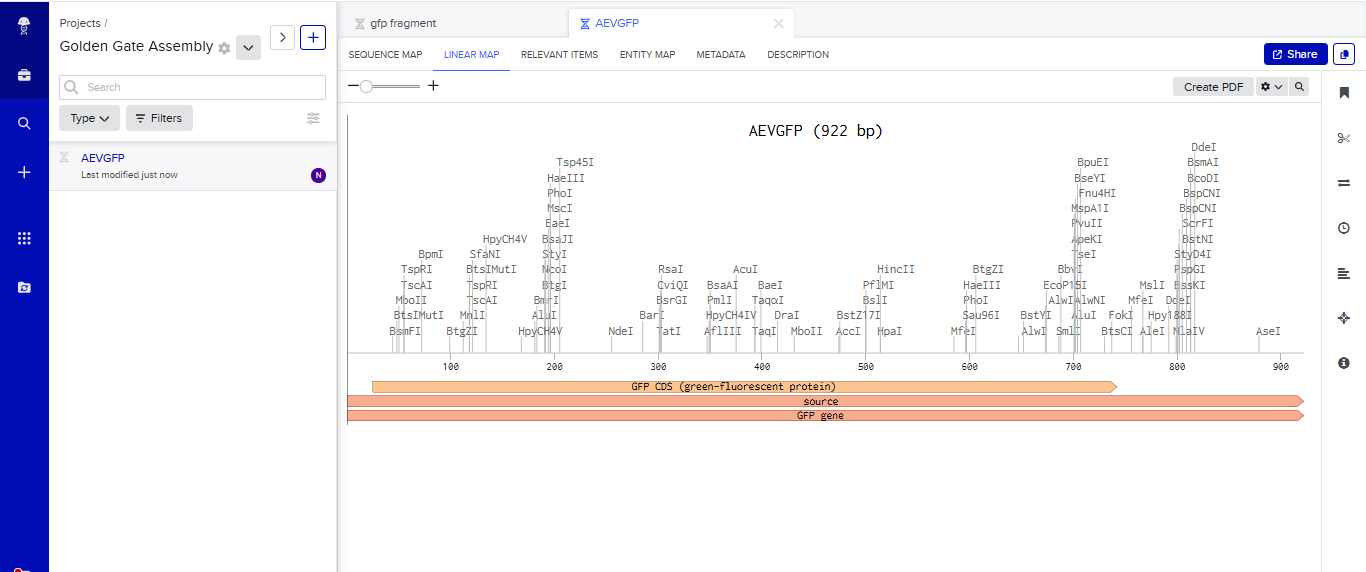
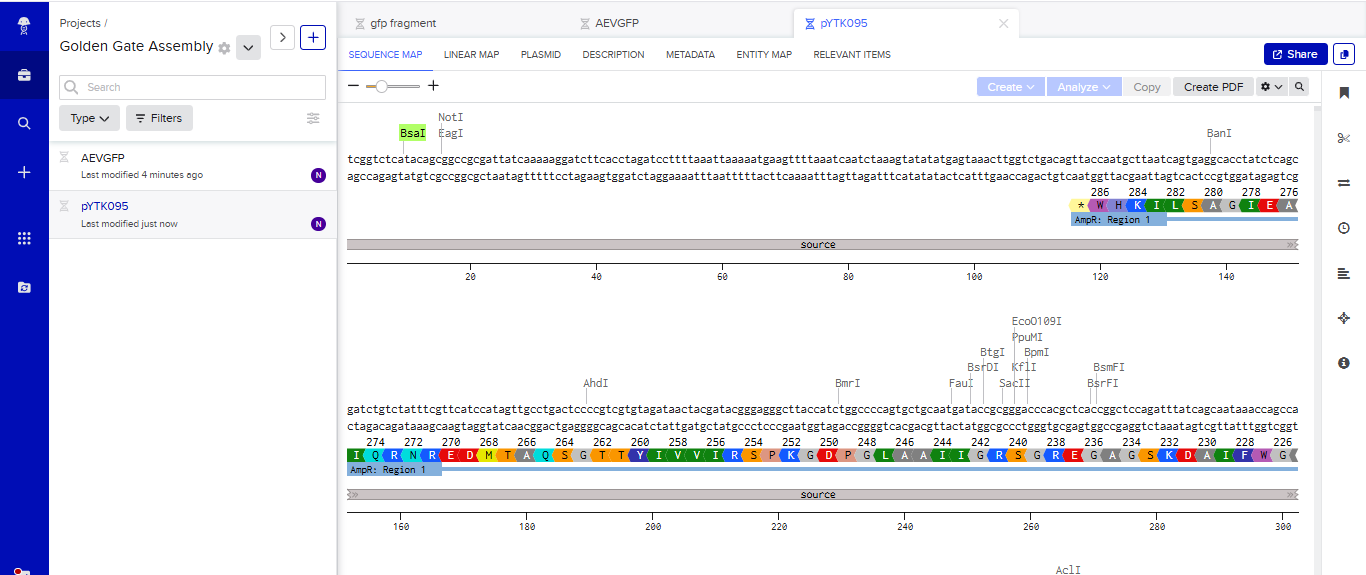
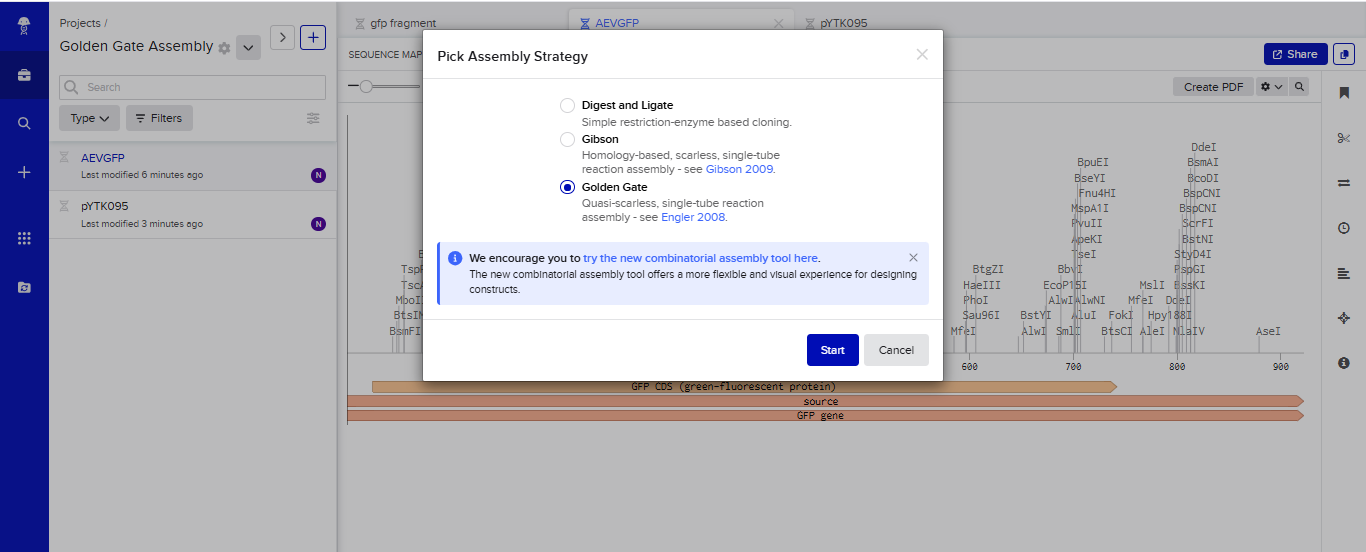
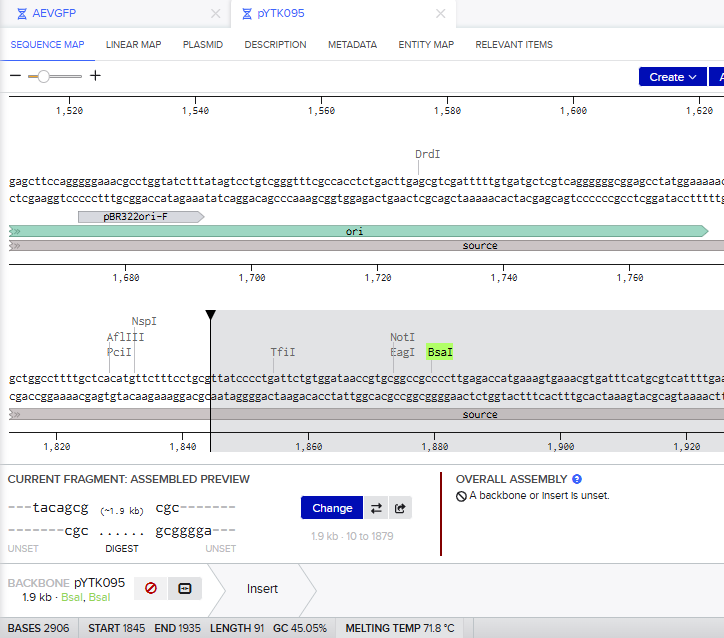
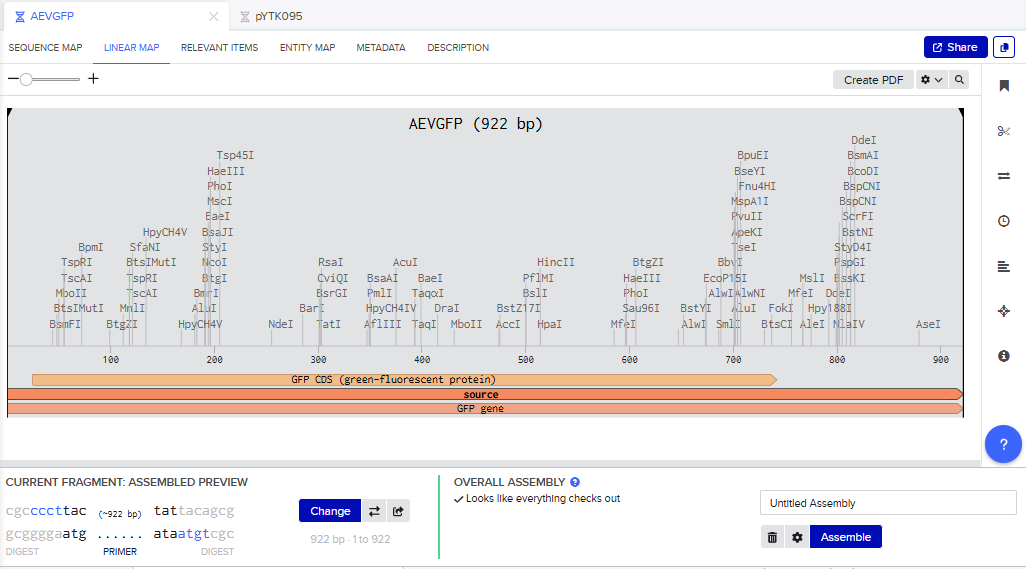
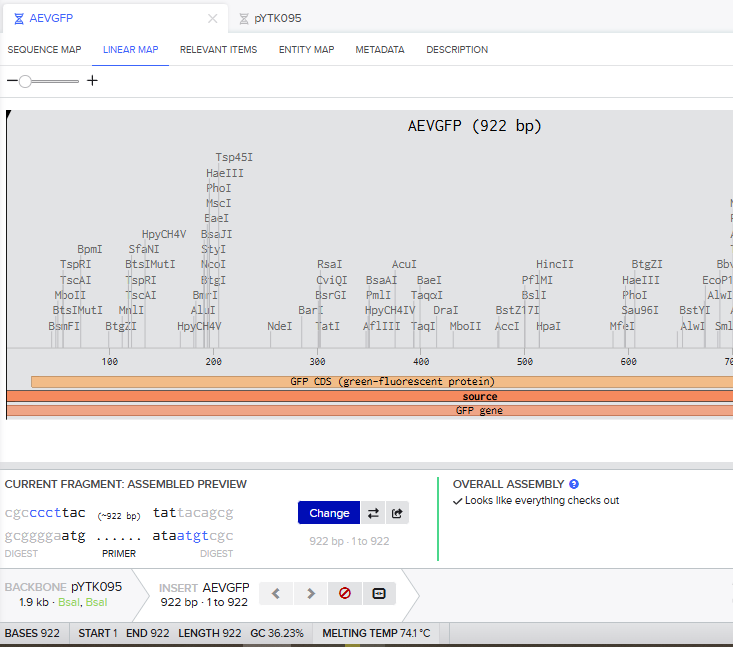
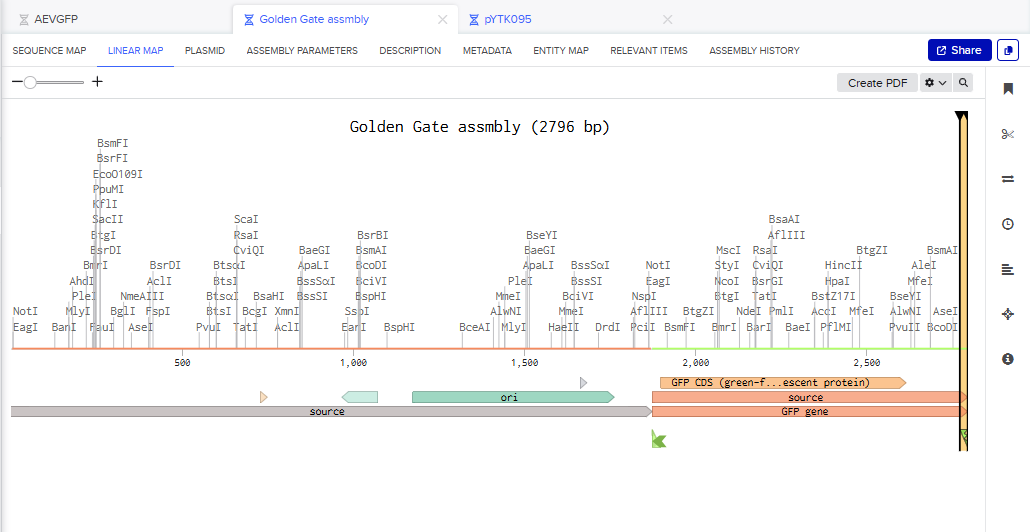
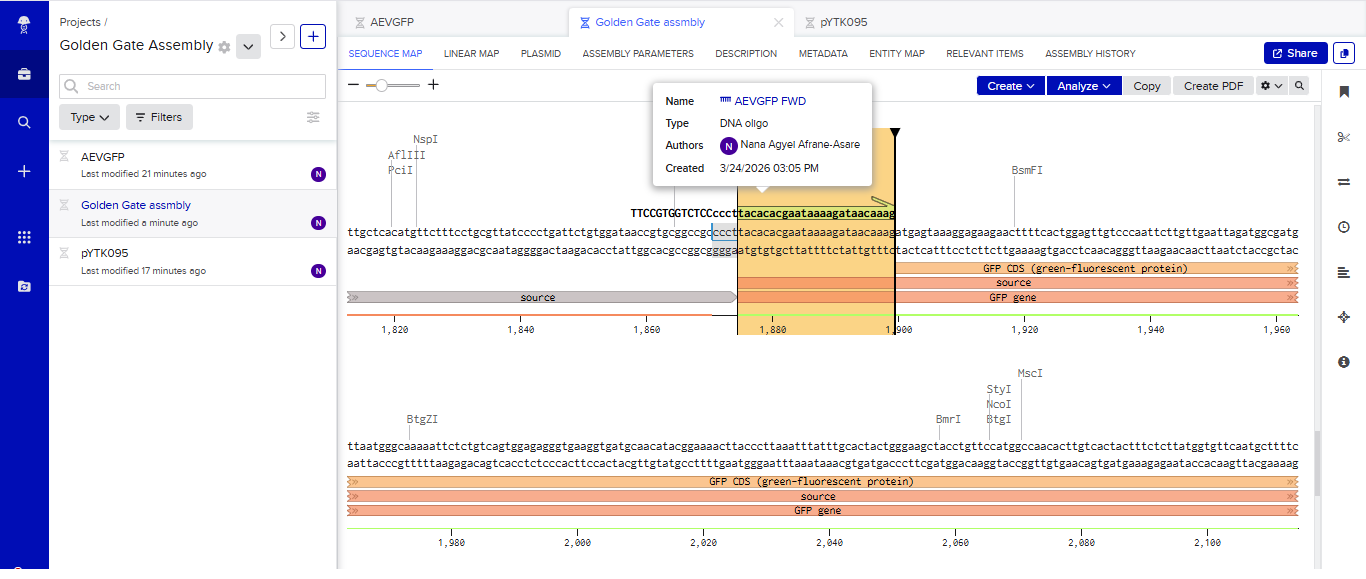
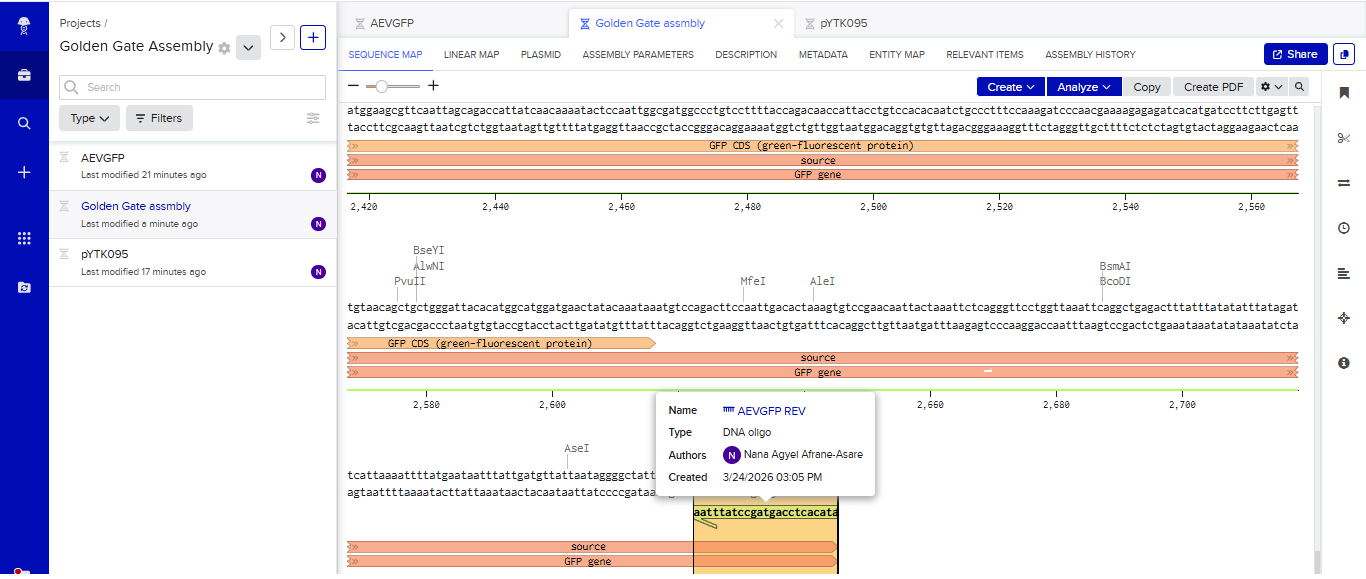
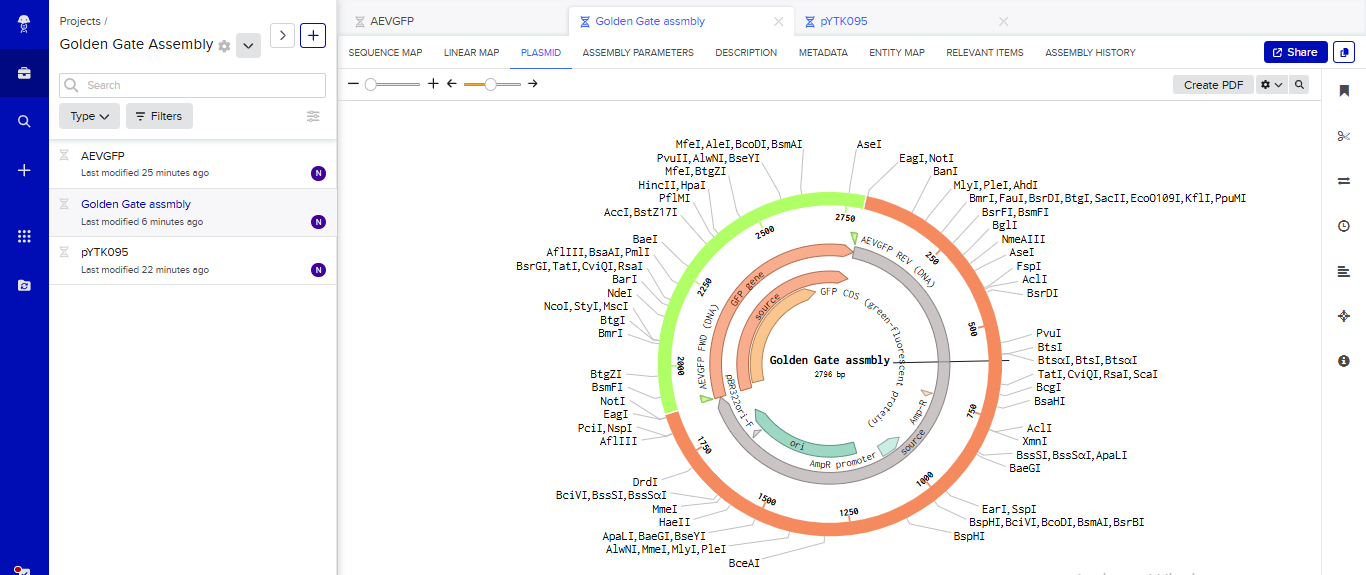
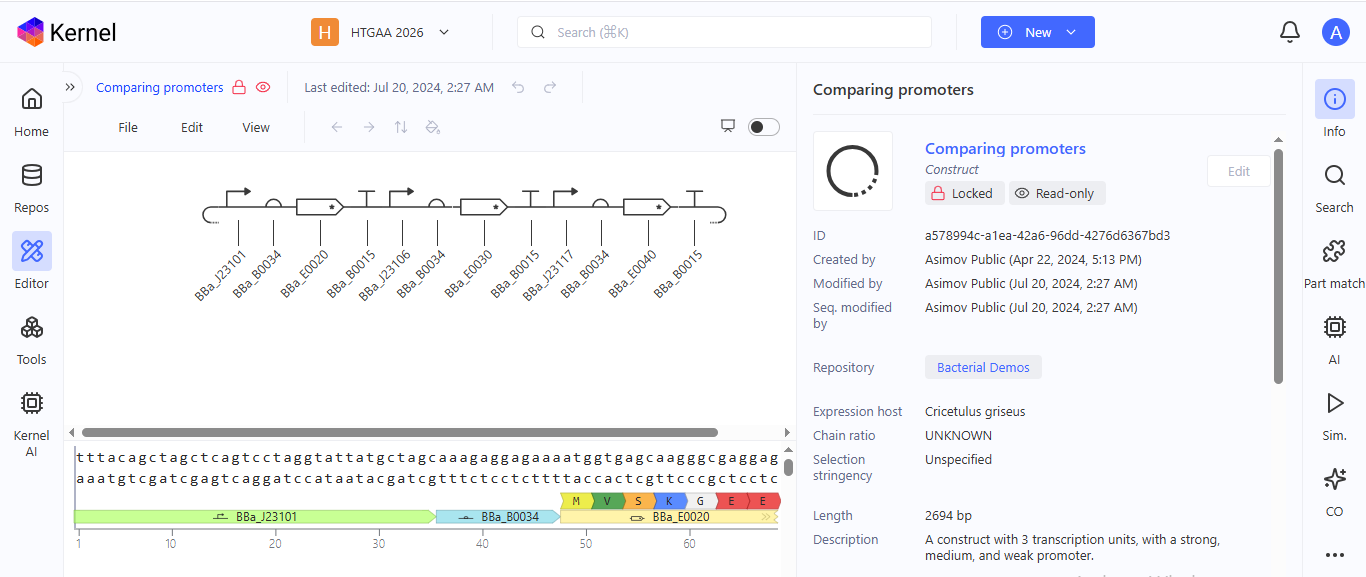
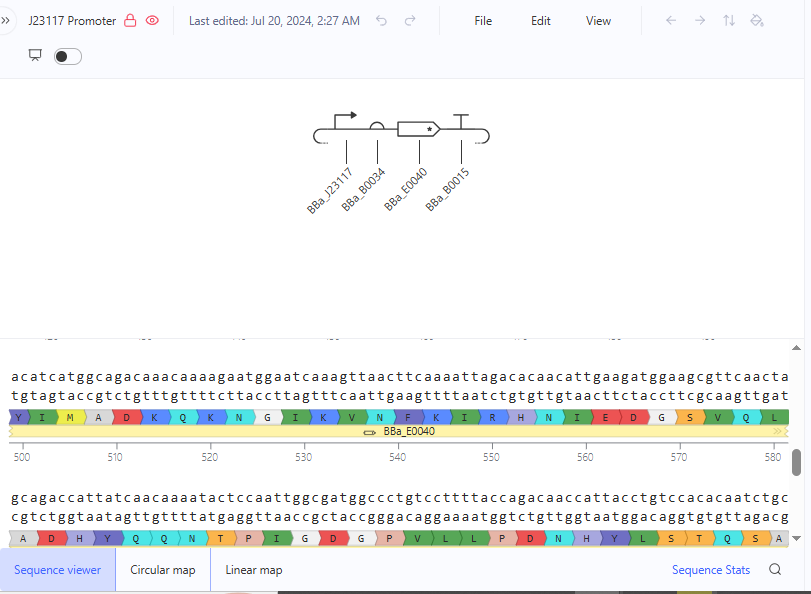
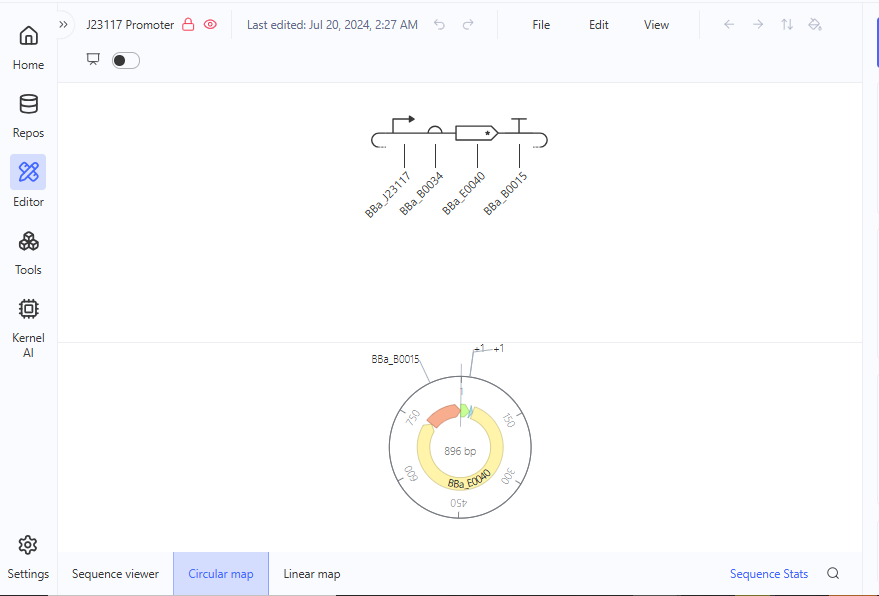
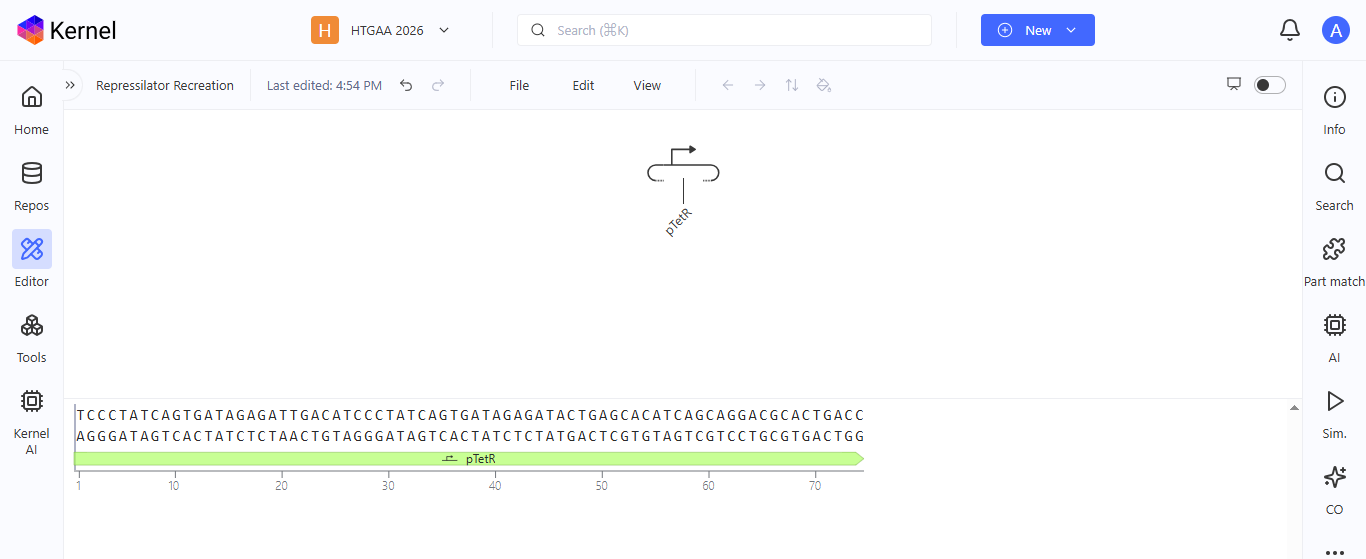
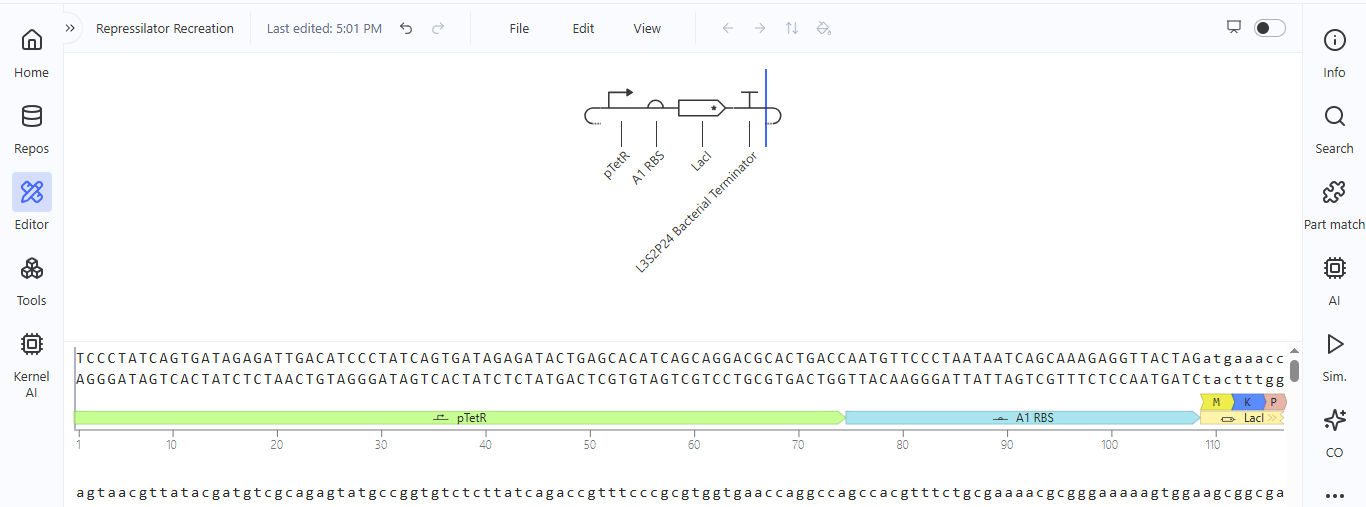
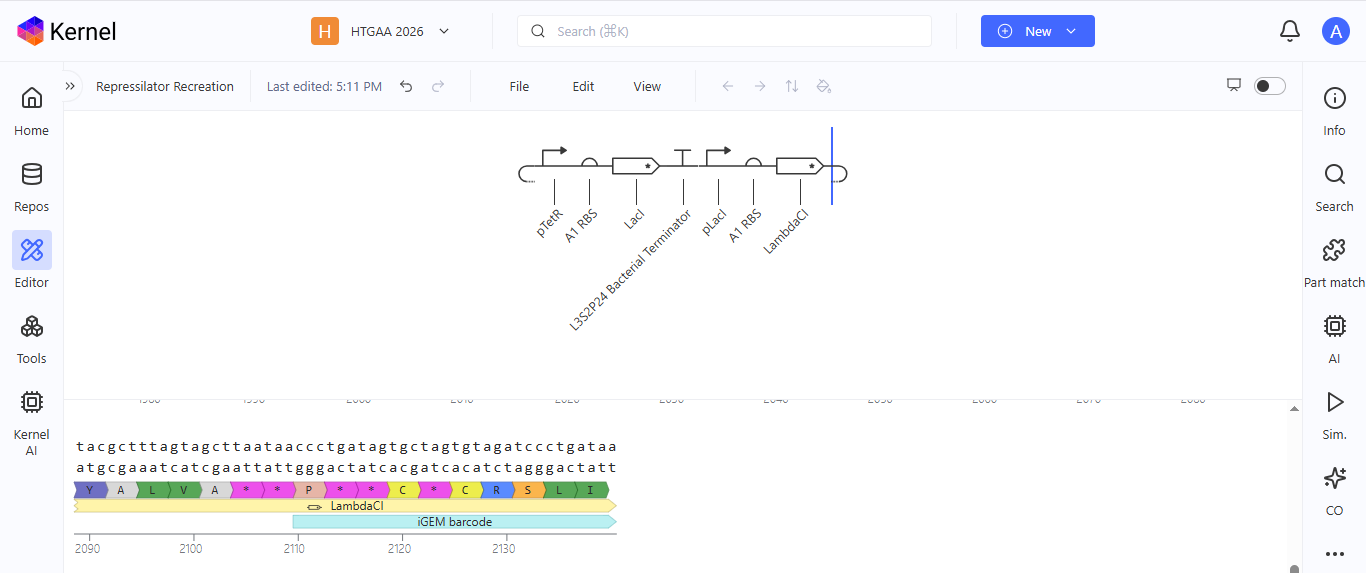
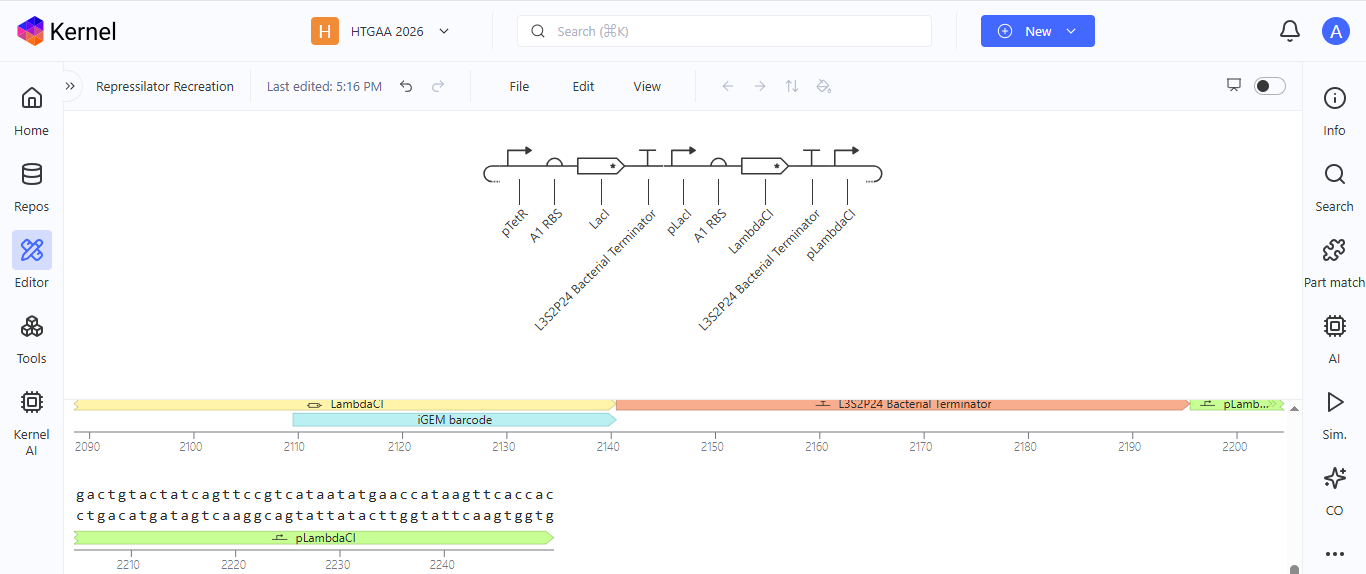
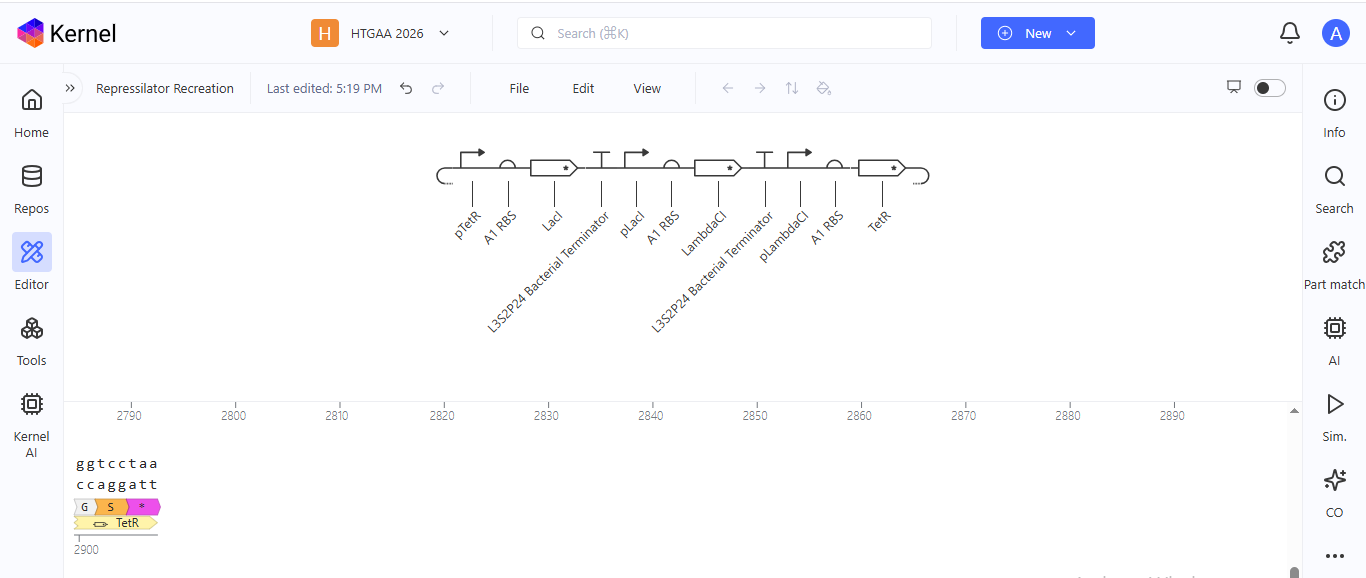
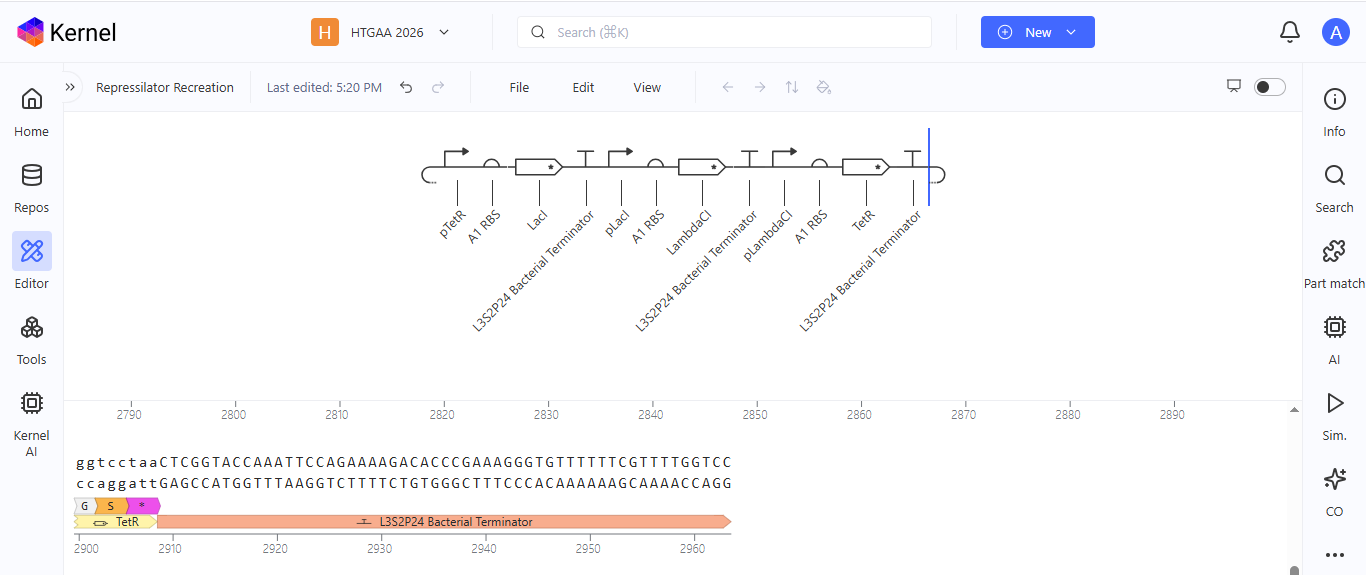
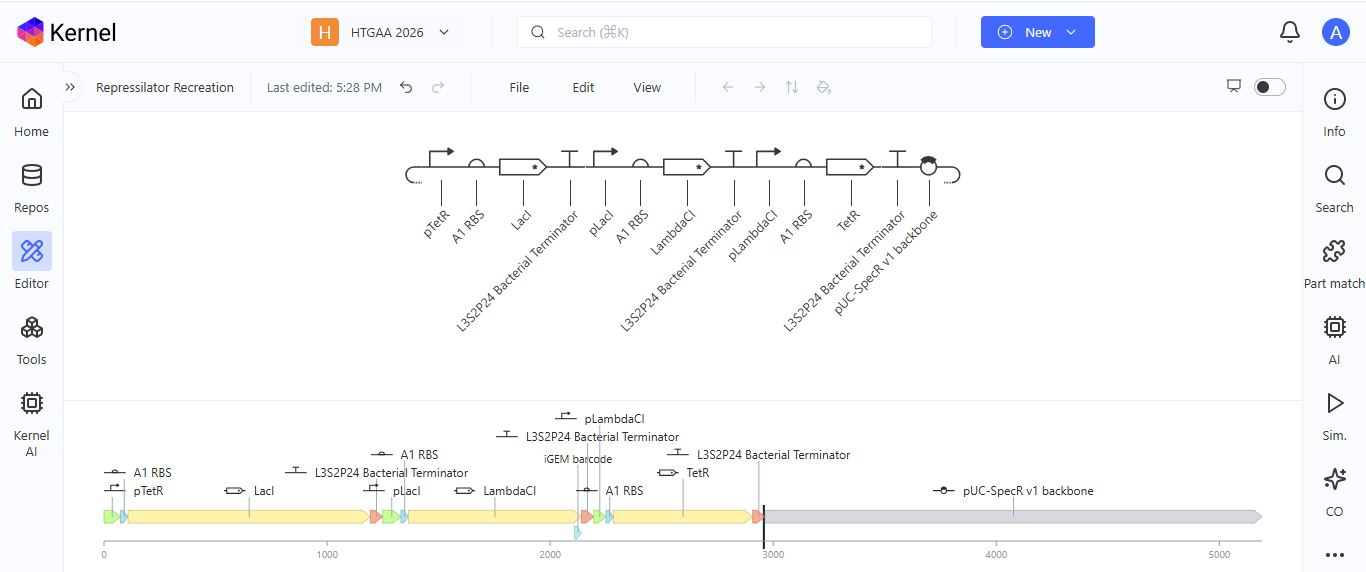
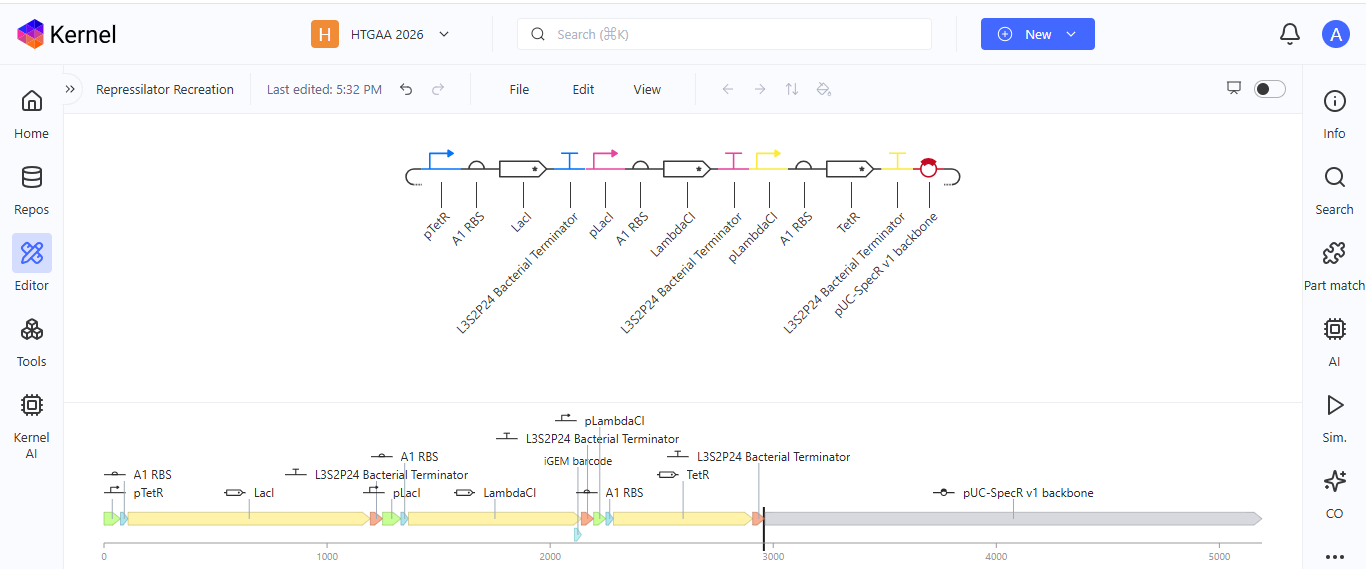
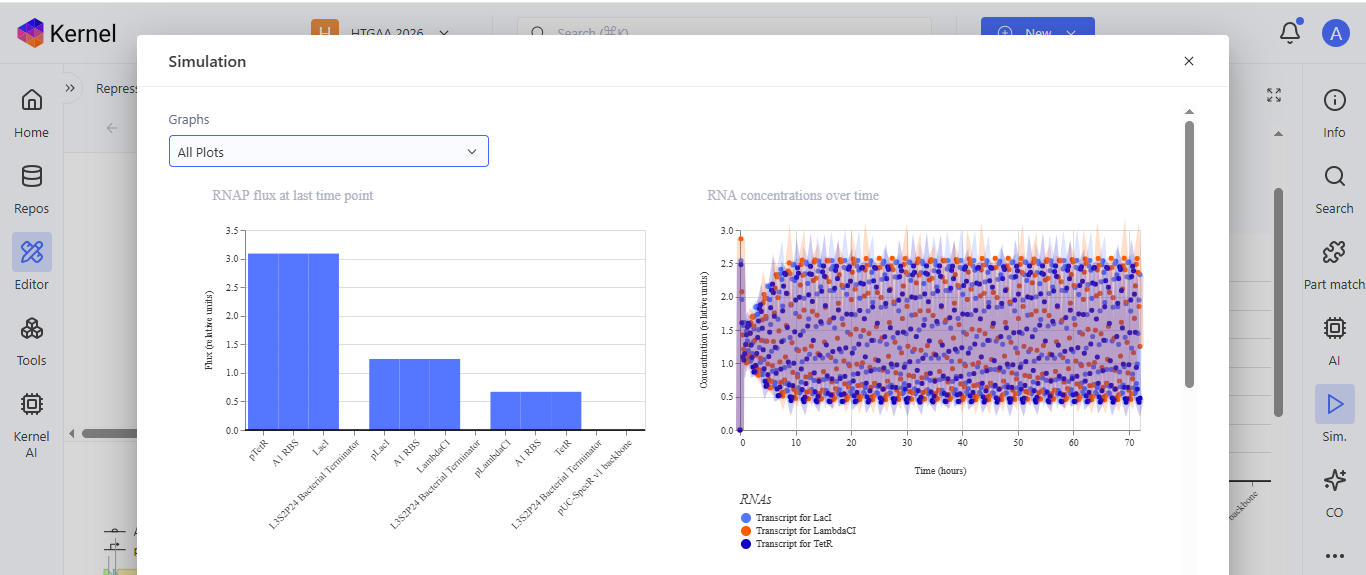
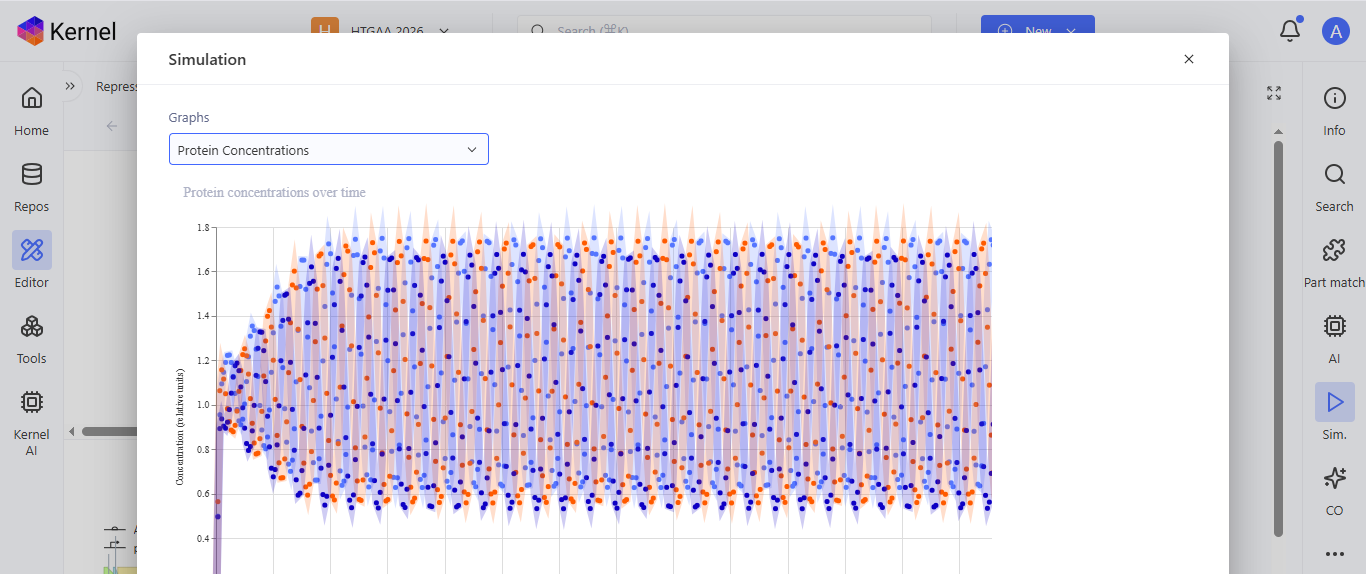
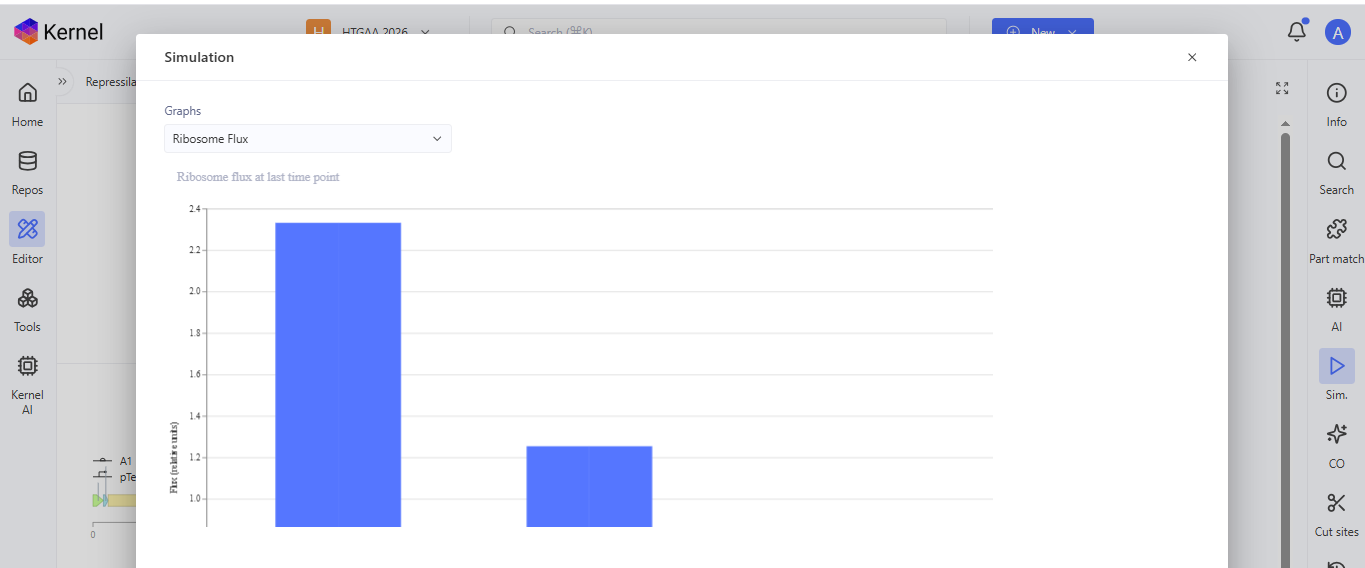
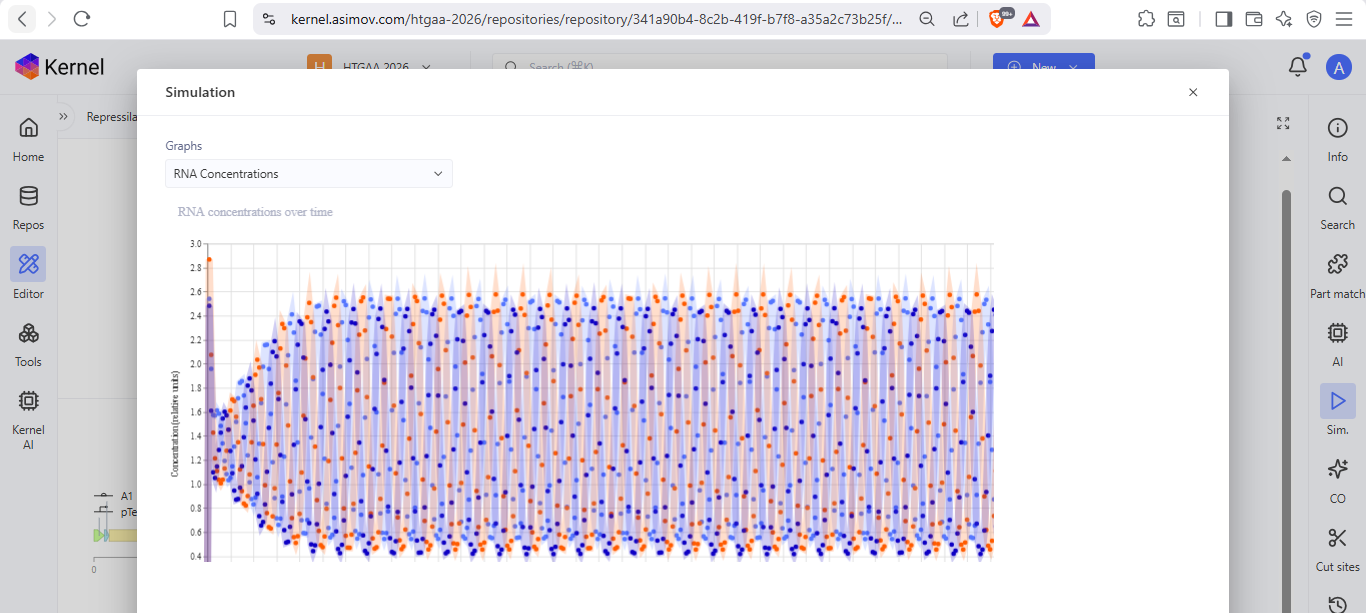
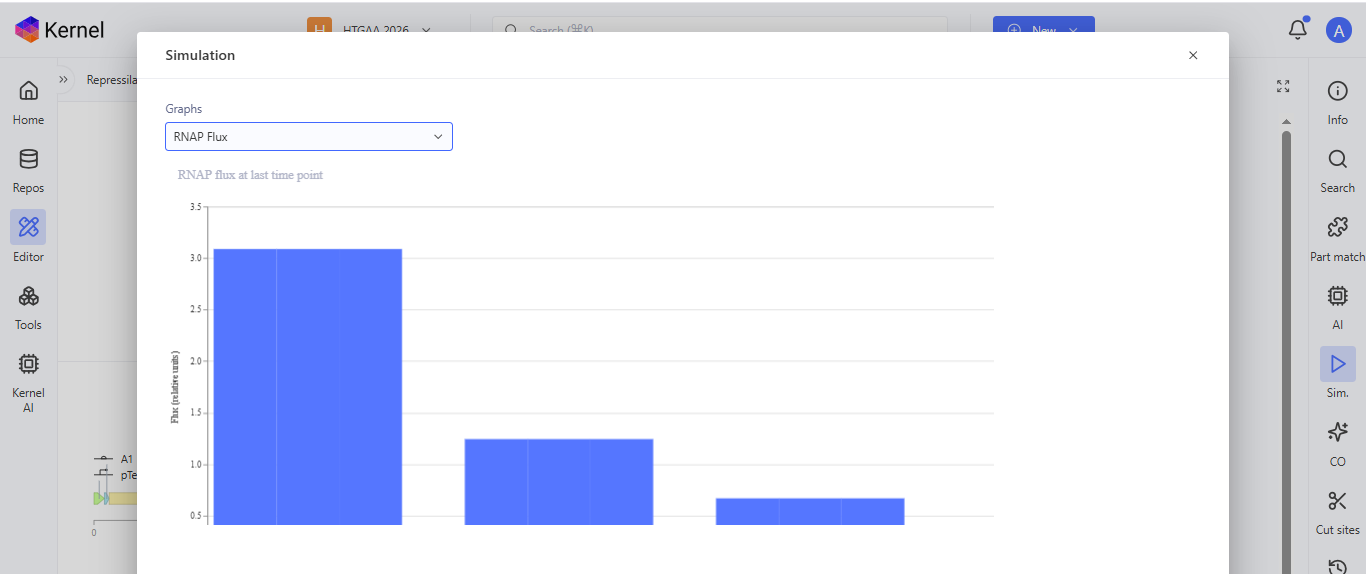
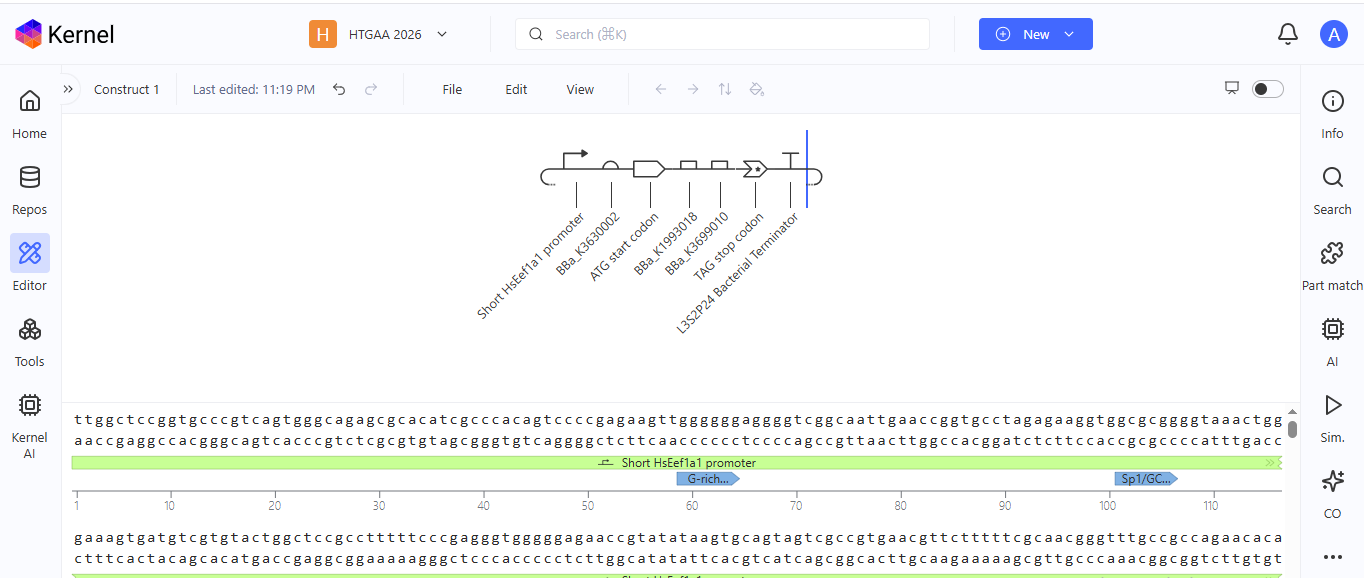
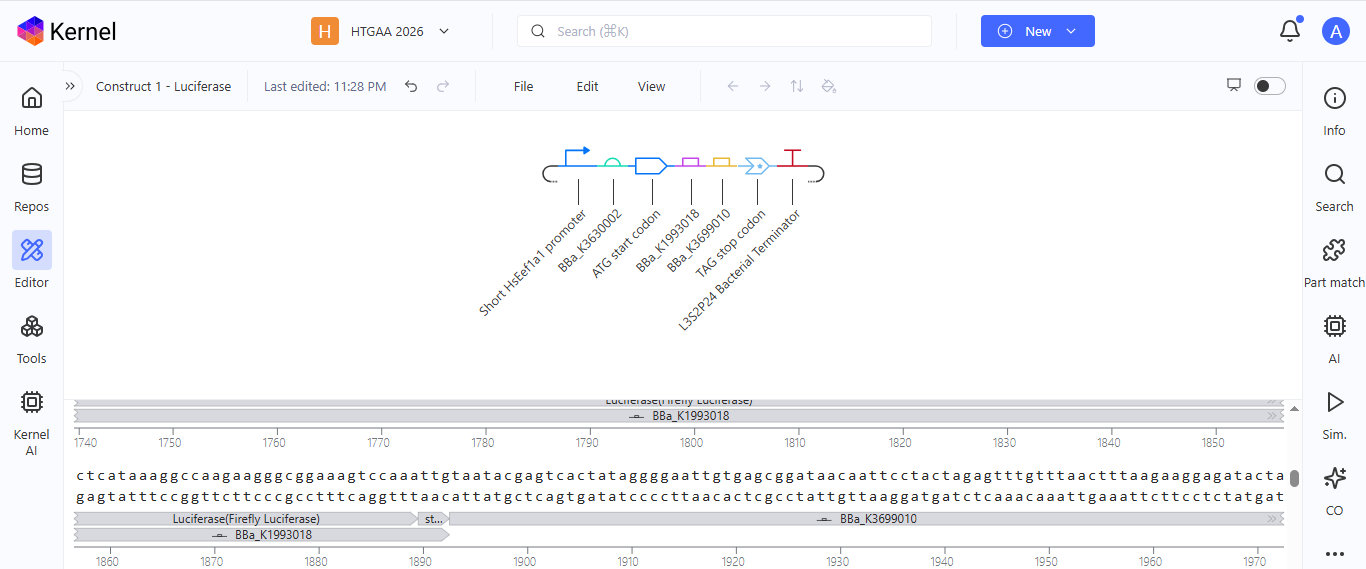
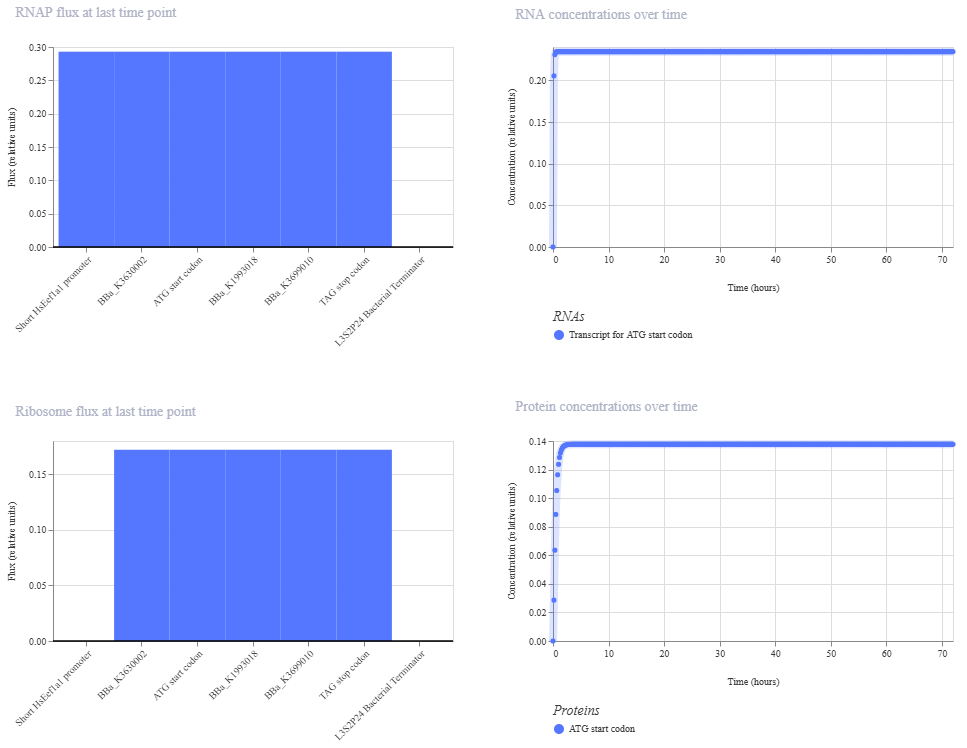
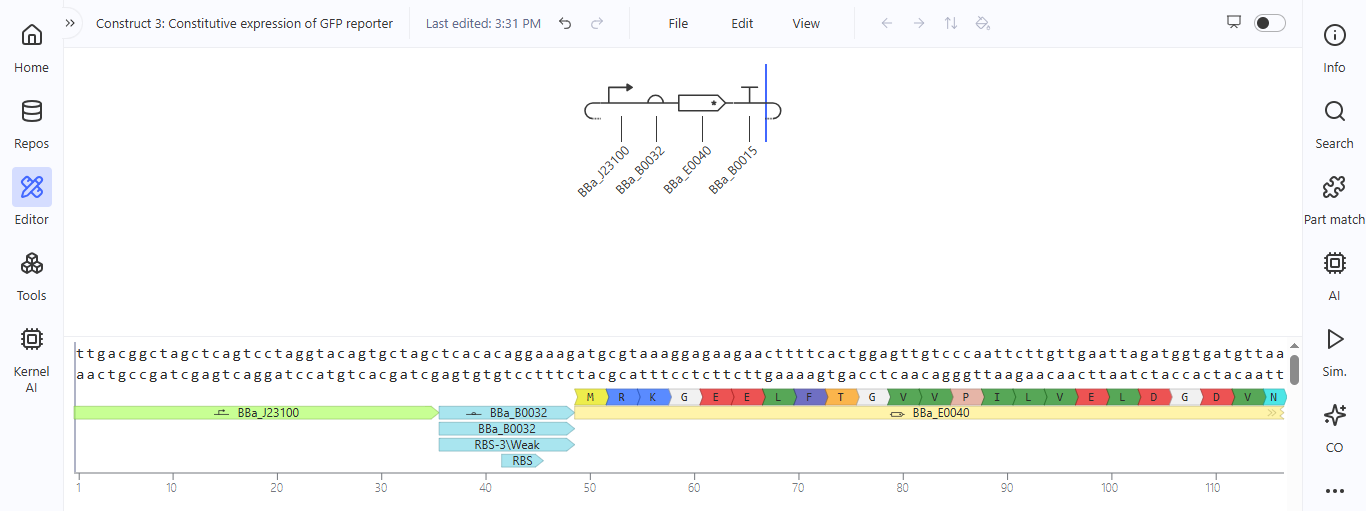
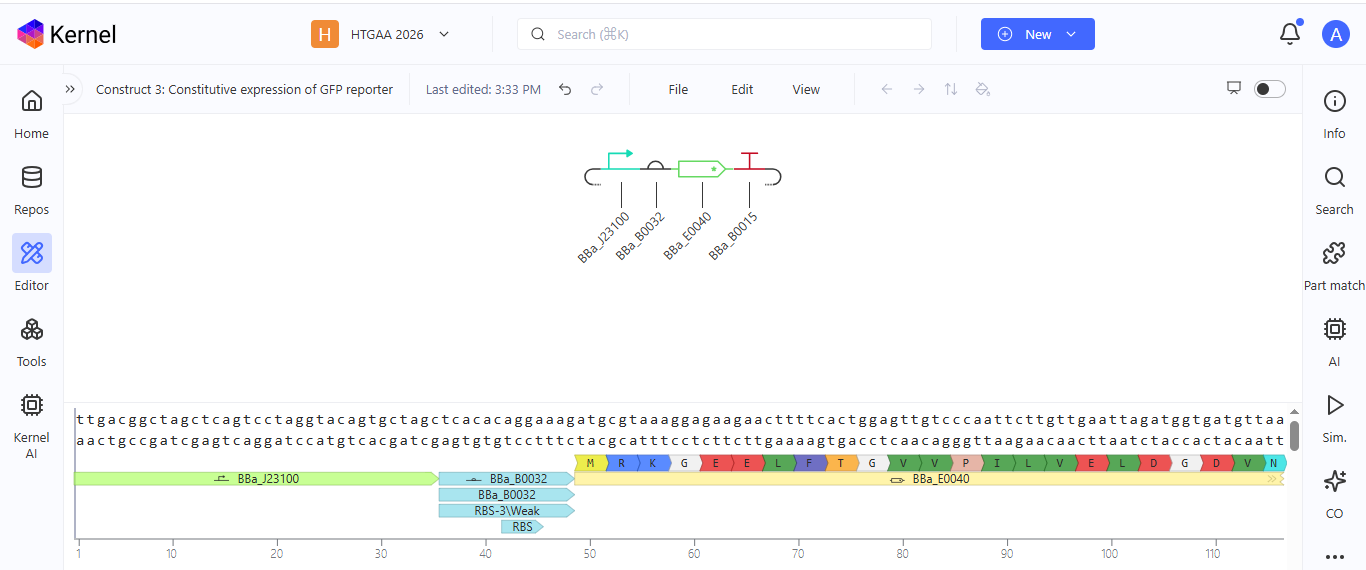
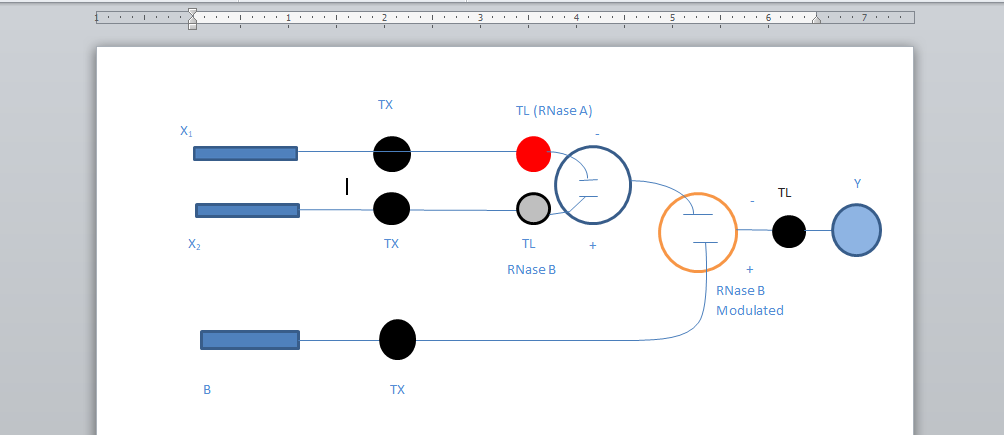
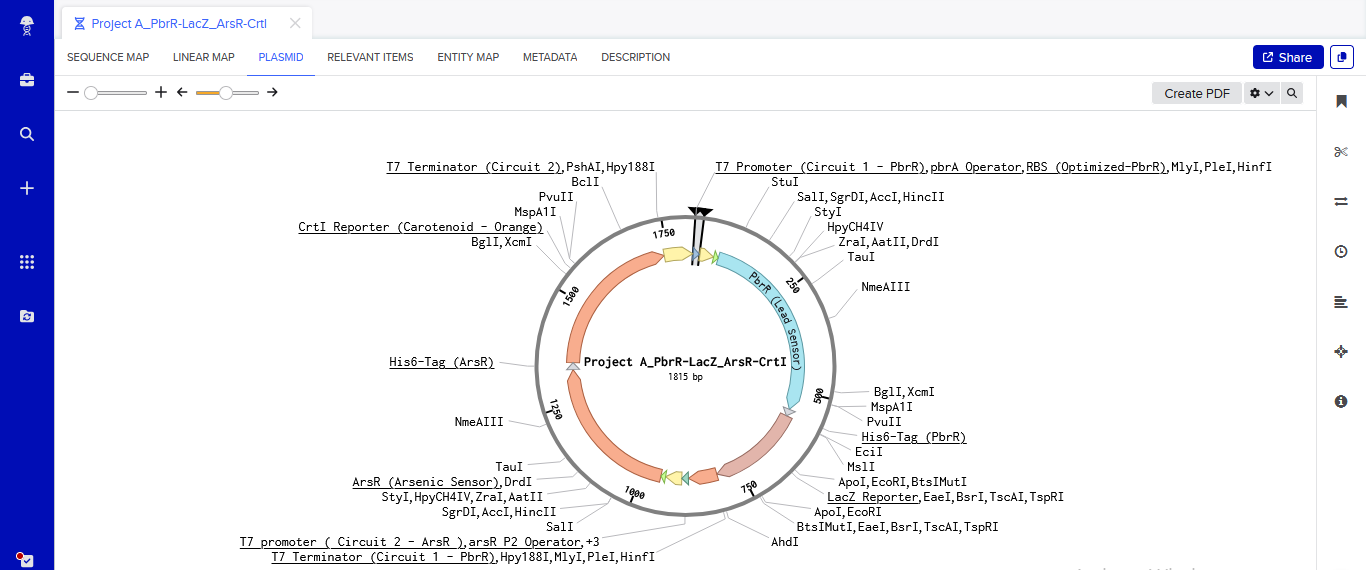
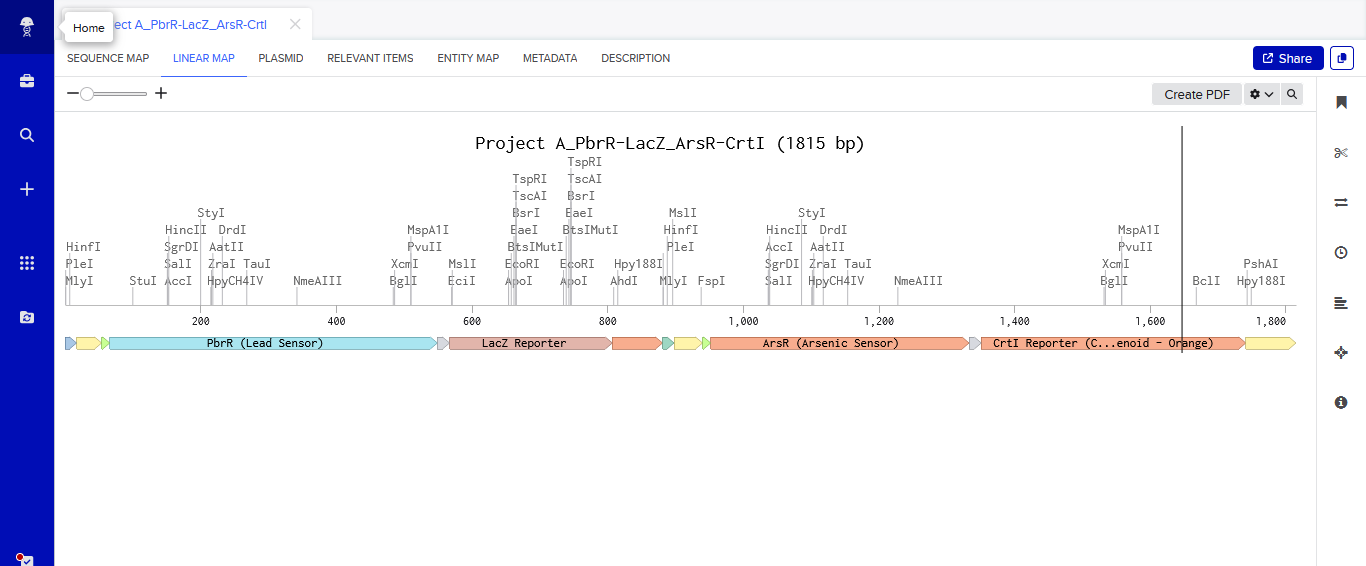
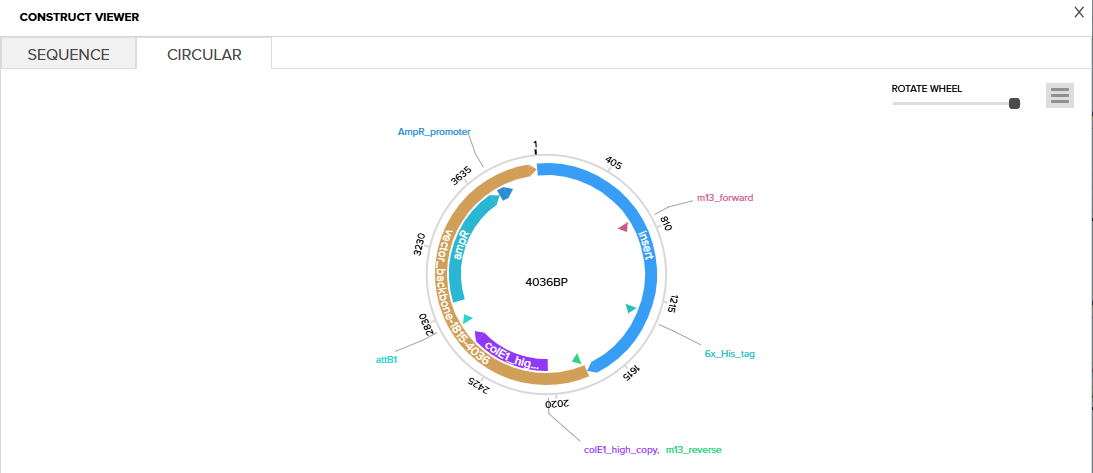
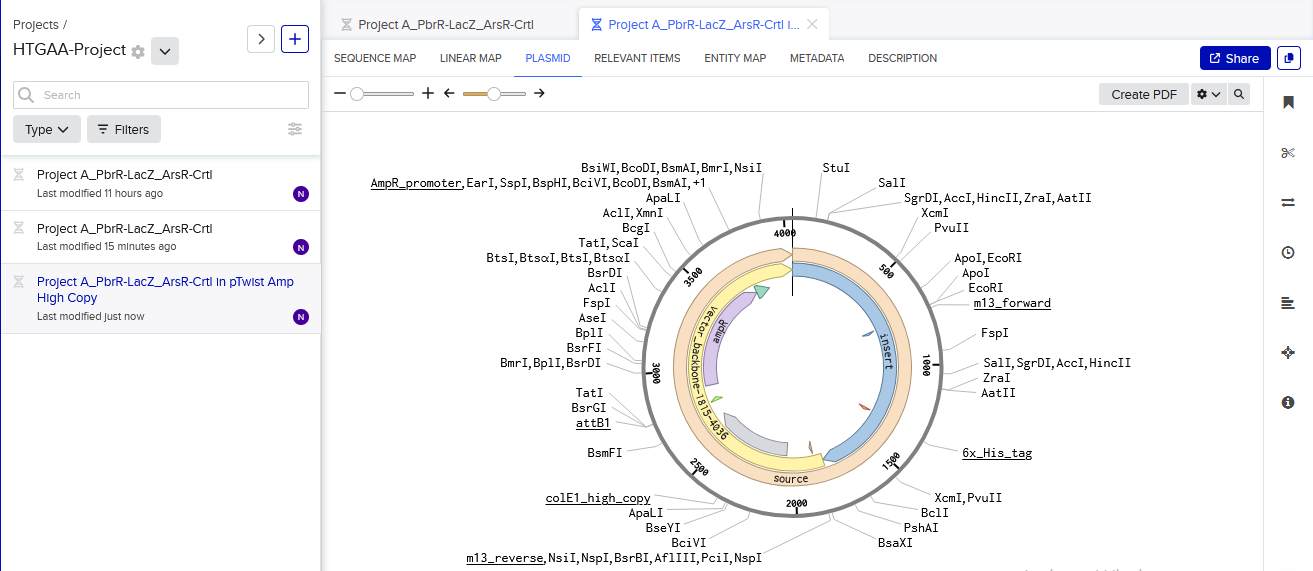


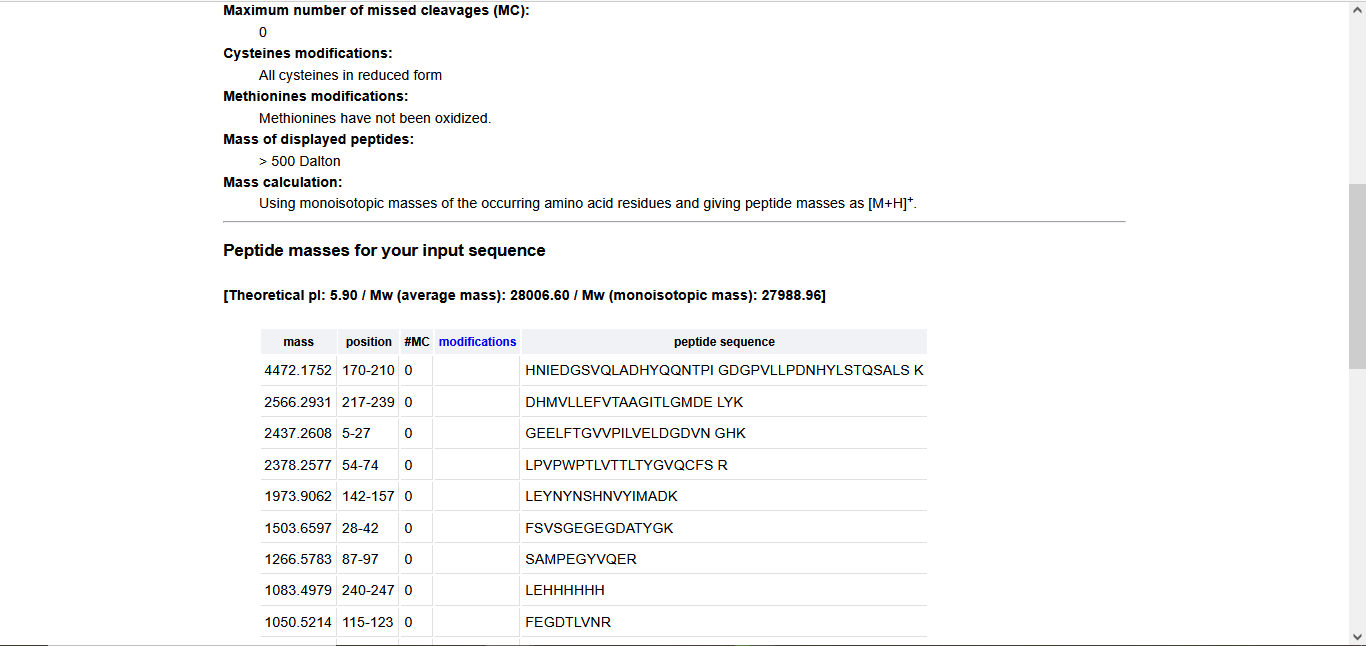
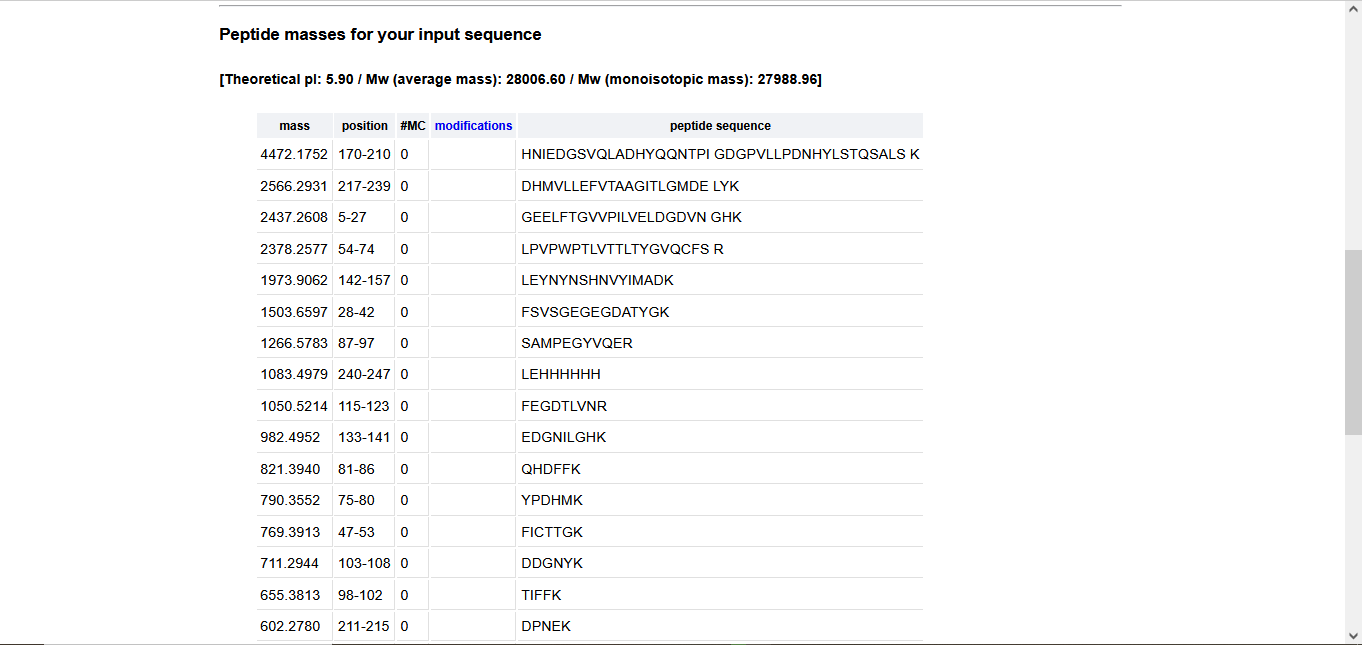
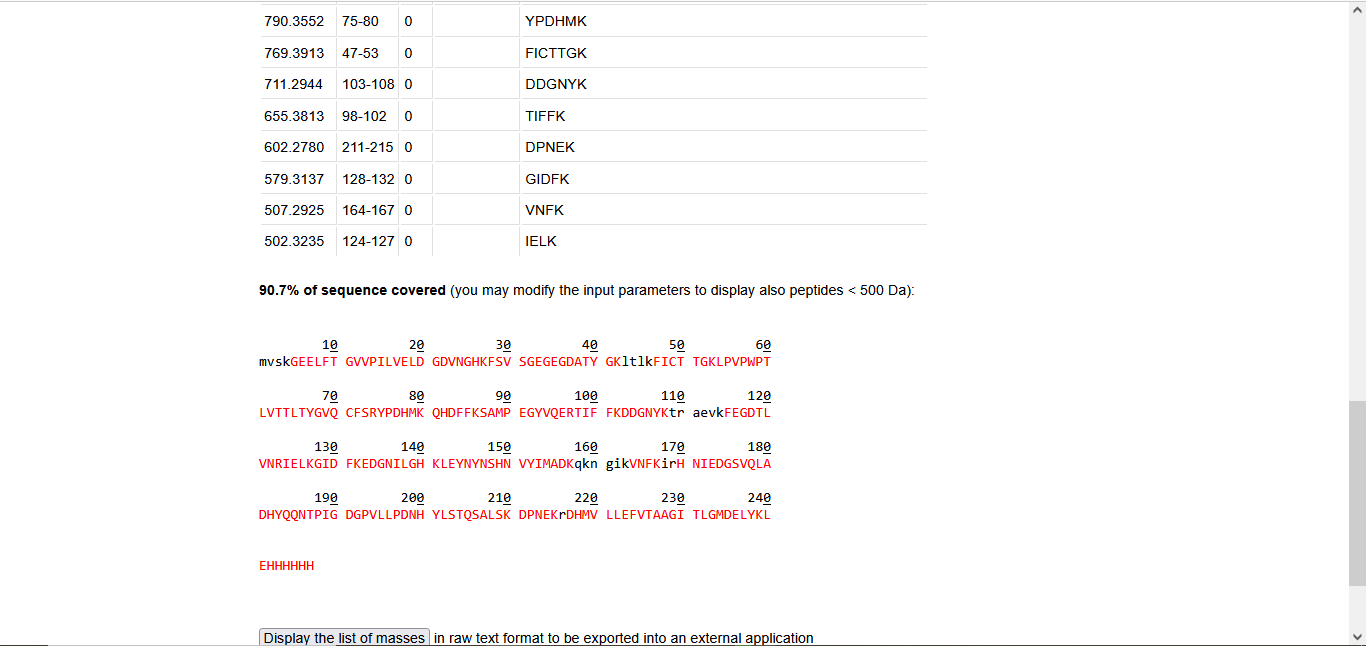


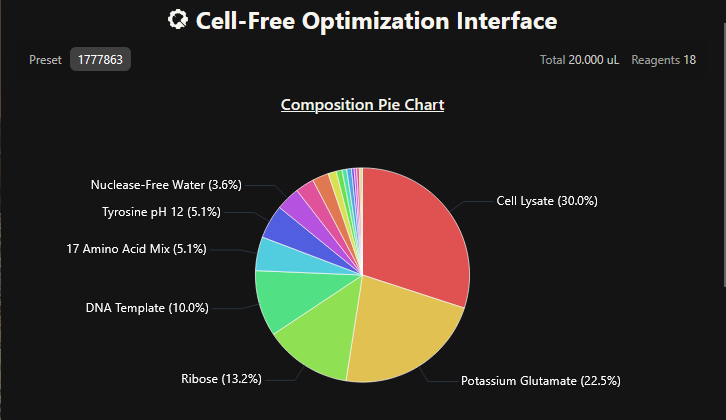
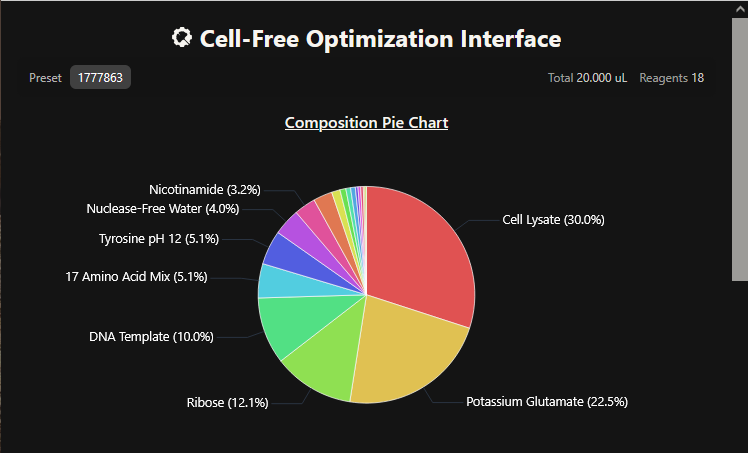
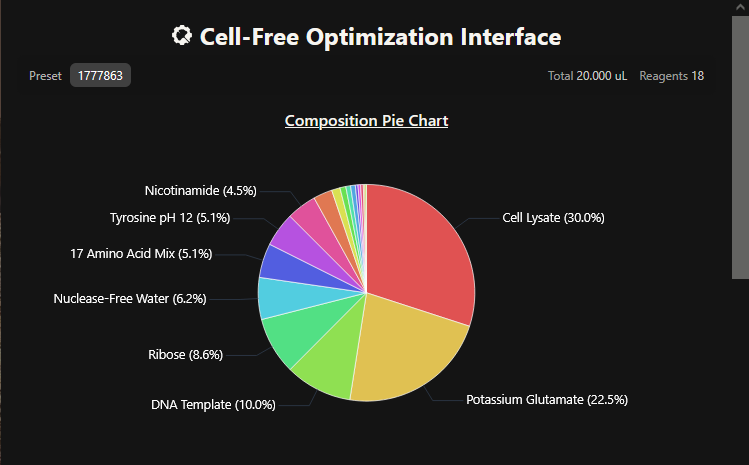
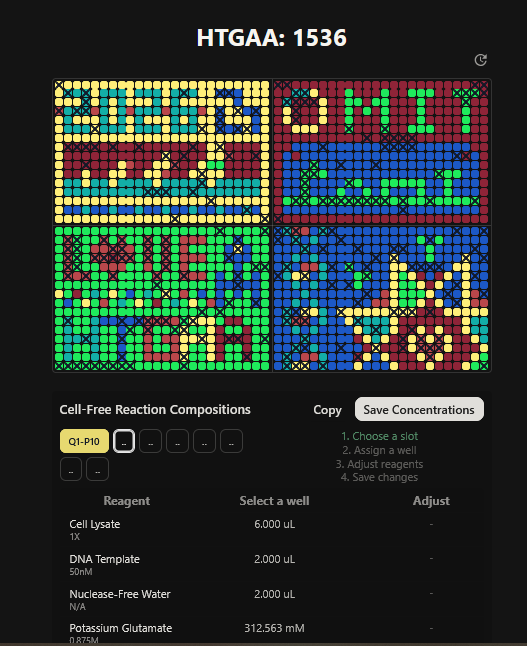
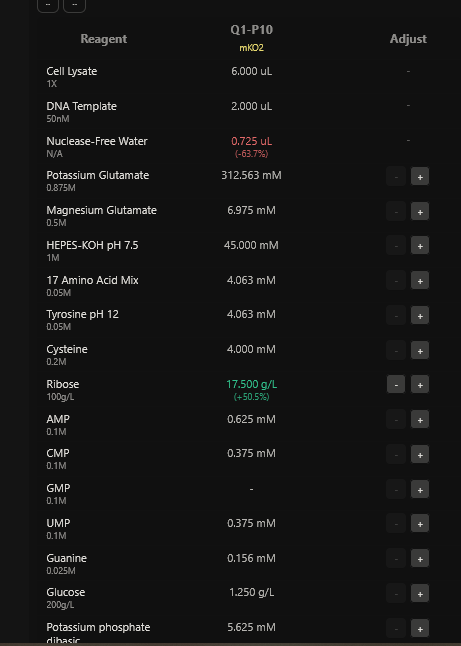
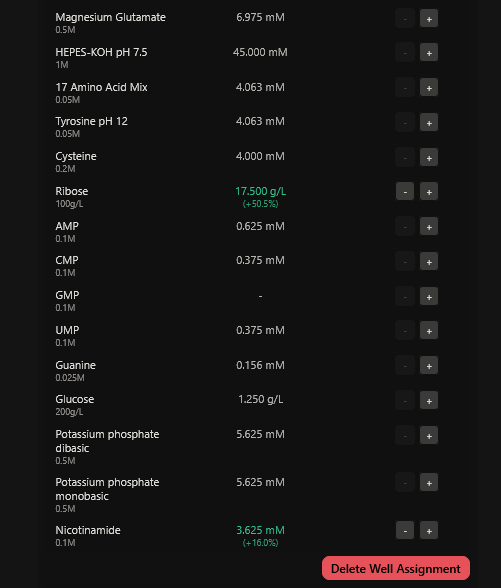
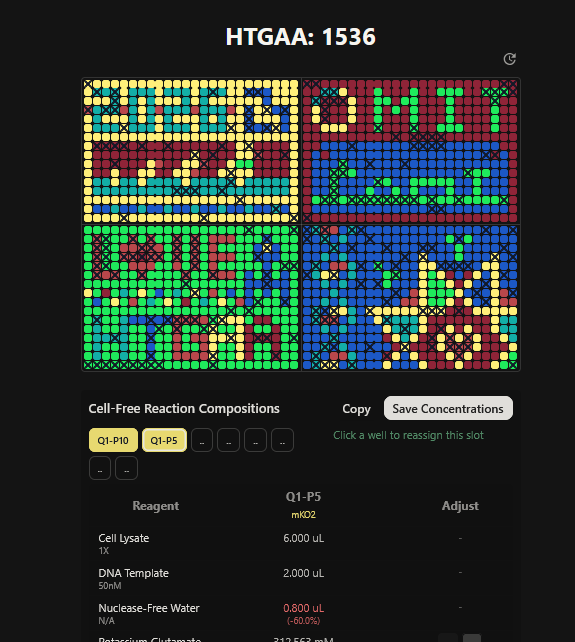
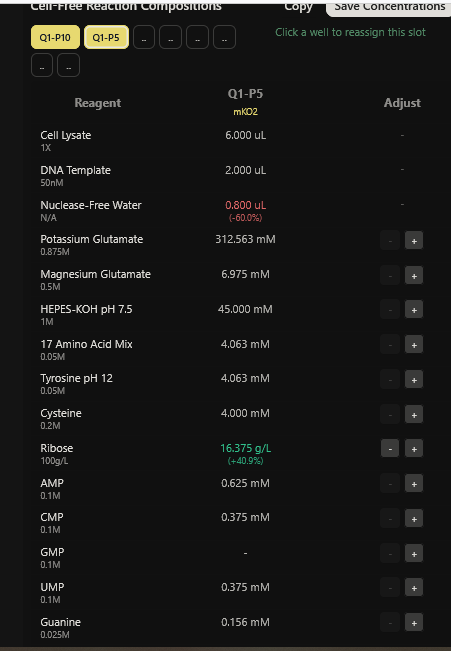
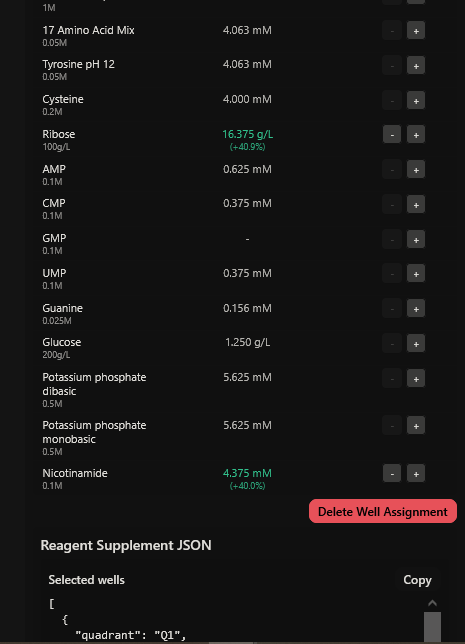
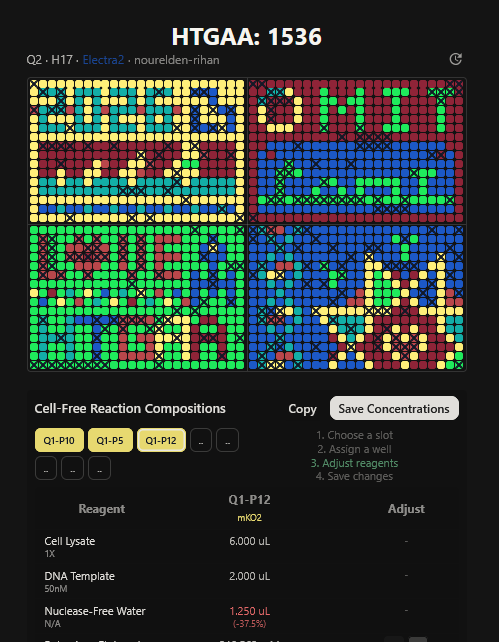
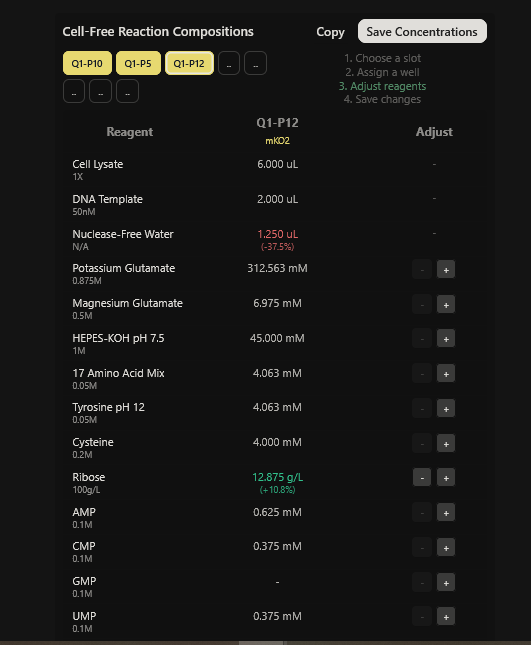
.PNG)
.PNG)
.PNG)