Week 4 HW: Protein Design - Part I

Part A. Conceptual Questions
Question 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Answer
A dalton is a unit of mass used to express the mass of atoms, molecules, and other subatomic particles.
The average percentage of protein in meat is about 20-30% of its total weight.
Using 30% as the average amount of protein in meat.
Mass of protein = 500 × 30% = 150g
Moles of amino acids = mass/molar mass = 150/100 = 1.5mol
Calculating the number of amino acid molecules in the meat:
Number of molecules = moles × Avogadro’s constant
Avogadro’s constant = 6.022 × 1023 = 1.5 × 6.022 × 1023 = 9.033 × 1023
Therefore, there are 9.0× 1023 amino acid molecules in 500g of meat.
Question 2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Answer
This is due to digestion breaking down food such as meat and fish into basic molecules like amino acids, fatty acids, and sugars. The DNA present in Food is therefore broken down and rendered nonfunctional; as such, it is not directly incorporated into our genetic structure.
Question 3. Why are there only 20 natural amino acids?
Answer
There are only 20 natural amino acids due to evolutionary optimization and biochemical efficiency. The 20 amino acids offer a balanced range of properties such as polarity, charge, and hydrophobicity that allow proteins to fold properly and perform diverse biological functions. The limit on the amino acids due to the degeneracy of the genetic code protects against mutations, ensures accurate protein synthesis, and metabolic efficiency.
Question 5 Where did amino acids come from before enzymes that make them, and before life started
Answer
Amino acids were most likely formed before enzymes and life began through abiotic chemical processes, fuelled by the exposure of simple molecules such as methane, ammonia, hydrogen, water, and carbon dioxide to energy sources.
This was demonstrated by Stanley Miller and Harold Urey’s 1953 experiment, which showed that when gases are subjected to electrical sparks, amino acids such as glycine, aspartic acid, and alanine are formed. Additionally, amino acids have been detected in the Murchison meteorite, which suggests that amino acid precursors were formed through photochemical and catalytic reactions.
Question 6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Answer
An α-helix is a secondary protein structure in which the polypeptide chain is twisted into a coil. a-Helices are crucial for protein stability and function. There are 3.6 amino acids per turn of the helix, with each turn having a height of 0.54nm.
D and L configurations describe the stereochemistry of amino acids based on the position of the amino group in a Fischer projection. L-amino acids have the amino group (NH2) on the left, while D-amino acids have it on the right. This makes them mirror images of each other.
An α-helix made using D-amino acids instead of the naturally occurring L-amino acids is expected to form a left-handed α-helix. This is due to the chirality of amino acids. In natural biological systems, proteins are made almost entirely of L-amino acids, which form right-handed α-helices. Since D-amino acids are mirror images of L-amino acids, when D-amino acids form an α-helix, they produce a mirror image structure of the typical right-handed helix. Thereby producing a left-handed α-helix.
Question 7. Can you discover additional helices in proteins?
Answer
It is possible to discover additional helices in proteins using a combination of experimental and computational methods. In certain environmental conditions, proteins adopt other helical conformations. Some examples of these are: 310 helix, which has 3.0 residues per turn and hydrogen bonds between residues i and i+3. Additionally, π Helix with 4.4 residues per turn and hydrogen bonding patterns of i to i+5.
Several experimental and computational methods are used to discover helices in proteins. One key technique is X-ray crystallography, which enables the determination of the three-dimensional coordinates of atoms in proteins. This technique reveals backbone angles and hydrogen-bonding patterns. Researchers then analyse the revealed structure to determine if the chains form a known helix or an uncharacterized one.
Additionally, computational tools, such as the Protein Data Bank, store thousands of protein structures, allowing scientists to use algorithms to scan for recurring backbone structures that may not match any known helices. If consistent and stable patterns are observed repeatedly across unrelated proteins, these patterns may suggest the existence of a new type of helix. Molecular dynamics simulations can also be used to predict whether alternative backbone conformations are stable, and researchers can design proteins based on the simulations and test whether they fold into new helices.
Question 8. Why are most molecular helices right-handed?
Answer
Most molecular helicases are right-handed because it is the most thermodynamically stable and energetically favorable conformation. This is due to the chirality, where right-handed folding maximizes hydrogen bonding and minimizes steric hindrance, making them more stable than left-handed helices.
Question 9 Why do β-sheets tend to aggregate?
- What is the driving force for β-sheet aggregation?
Answer
A β-sheet is a fundamental form of a secondary protein structure consisting of β-strands that are linked laterally by hydrogen bonds, forming a zig-zag and twisted pleated sheet configuration.
β-sheets aggregate due to their inherent structural tendency to form intermolecular hydrogen bonds and bury hydrophobic residues, which results in the creation of amyloid fibrils.
- The driving force of beta sheet aggregation is is the combination of thermodynamic stabilization through dehydration and the maximization of weak interactions in environments with high concentrations of misfolded protein intermediates.
Question 10 Why do many amyloid diseases form β-sheets?
- Can you use amyloid β-sheets as materials?
Answer
Amyloid diseases are a group of rare diseases caused by the abnormal misfolding and aggregation of proteins into insoluble fibrils known as amyloids. These fibrils accumulate in tissue and organs, disrupting normal function and potentially leading to organ failure. Some amyloid diseases are Alzheimer’s disease and parkinson’s disease.
Amyloid diseases form β-sheets due to misfolded proteins caused by mutations, stress, or aging, which expose β-strand regions that can form hydrogen bonds with strands from other proteins. The exposed β-strands then form exceptionally stable and low-energy β-structures that run perpendicular to the fibril axis, forming insoluble, self-propagating fibrils via hydrophobic interactions.
- Amyloid sheets can be used as materials due to amyloid fibrils being extremely strong, resistant to heat and chemical degradation, and self-assembling in nature. Not all amyloids are pathological; organisms such as E.coli and yeast naturally produce functional amyloids for structural and biological roles. As such, short peptides that deliberately form controlled β-sheets fibrils can be used to produce materials.
Part B: Protein Analysis and Visualization
Question 1
I chose the aequorin protein. While thinking of what protein to select for this section of the assignment, I decided to pick an interesting protein found in a sea organism and stumbled upon the Crystal jelly (*Aequorea victoria), a bioluminescent hydrozoan jellyfish.

Aequorin is a photoprotein that emits blue light in direct response to binding to calcium ions (Ca 2+), making it a calcium-sensitive photoprotein.
Honestly, I first chose aequorin because it is a bioluminescent protein and I love glowing stuff :). However, after reading more on it, I discovered it has played an important role in paving the way for the discovery and application of green fluorescent protein (GFP) and transformed the observation of molecular proteins inside cells. Groundbreaking work on awquorin and its related fluorescent proteins contributed to Osamu Shimomura, Martin Chalfie, and Roger Y. Tsien winning the Nobel Prize in Chemistry in 2008.
I selected the Aequorin-1 protein with an annotation score of 4/5 from Uniprot.
https://www.uniprot.org/uniprotkb/P07164/entry
Question 2
The amino acid sequence for the Aequorin-1 protein:
MTSEQYSVKLTPDFDNPKWIGRHKHMFNFLDVNHNGRISLDEMVYKASDIVINNLGATPEQAKRHKDAVEAFFGGAGMKYGVETEWPEYIEGWKRLASEELKRYSKNQITLIRLWGDALFDIIDKDQNGAISLDEWKAYTKSDGIIQSSEDCEETFRVCDIDESGQLDVDEMTRQHLGFWYTMDPACEKLYGGAVP
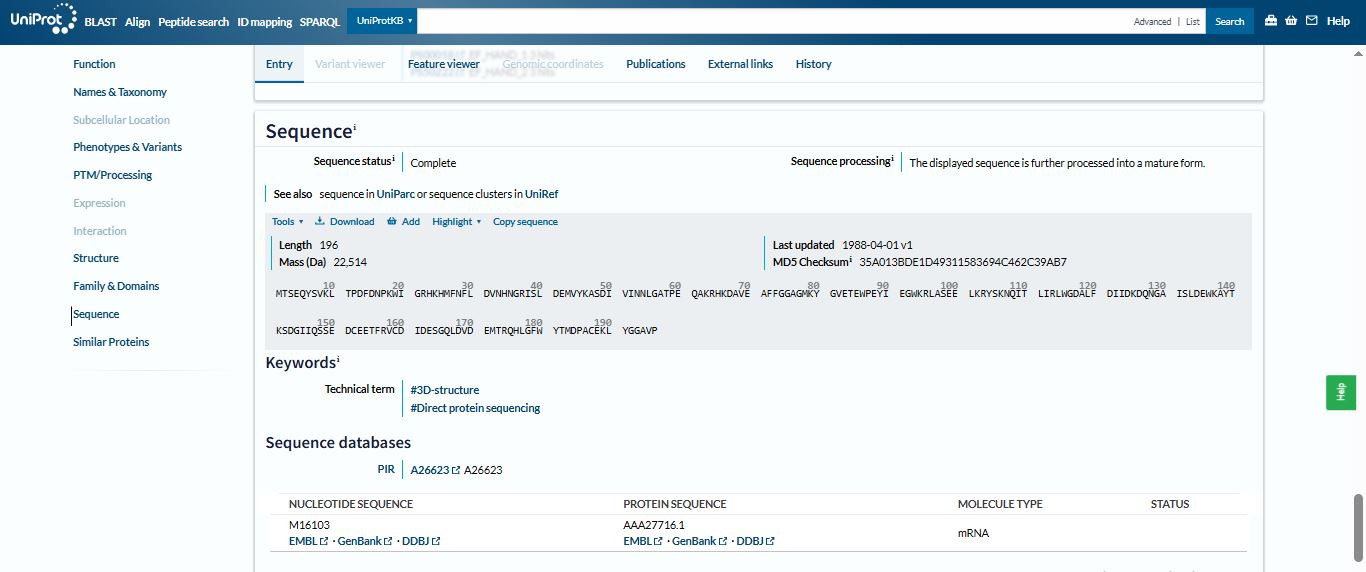
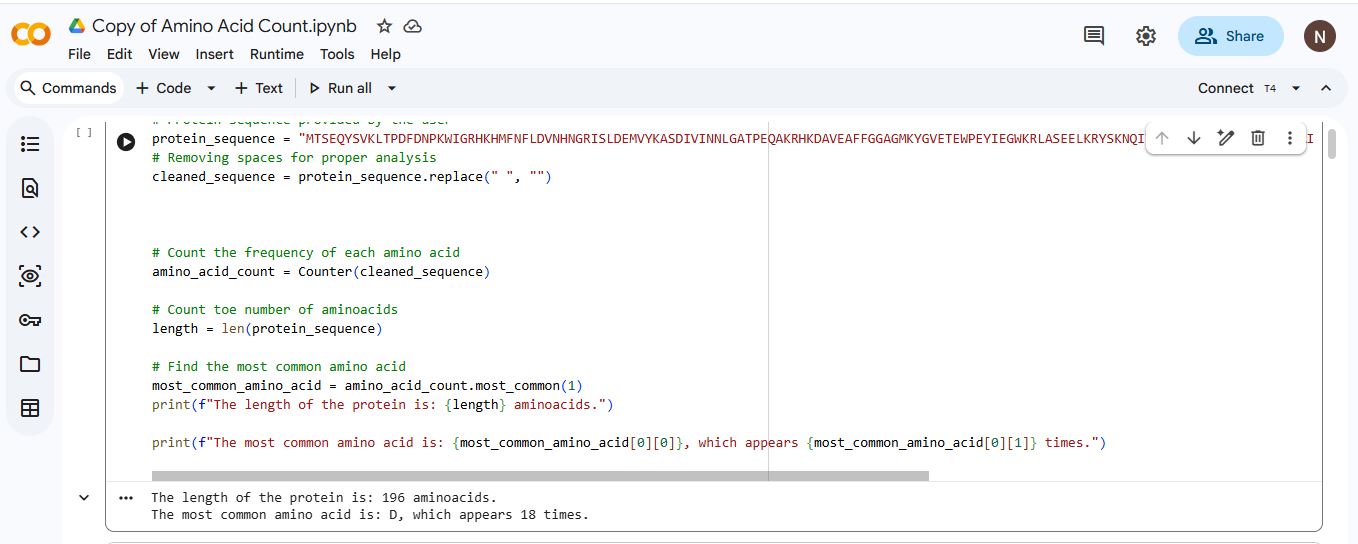
The length of the Aequorin-1 is 196 amino acids, and the most frequent amino acid is D (Aspartic acid), which appears 18 times throughout the sequence.
I run a blast for the homologs of the Aequorin-1 protein using Uniprot
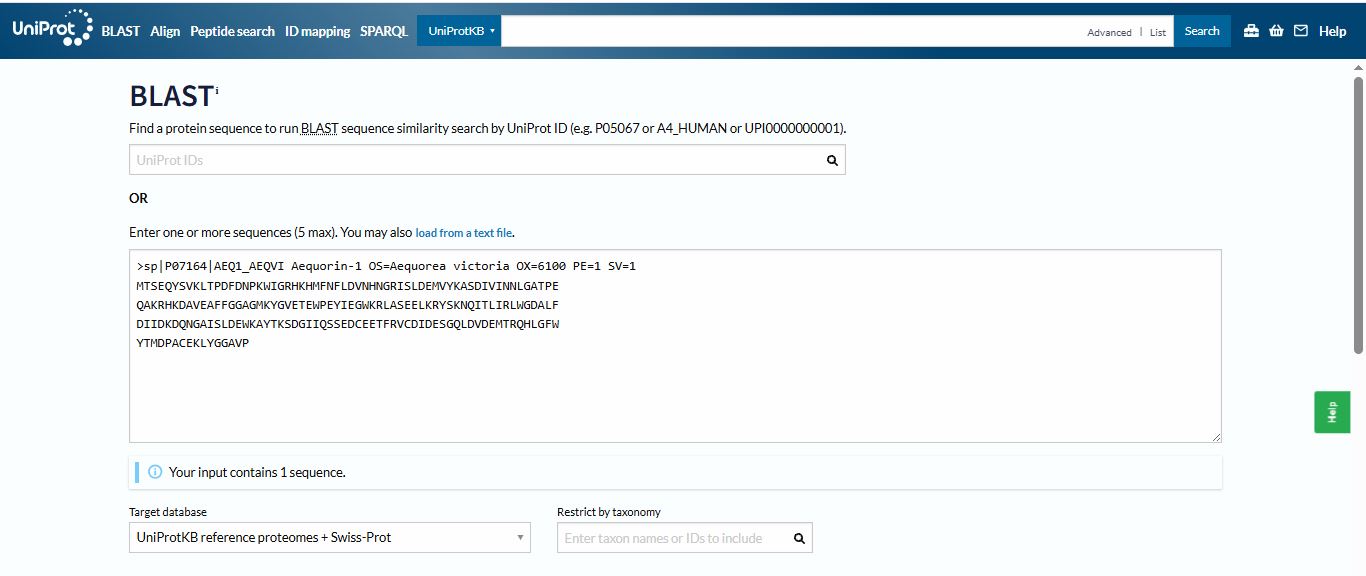
I found 250 homologs of the Aequorin-1 protein
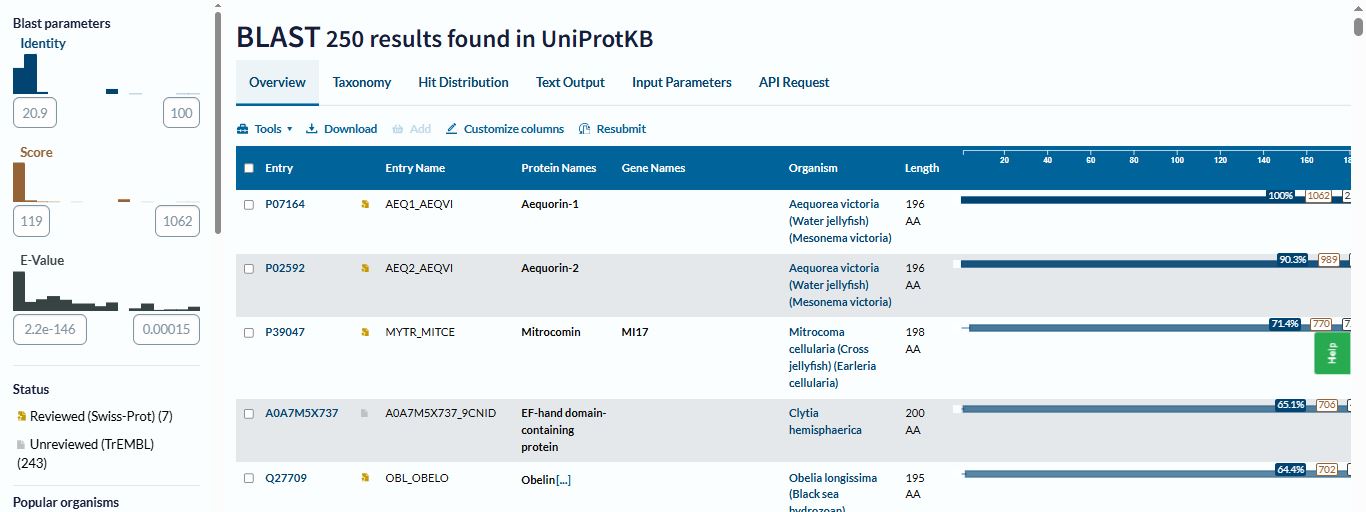
Aequorin-1 belongs to the EF-hand calcium-binding protein superfamily, which is a group of eukaryotic proteins that play a central role in cellular signaling, regulation, and homeostasis.
Question 3
The structure of the Aequorin-1 protein was deposited on 2004-03-05 and released on 2004-12-28 by Deng, L., Markova, S.V., Vysotski, E.S., Liu, Z.J., Lee, J., Rose, J., Wang, B.C. X-ray Diffraction was used to determine the 3d strcuture of the Aequorin protein.
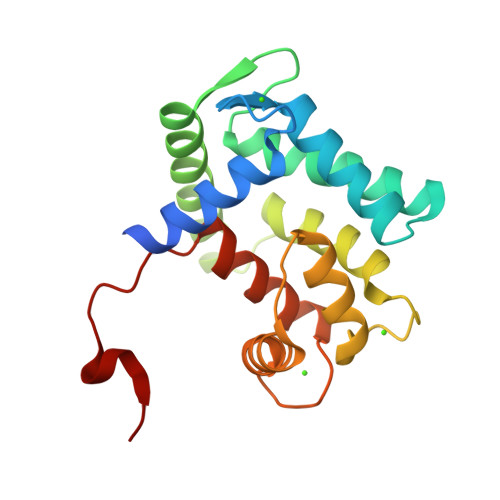
The protein has a good quality structure and a resolution of 1.70 Å. Using SCOP, I found out that Aequorin-1 belongs to the calmodulin-like structural family.
It has calcium ions (Ca2+) that form part of its structure
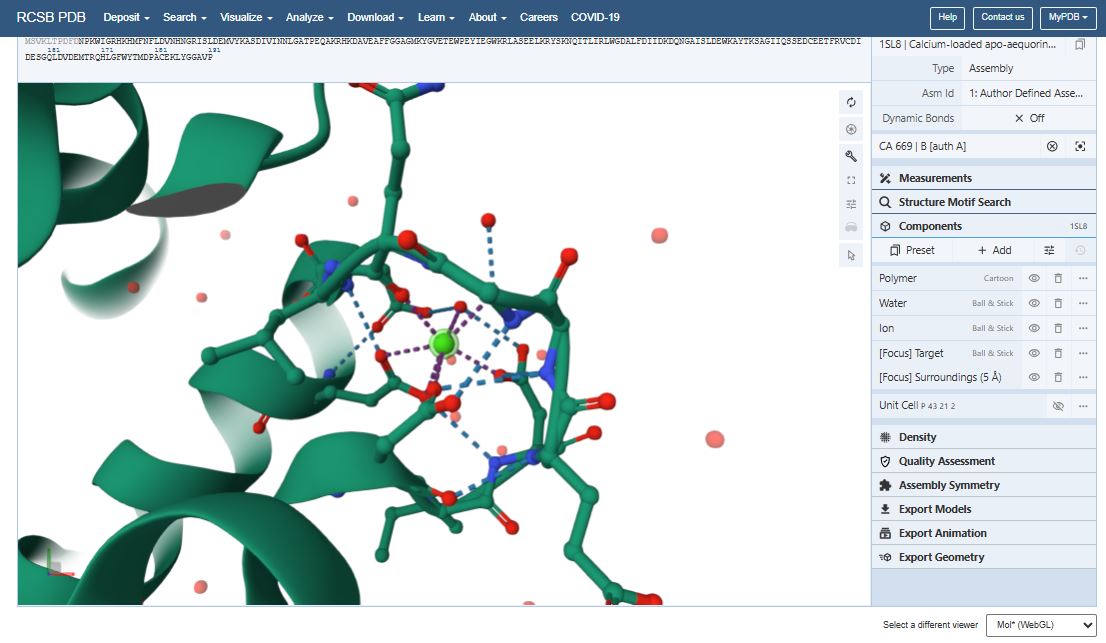
Question 4 I used PyMol to visualize the Aequorin-1 protein.
Aequorin-1 protein as a Cartoon
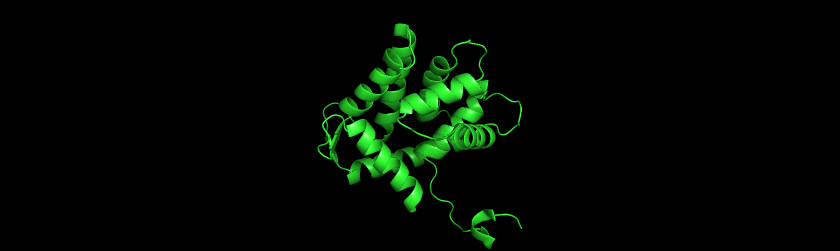
Aequorin-1 protein as a Ribbon

Aequorin-1 protein as a Ball and Stick
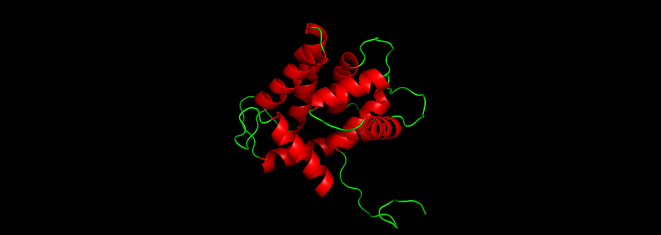
I colored the cartoon structure of Aequorin-1, highlighting the helices in red, the beta sheets in yellow, and the loops in green. Based on the image generated, Aequorin-1 has a significant amount of alpha helices and no beta sheets.
I tried to use a code to count the number of helices and sheets
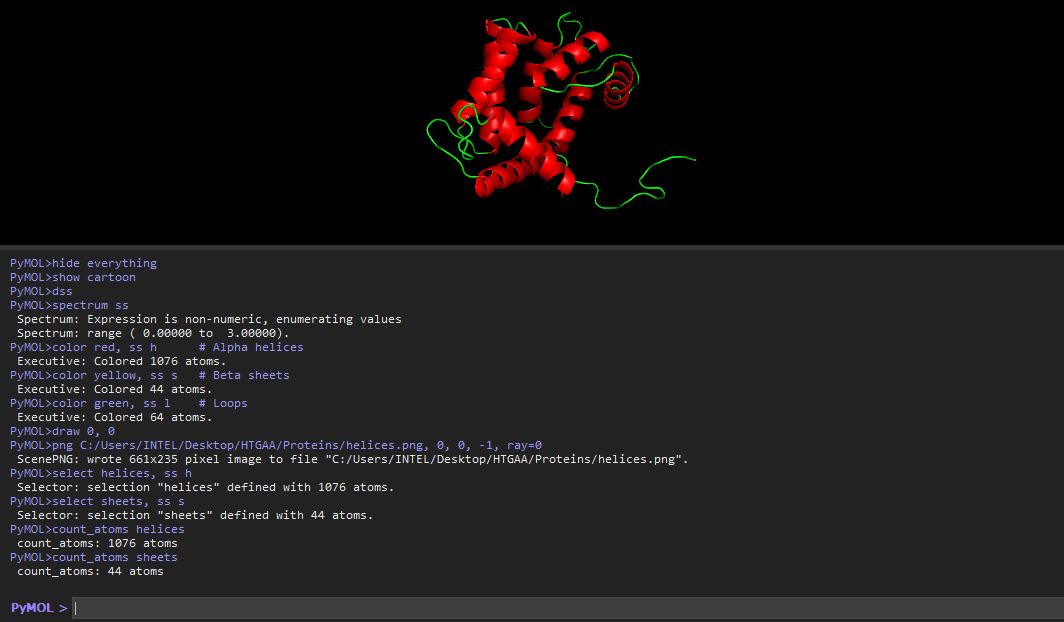
The helices atom count was 1076 atoms, and the atom sheet count was 44 atoms
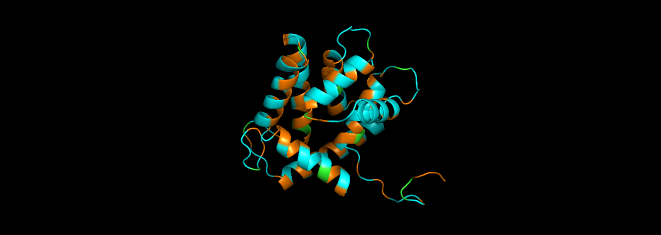
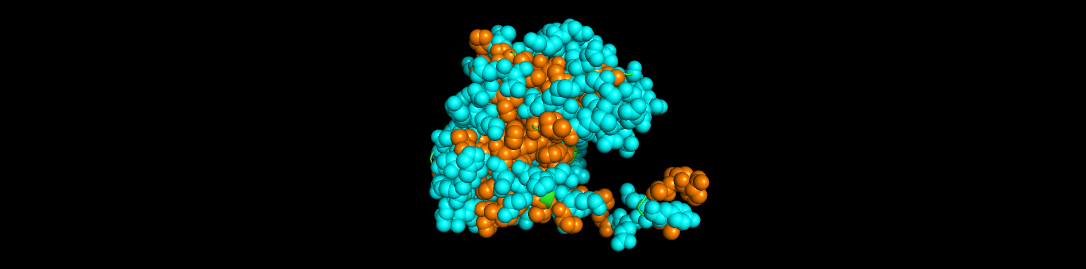
I highlighted the hydrophobic elements in orange and the hydrophilic elements in cyan.
Based on the color distribution, there are more hydrophilic elements than hydrophobic elements.
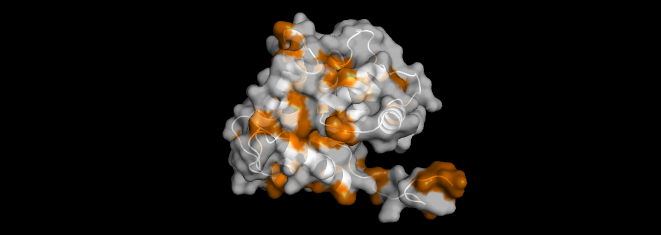
I visualized the surface of the protein, and it didn’t have any holes in it.
Part C. Using ML-Based Protein Design Tools
Question 1 - Deep Mutational Scans
Just like in part B, I chose the Aequorin-1 protein. I then inserted the mutation scan in Google Colab and ran it. I used the relative mode when I ran the mutation scan for the protein sequence.
This was the Mutation Scan Heatmap of the Aequorin-1 protein.
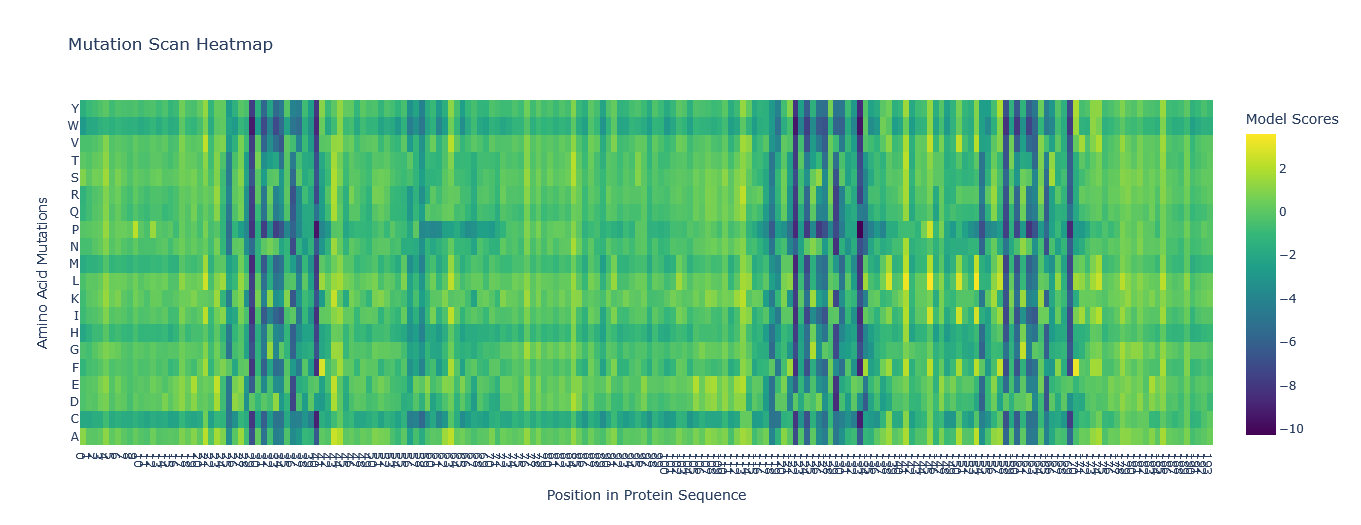
I tried to read the mutation scan heatmap and asked Gemini for a guide on how to read the map.
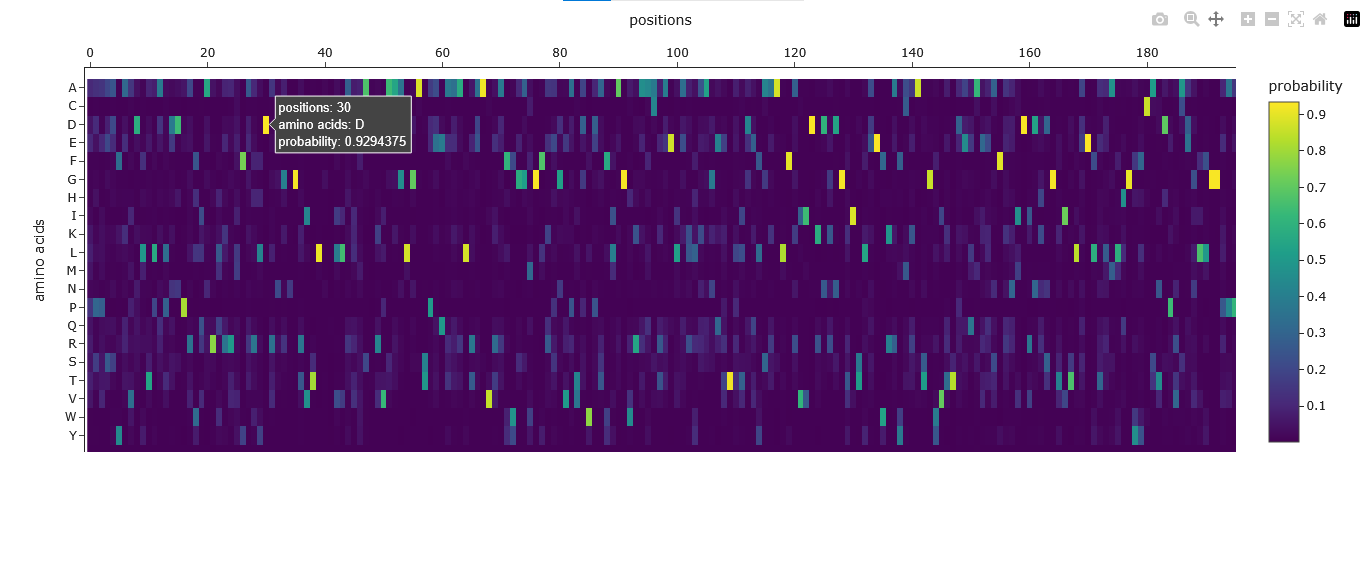
Based on the information it gave me, the X-axis showed the positions in the protein sequence, and the Y-axis represented the 20 standard amino acids that the wild-type residue at a given position Additionally, a high positive score (e.g., a bright yellow cell) indicates that the language model predicts the mutated amino acid is significantly more likely to occur at that position than the original wild-type amino acid. Conversely, a highly negative score (e.g., a dark purple cell) suggests the mutation is much less likely.
Using this information to read the map and pick a bright yellow residue at X = 84, Y = S, and Z = 1.941 (mutation score).
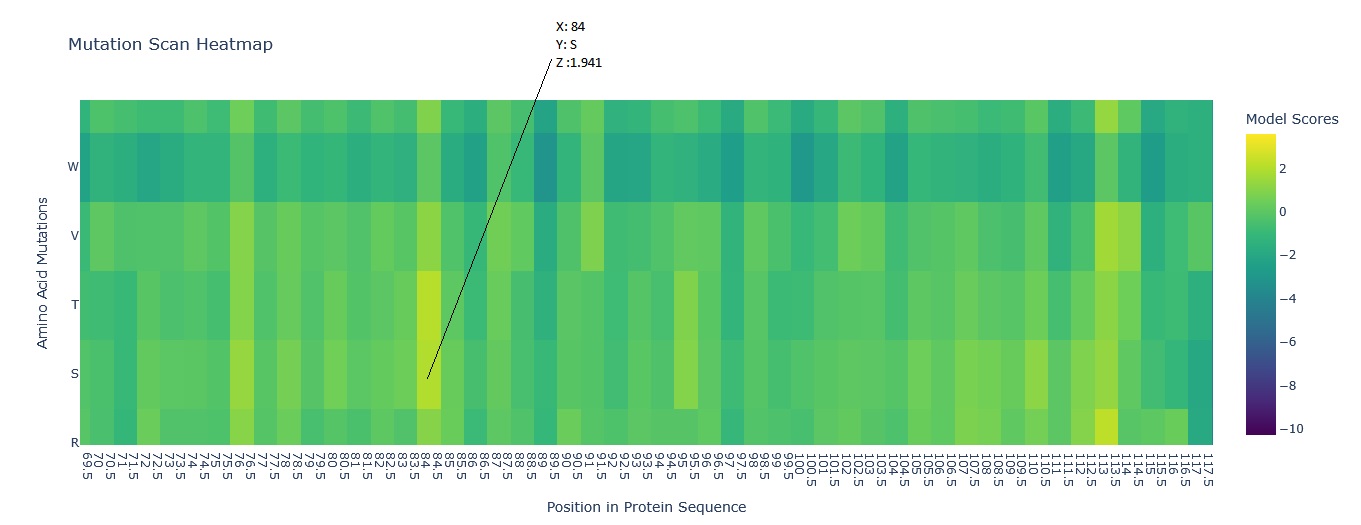
E (Glutamic acid) is at position 84 of the Aequorin-1 protein sequence, with a score of 1.941, which is a high positive score, and the cell could be bright red. This means it is highly likely that E (Glutamic acid) will mutate into S (Serine) at position 84 of the Aequorin-1 protein sequence.
Question 2 - Latent Space Analysis
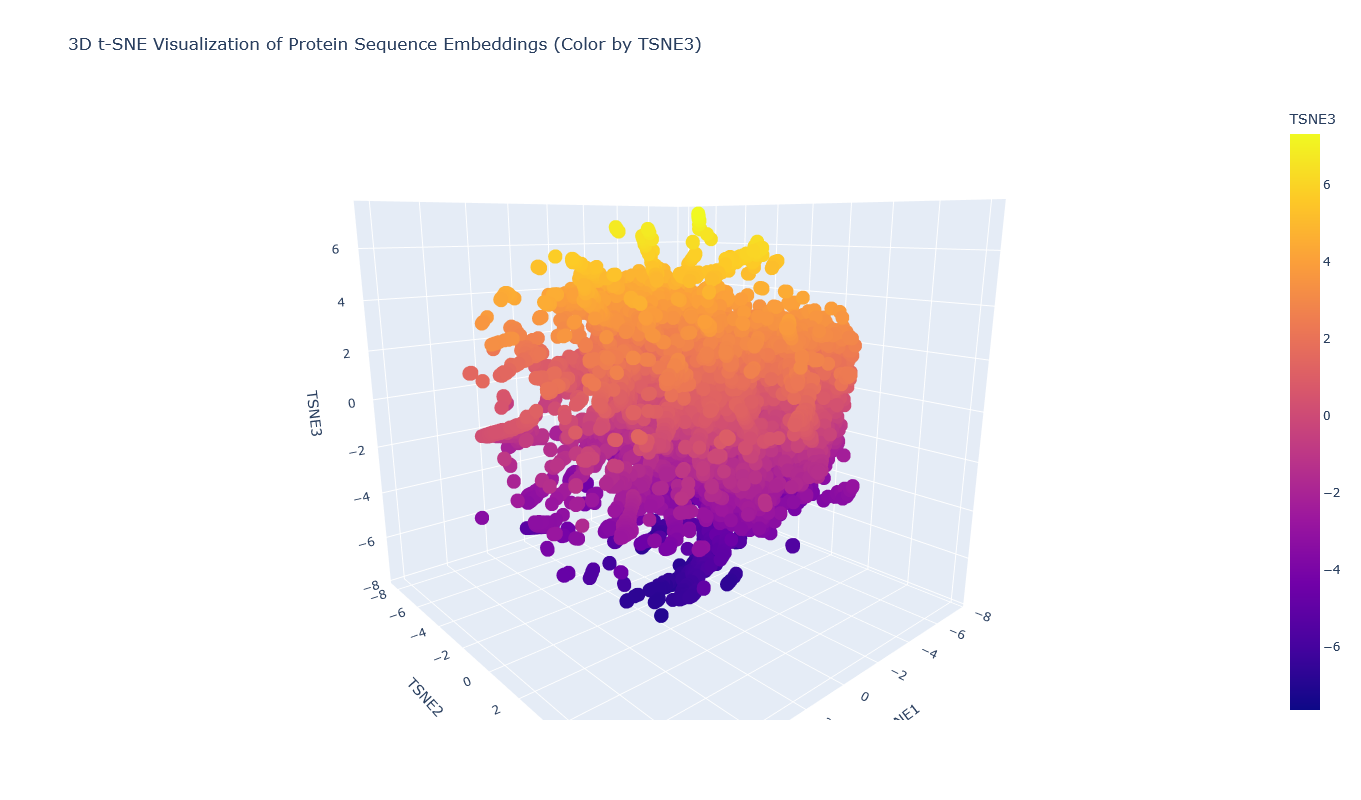
I used the provided sequence dataset to embed proteins in reduced dimensionality.
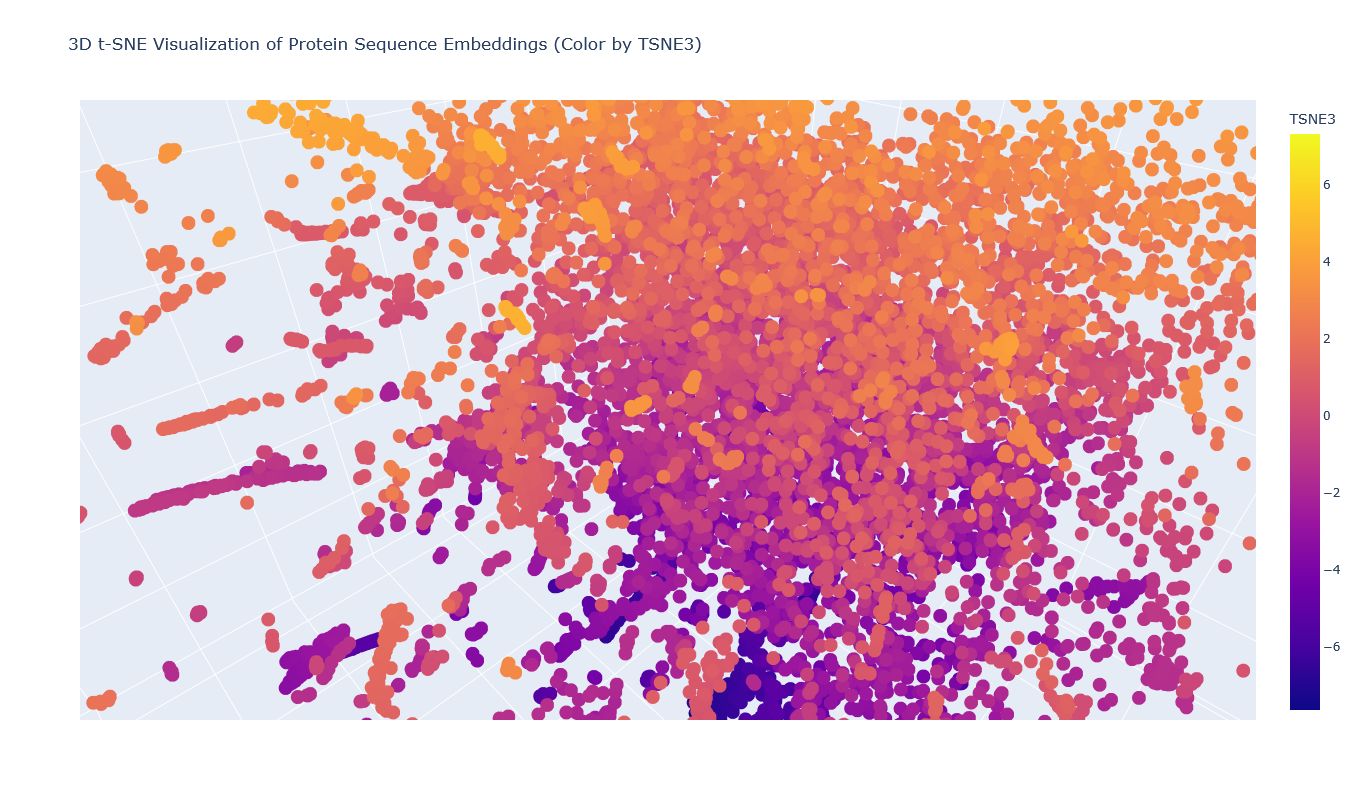
I placed the Aequorin-1 protein in the resulting map and coloured it red, while making all the other proteins blue.
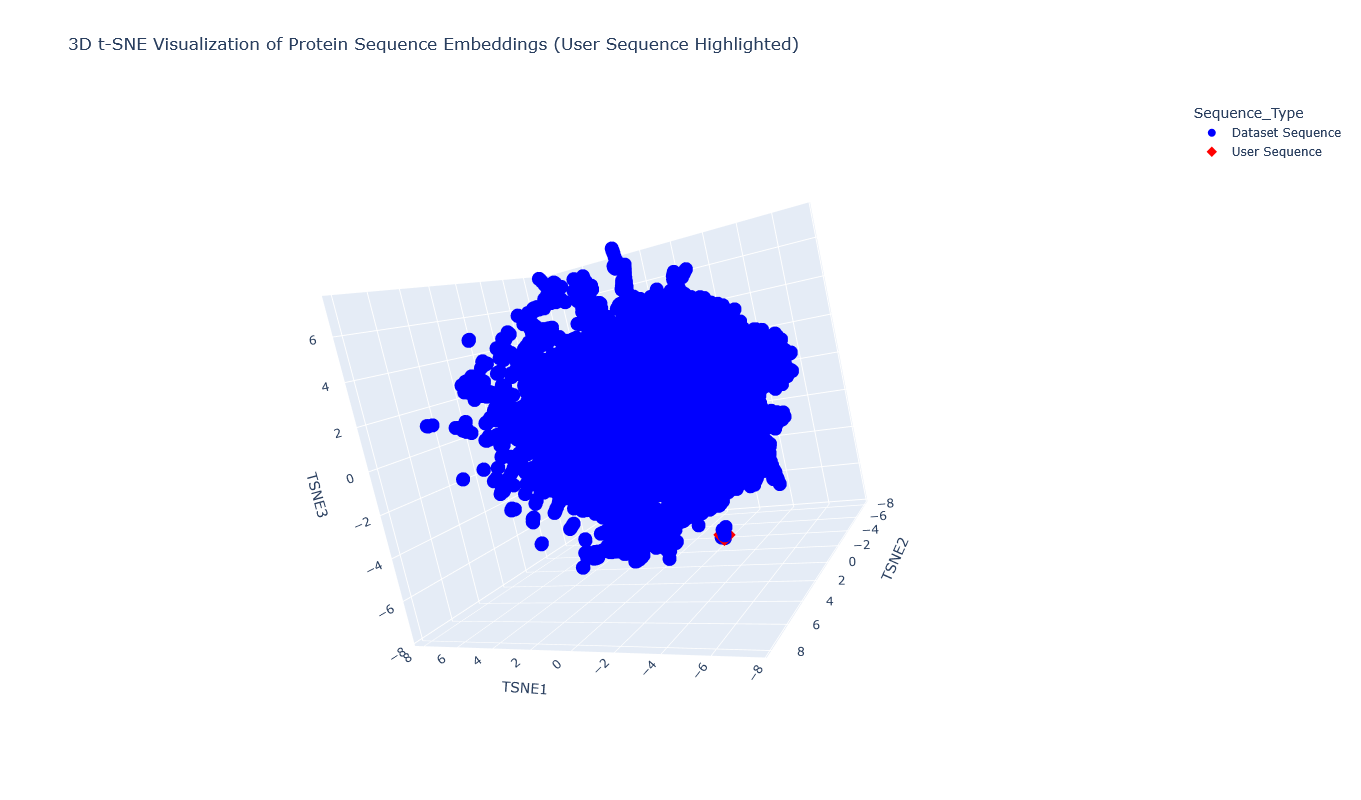
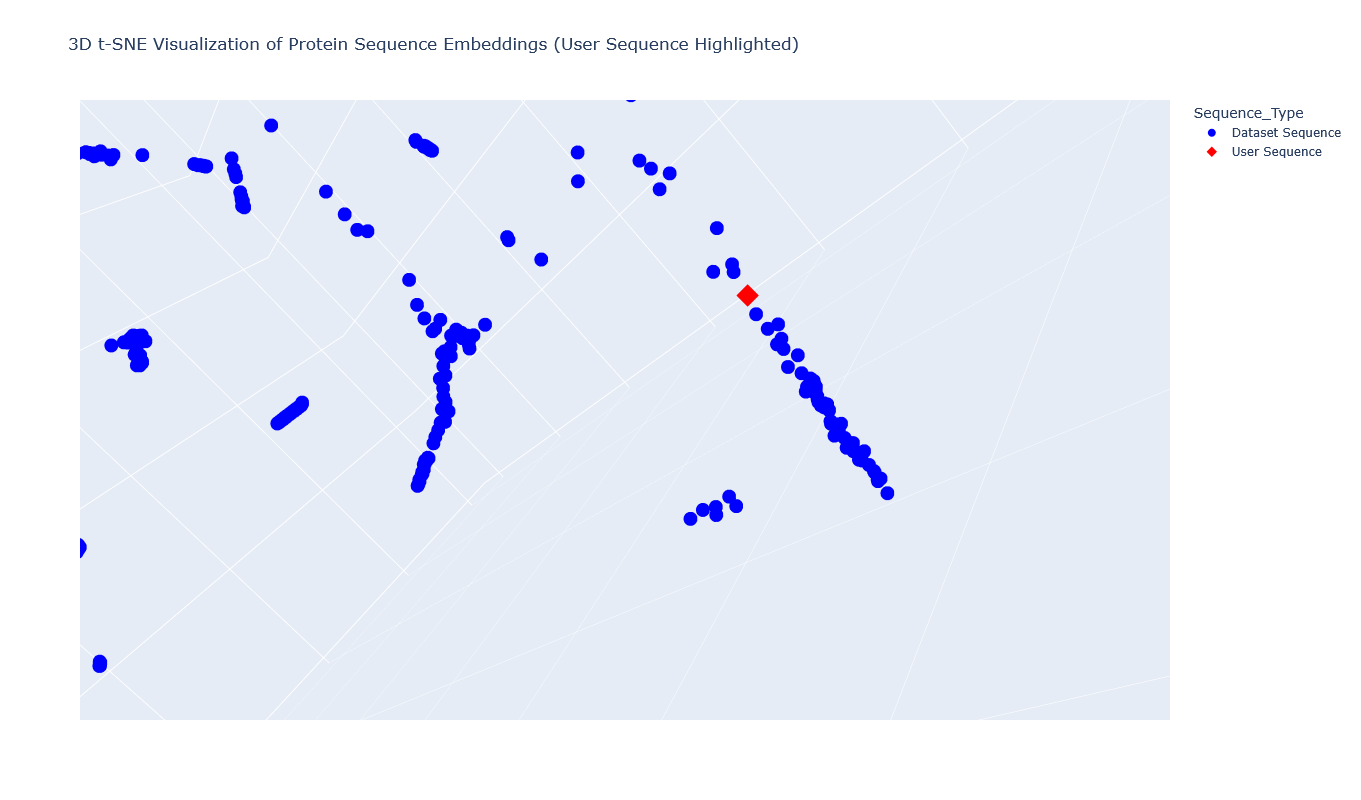
The Aequorin-1 was located at the following 3D t-SNE coordinates: TSNE1: -3.6272, TSNE2: 0.3001, TSNE3: -6.5145

The proteins that were close to the Aequorin-1 protein in the Latent space analysis were:
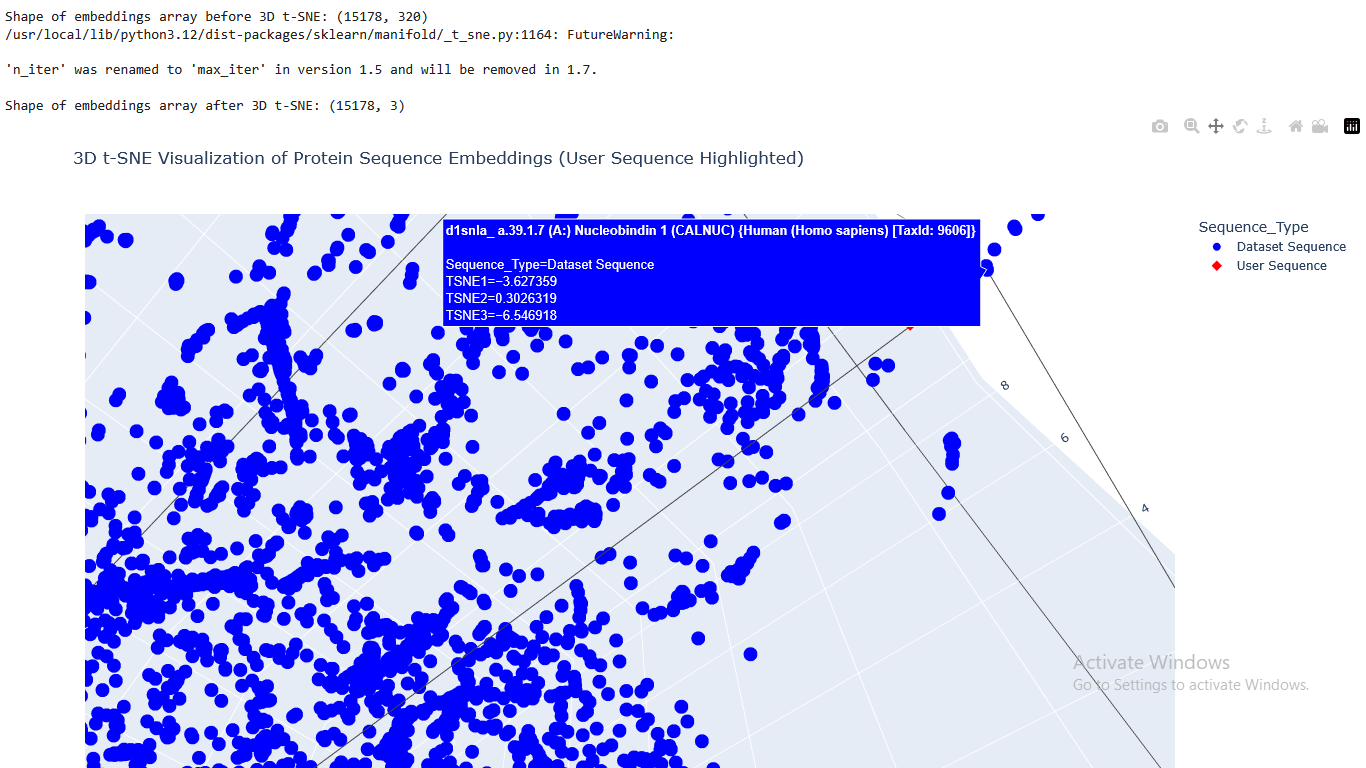
When I tried to determine the relation between them, I discovered they all possess the EF-hand calcium-binding motif except for Resp1.
C2. Protein Folding
I used ESMFOLD to fold the Aequorin-1 protein
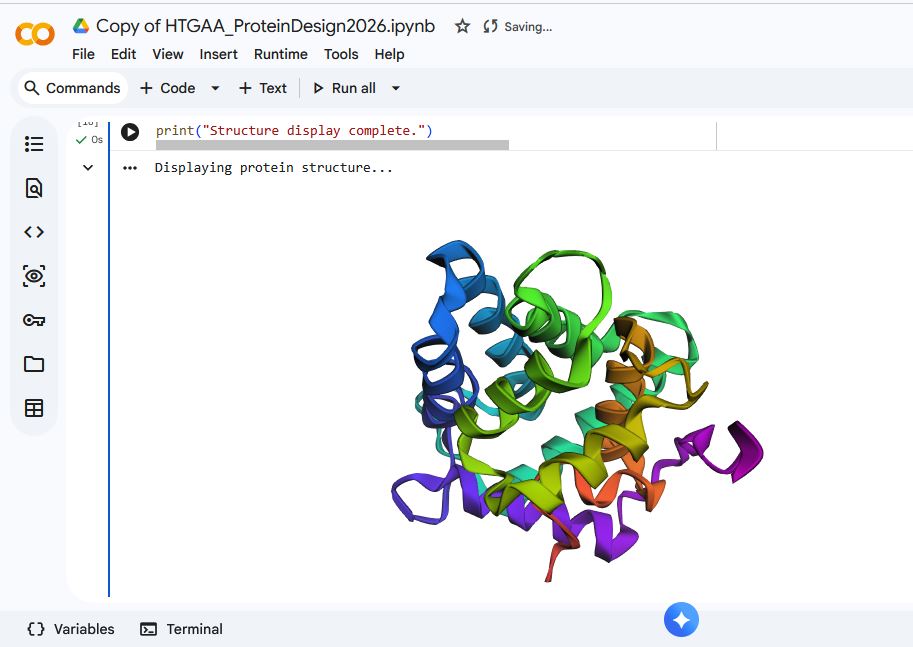
When I compared it to the structure of the Aequorin-1 protein in RCSB, I noticed there were differences in both structures
I think the differences could be due to conformation variability or accuracy limitations. I verified that I correctly copied and inputted the correct Aequorin-1 protein sequence, so the discrepancy in the structure is not due to wrong protein sequence inputs. Additionally, the structure displayed by ESMFold did have a plddt value of 80.295, which means there is a high likelihood that the general backbone structure is accurate.
The Aequorin-1 protein Sequence:
MTSEQYSVKLTPDFDNPKWIGRHKHMFNFLDVNHNGRISLDEMVYKASDIVINNLGATPEQAKRHKDAVEAFFGGAGMKYGVETEWPEYIEGWKRLASEELKRYSKNQITLIRLWGDALFDIIDKDQNGAISLDEWKAYTKSDGIIQSSEDCEETFRVCDIDESGQLDVDEMTRQHLGFWYTMDPACEKLYGGAVP
I tried playing around with the Aequorin-1 protein Sequence and introduced mutations by changing parts of it to see how they affected protein folding.
The bold sections are the parts of the sequence I changed.
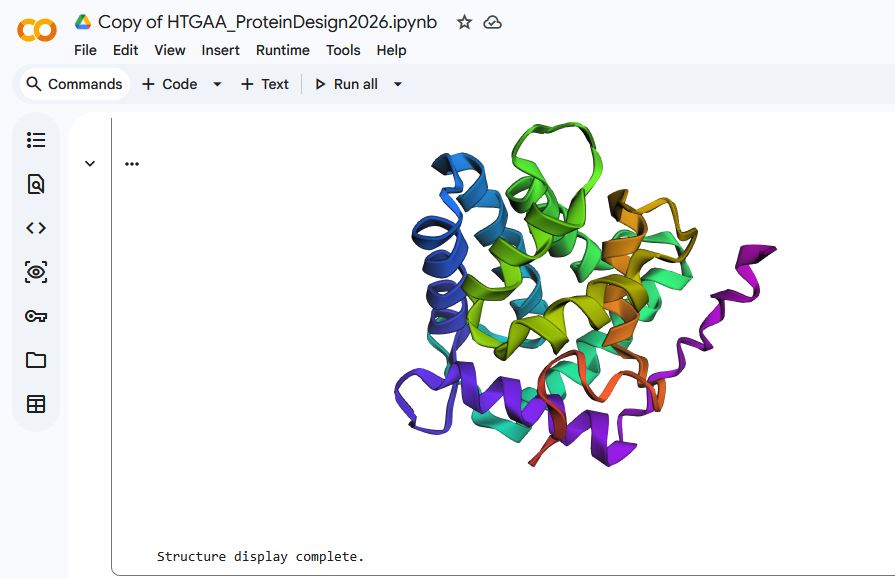
The Aequourin-1 protein Mutant_1 Sequence:
MTSEQYSVKLTPDFDNPKWIGRHKHMFNFLDVNHNGRISLDEMVYKASDIVINNLGATPEQAKRHKDAVEAFFGGAGMKYGVETEWPEYIEGWKRLASEELKRYSKNQITLIRLWGDALFDIIDKDQNGAISLDEWKAYTKSDGIIQSSEDCEETFRVCDIDESGQLDVDEMTRQHL TLJWIEKDNL EKLYGGAVP
The Aequourin-1 protein Mutant_2 Sequence:
MTSEQYSVKLTPDFDNPKWIGRHKHMFNFLDVNHNGRISLDEMVYKASDIVINNLGATPEQAKRHKDAVEAFFGGAGMKYGVETEWPEYIEGKMSEQTVPDFEYHKKHMFBFLHNGRQITVIIDKDQNGAISLDEWKAYTKSDGIIQSSEDCEETFRVCDIDESGQLDVDEMTRQHLGFWYTMDPACEKLYGGAVP
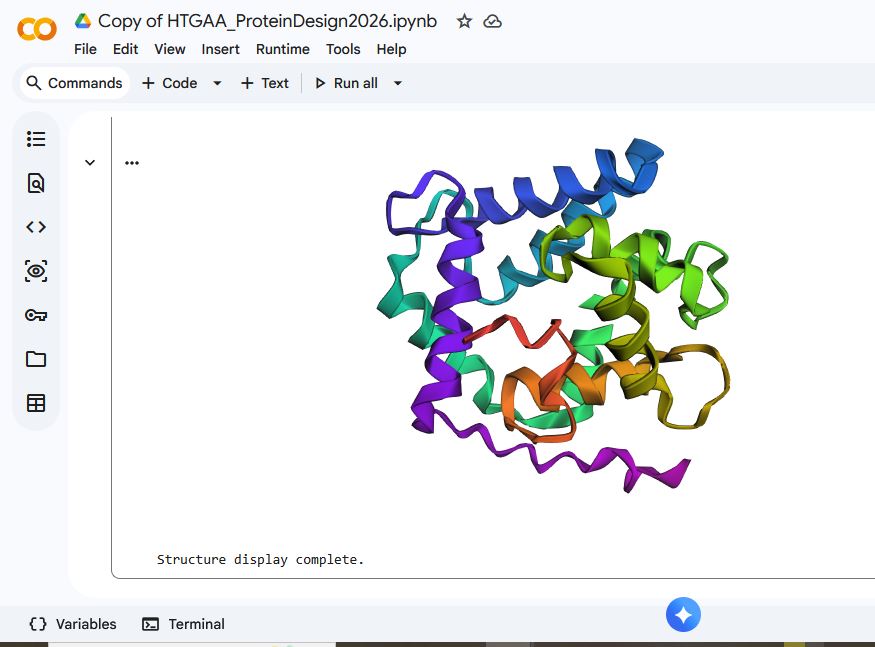
For mutant_1, I made some random changes in a small portion of the protein sequence, and for mutant_2, I made random changes to a larger part of the protein sequence.
There was not a drastic difference in the folding structures of Aequorin-1 protein in ESMFold and the Aequorin-1 protein Mutant_1, as well as the Aequorin-1 protein Mutant_2. Based on this, I can conclude that the protein is quite resilient to mutations.
C3. Protein Generation
I used the PDB file generated by ESMfold to inverse-fold the protein using ProteinMPNN.
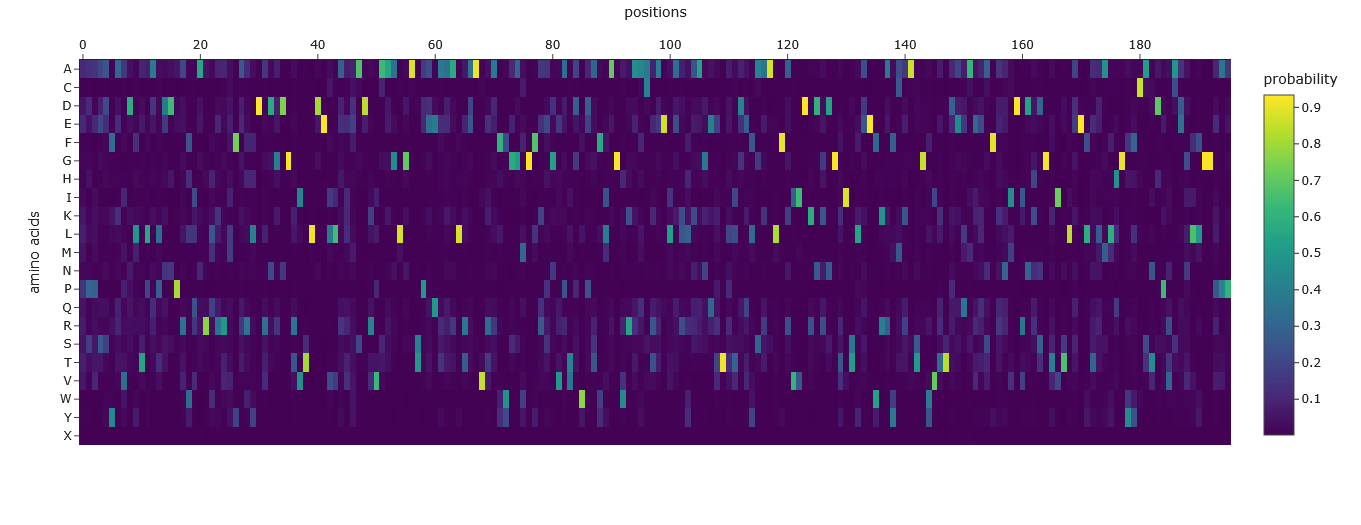
The Amino Acid Probabilities heatmap generated after the inverse folding with ProteinMPNN predicts the likelihood of each amino acid occurring at every position along my input protein backbone structure.
Original Protein Sequence:
MTSEQYSVKLTPDFDNPKWIGRHKHMFNFLDVNHNGRISLDEMVYKASDIVINNLGATPEQAKRHKDAVEAFFGGAGMKYGVETEWPEYIEGWKRLASEELKRYSKNQITLIRLWGDALFDIIDKDQNGAISLDEWKAYTKSDGIIQSSEDCEETFRVCDIDESGQLDVDEMTRQHLGFWYTMDPACEKLYGGAVP
New Protein Sequence from ProteinMPNN:
PPPSAYAVDLTLAPDDPRWQARQRRMFAFLDTDGDGKVSLDEVLAVMADRVAAGLGATPEEAAALREAVRAFFGAMGLREGVATDWDAFLAGWKAAARRELALLAAGEETLLDRLARALFAVIDRDRDGRISYDEWLAWAKATGLVKTEEQAREAFKNVDVDHDGYITLAELTAALRGYFCATAPVADILFGGPAP
By comparing the original Aequorin-1 protein to the new protein sequence generated by the inverse folding using ProteinMPNN, I noticed there was a significant difference between the two sequences. The sequence recovery rate was 41.33% this means that almost 60% of the protein residues were changed by ProteinMPNN. These changes might be due to ProteinMPNN’s design process, optimizing the sequence to best fit the provided 3D backbone structure. This means that some of the changes in the sequence were due to certain amino acids being substituted for others that are energetically or structurally more favorable in that specific local environment. I also noticed that both sequences maintained the same length of 196 residues despite the changes.
I entered the new protein sequence from ProteinMPNN into ESMFold, and the displayed structure resembled the one generated when I used ESMFold for the Aequorin-1 protein.
The documentation of how I used Gemini 2.5 flash in Google Colab.
| Prompt | Error | Fix / Explanation |
|---|---|---|
| the protein structure that was shown after using EMSFOLD was different from the structure in RCSB. What could be the reason for the difference in structure | None | Explained differences between predicted (ESMFold) and experimental (RCSB) structures, including prediction vs experimental methods, accuracy limitations, missing ligands, conformational flexibility, and environmental conditions. |
| is there a way to tell if a protein is resilient to mutations based on its structure? Using emsfold | None | Clarified that ESMFold predicts structure but does not directly measure mutation resilience. Suggested using mutation scanning, stability prediction tools, and structural analysis for assessing resilience. |
| how can you interpret the pLDDT score | None | Explained that pLDDT ranges from 0–100 and indicates model confidence per residue. Provided interpretation ranges (very high, high, low, very low confidence) and how to use them to evaluate the reliability of predicted regions. |
| how do I get the backbone of your chosen PDB to run Inverse folding with ProteinMPNN | None | Explained that the pdb variable must be updated to the correct ESMFold-generated PDB file path before running ProteinMPNN. Provided instructions to modify the setup cell. |
| what does your fix do | None | Explained that the fix updated the pdb variable so ProteinMPNN uses the correct ESMFold-generated structure for inverse folding. |
| ok run your fix | FileNotFoundError: [Errno 2] No such file or directory: ’test_85b81/ptm0.805_r3_default.pdb.pdb' | Error caused by double .pdb extension being appended. Identified that get_pdb function incorrectly added .pdb to a filename that already had it. |
| (Implicit – after execution error) | None (after fix) | Modified the get_pdb function to properly handle local PDB paths without appending an extra .pdb extension. Ready for re-execution. |
| (Implicit – after successful execution) | None | Confirmed successful ProteinMPNN setup execution and instructed user to run the sequence generation cell. |
| explain the results of the Amino acid possibilities in the mutation heat scan after the inverse folding with proteinMPNN | None | Explained amino acid probability heatmap: axes (positions vs amino acids), color intensity representing probability, and interpretation for residue preference, conservation, and sequence diversity. |
Part D. Group Brainstorm on Bacteriophage Engineering
Three students from the William and Mary Node and I formed a group for this part of the assignment.
We chose the goal of increasing the thermodynamic and structural stability of the Lysis protein while preserving its native fold and lytic function.
Here is the link to our one-page proposal:
https://docs.google.com/document/d/1676c1tgFUlGaP-Bwp9_vDexbk3VsOJeuQeylNfvz76o/edit?usp=sharing
Here are some additional pitfalls :
- Is a limited understanding of the structural biology of lysis protein as a beginner to phage engineering
- Increasing stability may not lead to improved function of the protein.
Here is a picture of the schematic of the pipeline

Reference
Greenwald, J., & Riek, R. (2010). Biology of amyloid: structure, function, and regulation. Structure, 18(10), 1244-1260.
Glyakina, A. V., Likhachev, I. V., Balabaev, N. K., & Galzitskaya, O. V. (2014). Right‐and left‐handed three‐helix proteins. II. Similarity and differences in mechanical unfolding of proteins. Proteins: Structure, Function, and Bioinformatics, 82(1), 90-102.
JPT Peptide Technologies. (n.d.). What are L- and D- amino acids? JPT Peptide Technologies. Retrieved February 28, 2026, from https://www.jpt.com/blog/l-d-amino-acids/
Johns Hopkins Medicine. (n.d.). Amyloidosis. Johns Hopkins Medicine. https://www.hopkinsmedicine.org/health/conditions-and-diseases/amyloidosis
Mills, C. E. (1999–present). Bioluminescence of Aequorea, a hydromedusa. Faculty of Science, University of Washington. Retrieved March 1, 2026, from https://faculty.washington.edu/cemills/Aequorea.html
Makin, O. S., Atkins, E., Sikorski, P., Johansson, J., & Serpell, L. C. (2005). Molecular basis for amyloid fibril formation and stability. Proceedings of the National Academy of Sciences of the United States of America, 102(2), 315–320. https://doi.org/10.1073/pnas.0406847102
Niu, Z., Gui, X., Feng, S., & Reif, B. (2024). Aggregation Mechanisms and Molecular Structures of Amyloid‐β in Alzheimer’s Disease. Chemistry–A European Journal, 30(48), e202400277.
ScienceDirect. (n.d.). Alpha helix. In ScienceDirect Topics. Elsevier. Retrieved February 28, 2026, from https://www.sciencedirect.com/topics/immunology-and-microbiology/alpha-helix
Sinnige T. (2022). Molecular mechanisms of amyloid formation in living systems. Chemical science, 13(24), 7080–7097. https://doi.org/10.1039/d2sc01278b