Week 5 HW: Protein Design - Part II
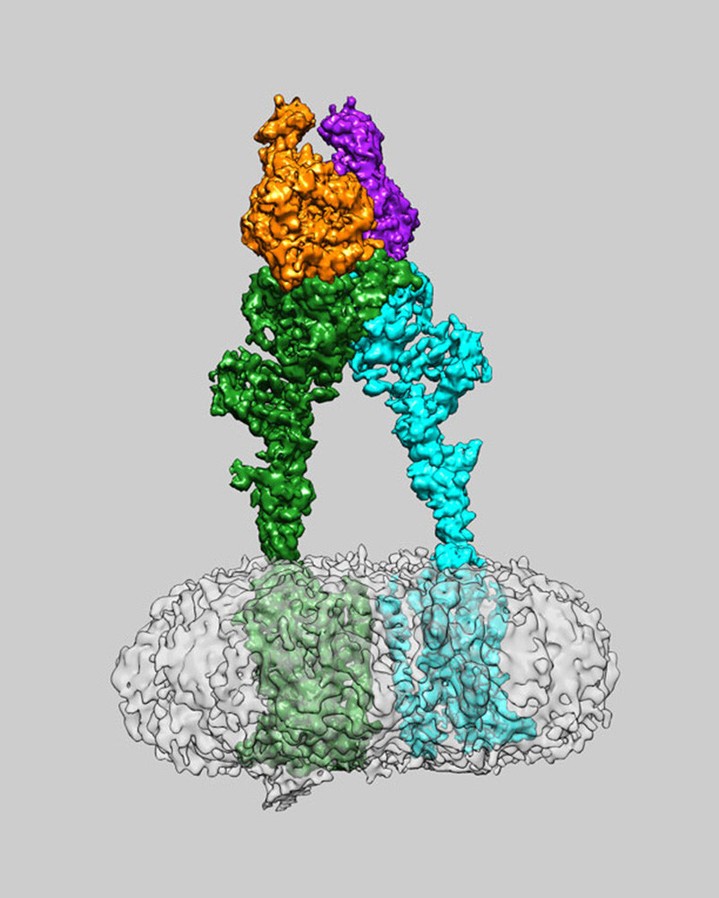
Part A: SOD1 Binder Peptide Design
A peptide binder is a short, engineered protein fragment usually <50 amino acids that binds to specific targets. It functions as a powerful, cost-effective, and stable alternative to larger antibiotics or small-molecule drugs. A peptide binder is used to modulate, degrade, or inhibit disease-related proteins, especially those that are deemed undruggable due to the absence of clear binding pockets.
My task this week is to design peptide binders for the SOD1 mutant.
Part 1: Generate Binders with PepMLM.
Question 1.
I retrieved the human SOD1 sequnce from Uniprot(P00441):
https://rest.uniprot.org/uniprotkb/P00441.fasta
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
I then introduced the SOD1 A4V mutation sequence:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
The A4V mutation is a mutation in the SOD1 gene that causes a rapidly progressive and aggressive form of familial amyotrophic lateral sclerosis(ALS). The mutation involves the substitution of A(alanine) for V (valine) at the 4th codon of the SOD1 sequence.
A (Alanine) is viewed as the 4th codon despite being in the 5th position in the protein sequence, due to sequences being read/counted after the start codon M (Methionine). Which is often cleaved after translation.
Question 2 & 3.
I generated four peptides, each of length 12 amino acids and with a k value of 3, conditioned on the SOD1 mutant sequence using PepMLM.
The K value determines the number of the most probable tokens(amino acids) considered at each step when generating the peptide sequence. A low K value leads to the model almost always picking the most probable amino acids, resulting in peptides that are highly coherent but lack diversity, while a high K value leads to the model having a wider range of choices by including more amino acids in the selection pool. Which could lead to novel peptide sequences that might explore less common but potentially effective binding sites. However, there is also a higher risk of generating less optimal binding peptides.
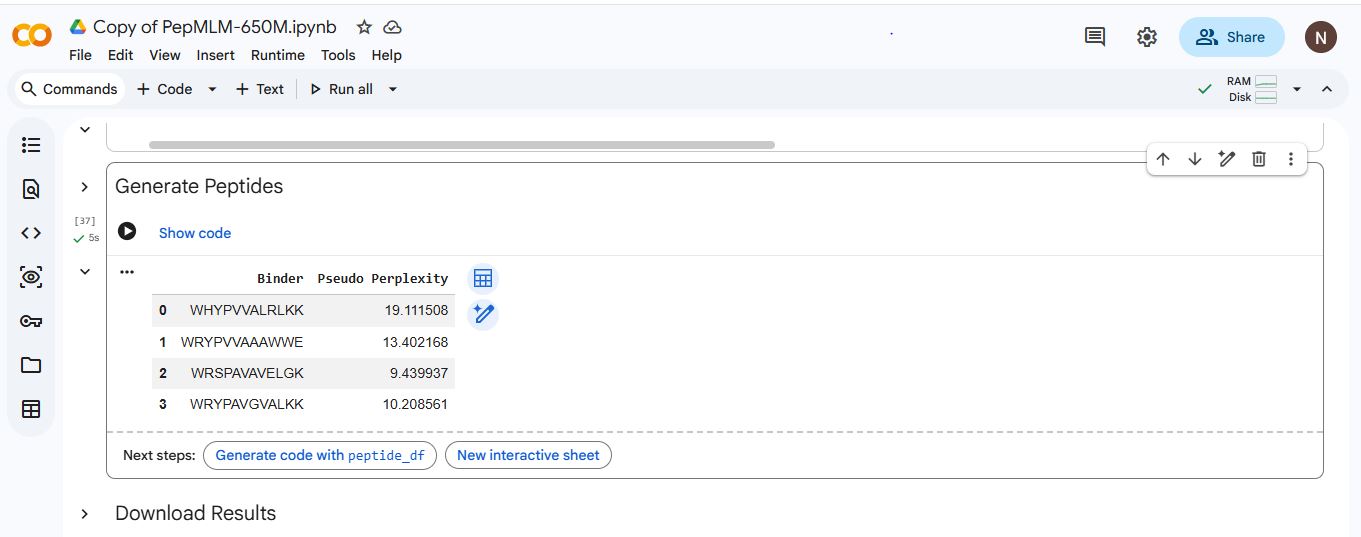
| Index | Binder | Pseudo Perplexity |
|---|---|---|
| 1 | WHYPVVALRLKK | 19.111508 |
| 2 | WRYPVVAAAWWE | 13.402168 |
| 3 | WRSPAVAVELGK | 9.439937 |
| 4 | WRYPAVGVALKK | 10.208561 |
Question 4
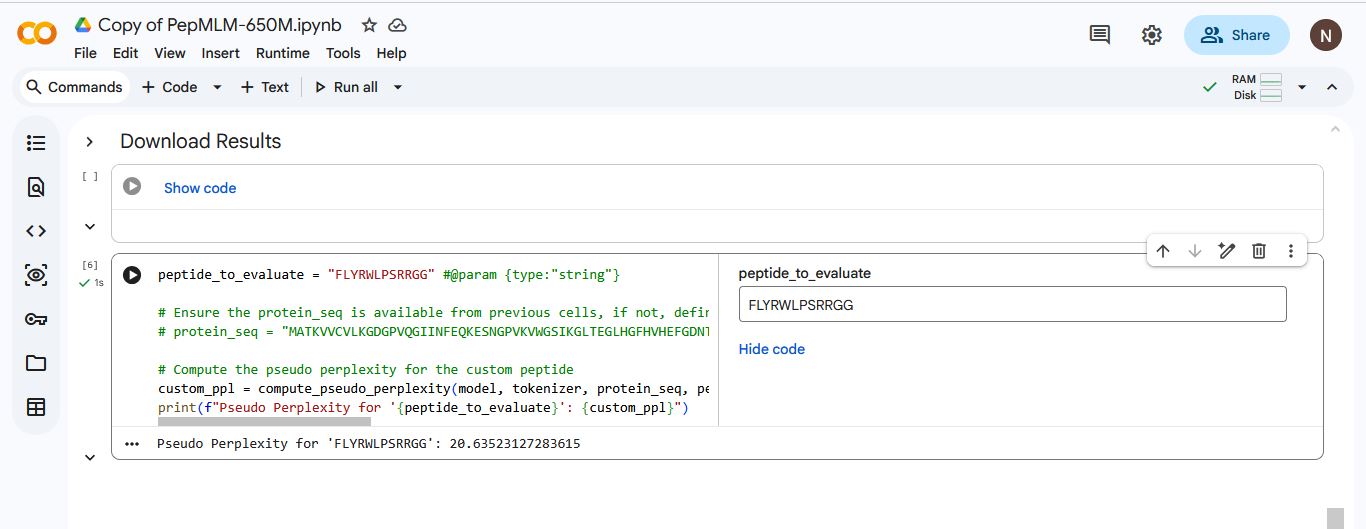
I added the known SOD1-binding peptide “FLYRWLPSRRGG” to the list of generated amino acids. I used Gemini to write a code that would generate a pseudo perplexity score for “FLYRWLPSRRGG” based on the SOD1 Mutant sequence.
| Index | Binder | Pseudo Perplexity |
|---|---|---|
| 1 | WHYPVVALRLKK | 19.111508 |
| 2 | WRYPVVAAAWWE | 13.402168 |
| 3 | WRSPAVAVELGK | 9.439937 |
| 4 | WRYPAVGVALKK | 10.208561 |
| 5 | FLYRWLPSRRGG | 20.635231 |
Question 5
The pseudo perplexity score measures how expected or natural a peptide sequence looks to the PepMLM model when interacting with a protein. Lower scores usually indicate better potential binders, while higher scores indicate a potentially poor binder.
Based on their pseudo perplexity scores, I indicate PepMLM’s confidence in the binders.
High confidence indicates a potentially better binder. Low coincidence indicates a potentially poor binder.
| Index | Binder | Pseudo Perplexity | Confidence Level |
|---|---|---|---|
| 1 | WHYPVVALRLKK | 19.111508 | Low confidence |
| 2 | WRYPVVAAAWWE | 13.402168 | Low confidence |
| 3 | WRSPAVAVELGK | 9.439937 | High confidence |
| 4 | WRYPAVGVALKK | 10.208561 | High confidence |
| 5 | FLYRWLPSRRGG | 20.635231 | Low confidence |
Part 2: Evaluate Binders with AlphaFold3
SOD1 A4V mutation sequence:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
I used the alphafold server to evaluate the protein-peptide complex of the SOD1 mutant and the binding peptides.
WHYPVVALRLKK peptide
It has an ipmTM score of 0.37, and the peptide appears to localize near the β-barrel. It does not appear to be buried in the protein’s structure.
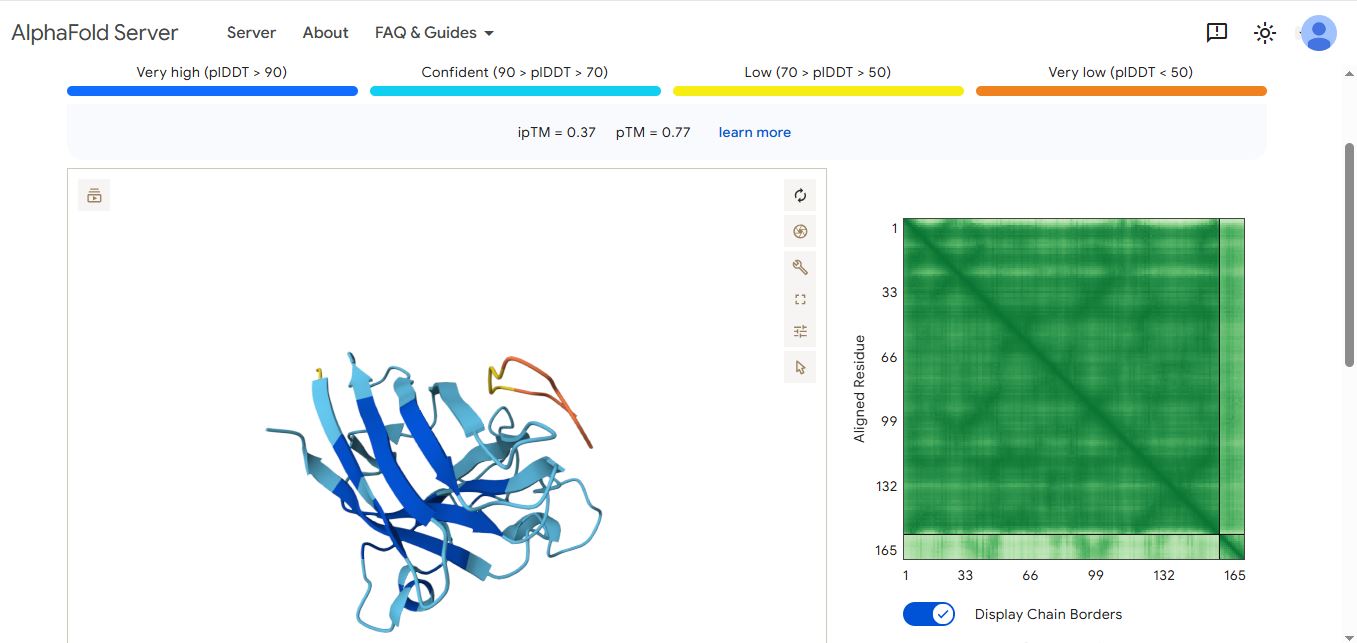
WRYPVVAAAWWE peptide
It has an ipmTM score of 0.22, and the peptide appears to localize near the dimer interface. It also does not appear to be buried in the protein’s structure
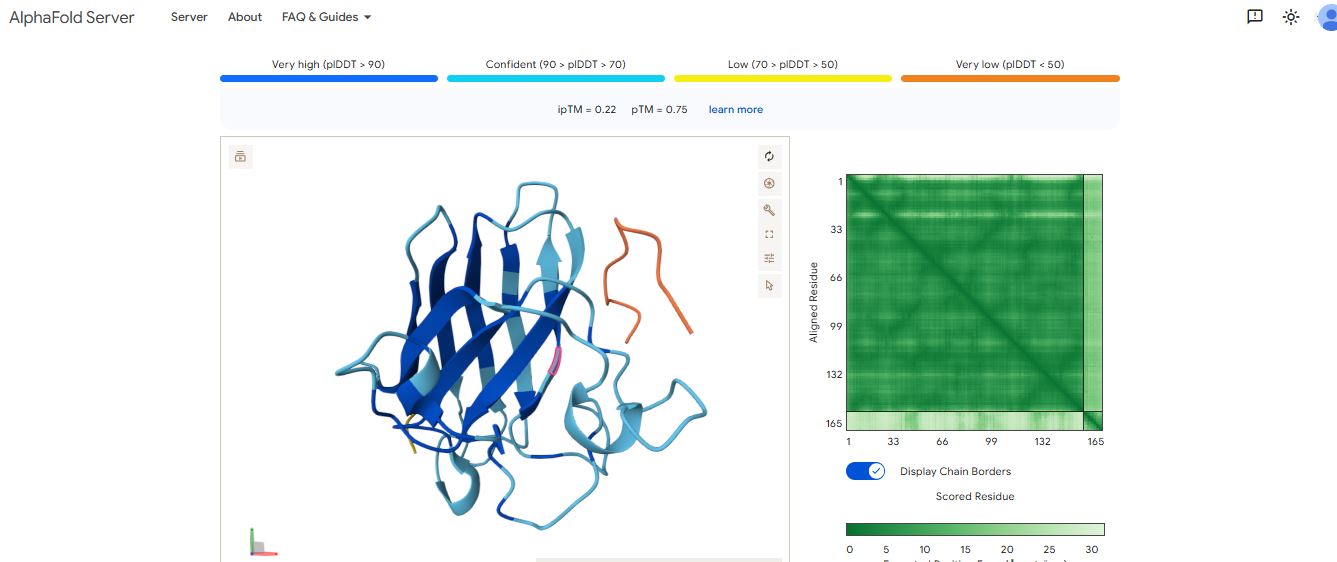
WRSPAVAVELGK peptide
It has an ipTM score of 0.24, and the peptide appears to localize near the β-barrel. It does not appear to be buried in the protein’s structure but is loosely associated with the surface of the protein.
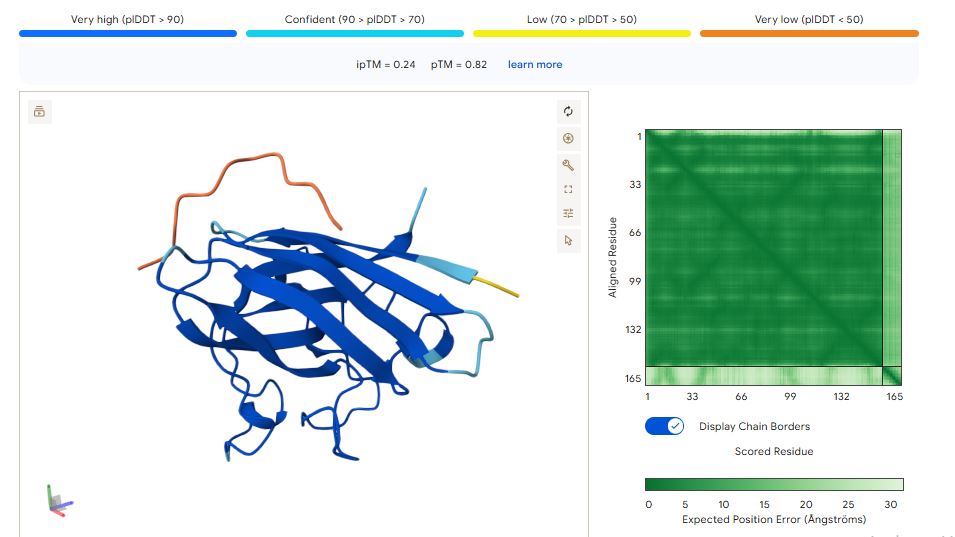
WRYPAVGVALKK peptide
It has an ipTM score of 0.25, and the peptide appears to localize near the dimer interface. It also appears to be loosely associated with the surface of the protein.
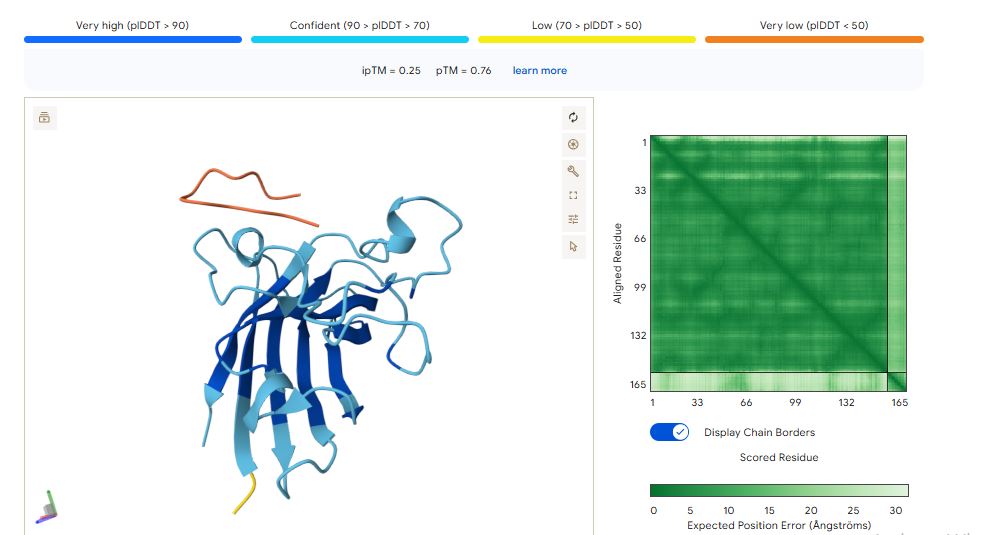
FLYRWLPSRRGG peptide
It has an ipTM score of 0.35, and the peptide appears to localise near the dimer interface. It also appears to be loosely associated with the surface of the protein
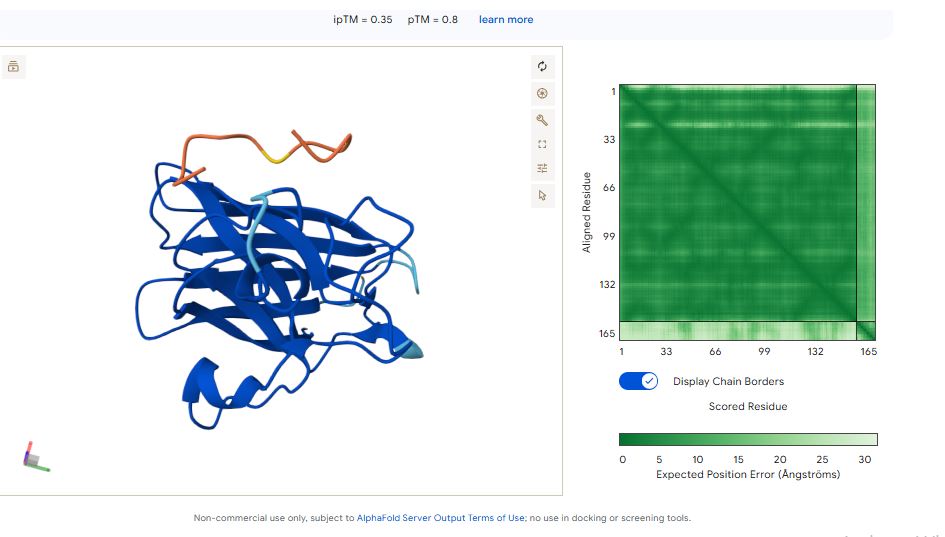
The ipTM values indicate how confident AlphaFold is in the interaction between different protein chains in a complex.
The pTM scores also indicate the confidence in the overall shape of the whole complex.
The ipTM scores for the peptides were low. This means AlphaFold was not confident in the interactions between the peptides and the SOD1 A4 mutant. I ranked the peptides from the highest score (Highest confidence level) to the lowest score (Lowest confidence level):
WHYPVVALRLKK (ipTM = 0.37) > FLYRWLPSRRGG (ipTM = 0.35) > WRYPAVGVALKK (ipTM = 0.25) > WRSPAVAVELGK (ipTM = 0.24) > WRYPVVAAAWWE (ipTM = 0.22)
Peptide WHYPVVALRLKK had the highest ipTM score and was the only peptide generated by PepMLM that surpassed the score of the known SOD1 mutant binder FLYRWLPSRRGG.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
I used the Peptiverse to evaluate the generated peptides based on predicted binding affinity, solubility, hemolysis probability, net charge, and molecular weight.
Peptide WHYPVVALRLKK
It is soluble, non-hemolytic, has weak binding affinity, a net positive charge of 2.84, and a molecular weight of 1509.8 Daltons.
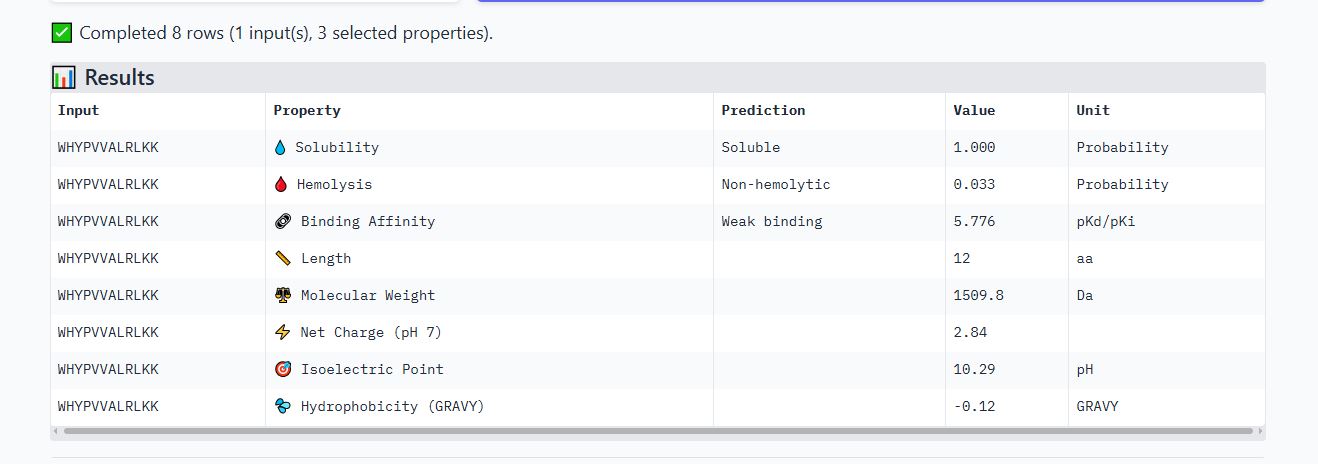
Peptide WRYPVVAAAWWE
It is soluble, non-hemolytic, has medium binding affinity, a net negative charge of -0.23, and a molecular weight of 1533.7 Daltons.
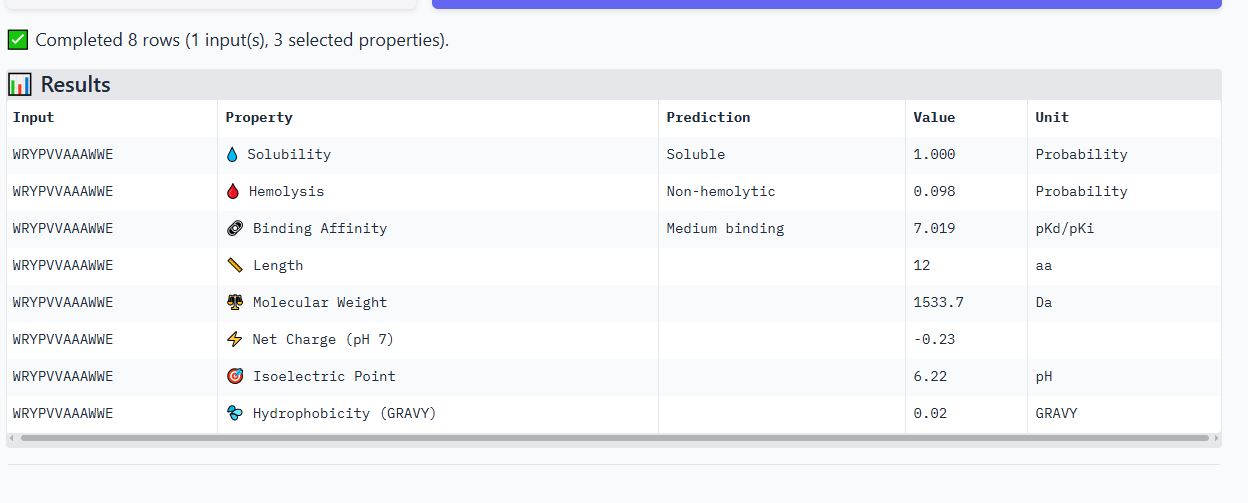
Peptide WRSPAVAVELGK
It is soluble, non-hemolytic, has weak binding affinity, a net positive charge of 0.76, and a molecular weight of 1312.5 Daltons.
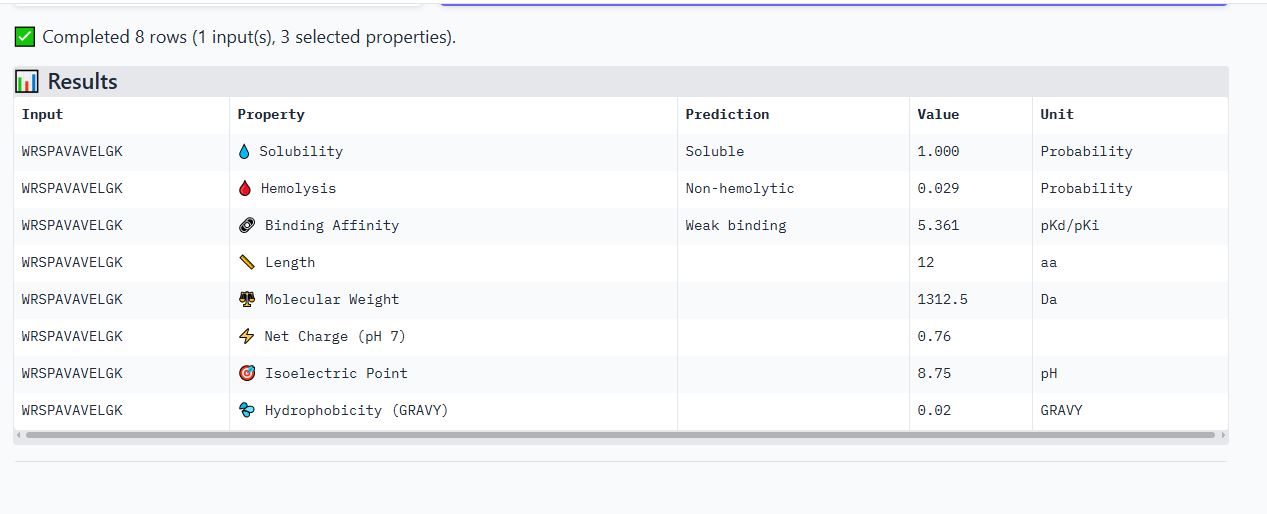
Peptide WRYPAVGVALKK
It is soluble, non-hemolytic, has weak binding affinity, a net positive charge of 2.76, and a molecular weight of 1387.7 Daltons.
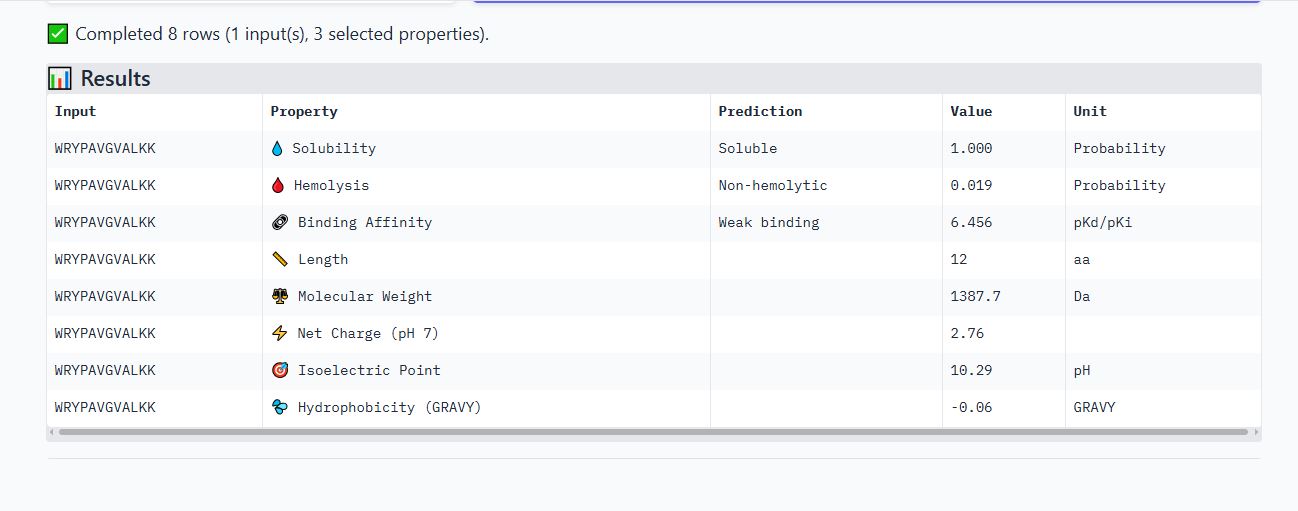
Peptide FLYRWLPSRRGG
It is soluble, non-hemolytic, has a weak binding affinity, a net positive charge of 2.76, and a molecular weight of 1507.7 Daltons.
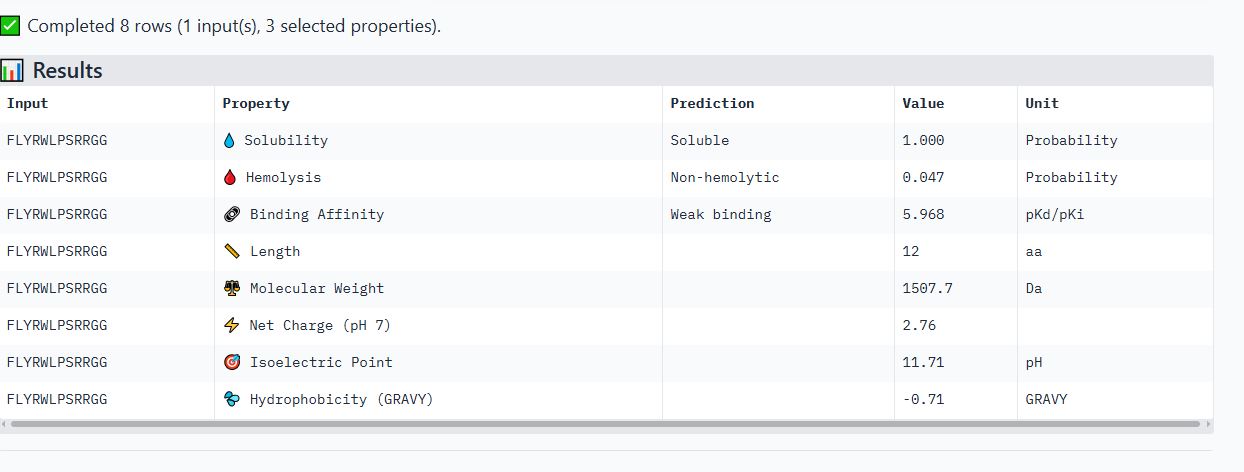
I noticed all the peptides had solubility of 1 and were non-hemolytic. Their hemolytic probability ranges from 0.019 to 0.098.
When comparing ipTM scores with binding affinity, I observed that there was not a directly proportional relationship between the two. Some peptides with higher ipTM scores did not demonstrate stronger predicted binding affinity. For instance, peptide WRYPAVGVALKK had the highest ipTM score among the tested peptides but did not have the highest binding affinity. However, peptide WRYPVVAAAWWE, which had the lowest ipTM score, exhibited the highest binding affinity, with a medium binding affinity of 7.819.
Based on their properties, I would advance peptide WRYPAVGVALKK. This is because it has a good balance of properties as compared to the other peptides. It has the lowest hemolytic probability (0.019), the second highest binding affinity (6.456), a good net charge of 2.76, and a low molecular weight (1387.7 Da), making it the ideal choice. Unlike peptide WRYPVVAAAWWE, which has the highest binding affinity but a net negative charge(-0.23) that would likely reduce its membrane penetration.
Part 4: Generate Optimized Peptides with moPPIt
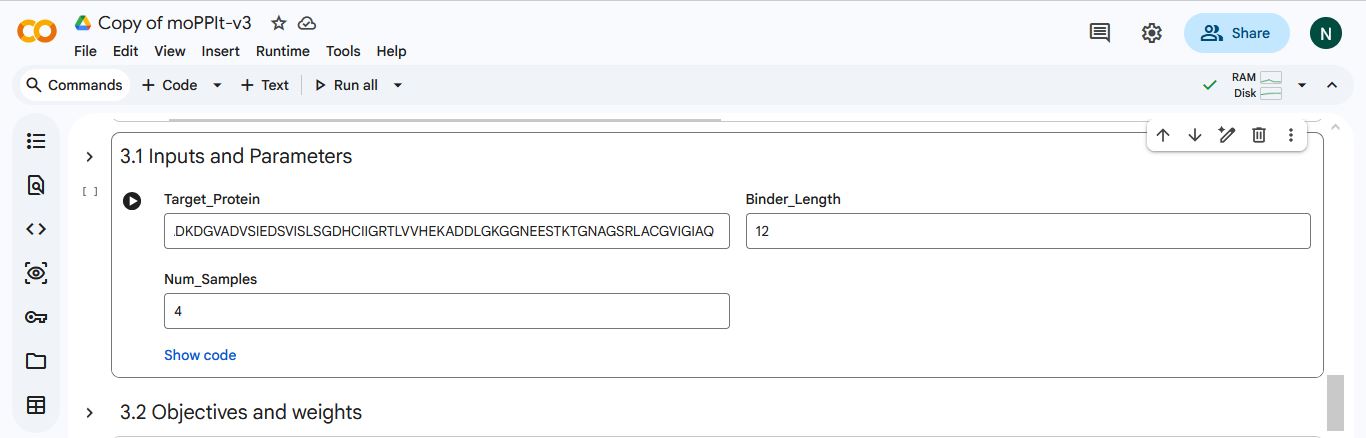
I made a copy of the momPPIt Colab and inserted the SOD1 AV4 mutant sequence. I set the binder length to 12 amino acids and the number of samples to 4.
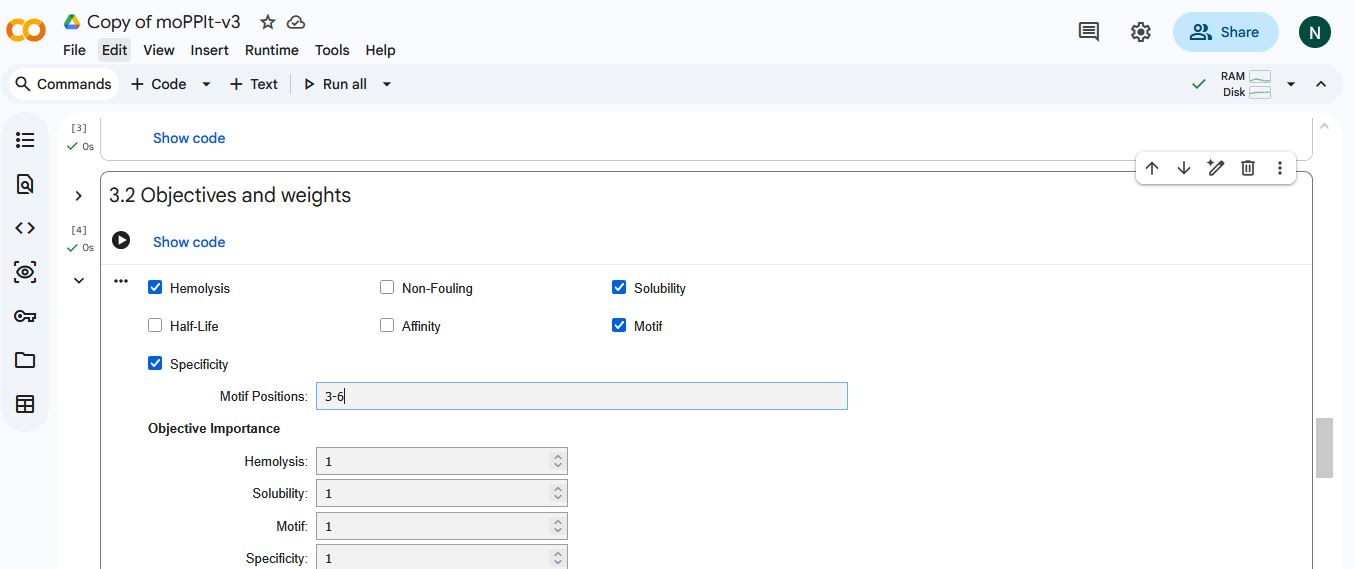
I selected hemolysis, solubility, specificity, and motif binding as the objectives. I select 3-6 as the motif binding regions to let the peptides bind to residues near the AV4 position.
MoPPIt generated the following peptide sequences
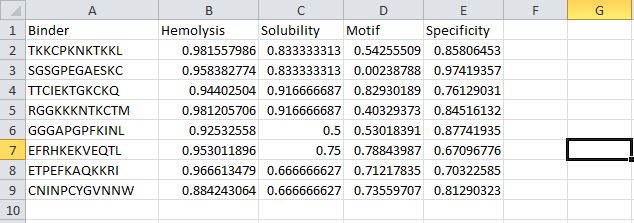
After comparing the peptides generated by moPPIt and my PepMLM peptides. I noticed the peptides generated by moPPIt had higher motif-binding scores and specificity. However, they had lower solubility scores and higher hemolytic scores, meaning they might be more prone to damage or rupture red blood cells.
I would evaluate the peptides generated by moPPIt before advancing them for clinical studies by first using computational tools such as GROMACS and alphafold to further validate their structure, target binding strength, stability, physicochemical properties, and potential toxicity. After further validation, I would then select peptides that demonstrate high binding strength, stability, and no toxicity to advance to the next phase, which would involve animal studies to determine their therapeutic effects in living systems. Finally, I would ensure all safety and quality standards set by the regulatory agency are met before advancing the peptide for clinical(Human) trials.
Part B: BRD4 Drug Discovery Platform Tutorial
Part C: Final Project: L-Protein Mutants
The objective for this section of the assignment is to improve the stability and auto-folding of the lysis protein of an MS2-phage. Which might be the key to understanding how phages can potentially solve antibiotic resistance.
L-Protein Engineering | Option 1: Mutagenesis
I formed a group with a couple of students from the William and Mary Node. We decided to tackle Option 1: Mutagenesis of the L-Protein.
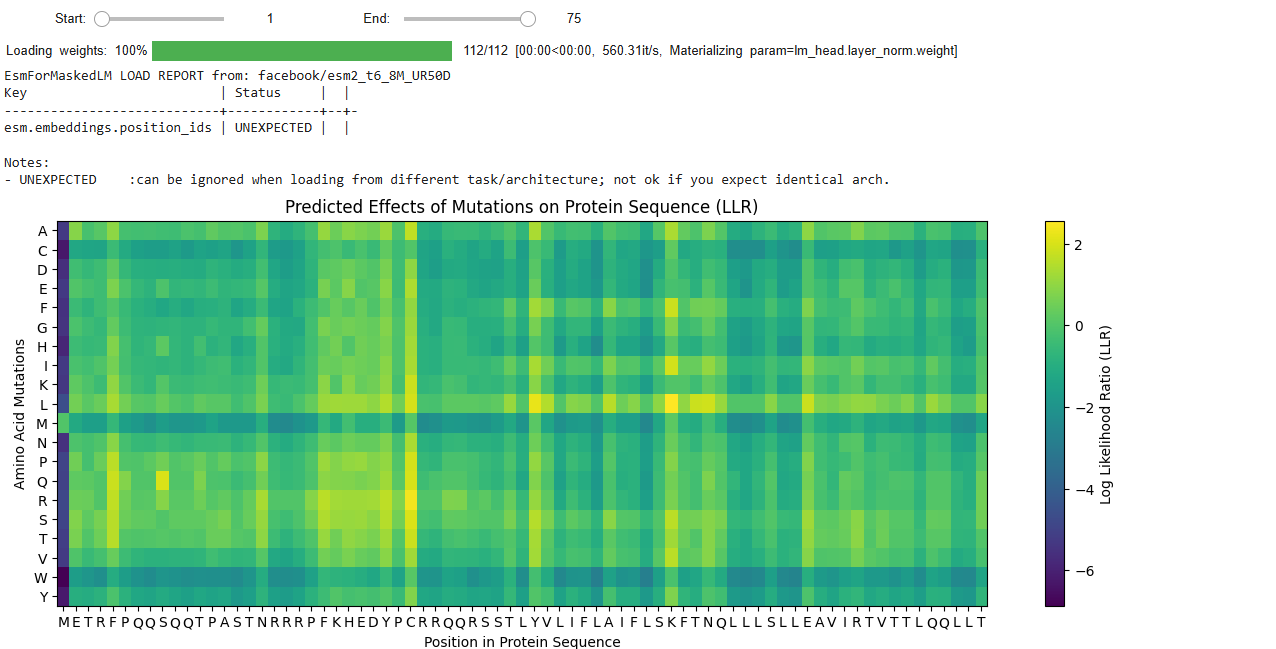
We run the Colab notebook to generate likely mutation positions in the L-protein. We used the generated Mutation heat map to identify possible positive mutations.
We also used the BLAST results for the L-protein provided in the Google Drive and performed sequence alignment using Clustal Omega to determine the conserved regions of the L-Protein to guide the mutant selection process.
BLAST results for the L-protein: https://drive.google.com/drive/folders/1eQeuwL9WiO16bw6Lb8z-TVpbWIoAF4EH?usp=share_link
Clustal Omega Sequence alignment results for the L-protein:
clustalo-I20260309-215356-0951-56325226-p2m
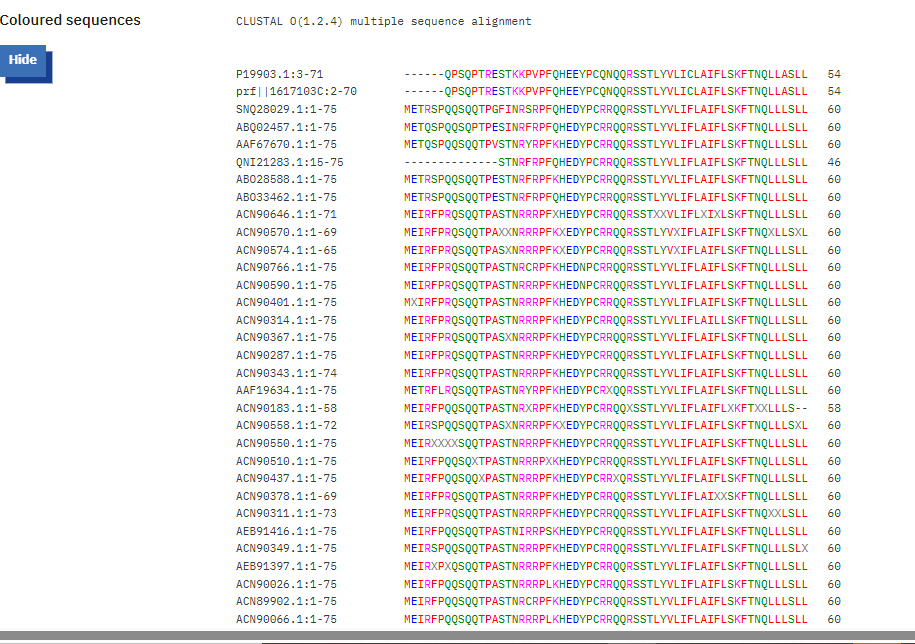
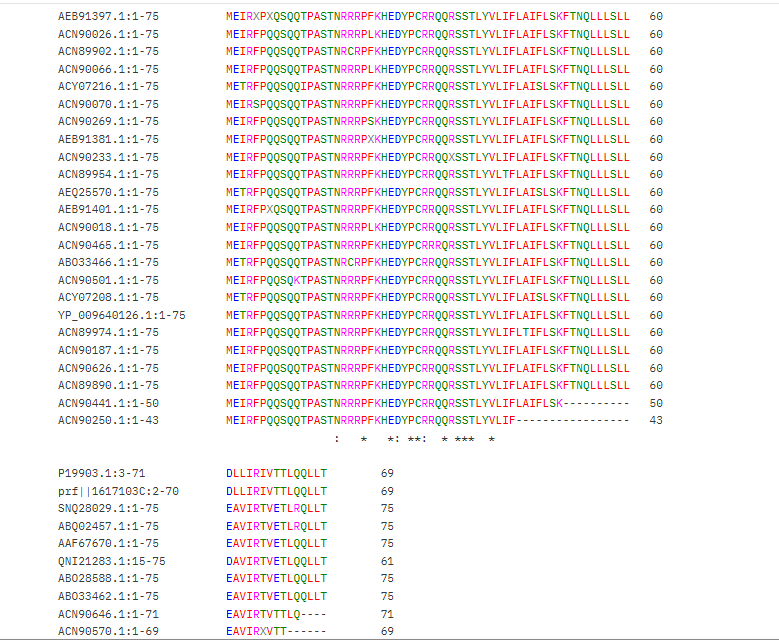
Conserved residues of the L-protein

The table below shows the meaning of the level of conservation of the L-protein. We avoided selecting mutants that occurred at residues of high conservation to avoid negatively affecting the function of the L-protein.
| Symbol | Meaning | Conservation Level |
|---|---|---|
* | Fully conserved residue (all sequences identical at that position) | ⭐⭐⭐ Highest |
: | Strongly conserved substitution (similar chemical properties) | ⭐⭐ High |
. | Weakly conserved substitution (some similarity) | ⭐ Moderate |
(space) | No conservation; residues differ significantly | ❌ Low |
We each came up with 5 mutants; 2 of my mutants had mutations in the soluble region of the L-protein, and the rest had mutations in the transmembrane region.
Soluble Region Mutations
Mutant 1 :
Position 5: F -> Q
METRQPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Mutant 2:
Position 17: N -> R
METRFPQQSQQTPASTRRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Transmembrane region Mutations
Mutant 3:
Position: 40 V -> L
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYLLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Mutant 4:
Position 50 K -> L
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
Mutant 5:
Position 65 R -> L
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVILTVTTLQQLLT
I selected these mutants based on the LLR (Log Likelihood Ratio) Score from the ESM Model Colab notebook.
The LLR scores quantify how much more or less likely a mutated amino acid is at a specific position compared to the wild-type (original) amino acid, according to the protein language model. A positive LLR score suggests that the mutated amino acid is more likely to appear at that position than the wild-type amino acid, implying the mutation might be beneficial or stabilizing. However, a negative LLR score indicates that the mutated amino acid is less likely, suggesting the mutation might be detrimental or destabilizing to the protein’s function or structure.
LLR Scores for My Selected Mutants :
Soluble Region
- Position 5: F -> Q ( LLR Score = 1.79524445533752)
- Position 17: N -> R ( LLR Score = 1.32365107536315)
Transmembrane region
- Position: 40 V -> L ( LLR Score = 1.79524445533752)
- Position 50 K -> L (LLR Score = 2.56146419048309)
- Position 65 R -> L ( LLR Score = 1.0260357856750488)
I chose these mutations because they have a high positive LLR score and are outside the highly conserved region of the L-protein genome; as such, they are more likely to have a positive effect on L-protein stabilization and autofolding.
We used AF2_Multimer to generate a Multimeric Assembly for each of our mutants.
Multimeric assembly refers to the process by which individual protein subunits, known as monomers, associate through non-covalent interactions to form larger, functional complexes.
Here is a multimeric assembly for my five mutants:
METRQPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTRRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYLLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVILTVTTLQQLLT
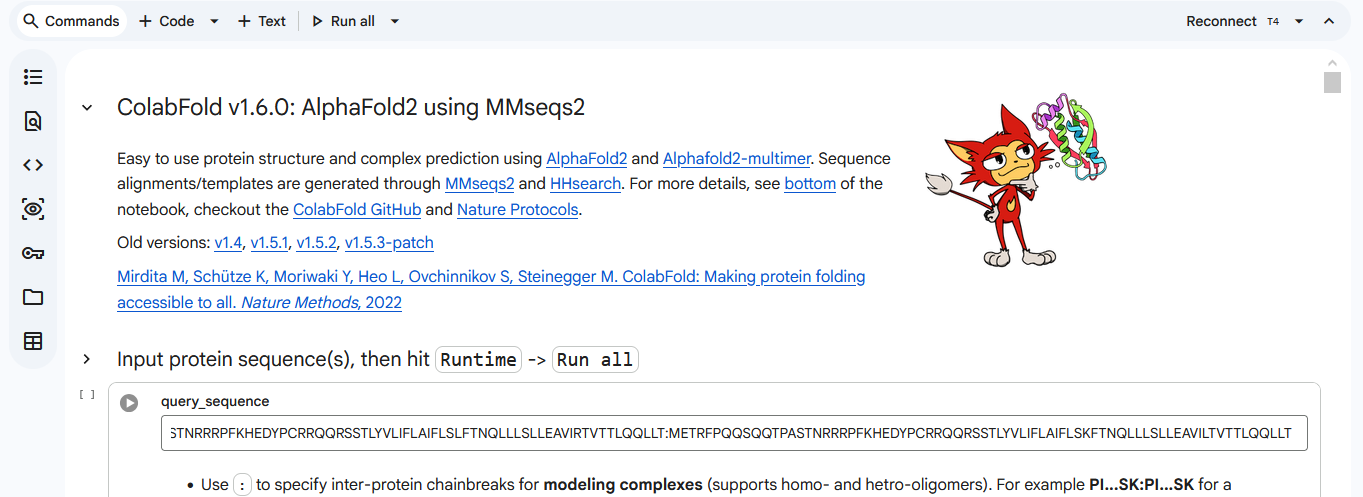



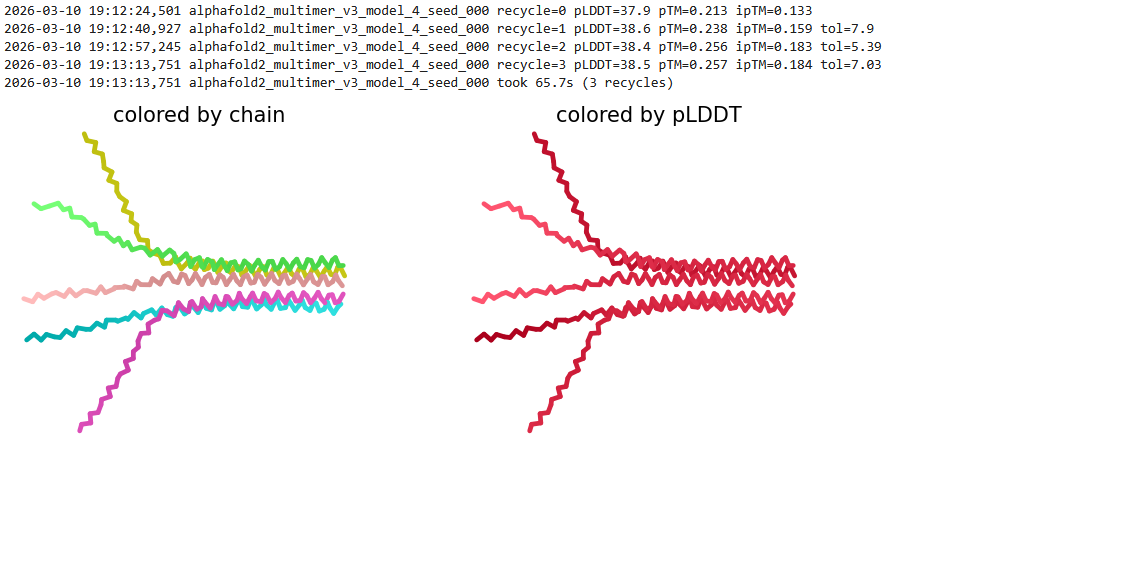
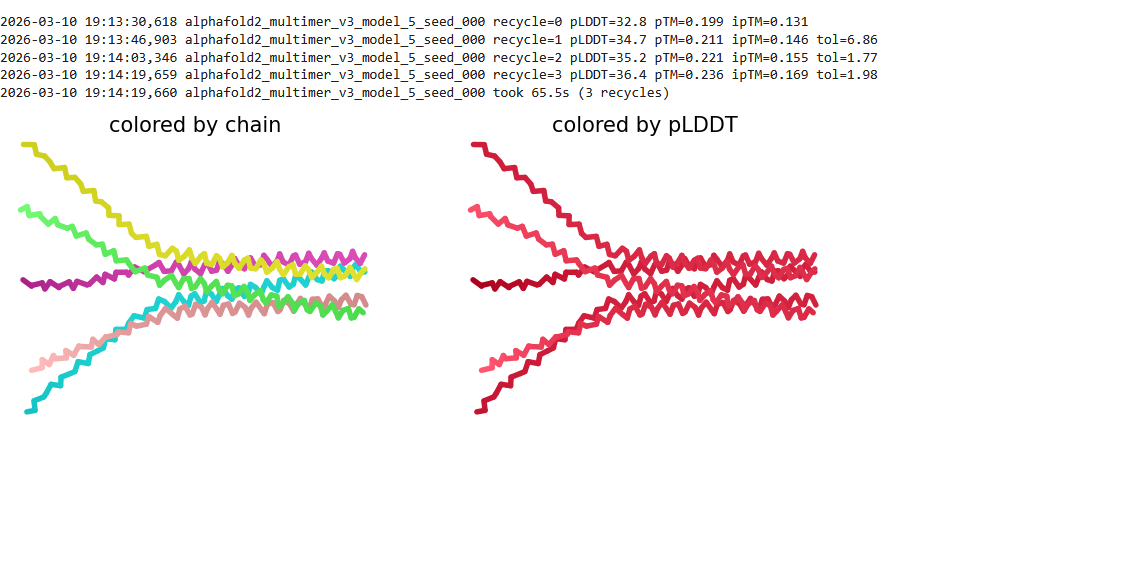
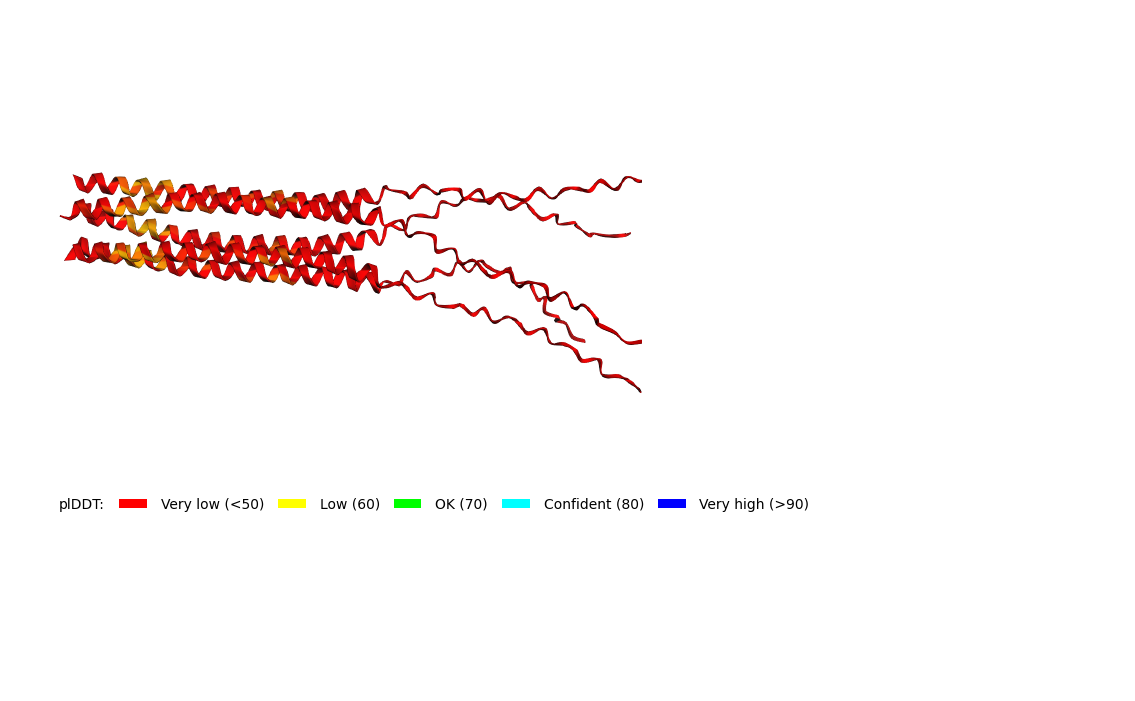
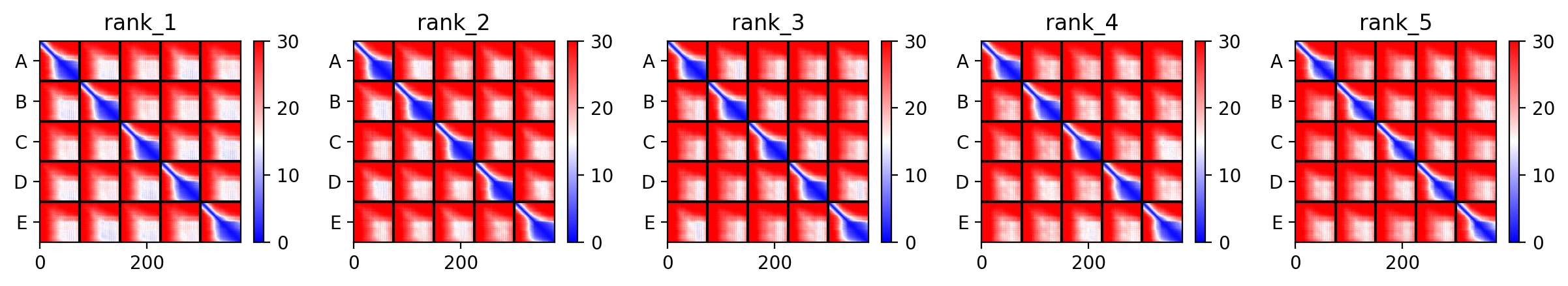
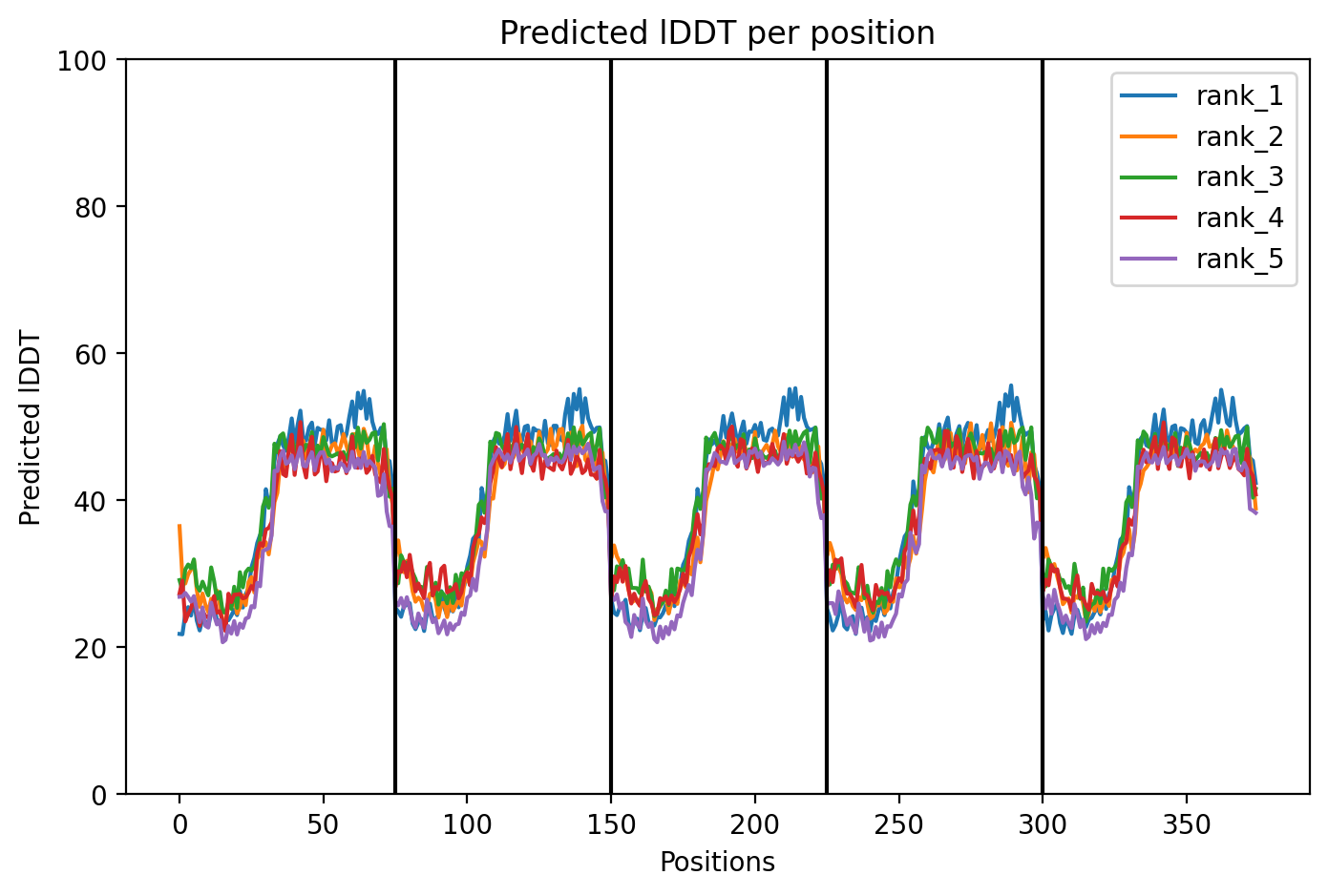
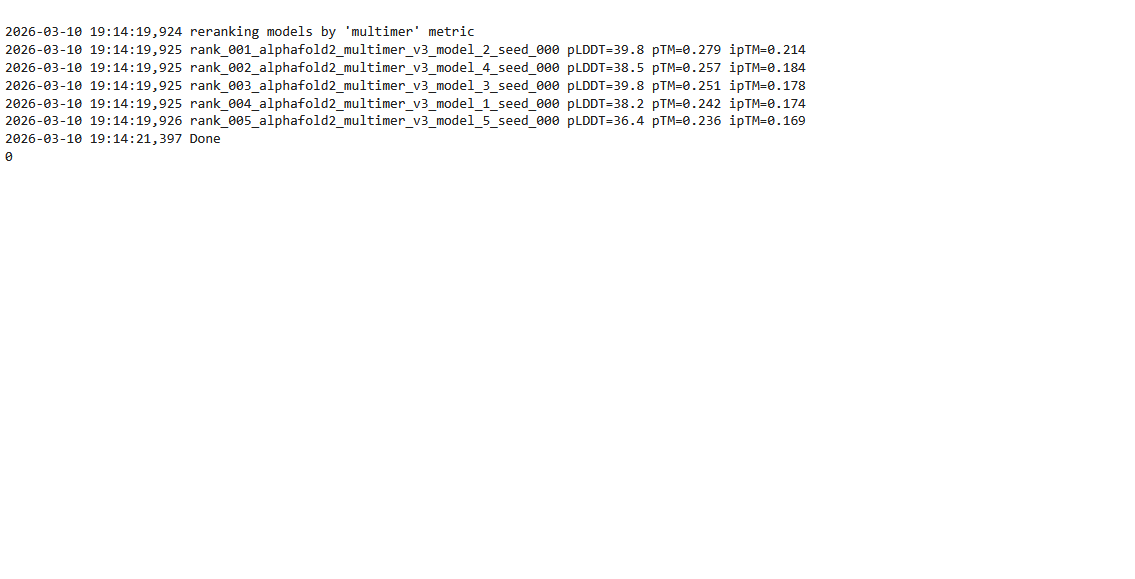
Based on the pLDDT, PTM, and ipTM scores, all the models showed poor scores, meaning Alpha Fold did not have confidence in their structure fold accuracy and prtein complex interfaces. However, model 2 stood out with the highest confidence in terms of fold accuracy, protein complex interfaces, and residue positioning.