Week 6 HW: Genetic Circuit - Part I
Assignment: DNA Assembly
Question 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Answer
A PCR master mix is a pre-formulated, ready-to-use solution containing all the components required for PCR, except the DNA template and primers. It usually includes Taq DNA polymerase, deoxynucleotide triphosphates (dNTPs), magnesium ions, and an optimized reaction buffer at precise concentrations to ensure efficient and reproducible DNA amplification.
The Phusion High-fidelity PCR Master Mix is a ready-to-use 2X mastermix designed for high-speed, high-fidelity PCR applications. It was developed by Finnzymes Oy and manufactured and controlled by New England Biolabs (NEB) under Thermo Fisher Scientific’s Band.
The components in the Phusion High-Fidelity PCR Master Mixcomponents are:
Phusion High-Fidelity DNA Polymerase: It is a specialized thermostable enzyme engineered for high speed, accuracy, and processivity. It is responsible for synthesizing new DNA strands during PCR. It also possesses 5’ to 3’ polymerase activity and 3’ to 5’ exonuclease proofreading activity, which reduces error rates.
2X Phusion HF or GC Buffer: It maintains the optimal chemical environment for enzyme activity by stabilizing polymerase and DNA, correcting pH and the ionic strength needed for effective amplification. GC buffer is used when working with templates with high GC content, as it aids the amplification of GC-rich templates that are difficult to denature.
dNTPs (Deoxynucleotide triphosphates) They are the building blocks of DNA synthesis, the mix contains dATP, dTTP, dCTP, and dGTP. They are incorporated by DNA polymerase during PCR to extend the new DNA strand complementary to the template.
MgCL2 (Magnesium Ions) It acts as a necessary cofactor for DNA polymerase enzyme activity.
DMSO (Dimethyl sulfoxide) It is used to improve the amplification of difficult GC-rich templates by reducing secondary structures. It is not always included in the master mix.-
Question 2. What are some factors that determine primer annealing temperature during PCR?
Answer
Annealing temperature in PCR is the temperature at which primers hybridize/bind to their complementary sequences on the single-stranded DNA template during a PCR cycle. Typically, annealing temperature ranges from 50 to 65 degrees celcuis. Annealing temperature is very important because it determines the specificity and efficiency of amplification.
Annealing temperature is directly determined by factors such as primer length and sequence composition. Primers with longer lengths form more hydrogen bonds with the template DNA, increasing the stability of the primer-template complex, and thus require a higher annealing temperature to ensure binding. However, shorter primers bind less strongly and require lower annealing temperatures. Additionally, primers that contain a higher proportion of guanine(G) and cytosine(C) form stronger bonds with the DNA template due to GC base pairs forming three hydrogen bonds, which require a higher annealing temperature when compared with the weaker two hydrogen bonds formed by adenine(A) and thymine(T).
Primer melting temperature, which is the temperature at which 50% of a DNA primer is separated from the primer-template duplex. Also plays a role in determining annealing temperature, as annealing temperature is usually 3-5 degrees Celsius below the melting temperature.
Question 3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other
Answer
A restriction enzyme digest is a molecular biology technique that uses restriction enzymes to cut DNA at specific recognition sequences known as restriction sites. However, the polymerase chain reaction (PCR) is a laboratory technique used to amplify specific segments of DNA, to generate millions to billions of copies of a target DNA sequence from a very small initial sample. They are both molecular biology techniques used to produce linear DNA fragments, but have different protocols, mechanisms, and ideal applications.
In terms of protocol, restriction enzyme digests involve incubating DNA with one or more restriction enzymes in a suitable buffer at 37 degrees celcuis for 1 to 2 hours, resulting in the cleavage of the DNA at defined sites to produce linear fragments with either blunt or sticky ends. However, PCR takes place in a Thermocycler and involves repeated cycles of denaturation, primer annealing, and extension, which exponentially amplify the target region of a DNA using primers, nucleotides, buffer, DNA template, and thermostable DNA polymerase.
PCR and restriction enzyme digests also differ in the mechanism by which DNA fragments are generated. PCR creates DNA fragments by synthesizing new DNA strands based on primer-defined boundaries, allowing the precise control of the start and end of the amplified fragment. While in restriction enzyme digests, DNA fragments are created by cutting existing DNA molecules at naturally occurring sites, with the size of a fragment depending on the location of the restriction site in the DNA sequence.
It is preferable to use PCR when your goal is to amplify and generate a specific DNA fragment from unknown or low-abundance DNA. It is useful when working with degraded or low-quality DNA needed for downstream applications. However, restriction enzyme digests are best for analyzing or modifying DNA based on sequence-specific cleavage sites. It is used when verifying plasmid constructs, cloning DNA, DNA fingerprinting, and linearizing circular DNA for gel electrophoresis.
Question 4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Answer
Gibson Assembly is a molecular cloning technique that enables the seamless joining of multiple DNA fragments in a single, isothermal reaction without the need for restriction enzymes.
To ensure that DNA sequences that have been digested and PCR-ed will be appropriate for Gibson cloning, you would have to clean the DNA using a purification protocol. This will remove buffer components that inhibit the activity of Gibson enzymes while concentrating the DNA fragments so they can assemble more efficiently. Additionally, the DNA sequences must have overlapping regions, which would have been introduced when designing the PCR primers. The primers would both require homologous sequences at their 5′ends. The overlap on the forward primer should be complementary to the backbone insertion site, and the reverse primer should also contain a complementary overlap to the forward primer. These overlaps will ensure the 5’ exonuclease creates single-stranded overhangs and complementary overlaps anneal together during Gibson assembly.
Question 5. How does the plasmid DNA enter the E. coli cells during transformation?
Answer
Plasmid DNA is usually introduced into E. coli cells during transformation via either heat shock or electroporation.
Transformation via a heat shock involves using a sudden change in temperature to temporarily create pores in the cell walls of E. coli, which enables the plasmid DNA to enter the E.coli cells. The change in temperature is achieved by briefly heating the E.coli cells to 42 degrees celcusi for 30 to 60 seconds. Before heat shock, E. coli cells are treated with salts such as calcium chloride to neutralize the negative charges on the cell wall and membrane. The calcium ions reduce the electrostatic repulsion between DNA and the bacterial cell membrane. However, electroporation utilizes a short high voltage electric pulse to temporarily create pores in the cell wall and membrane of E.coli to allow the plasmid DNA to enter the E.coli cells. Electroporation has a higher transformation efficiency and works effectively with large plasmid DNA. After transformation via either electroporation or heat shock, the E.coli cells are given a recovery period and incubated in nutrient-rich media so they can repair the pores in their membranes before they are cultured on agar containing antibiotics for selection.
Question 6. Describe another assembly method in detail (such as Golden Gate Assembly)
- Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
- Model this assembly method with Benchling or Asimov Kernel!
Answer
- Golden Gate assembly is a molecular cloning method that enables the seamless joining of multiple DNA fragments into a single construct in a single reaction using Type IIS restriction enzymes and T4 DNA ligase. It uses Type IIS restriction enzymes such as Bsal, BsmBI, and BbsI to cleave DNA at positions outside the restriction sites to create unique custom non-palindromic overhangs. The overhangs created are designed to be unique and complementary, ensuring the DNA fragments ligate together in a specific order. Since the recognition sites are removed during Golden Gate assembly, the final construct is scarless and has no extra sequences left behind, making the method highly efficient.
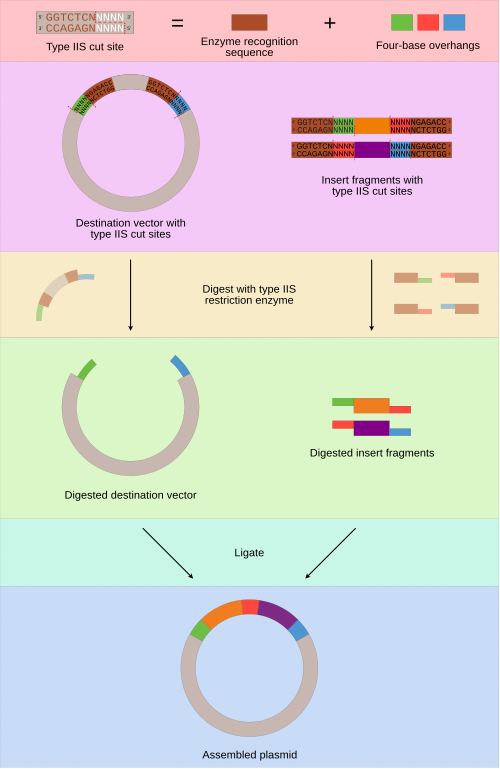
The image is by Rob Hurt - Own work, CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=88975448
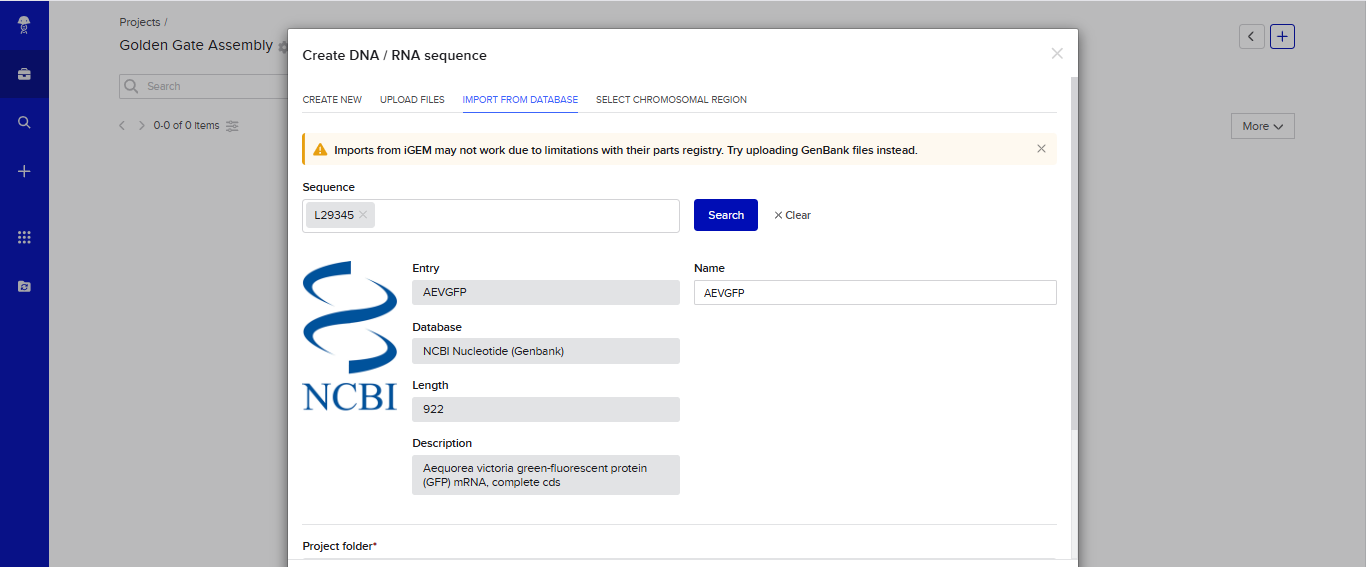
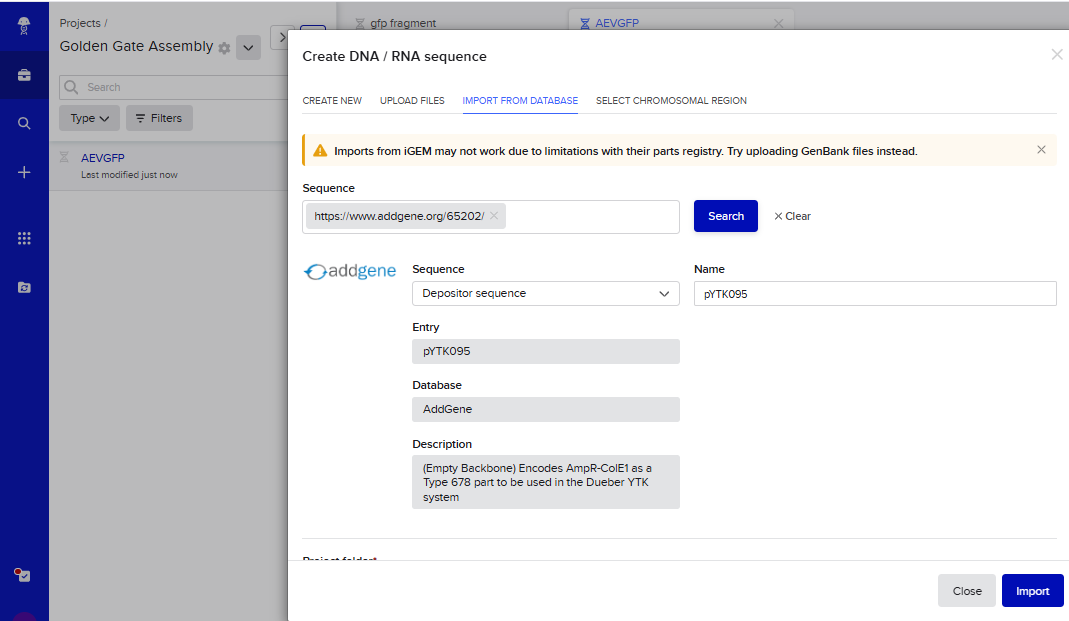
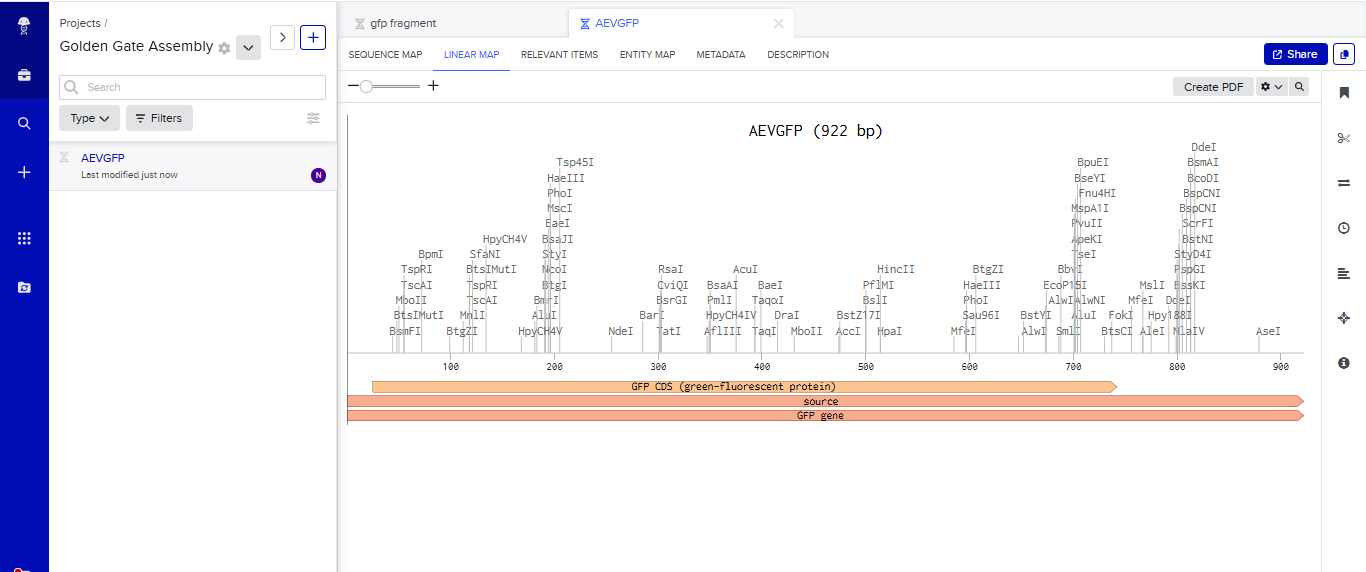
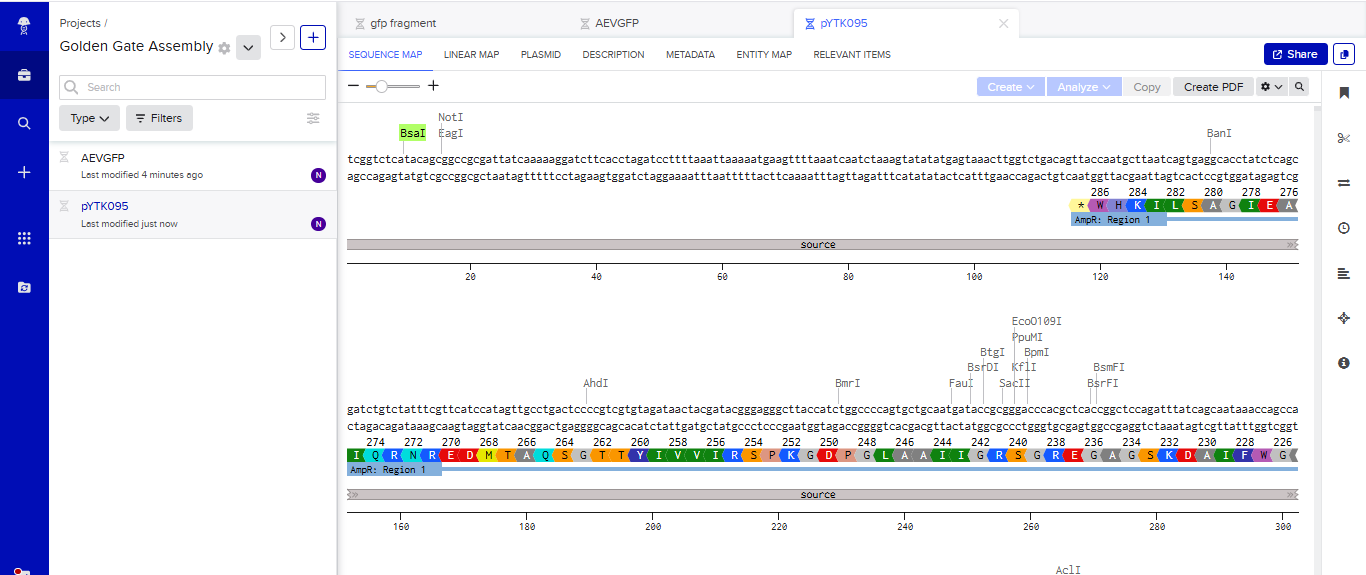
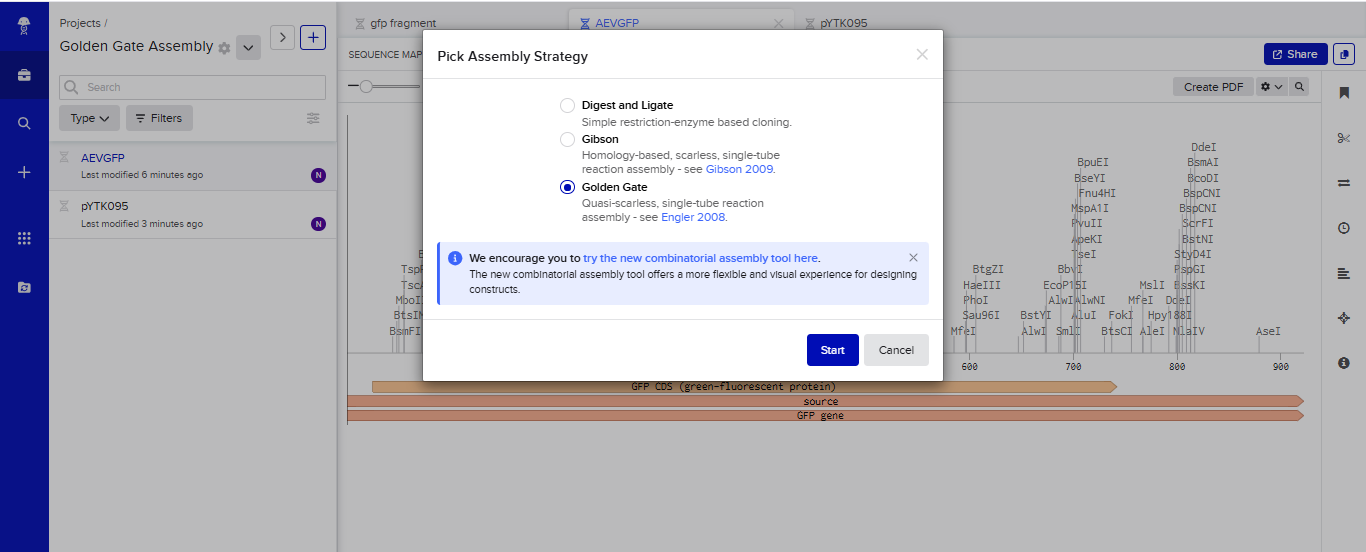
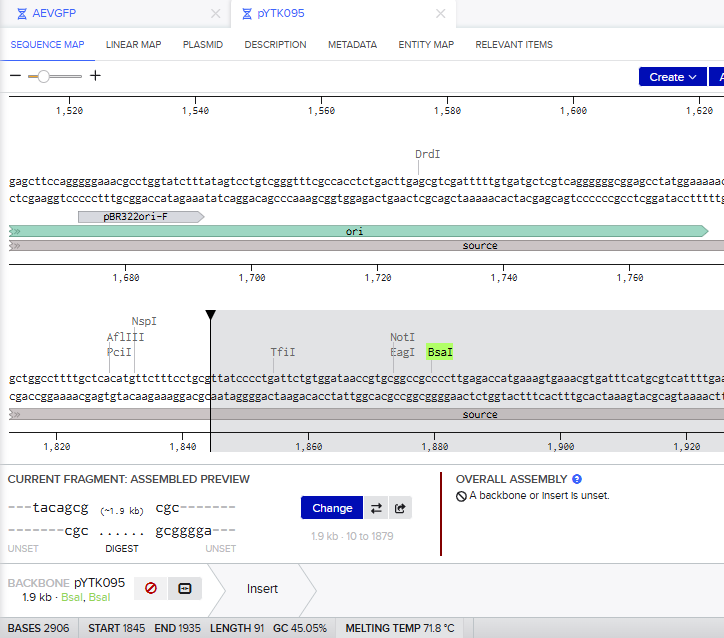
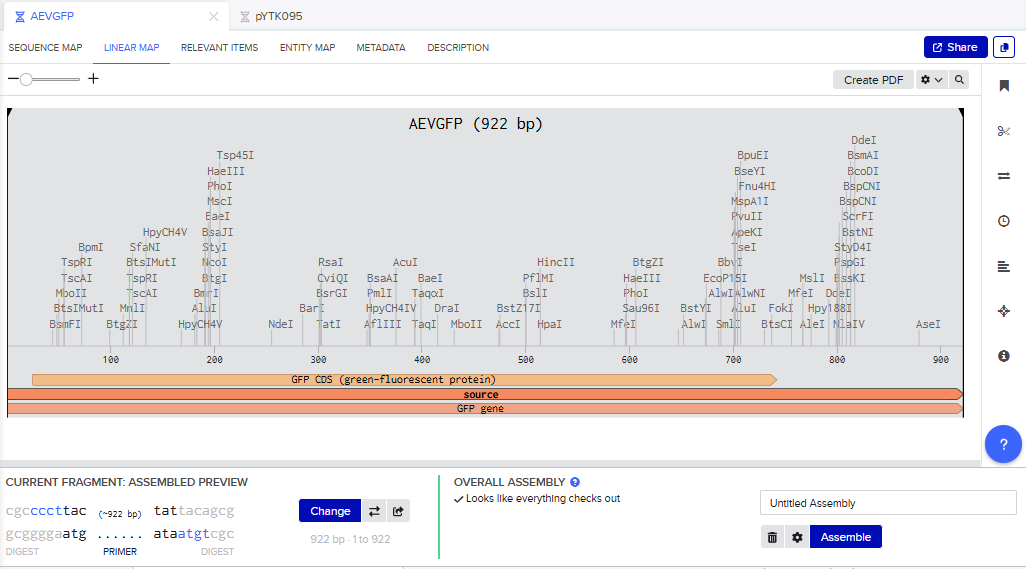
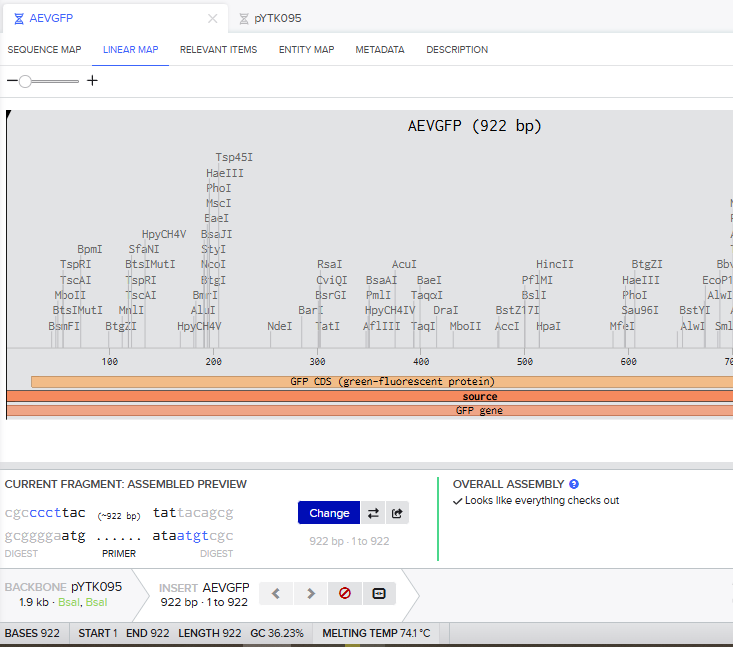
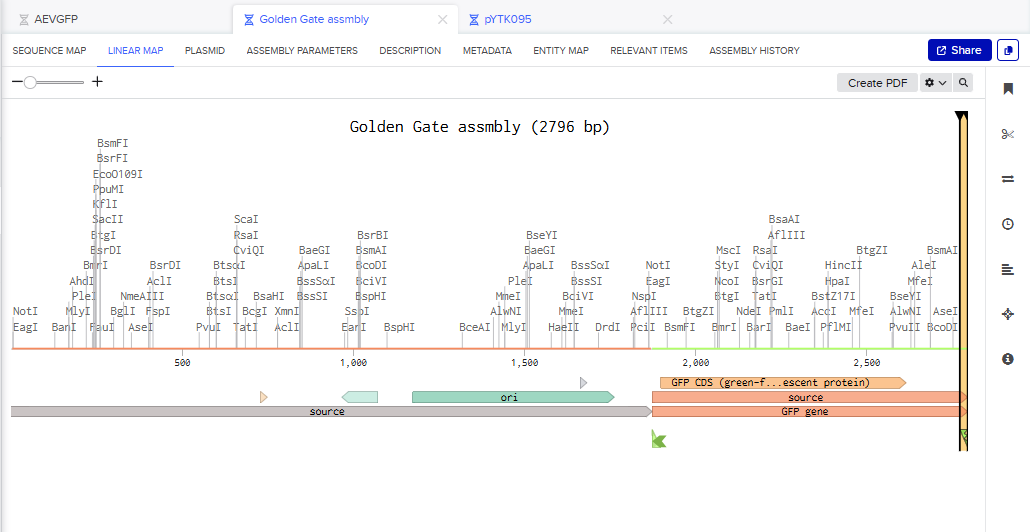
- I tried to model the Golden Gate assembly in Benchling. I created a new project in Benchling called “Golden Gate Assembly”. I used Aequorea victoria green fluorescent protein (GFP) mRNA complete cds, which I obtained from NCBI as the insert, and the pYTK095 plasmid from Addgene (https://www.addgene.org/65202/), which encodes AmpR-ColE1 as the backbone. I used Benchling’s Molecular Biology tools to simulate the Golden Gate Assembly. Finally, I validated the construct, confirming the proper insertion of the green fluorescent protein.
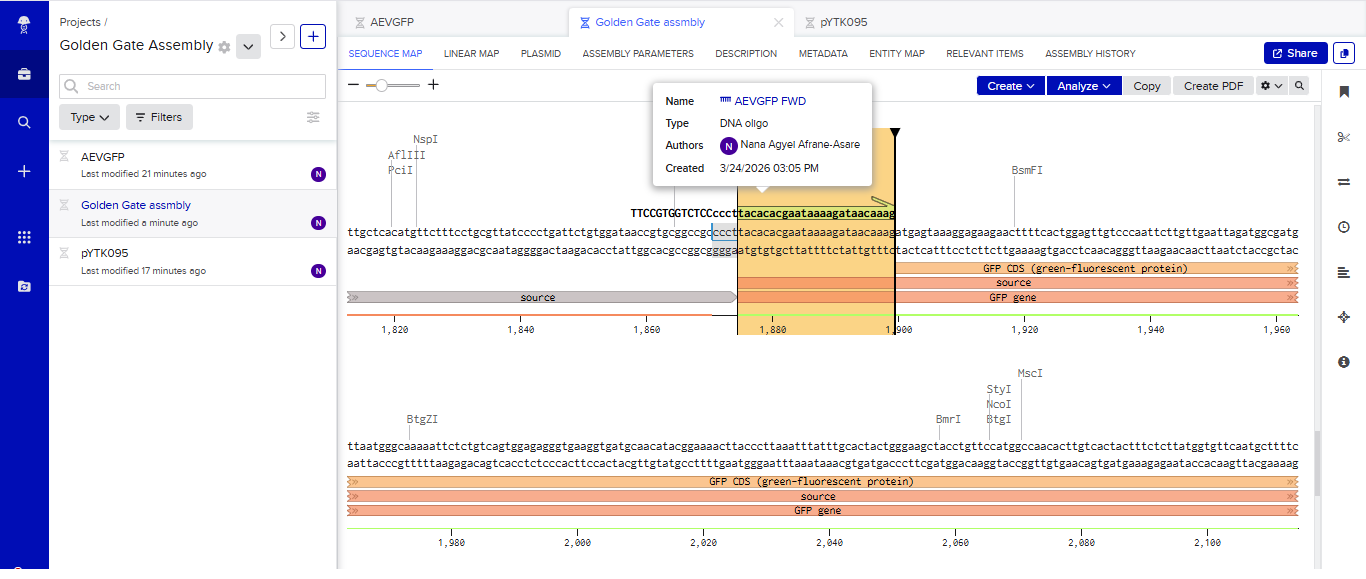
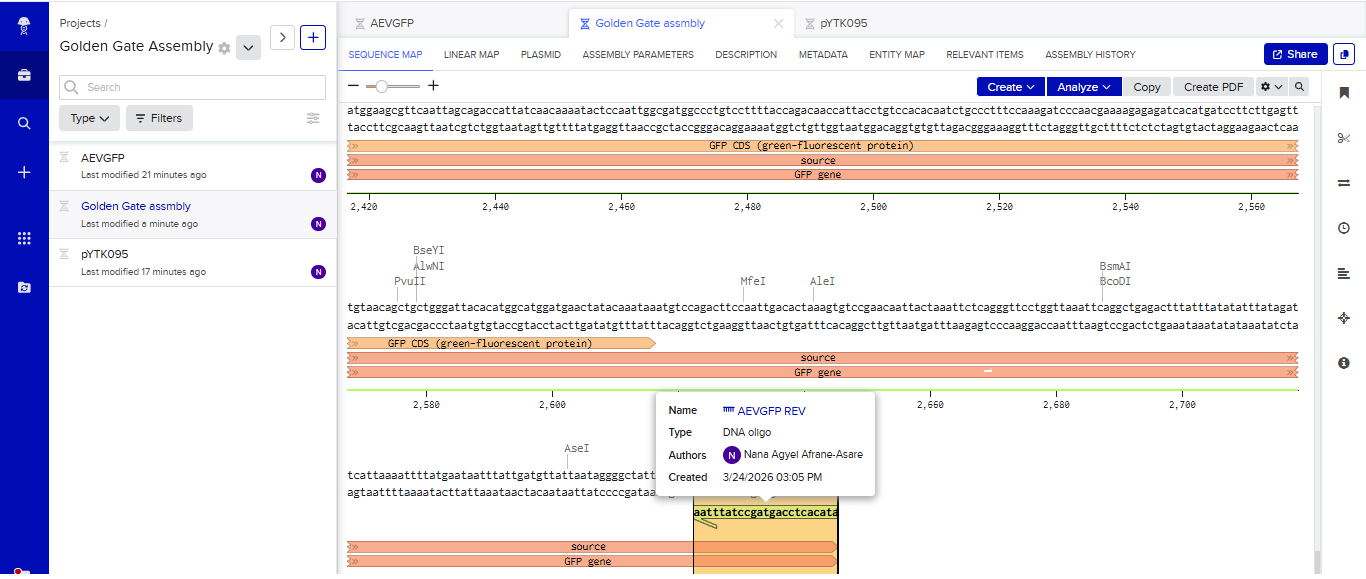
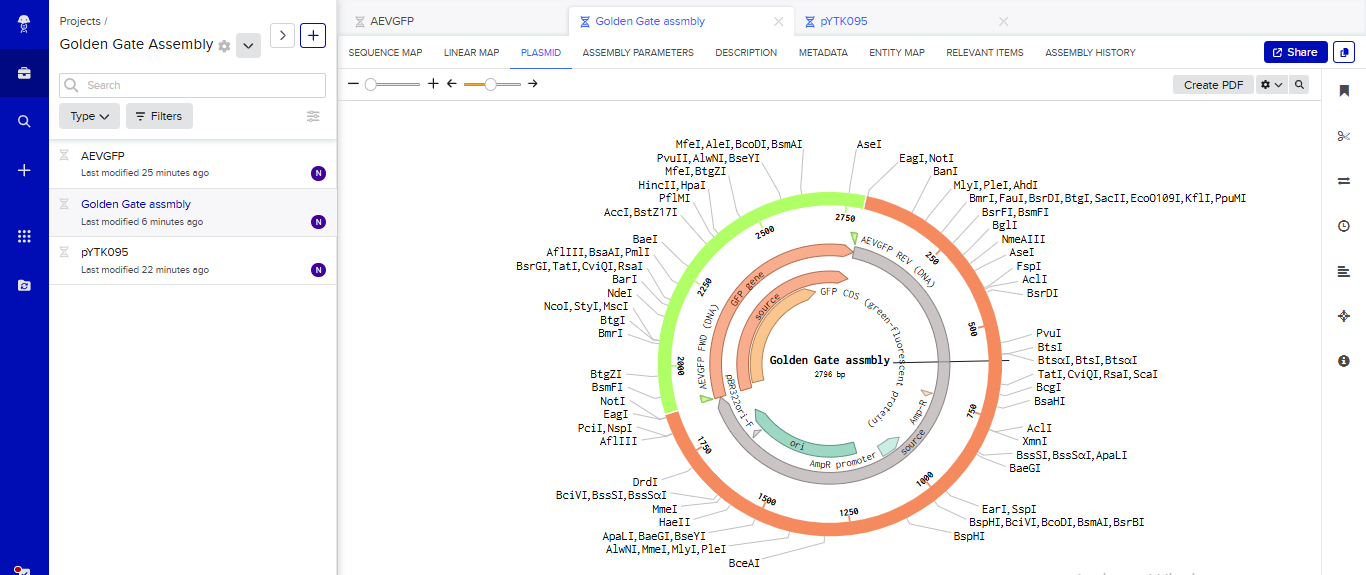
Assignment: Asimov Kernel
Question 1 & 2
I created a new repository named “William & Mary_Nana Agyei” and created a new notebook titled “Nana Agyei’s HW 6 Entry" to document my use of Asimov Kernel.
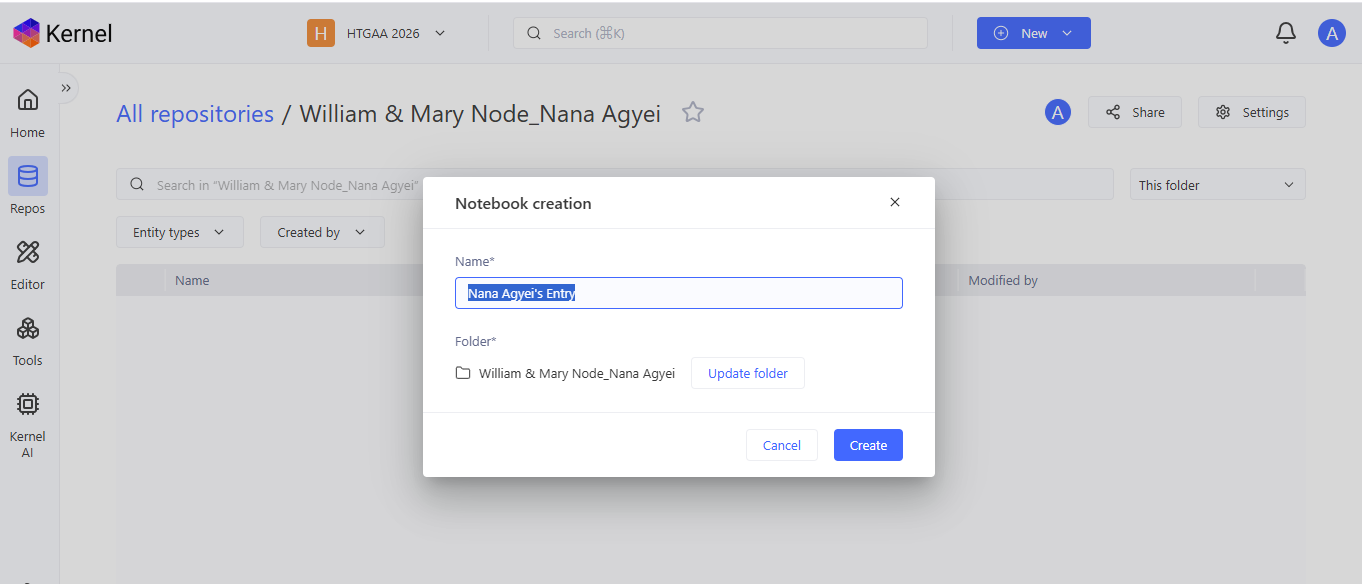
Question 3
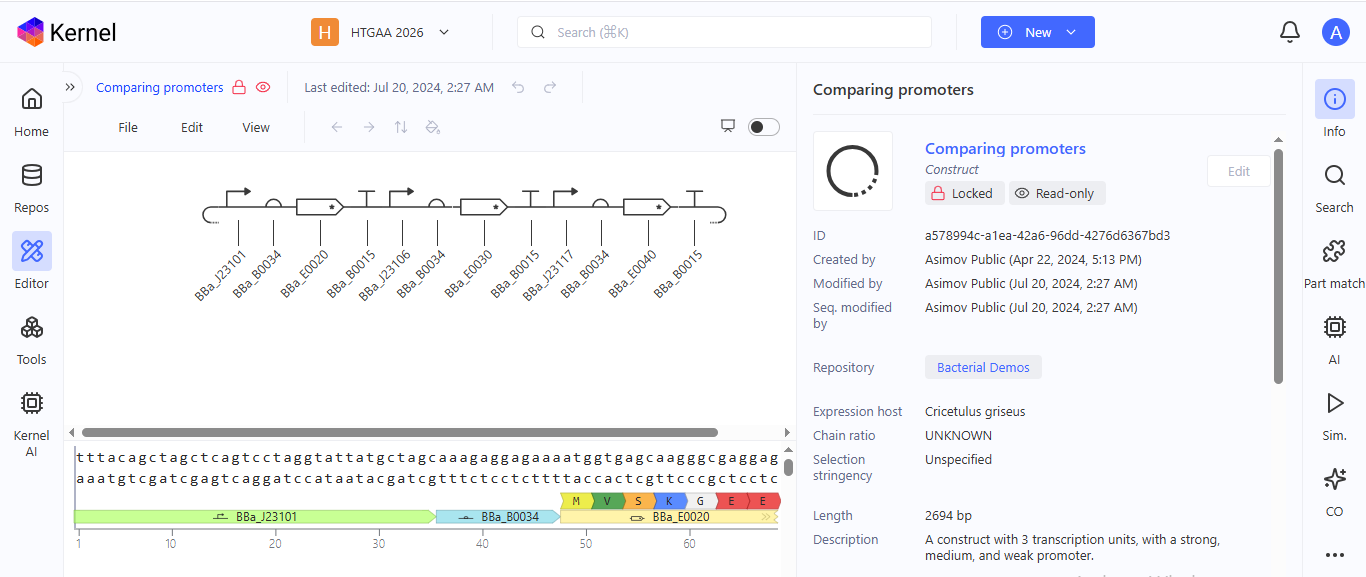
I explored the Comparing promoters, Repressilator, and J23117 Promoter constructs in the Bacterial Demos Repository to familiarize myself with the parts of the various constructs and how they work. The UI of Asimov Kernel is pretty easy to use and visually appealing, making the exploration and tests easy and interesting.
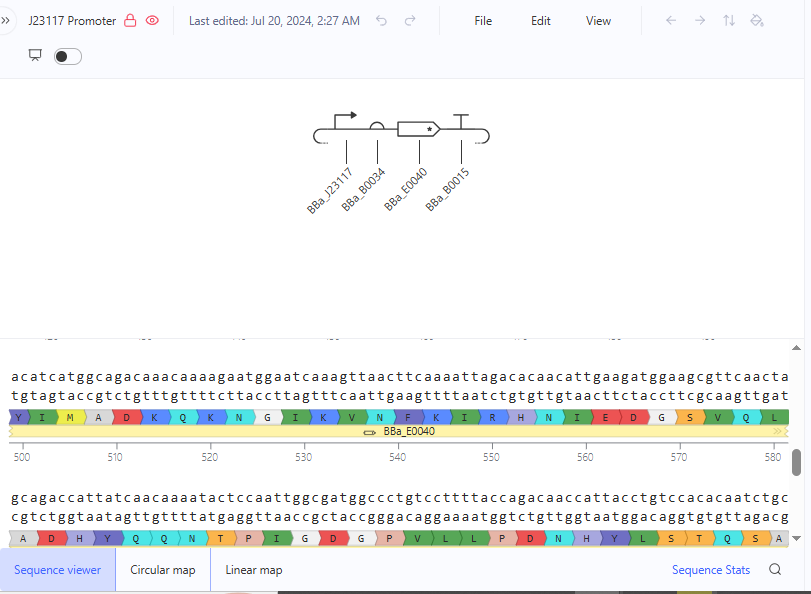
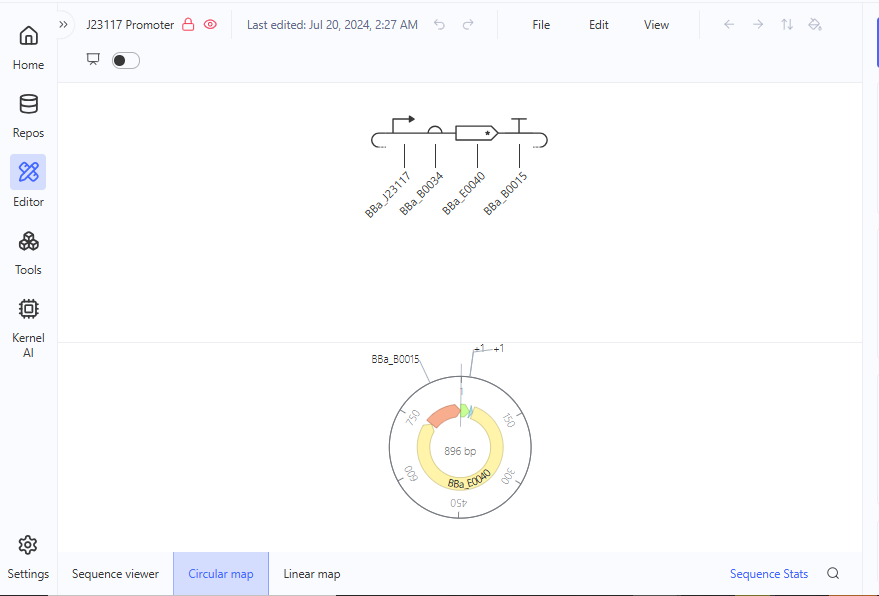
Question 4
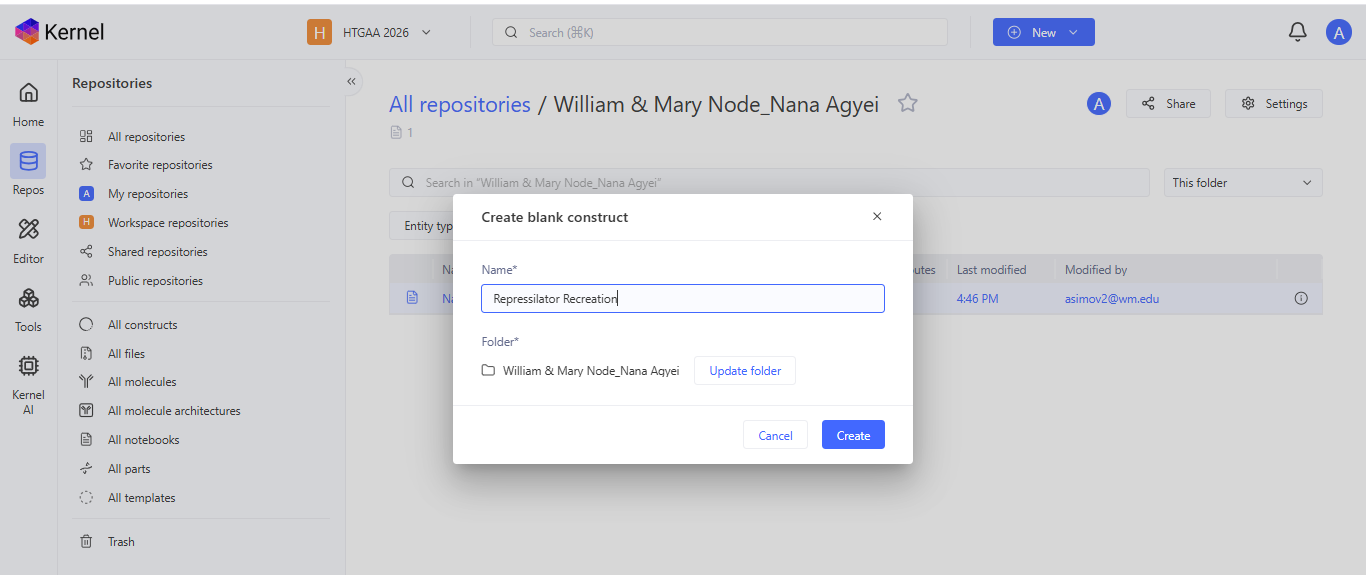
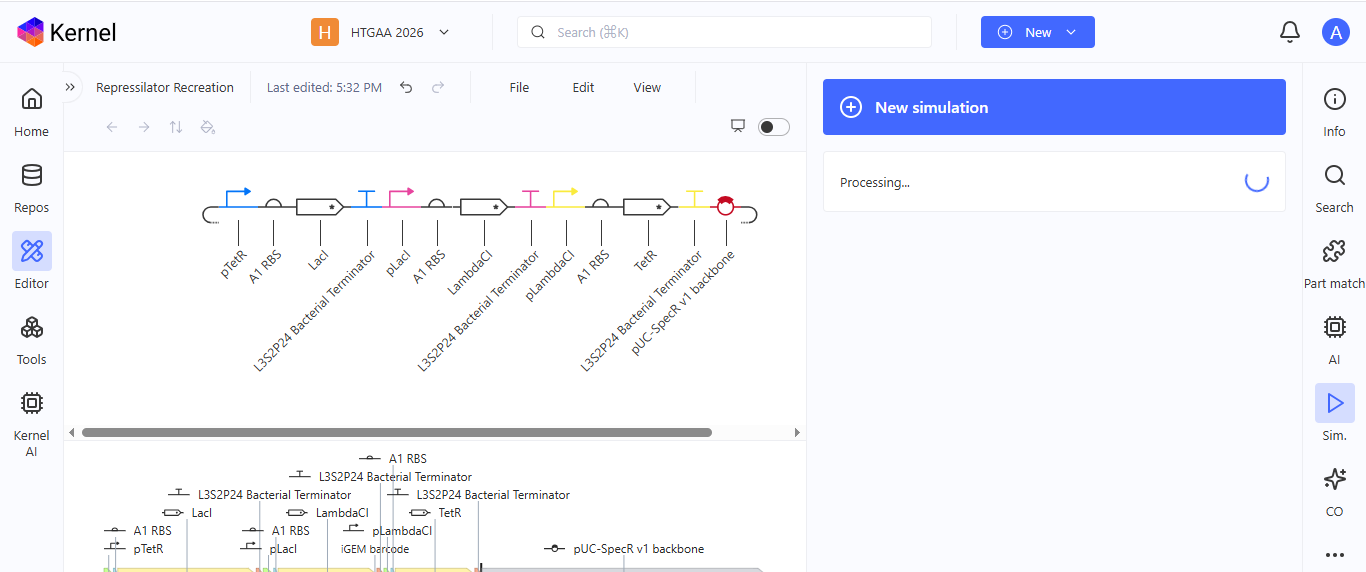
I noted down all the parts used to create the Repressilator in the Bacterial Demos repository and created a blank construct to recreate the Repressilator, which I named “Repressilator recreation.” I used the search function to find all the parts I had noted down.
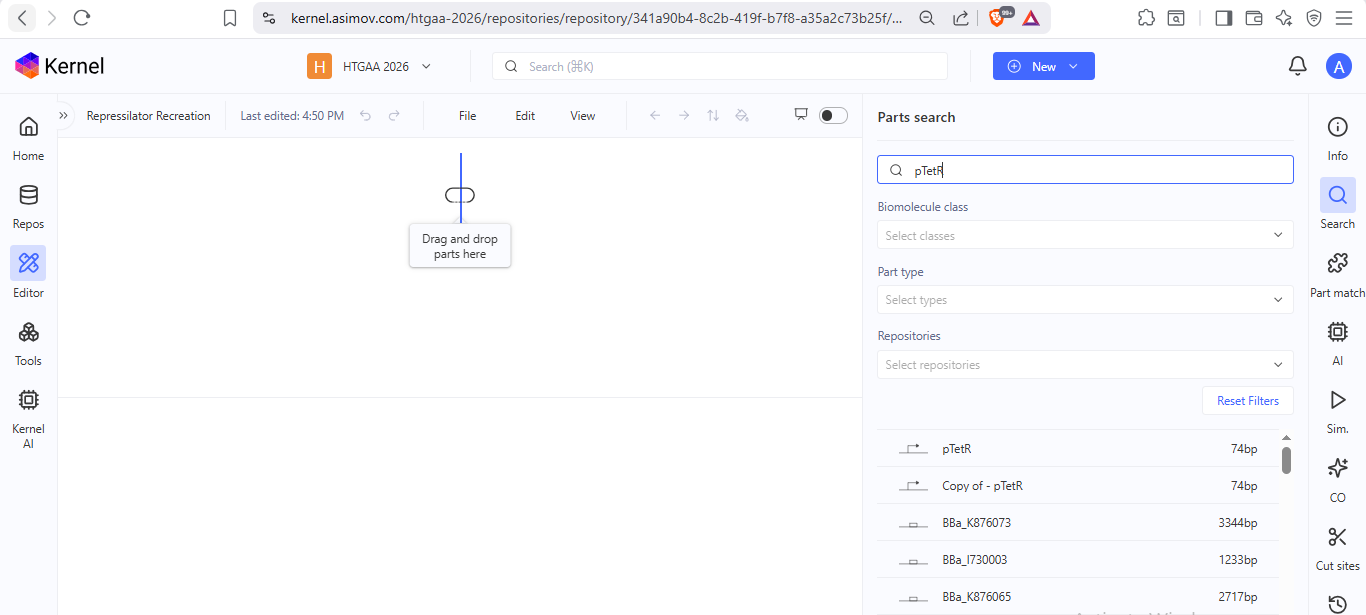
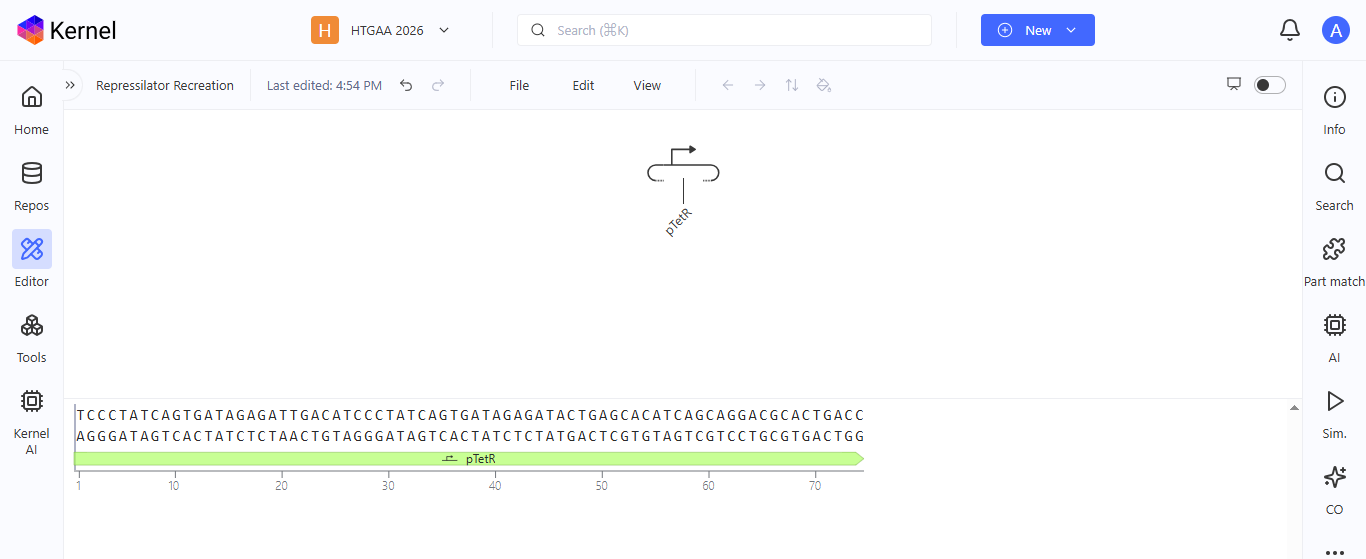
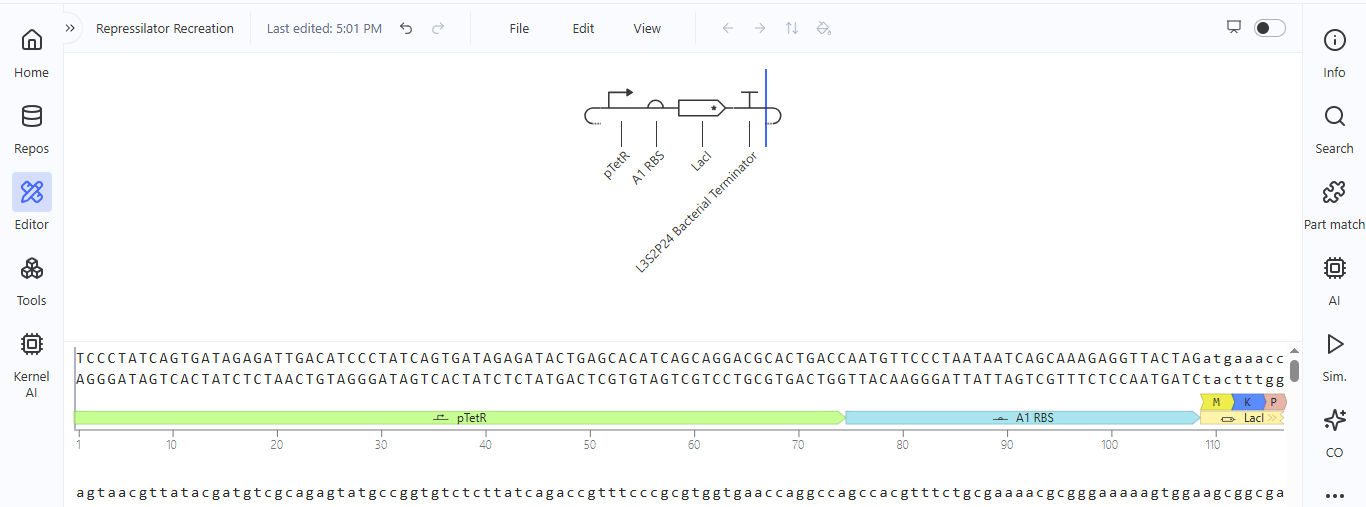
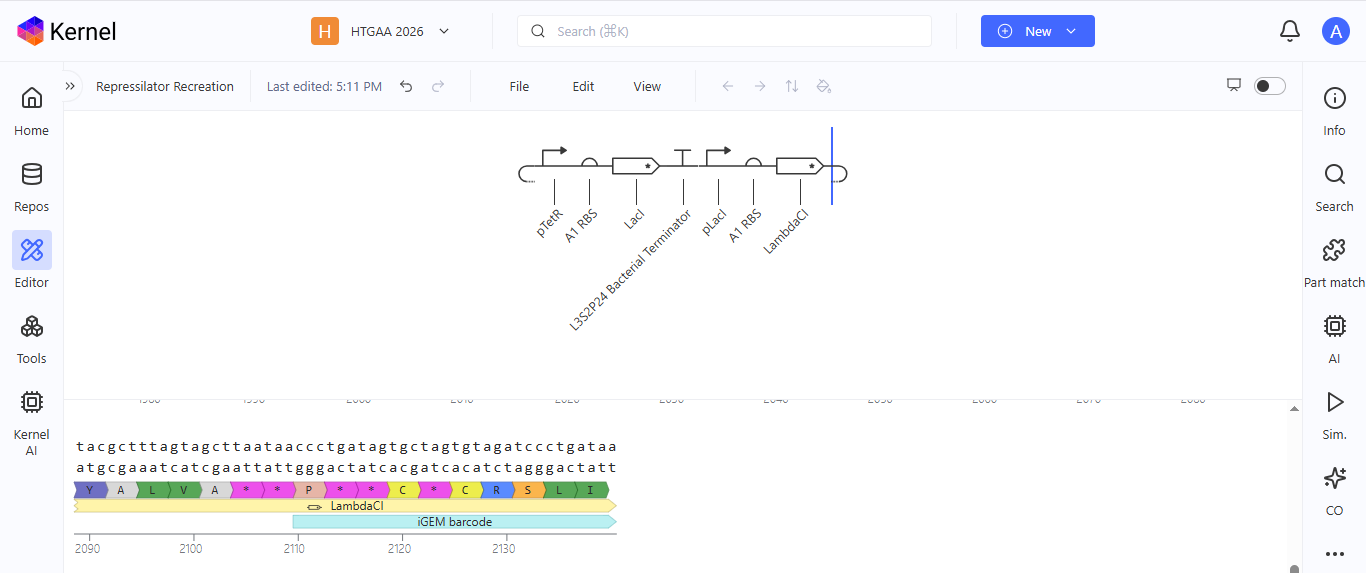
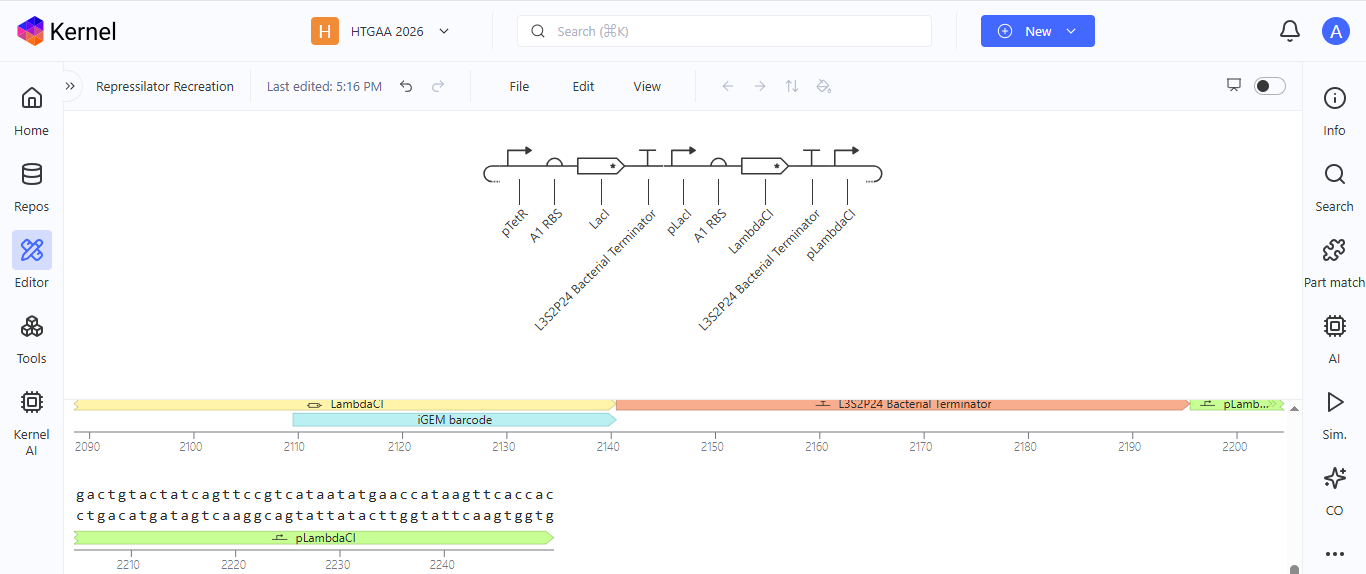
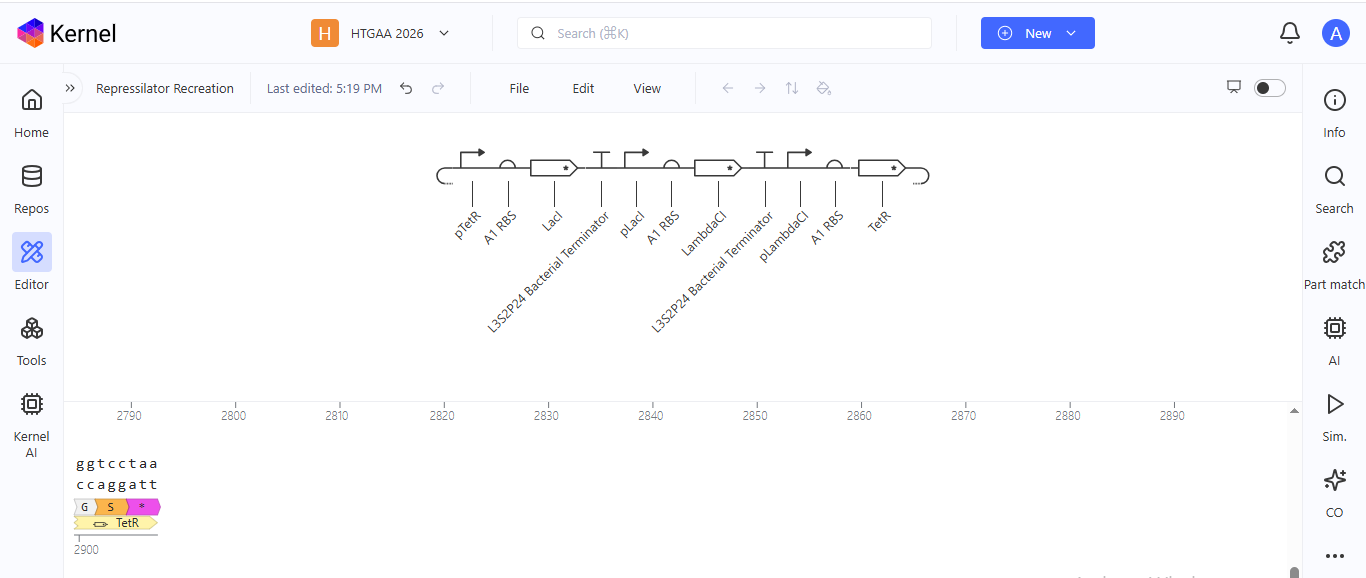
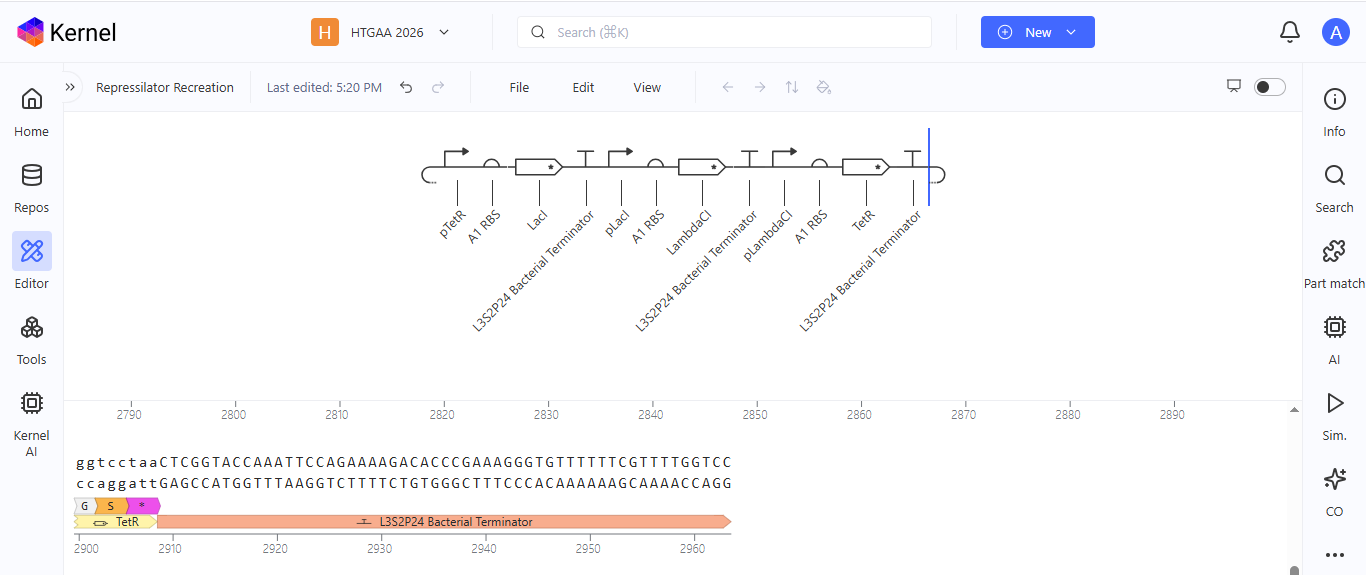
I ran into a problem while searching for the pUC-SpecR v1 backbone. I could only find the pUC-SpecR v2 backbone, so I just copied the sequence for the pUC-SpecR v1 backbone from the Repressilator device and pasted it in the appropriate position of my construct.
After recreating the Repressilator, I tried to color the different components of the circuits
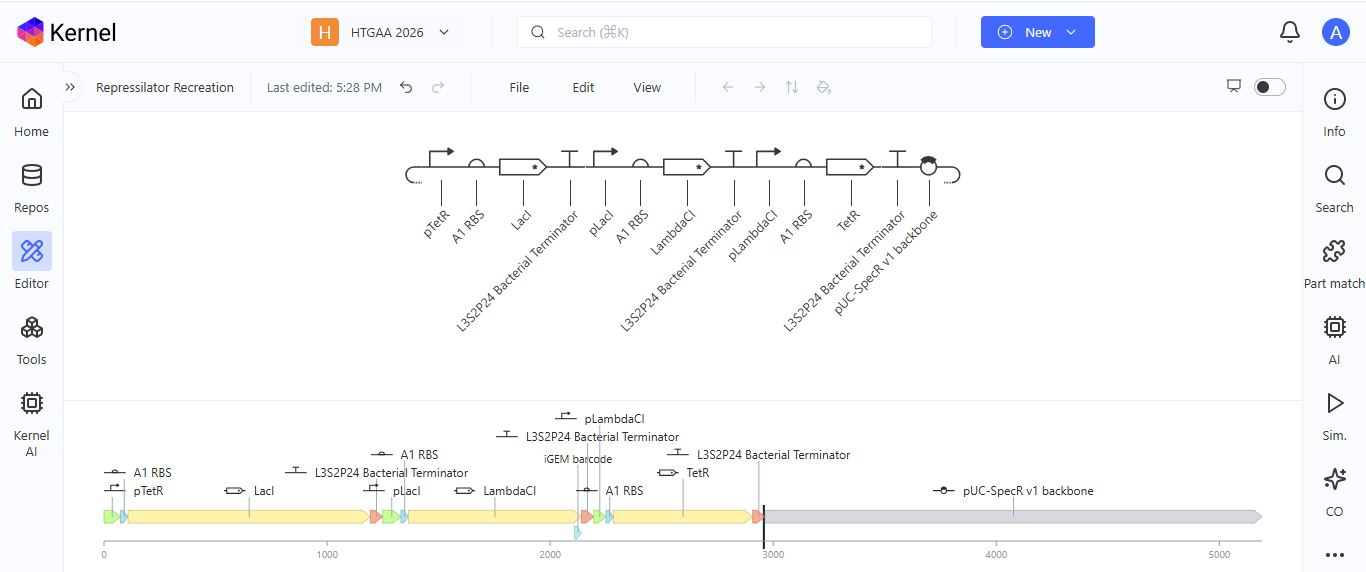
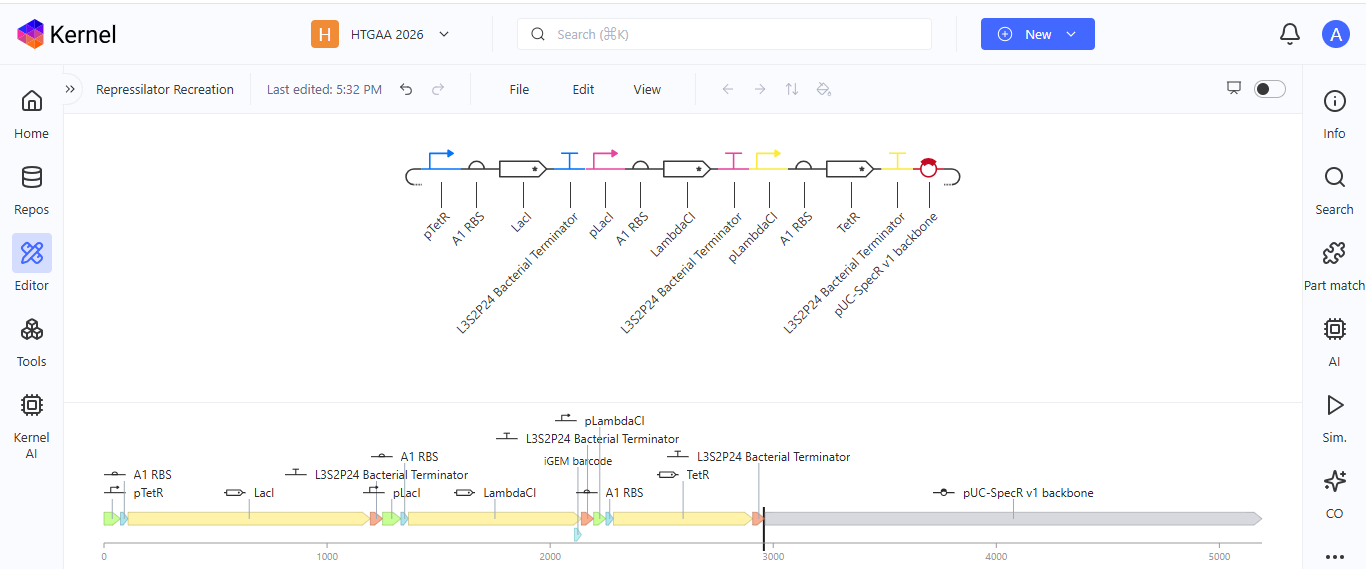
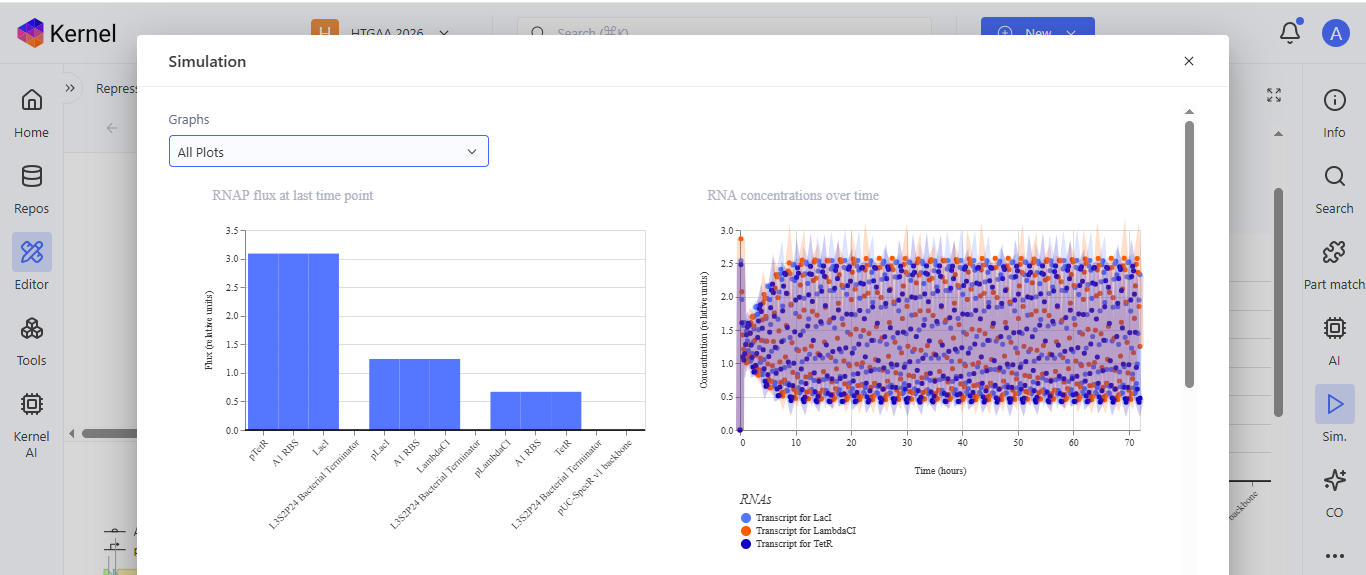
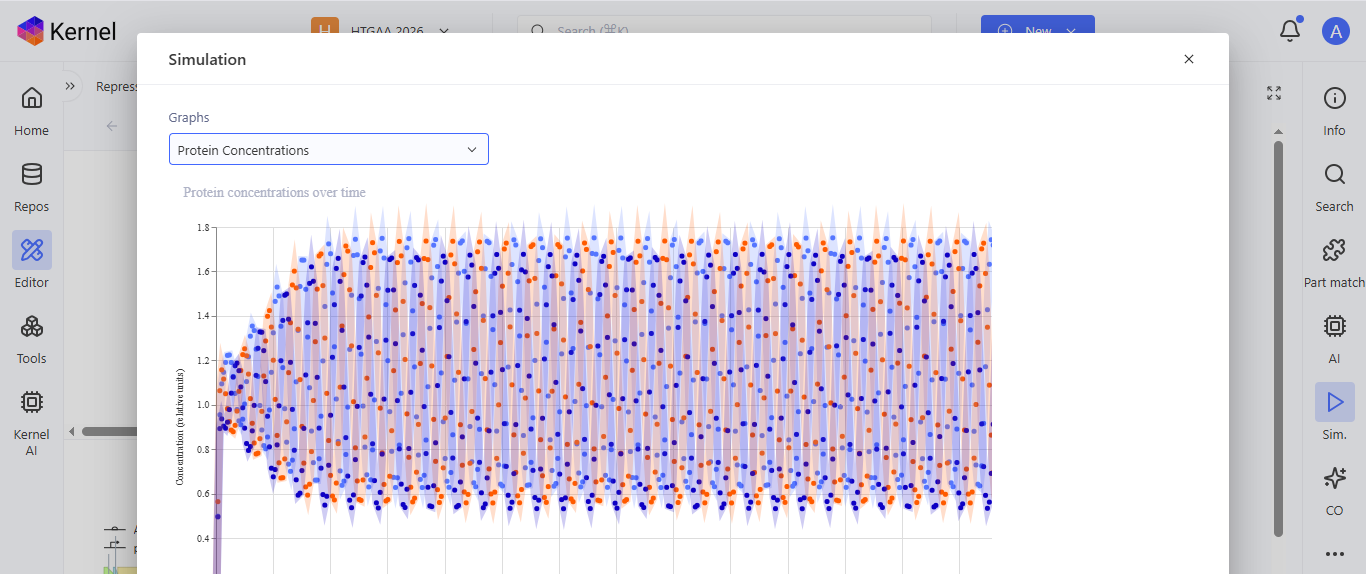
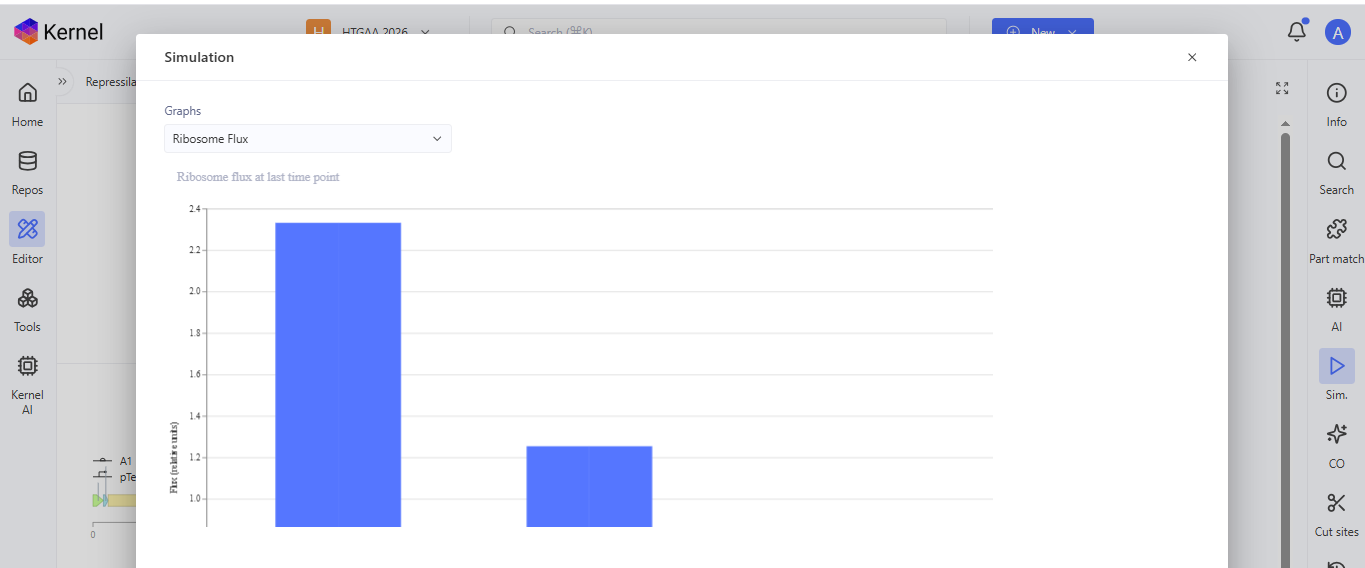
I run a simulation on my recreation of the Repressilator, and it seemed to work as expected
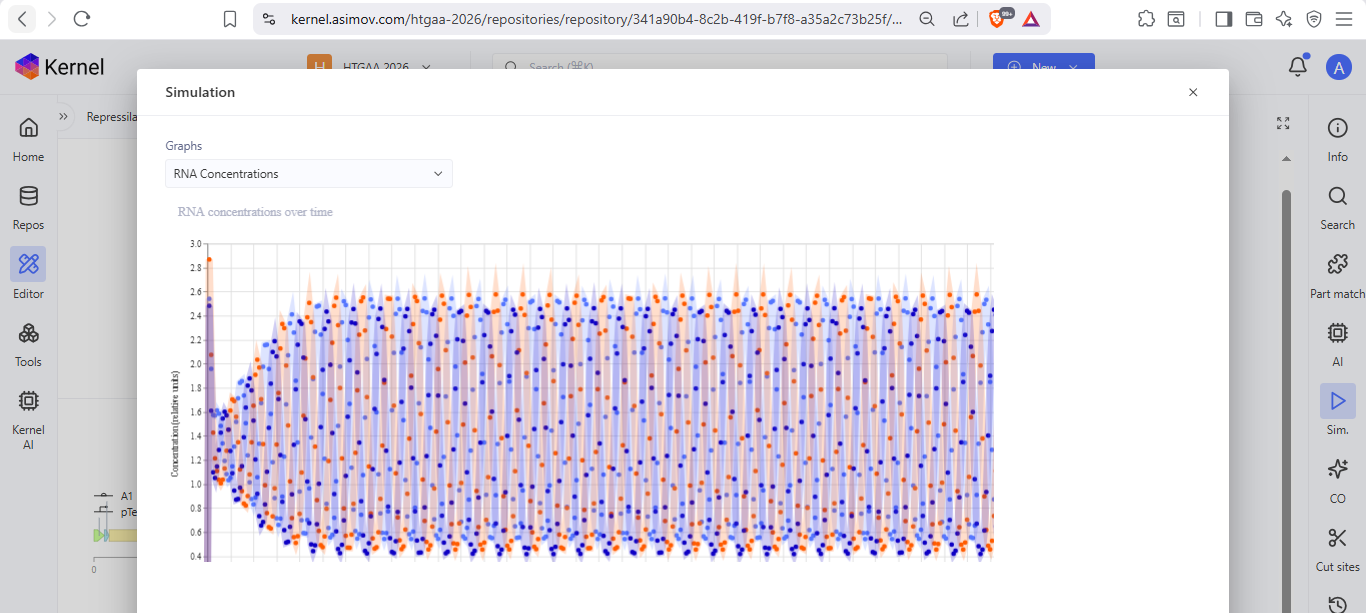
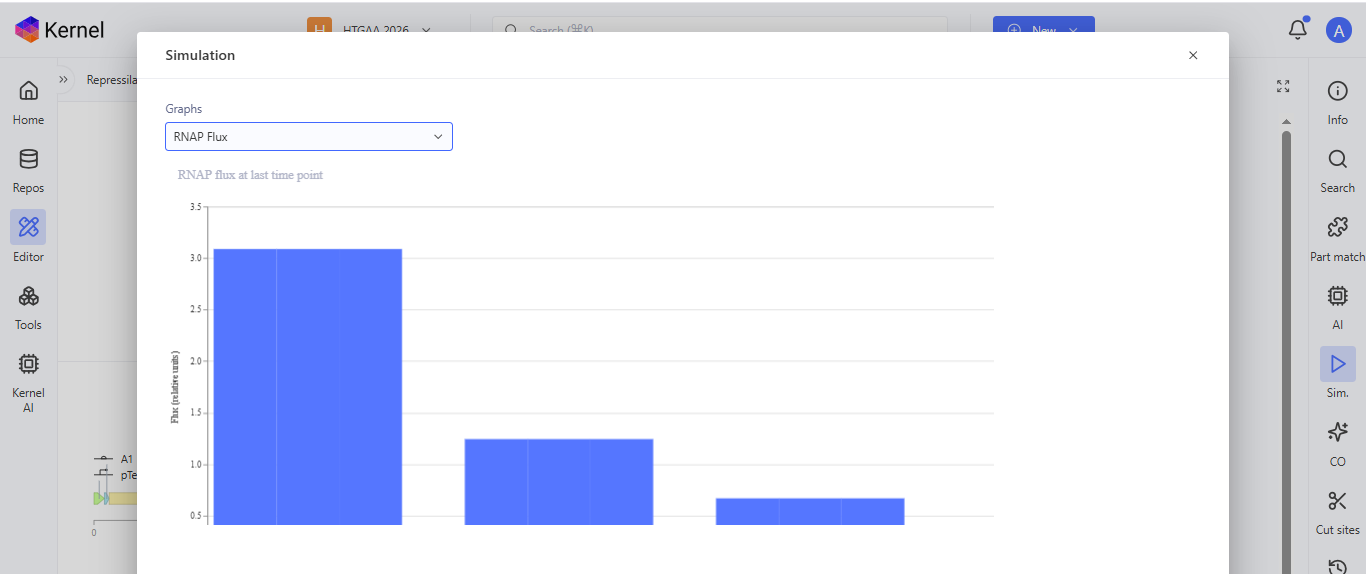
Question 5
I tried to build my own constructs using the parts in the Characterized Bacterials Part repository.
Construct 1. Luciferase gene expression
I create a genetic circuit construct to control the expression of the Luciferase gene. I based it on the luciferase gene construct I built in Benchling during week 2.
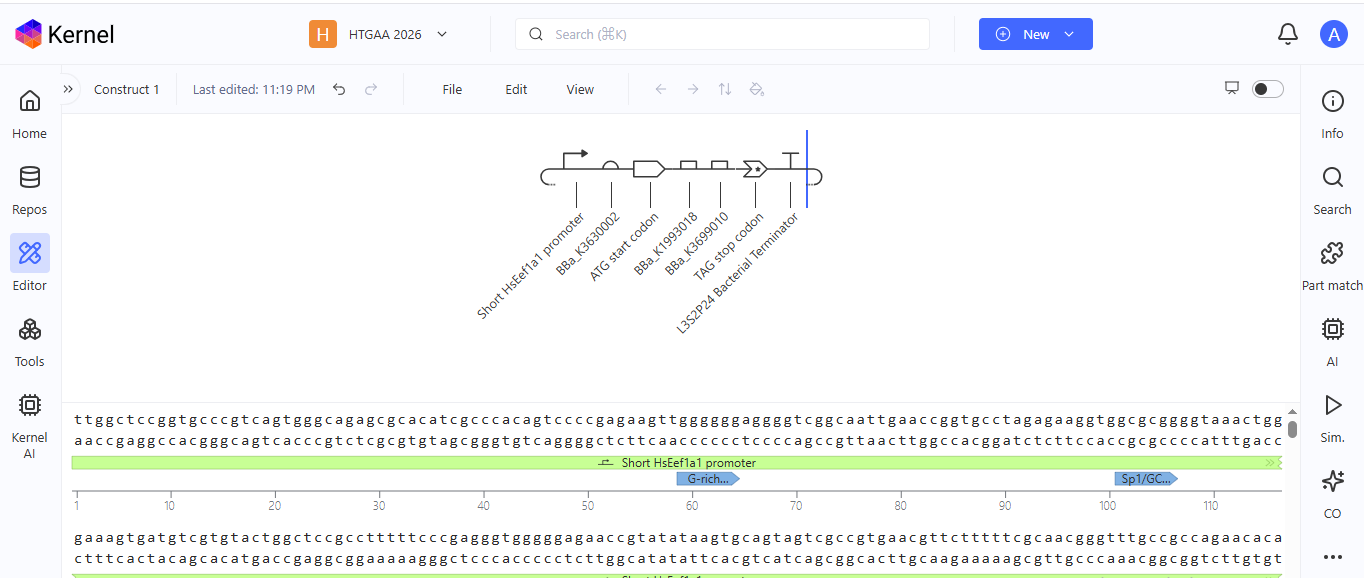
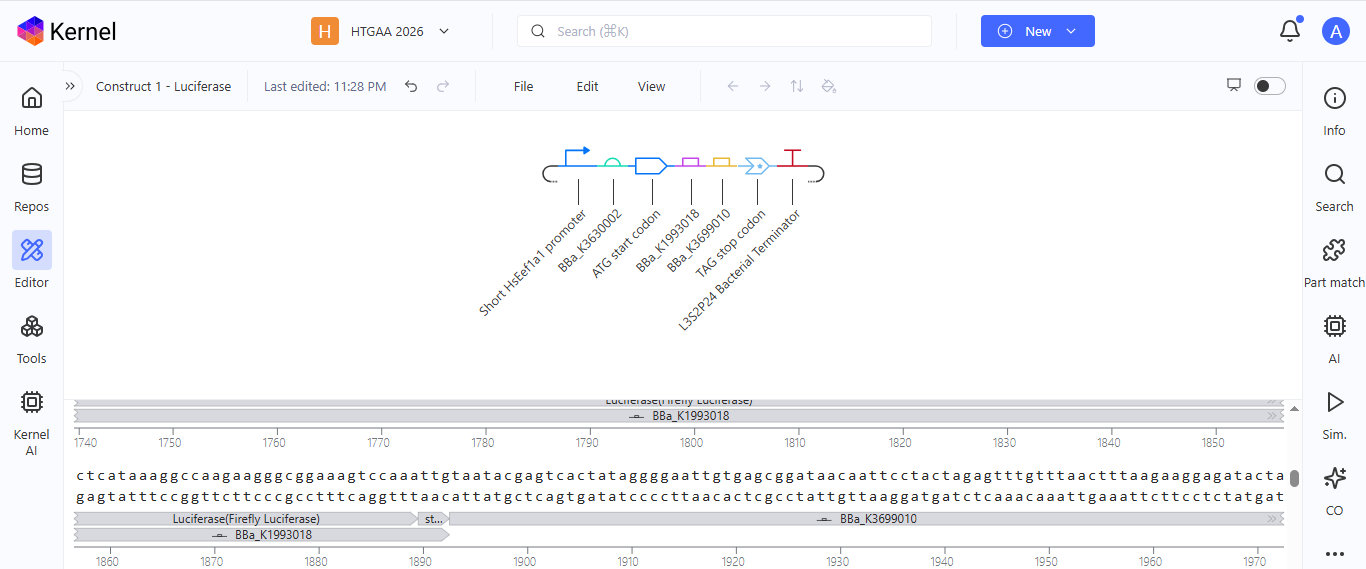
Based on the results for construct 1 below, the circuit is working properly, with each component turning on as expected.
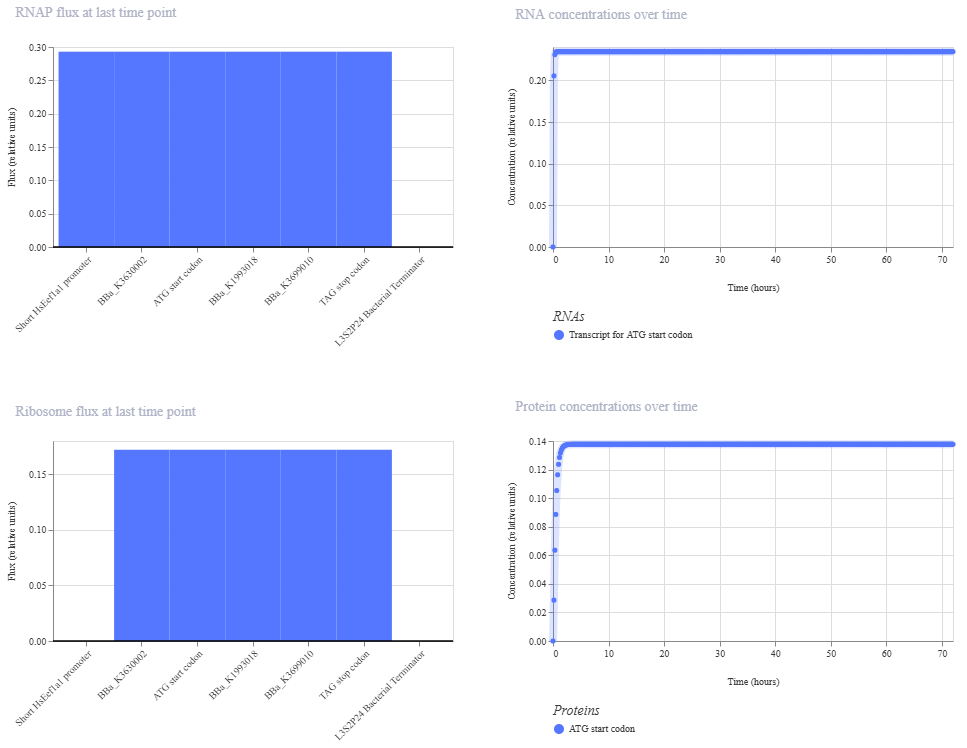
Construct 2. Inducible Arabinose GFP reporter
I created a genetic circuit construct that would express green fluorescent protein (GFP) when arabinose is present.
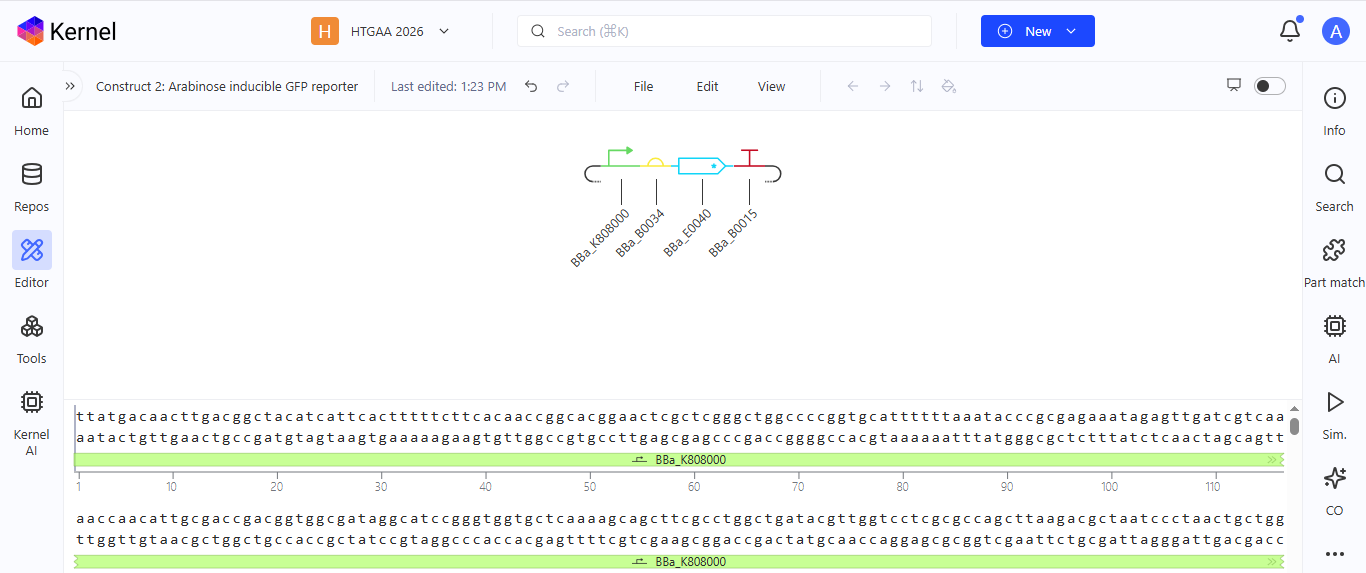
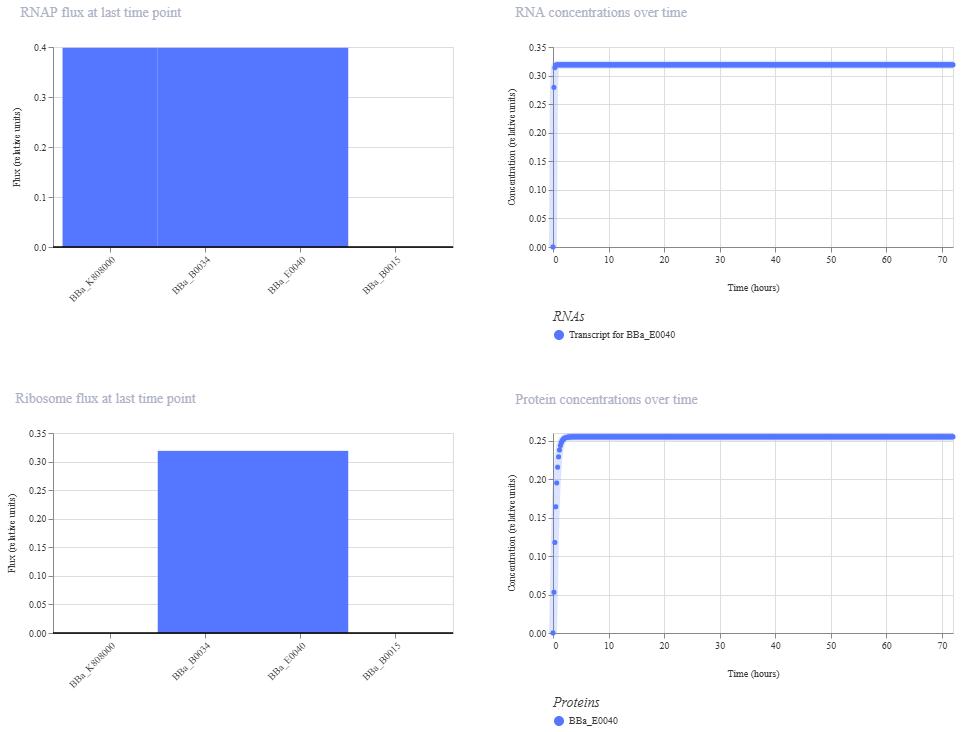
Based on the results for construct 2, the genetic circuit is working properly. All its components turn on, and GFP is produced, with its concentration increasing over time before stabilizing at a steady level.
**Construct 3. Constitutive expression of the GFP gene
I created a genetic circuit construct that would constantly express green fluorescent protein (GFP) without any external input needed.
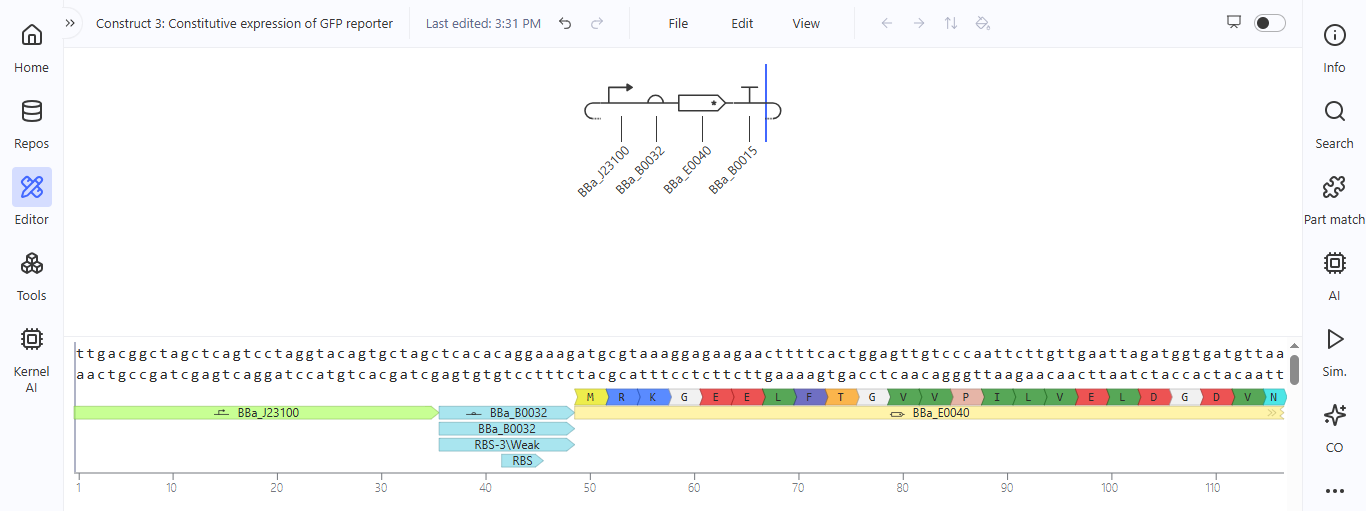
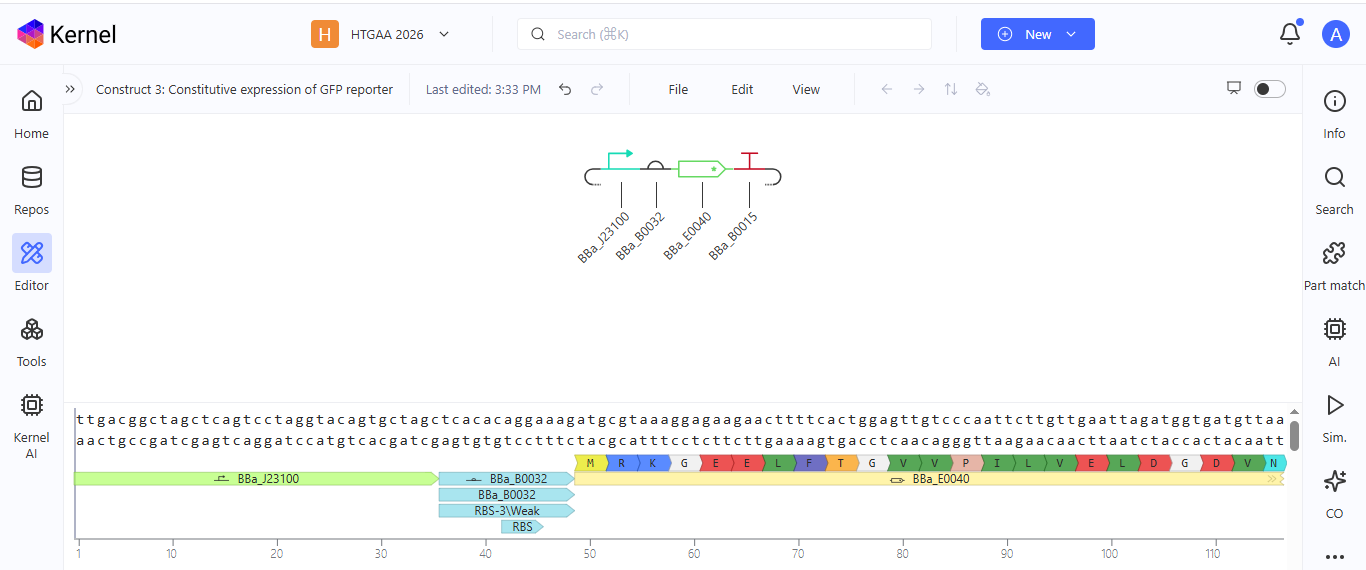
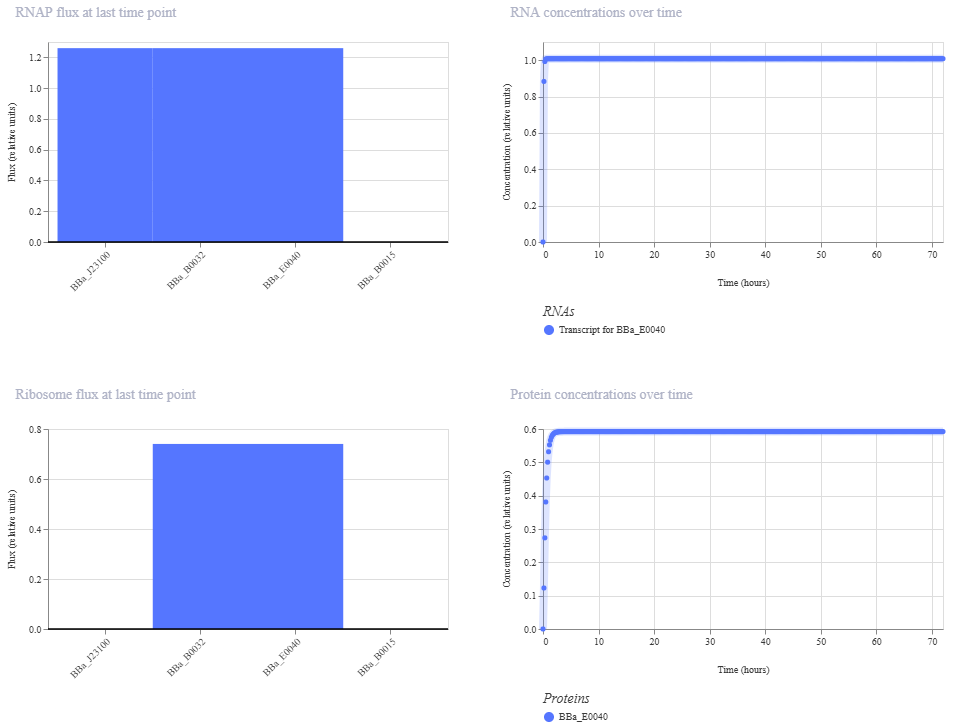
Based on the results for construct 3, the genetic circuit is working properly. All its components turn on, and GFP is produced.
Reference
3CR Bioscience. (n.d.). What is a PCR master mix? Components and functions. Retrieved March 13, 2026, from https://3crbio.com/blog/what-is-a-pcr-master-mix/
National Institute of Justice. (2023, July 31). Primer annealing temperature. In DNA amplification for forensic analysts. https://nij.ojp.gov/nij-hosted-online-training-courses/dna-amplification/overview/primer-design/primer-annealing-temperature
New England Biolabs. (n.d.). Golden Gate assembly.Retrieved March 20, 2026, from https://www.neb.com/en/applications/cloning-and-synthetic-biology/dna-assembly-and-cloning/golden-gate-assembly?
New England Biolabs. (n.d.). Restriction enzyme digestion. Retrieved March 14, 2026, from https://www.neb.com/en/applications/cloning-and-synthetic-biology/dna-preparation/restriction-enzyme-digestion
New England Biolabs. (n.d.). Phusion® High-Fidelity PCR Master Mix with HF buffer (M0531). Accessed on March 13, 2026, from https://www.neb.com/en/products/m0531-phusion-high-fidelity-pcr-master-mix-with-hf-buffer
QB3-Berkeley. (n.d.). Gibson assembly. QB3 Berkeley Lab Fundamentals Bootcamp Manual.Retrieved on March 15, 2026, from https://qb3.berkeley.edu/education/lab-fundamentals-bootcamp/manual/cloning/gibson-assembly/
QIAGEN. (n.d.). PCR conditions. Retrieved March 13, 2026, from https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/introduction/pcr-conditions
Starčič Erjavec, M. (2020). Annealing Temperature of 55°C and Specificity of Primer Binding in PCR Reactions. In Synthetic Biology - New Interdisciplinary Science. IntechOpen. https://doi.org/10.5772/intechopen.85164