Week 2: Bioart & Genetics
Name: Negin Aghayan
Topic: From Reading DNA to Designing Antibiotic Resistance
Part 1 & 2: DNA Analysis and Gel Art
1.1. Virtual Digest of Lambda Phage DNA
In this lab, I utilized Benchling to simulate a restriction digest of the Lambda DNA (48,502 bp). Lambda DNA is a classic substrate in molecular biology because its entire sequence is known, making it the perfect “map” for testing Type II restriction enzymes like EcoRI, HindIII, and BamHI. By simulating these digests, I visualized how specific palindromic sequences are recognized and cleaved, producing a unique “DNA fingerprint.”
1.2. Verification of Enzymatic Cleavage Patterns
Before proceeding to the creative design phase, I performed a standard benchmark digestion of the Lambda phage genome. This step is crucial to verify the specificity of Type II restriction enzymes and their respective recognition sites across the linear DNA molecule.
Establishing a reliable DNA ladder and reference digest is the foundation of any molecular cloning project, a practice consistent with rigorous laboratory standards.
Enzymes Characterized:
- EcoRI, HindIII, BamHI: Standard enzymes used for mapping.
- KpnI, EcoRV, SacI, SalI: Additional Type II enzymes to observe diverse fragment distributions.
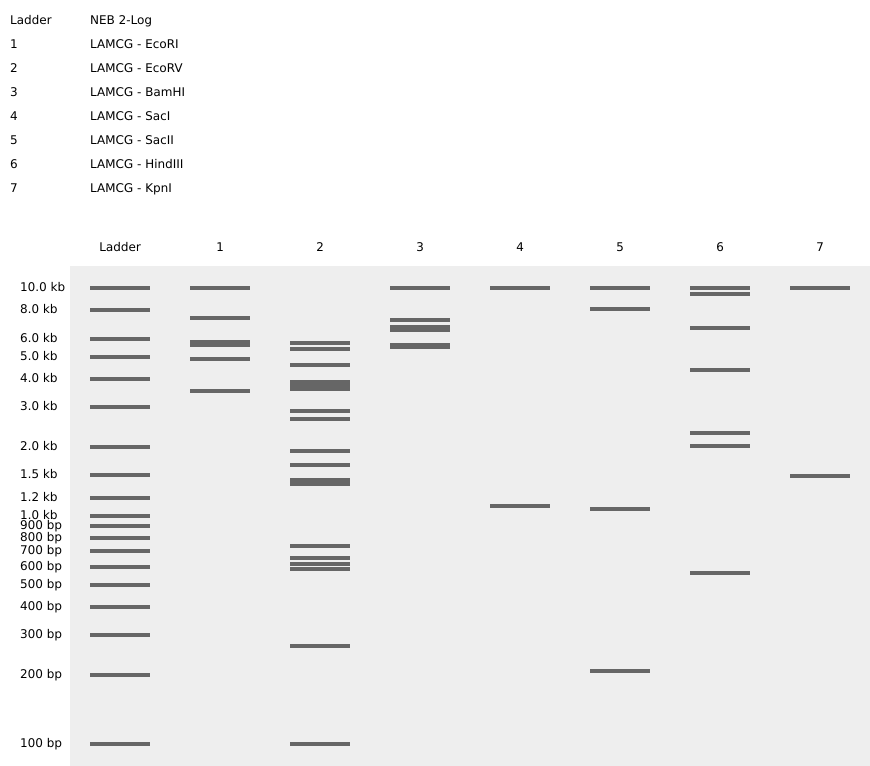
Figure 1: Virtual electrophoresis showing individual digestion patterns for the seven required enzymes.
2.1. Gel Art: “The DNA Butterfly” - A 10-Lane Symmetrical Exploration
As a microbiology student with experience in molecular techniques, I aimed to bridge the gap between rigorous genomic analysis and aesthetic expression. The butterfly is more than just a visual choice; in the context of microbiology and biotechnology, it represents metamorphosis—the profound phenotypic transformation driven by underlying genetic information. This mirrors the Central Dogma, where static digital sequences are “transformed” into functional biological entities.For this assignment, I designed a 10-lane symmetrical pattern titled “The DNA Butterfly”, utilizing the 48.5 kb Lambda DNA as my molecular canvas.
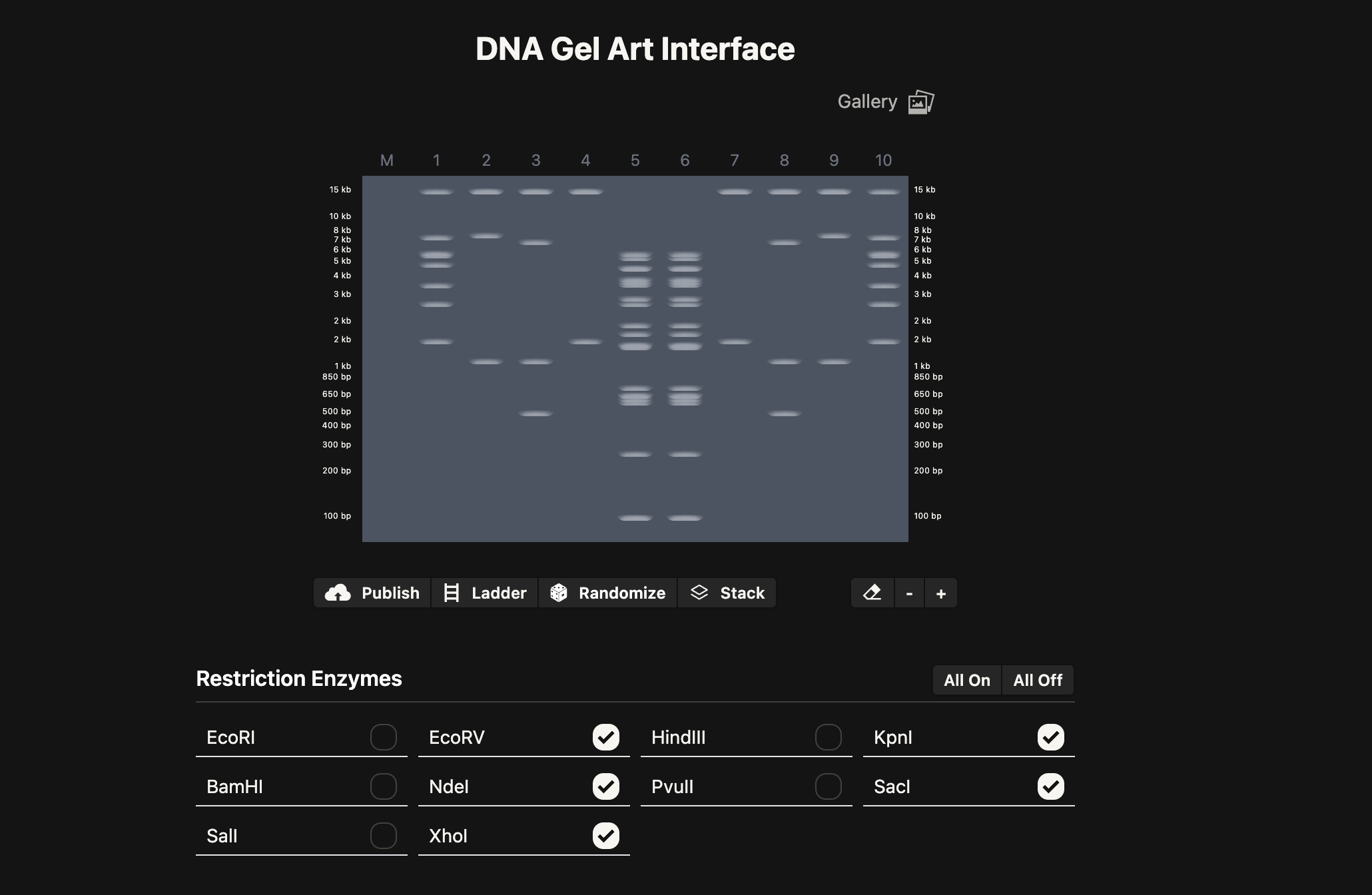
The Design Protocol (10-Lane Symmetry): To achieve a high-fidelity mirror image, I carefully selected Type II restriction enzymes based on their cleavage frequency to create a balanced visual weight across the gel:
- Lanes 5 & 6 (The Axis of Symmetry): Digested with EcoRV. These lanes form the “body” of the butterfly. The high density of bands in the mid-range (approx. 3-6 kb) creates a solid central vertical axis.
- Lanes 4 & 7 (Structural Framework): Digested with KpnI. These lanes provide the “scaffolding” for the wings, featuring a prominent high-molecular-weight band at ~17 kb, representing the strong upper edges of the butterfly’s wings.
- Lanes 3/8 & 2/9 (Internal Wing Textures): Utilizing double digests (SacI + SalI and SacI + XhoI). By combining these enzymes, I generated specific low-molecular-weight fragments (1-3 kb) that simulate the intricate, delicate patterns found on the interior of a butterfly’s wing.
- Lanes 1 & 10 (External Wing Margin): A double digest of EcoRI + KpnI. These lanes frame the entire artwork, providing a diverse range of band sizes that define the outer boundaries of the biological form.
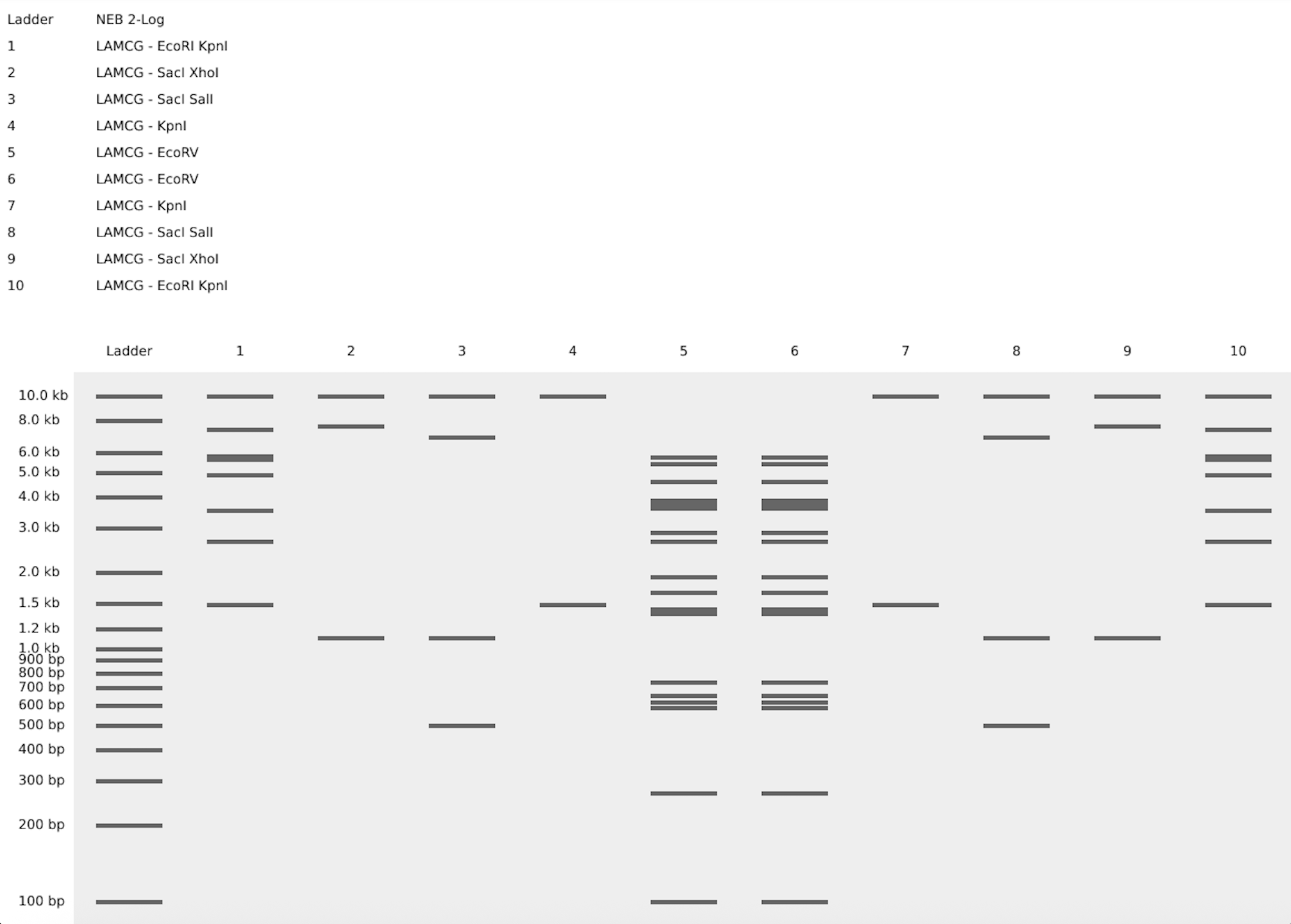
This exercise demonstrates that DNA is not just a carrier of information but a medium for structural design. The precision of the band migration confirms the successful mapping of the Lambda genome through enzymatic “sculpting.”
Part 3: DNA Design Challenge (The “Write” Phase)
3.1. Chosen Protein: TEM-1 Beta-lactamase (P62593)
For my protein design, I chose TEM-1 Beta-lactamase. Reasoning: As a microbiology student, I am focused on the global crisis of Antibiotic Resistance. TEM-1 is the most common beta-lactamase found in Gram-negative bacteria (like E. coli). It provides resistance against Penicillin and early Cephalosporins by hydrolyzing the beta-lactam ring. Designing this protein from scratch allows me to study the fundamental “software” that enables bacterial survival against clinical interventions. Protein Sequence (UniProt): The following is the amino acid sequence for the TEM-1 Beta-lactamase (286 AA), which I will be using for the reverse translation and codon optimization phases:
>sp|P62593|BLAT_ECOLI Beta-lactamase TEM-1 OS=Escherichia coli OX=562 PE=1 SV=1MSIQHFRVALIPFFAAFCLPVFAHPETLVKVKDAEDQLGARVGYIELDLNSGKILESFRPEERFPMMSTFKVLLCGAVLSRIDAGQEQLGRRIHYSQNDLVEYSPVTEKHLTDGMTVRELCSAAITMSDNTAANLLLTTIGGPKELTAFLHNMGDHVTRLDRWEPELNEAIPNDERDTTMPVAMATTLRKLLTGELLTLASRQQLIDWMEADKVAGPLLRSALPAGWFIADKSGAGERGSRGIIAALGPDGKPSRIVVIYTTGSQATMDERNRQIAEIGASLIKHW
3.2. Reverse Translation: Protein (amino acid) sequence to DNA (nucleotide) sequence
The Central Dogma of molecular biology serves as the fundamental framework for this process. In nature, information flows from DNA to RNA (transcription) and then to protein (translation). However, in Synthetic Biology, we often operate in reverse: we start with a functional protein—in this case, the TEM-1 Beta-lactamase—and work backward to determine the exact nucleotide sequence required to encode it.
I understand that while the genetic code is universal, it is also degenerate, meaning most amino acids are encoded by multiple codons. This reverse translation step is the first stage in “writing” biological software, allowing us to move from a structural protein sequence back to a digital DNA format ($A, T, C, G$). This process is critical for my future goals in biotechnology, as it enables the custom synthesis of genes for expression in various hosts.
Using Benchling’s reverse translation tool, I have derived the following 861 bp nucleotide sequence from the 286 amino acid sequence of TEM-1:
TEM-1 Beta-lactamase DNA sequence (Derived):
atgagtattcaacatttccgtgtcgcccttattcccttttttgcggcattttgccttcctgtttttgctcacccagaaacgctggtgaaagtaaaagatgctgaagatcagttgggtgcacgagtgggttacatcgaactggatctcaacagcggtaagatccttgagagttttcgccccgaagaacgttttccaatgatgagcacttttaaagttctgctatgtggcgcggtattatcccgtattgacgccgggcaagagcaactcggtcgccgcatacactattctcagaatgacttggttgagtactcaccagtcacagaaaagcatcttacggatggcatgacagtaagagaattatgcagtgctgccataaccatgagtgataacactgcggccaacttacttctgacaacgatcggaggaccgaaggagctaaccgcttttttgcacaacatgggggatcatgtaactcgccttgatcgttgggaaccggagctgaatgaagccataccaaacgacgagcgtgacaccacgatgcctgtagcaatggcaacaacgttgcgcaaactattaactggcgaactacttactctagcttcccggcaacaattaatagactggatggaggcggataaagttgcaggaccacttctgcgctcggcccttccggctggctggtttattgctgataaatctggagccggtgagcgtgggtctcgcggtatcattgcagcactggggccagatggtaagccctcccgtatcgtagttatctacacgacggggagtcaggcaactatggatgaacgaaatagacagatcgctgagataggtgcctcactgattaagcattggtaa
3.3. Codon Optimization
Once the nucleotide sequence of the protein is determined, it is essential to perform Codon Optimization before DNA synthesis.
describe why you need to optimize codon usage: Although the genetic code is universal, it is also degenerate, meaning that most amino acids are encoded by multiple synonymous codons. However, different organisms do not use these codons with equal frequency, a phenomenon known as Codon Bias.
From my perspective as a microbiology student, optimization is necessary for the following reasons: Translation Efficiency: Each organism has a specific pool of available transfer RNAs (tRNAs). By using codons that match the most abundant tRNAs in the host, we prevent “ribosomal stalling” and ensure rapid and efficient protein synthesis. Maximizing Yield: For professional biotechnology applications, such as those performed at companies, achieving high-level protein expression is a primary goal for scalability and cost-effectiveness. mRNA Stability and Folding: Optimization tools help eliminate unwanted secondary structures in the mRNA that could interfere with translation or lead to improper protein folding. Sequence Sanitation: During this process, I ensured the removal of “forbidden” sequences, specifically Type IIs enzyme recognition sites such as BsaI, BsmBI, and BbsI. This makes the sequence compatible with modern assembly methods like Golden Gate Cloning and DNA synthesis requirements from providers like Twist Bioscience.
The Methodology: How was this achieved? To perform this refinement, I utilized algorithmic tools (benchmarked against Codon Usage Tables like Kazusa) that analyze the frequency of codon usage in E. coli. The algorithm performs a synonymous substitution:
- It identifies every amino acid in the TEM-1 sequence.
- It replaces the existing codon with the one that has the highest frequency of use in the E. coli genome.
- It performs Sequence Sanitation by screening the entire 861 bp sequence to eliminate Type IIs restriction sites—specifically BsaI, BsmBI, and BbsI. This step is vital for ensuring that the DNA is compatible with Golden Gate Assembly, a method I am familiar with through my research interests in biotechnology and synthetic biology.
Which organism have you chosen to optimize the codon sequence for and why? I have chosen Escherichia coli as my expression host.
Reasoning:
- Academic Background: Throughout my 7th-semester Microbiology curriculum, E. coli has been the primary model organism for studying microbial genetics and molecular biology.
- Industry Standard: It remains the most widely used and well-characterized bacterial host for the commercial production of recombinant proteins in the biotechnology industry.
- Practical Familiarity: My internship experience at the National Institute of Genetic Engineering and Biotechnology (NIGEB) and my lab work in microbial culturing and DNA extraction have provided me with the necessary hands-on skills to work with this specific host efficiently.
TEM-1 Beta-lactamase DNA sequence with Codon-Optimization (for E. coli):
ATGAGCATTCAACATTTCCGTGTCGCTCTGATCCCGTTCTTCGCTGCCTTTTGCCTGCCGGTATTCGCTCACCCGGAAACCCTGGTTAAAGTTAAAGACGCCGAAGATCAGCTGGGTGCACGTGTTGGTTACATCGAACTGGATCTGAACAGCGGTAAAATCCTGGAAAGCTTCCGTCCGGAAGAACGTTTCCCGATGATGAGCACCTTTAAAGTTCTGCTGTGTGGTGCAGTTCTGAGCCGTATTGACGCAGGTCAAGAACAGCTGGGTCGTCGTATCCACTACAGCCAGAACGATCTGGTTGAATACAGCCCGGTTACCGAAAAACATCTGACCGACGGTATGACCGTTCGTGAACTGTGTAGCGCTGCTATCACCATGAGCGATAACACCGCTGCTAACCTGCTGCTGACCACCATTGGTGGCCCGAAAGAACTGACCGCCTTCCTGCACAACATGGGTGATCATGTTACCCGTCTGGATCGTTGGGAACCGGAACTGAACGAAGCTATCCCGAACGACGAACGTGATACCACCATGCCGGTTGCAATGGCTACCACCCTGCGTAAACTGCTGACCGGTGAACTGCTGACCCTGGCTAGCCGTCAGCAACTGATCGACTGGATGGAAGCTGATAAAGTTGCAGGTCCGCTGCTGCGTAGCGCTCTGCCGGCTGGTTGGTTCATTGCTGATAAAAGCGGTGCAGGTGAACGTGGTAGCCGTGGTATCATTGCTGCGCTGGGTCCGGATGGTAAACCGAGCCGTATTGTTGTTATCTACACCACCGGTAGCCAGGCTACCATGGATGAACGTAACCGTCAGATCGCTGAAATTGGTGCTAGCCTGATCAAACATTGGTAA
Analysis of Modifications: While it is impractical to list all 800+ nucleotide substitutions, I performed a comparative analysis between the original and optimized sequences.
- Synonymous Changes: The majority of changes involved switching from rare codons (e.g., using
AGAfor Arginine) to the preferred E. coli codons (e.g.,CGTorCGC). - Sequence Sanitation: I specifically verified the removal of internal BsaI (GGTCTC) and EcoRI (GAATTC) sites. In the original sequence, these might have interfered with downstream cloning protocols, but in this synthetic version, they have been successfully eliminated without altering the amino acid sequence of the Beta-lactamase.
3.4. Production Method: Transforming Digital Code into Matter
Having finalized the digitally optimized DNA sequence for TEM-1 Beta-lactamase, the challenge shifts from computational design to physical realization. To transform this 861 bp sequence into a functional enzyme, we must navigate through DNA synthesis, host selection, and the molecular execution of the Central Dogma.
1. DNA Synthesis: From Digital Information to Physical Matter
The first step is De Novo DNA Synthesis. Since this sequence is a synthetic product specifically optimized for E. coli, it cannot be extracted from a natural source; it must be chemically manufactured.
- Synthesis Technology: Utilizing high-throughput silicon-based platforms, such as those provided by Twist Bioscience or IDT, the digital sequence is converted into physical DNA through phosphoramidite chemistry.
- Verification: The resulting fragment is typically sequence-verified using Next-Generation Sequencing (NGS) to ensure 100% accuracy before being cloned into an expression vector (e.g., a pET-series plasmid) containing necessary regulatory elements like promoters.
2. Production Technologies: Choosing the Biological Factory
I have identified two primary technological routes for producing the protein from the synthetic DNA:
- Cell-Dependent (In Vivo) Expression: This is the industry standard for large-scale production, widely utilized in biotechnology companies. The synthetic DNA is transformed into a specialized host, such as Escherichia coli (BL21 DE3). These bacteria serve as living bioreactors. Upon induction with a molecule like IPTG, the cellular machinery is “hijacked” to produce the Beta-lactamase. This method is highly scalable and benefits from the host’s natural chaperones that aid in proper protein folding.
- Cell-Free Protein Synthesis (CFPS): This modern approach involves using cell lysates that contain all the necessary molecular components—ribosomes, RNA polymerase, and tRNAs—without the cell membrane. This allows for rapid “Transcription-Translation” (TX-TL) in a test tube. For research settings, such as those at institutes like NIGEB, this method is invaluable for quickly screening enzyme variants without the time-consuming steps of cell culture and transformation.
3. The Molecular Execution: Transcription and Translation
Regardless of the production method, the DNA sequence must be executed through the two-step process of the Central Dogma to become a protein:
- Transcription (DNA → mRNA): The enzyme RNA Polymerase identifies the Promoter (the “start” signal) upstream of the Beta-lactamase gene. It reads the DNA template and assembles a complementary messenger RNA (mRNA) strand. This mRNA carries the genetic instructions from the stable DNA storage to the active protein-making machinery.
- Translation (mRNA → Protein): The Ribosome identifies the Ribosome Binding Site (RBS) on the mRNA and initiates synthesis at the Start Codon (ATG). Guided by the optimized codons designed in the previous step, tRNAs deliver amino acids to the ribosome with high efficiency. Because the sequence was tailored to the host’s tRNA pool, the ribosome can move smoothly without stalling, linking amino acids into a polypeptide chain until it reaches a Stop Codon.
4. Achieving Functionality: Protein Folding
The final, crucial stage is Protein Folding. For TEM-1 Beta-lactamase, the polypeptide chain must fold into a precise three-dimensional conformation to form its active site. This folded structure is what enables the enzyme to bind to and hydrolyze the beta-lactam ring of antibiotics, providing the bacterial cell with the resistance mechanism that is a central focus of my current studies and research interests.
3.5. How does it work in nature/biological systems?
In classical genetics, we often think of “one gene, one protein.” However, natural systems—especially viruses and bacteria—are far more efficient. A single DNA locus can code for multiple proteins through several fascinating mechanisms:
1. Overlapping Genes (Frameshifting)
As seen in the Phage MS2 example provided in my research, biological systems can use Overlapping Reading Frames (ORFs). Because the genetic code is read in triplets, a single DNA sequence can encode different proteins depending on the “starting point” of the ribosome. By shifting the reading frame (e.g., a +1 or -1 shift), the same sequence of nucleotides is interpreted as a completely different set of codons, producing entirely distinct polypeptide chains.
2. Alternative Splicing (Eukaryotic Complexity)
In eukaryotic organisms, a single primary transcript (pre-mRNA) can undergo Alternative Splicing. By selectively joining different combinations of exons, a cell can generate multiple mRNA isoforms from a single gene. This allows for the production of protein variants with different functions or localizations within the cell.
3. Polycistronic mRNA (The Operon Model)
In many bacterial systems, multiple genes are grouped into a single Operon and transcribed into one long mRNA molecule. This polycistronic transcript contains multiple Ribosome Binding Sites (RBS), allowing ribosomes to initiate translation at several points and produce multiple independent proteins (like those in a metabolic pathway) simultaneously from a single transcriptional event.
Molecular Alignment: The Flow of Information (TEM-1 Case Study)
Following the example of the MS2 L-protein, I have aligned the first 30 nucleotides of my synthetic TEM-1 Beta-lactamase gene to demonstrate the molecular flow from DNA to Protein. Note that during transcription, Thymine (T) is replaced by Uracil (U), and during translation, each triplet (codon) is converted into a specific amino acid.
DNA (Coding Strand):
5' - A T G A G C A T T C A A C A T T T C C G T G T C G C T - 3'
RNA (mRNA Transcript):
5' - A U G A G C A U U C A A C A U U U C C G U G U C G C U - 3'
Protein (Amino Acid Sequence):
M S I Q H F R V A L ...
Analysis: This alignment illustrates the “digital-to-analog” conversion of life. A simple change in the DNA sequence—such as the codon optimization I performed in section 3.3—directly influences the efficiency of this flow without changing the final protein product. This dense packing of information is what allows a tiny phage or a complex bacterium to execute life-sustaining functions with such high precision.
Part 4: Building the Expression Cassette
4.2. Build Your DNA Insert Sequence
In this final design phase, I assembled a complete Expression Cassette for the TEM-1 Beta-lactamase gene. An expression cassette is a modular genetic unit consisting of a coding sequence and the necessary regulatory elements to direct the cell’s machinery to produce the desired protein.
Genetic Architecture and Components
The construct was assembled in Benchling using a linear topology. I manually integrated and annotated each component to ensure optimal expression in my target host, Escherichia coli.
| Component | Sequence ID / Source | Function |
|---|---|---|
| Promoter | BBa_J23106 | A strong constitutive promoter that ensures continuous transcription of the gene without the need for external induction. |
| RBS | BBa_B0034 (with spacers) | Optimized Ribosome Binding Site to ensure efficient translation initiation and prevent ribosomal stalling. |
| Start Codon | ATG | The universal initiation signal for protein synthesis. |
| Coding Sequence | Optimized TEM-1 (Section 3.3) | The core genetic instruction for Beta-lactamase, refined for E. coli codon bias to maximize yield. |
| 7x His Tag | C-terminal extension | Added to enable downstream protein purification using Immobilized Metal Affinity Chromatography (IMAC), a standard technique I am familiar with through my laboratory experience. |
| Stop Codon | TAA | Signal to the ribosome to terminate translation and release the polypeptide chain. |
| Terminator | BBa_B0015 | A robust double terminator that stops RNA polymerase to prevent transcriptional read-through into the vector backbone. |
Design Rationale
The integration of a 7x His Tag is a strategic choice for my professional goals in biotechnology. In industrial settings, such as the production of recombinant enzymes, efficient purification is as critical as high expression. By placing the tag at the C-terminus (before the final stop codon), I ensure that the functional enzyme can be easily isolated from the E. coli lysate while maintaining its catalytic activity against beta-lactam antibiotics.
Final Outputs
Benchling Design Link: [https://benchling.com/s/seq-yLbYKs2DfT4qqdEf6a1P?m=slm-ryzRMIvsrygNZeW9f1sq ]
Exported Sequence: >Untitled sequenceTEM1_Expression_Cassette. fasta TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGCCATTAAAGAGGAGAAAGGTACCATGAGCATTCAACATTTCCGTG TCGCTCTGATCCCGTTCTTCGCTGCCTTTTGCCTGCCGGTATTCGCTCACCCGGAAACCCTGGTTAAAGTTAAAGACGC CGAAGATCAGCTGGGTGCACGTGTTGGTTACATCGAACTGGATCTGAACAGCGGTAAAATCCTGGAAAGCTTCCGTCCG GAAGAACGTTTCCCGATGATGAGCACCTTTAAAGTTCTGCTGTGTGGTGCAGTTCTGAGCCGTATTGACGCAGGTCAAG AACAGCTGGGTCGTCGTATCCACTACAGCCAGAACGATCTGGTTGAATACAGCCCGGTTACCGAAAAACATCTGACCGA CGGTATGACCGTTCGTGAACTGTGTAGCGCTGCTATCACCATGAGCGATAACACCGCTGCTAACCTGCTGCTGACCACC ATTGGTGGCCCGAAAGAACTGACCGCCTTCCTGCACAACATGGGTGATCATGTTACCCGTCTGGATCGTTGGGAACCGG AACTGAACGAAGCTATCCCGAACGACGAACGTGATACCACCATGCCGGTTGCAATGGCTACCACCCTGCGTAAACTGCT GACCGGTGAACTGCTGACCCTGGCTAGCCGTCAGCAACTGATCGACTGGATGGAAGCTGATAAAGTTGCAGGTCCGCTG CTGCGTAGCGCTCTGCCGGCTGGTTGGTTCATTGCTGATAAAAGCGGTGCAGGTGAACGTGGTAGCCGTGGTATCATTG CTGCGCTGGGTCCGGATGGTAAACCGAGCCGTATTGTTGTTATCTACACCACCGGTAGCCAGGCTACCATGGATGAACG TAACCGTCAGATCGCTGAAATTGGTGCTAGCCTGATCAAACATTGGCATCACCATCACCATCATCACTAACCAGGCATC AAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAG TCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
The final construct is verified to be compatible with Golden Gate Assembly as all internal BsaI, BsmBI, and BbsI sites were removed during the optimization phase in section 3.3.
4.6. Final Plasmid Construction and Verification
The final engineering step involved the successful integration of the synthetic expression cassette into a circular plasmid backbone.
Final Construct Summary:
- Insert: Codon-optimized TEM-1 Beta-lactamase cassette (1,068 bp).
- Vector: pTwist Amp High Copy.
- Selection Marker: Ampicillin resistance (ampR).
- Replication Origin: High-copy colE1, ensuring maximum yield.
By re-importing the GenBank file from Twist into Benchling, I have verified the structural integrity of the plasmid. The circular map clearly shows the seamless transition from the vector backbone to our custom-designed insert, confirming that the genetic tool is ready for transformation into E. coli BL21 (DE3) for protein production.
https://benchling.com/s/seq-kngCff7XhEihMJr8nmSR?m=slm-a9cFRgWHjVUV0suFBbR9
Download TEM1_Expression_Cassette PDF
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) Target DNA for Sequencing
I would choose to sequence the whole-genome of a multi-drug resistant (MDR) clinical isolate of Escherichia coli.
Rationale: Having designed a synthetic TEM-1 Beta-lactamase gene in the previous sections, it is scientifically valuable to observe how such genes exist and evolve in nature. Clinical isolates often carry resistance genes on complex mobile genetic elements. Sequencing the entire genome allows me to:
- Identify the genomic context of resistance genes (e.g., chromosomal vs. plasmid-borne).
- Discover co-existing resistance markers that contribute to a multi-drug resistant phenotype, which is a key area of interest in my microbiology studies.
- Track the epidemiological spread of specific resistance alleles in hospital environments.
(ii) Selected Sequencing Technology
I would utilize Oxford Nanopore Technologies (ONT), specifically the MinION platform, to perform this sequencing.
Technology Breakdown:
| Question | Analysis & Answer |
|---|---|
| Generation | Third-Generation. This technology performs single-molecule, real-time sequencing without the need for DNA synthesis or PCR amplification. |
| Input & Preparation | Input: High-molecular-weight (HMW) genomic DNA extracted from the E. coli isolate. Preparation Steps: 1. Extraction: Pure genomic DNA recovery. 2. End-repair & A-tailing: Preparing DNA ends for adapter attachment. 3. Adapter Ligation: Attaching motor proteins and sequencing adapters to the DNA library. 4. Clean-up: Using magnetic beads to remove reagents. |
| Decoding Mechanism | Ionic Current Disruption. As a single strand of DNA is pulled through a protein nanopore, each nucleotide ($A, T, C, G$) creates a unique disruption in the electrical current across the membrane. |
| Base Calling | The raw electrical signals (squiggles) are decoded using Neural Network-based algorithms that translate the specific current patterns into a nucleotide sequence in real-time. |
| Output | Raw Data: Fast5 files (containing signal data). Processed Data: FASTQ files (containing the sequence and quality scores), which can then be used for genome assembly and resistance gene mapping. |
Why this technology? the Long-Read capability of Nanopore is its greatest advantage for me as a microbiology researcher. It allows for the complete assembly of bacterial plasmids and the identification of large structural variations that short-read technologies (like Illumina) often miss. This is particularly relevant for the high-level genomic analysis performed at institutes like NIGEB.
5.2 DNA Write
(i) Target DNA for Synthesis
I want to synthesize the TEM-1 Beta-lactamase expression cassette (1,068 bp) that I’ve been meticulously designing and optimizing.
My Motivation: There’s a special kind of satisfaction in seeing a digital design from Benchling turn into a physical piece of DNA. My goal here is to take what I’ve learned about antibiotic resistance and actually test it in the lab. Specifically, I want to:
- Test the Optimization: See if the codon changes I made actually lead to the high protein levels I’m expecting in E. coli.
- Study the Enzyme: Use the synthesized DNA to produce pure TEM-1 and analyze its kinetics against different antibiotics.
- Create a Modular Tool: This isn’t just a homework assignment; it’s a standardized part that could be a building block for future biosensors or biotech projects I might work on.
The Sequence:```text TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGCCATTAAAGAGGAGAAAGGTACCAGCATTCAACATTTCCGTGTCGCTCTGATCCCGTTCTTCGCTGCCTTTTGCCTGCCGGTATTCGCTCACCCGGAAACCCTGGTTAAAGTTAAAGACGCCGAAGATCAGCTGGGTGCACGTGTTGGTTACATCGAACTGGATCTGAACAGCGGTAAAATCCTGGAAAGCTTCCGTCCGGAAGAACGTTTCCCGATGATGAGCACCTTTAAAGTTCTGCTGTGTGGTGCAGTTCTGAGCCGTATTGACGCAGGTCAAGAACAGCTGGGTCGTCGTATCCACTACAGCCAGAACGATCTGGTTGAATACAGCCCGGTTACCGAAAAACATCTGACCGACGGTATGACCGTTCGTGAACTGTGTAGCGCTGCTATCACCATGAGCGATAACACCGCTGCTAACCTGCTGCTGACCACCATTGGTGGCCCGAAAGAACTGACCGCCTTCCTGCACAACATGGGTGATCATGTTACCCGTCTGGATCGTTGGGAACCGGAACTGAACGAAGCTATCCCGAACGACGAACGTGATACCACCATGCCGGTTGCAATGGCTACCACCCTGCGTAAACTGCTGACCGGTGAACTGCTGACCCTGGCTAGCCGTCAGCAACTGATCGACTGGATGGAAGCTGATAAAGTTGCAGGTCCGCTGCTGCGTAGCGCTCTGCCGGCTGGTTGGTTCATTGCTGATAAAAGCGGTGCAGGTGAACGTGGTAGCCGTGGTATCATTGCTGCGCTGGGTCCGGATGGTAAACCGAGCCGTATTGTTGTTATCTACACCACCGGTAGCCAGGCTACCATGGATGAACGTAACCGTCAGATCGCTGAAATTGGTGCTAGCCTGATCAAACATTGGCATCACCATCACCATCATCACCCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
5.3 DNA Edit
(i) What DNA i would want to edit and why.
After designing and synthesizing the TEM-1 Beta-lactamase gene, the next logical step in my research is to explore how we can “undo” this resistance. I want to edit the chromosomal or plasmid-borne blaTEM-1 gene in clinical isolates of Escherichia coli.
The “Why” behind this: While we spend a lot of time understanding how resistance works, the future of therapy might lie in reversing it. By editing this specific gene, I aim to:
- Restore Antibiotic Sensitivity: If we can successfully disrupt the TEM-1 gene, we can make previously resistant bacteria susceptible to penicillin-group antibiotics again. This is a huge area of interest in the fight against “superbugs.”
- Study Gene Function: By making precise edits (like single amino acid swaps) in the active site of the enzyme, I can study exactly which parts of the protein are responsible for its catalytic activity.
- CRISPR-Antimicrobials: This serves as a proof-of-concept for using CRISPR as a sequence-specific antimicrobial, which is much more targeted than traditional broad-spectrum antibiotics.
(ii) Selected Editing Technology: CRISPR-Cas9
To perform these edits, I’ll use the CRISPR-Cas9 system. It’s currently the most versatile and precise tool we have for bacterial genome editing, and it’s something I’ve been keen to explore more in a lab setting like NIGEB.
Technical Breakdown:
| Question | My Analysis & Answer |
|---|---|
| How does it edit DNA? | The system uses a Guide RNA (gRNA) to lead the Cas9 nuclease to a specific 20-bp sequence in the TEM-1 gene. Once there, Cas9 acts like “molecular scissors” and creates a Double-Strand Break (DSB). In bacteria, this break is often lethal or repaired by Non-Homologous End Joining (NHEJ), which introduces small errors (indels) that effectively break the gene’s function. |
| Essential Steps | 1. Targeting: Identifying a unique sequence within the TEM-1 gene. 2. Binding: The Cas9-gRNA complex scans the DNA for a PAM sequence ($5’-NGG-3’$) and binds to the target. 3. Cleaving: Cas9 cuts both strands of the DNA. 4. Repair/Disruption: The cell attempts to fix the break, leading to mutations that “knock out” the resistance gene. |
| Preparation & Inputs | Design: Designing a 20-nucleotide gRNA that is specific to TEM-1 to avoid off-target effects. Inputs: 1. Cas9 Enzyme: Delivered via a plasmid. 2. sgRNA (single guide RNA): Designed to match the target gene. 3. Competent Cells: E. coli cells ready to take up the CRISPR plasmids. 4. Donor Template (optional): If I wanted to “swap” a base instead of just breaking it (Homology-Directed Repair). |
| Limitations: Precision | The main risk is Off-target effects, where Cas9 might cut a similar-looking sequence elsewhere in the genome. While rare in small bacterial genomes, it’s still a concern. |
| Limitations: Efficiency | Not every bacteria will take up the CRISPR system (low transformation efficiency). Also, since a DSB is often lethal for E. coli, the survival rate of the “edited” cells can be low unless a repair template is provided. |
Conclusion of the Assignment
This assignment has been an incredible journey through the “Design-Build-Test-Learn” cycle of synthetic biology. From decoding the MS2 virus to optimizing an antibiotic resistance gene and finally learning how to read, write, and edit it, I feel much more equipped to handle complex genetic engineering tasks. It’s exciting to see how these digital tools can translate into real-world solutions for global health challenges.