Biological Engineering application Bioremediation with Developmental control General purposal: Engineered bacteria that sculpt the rhizosphere for enhanced bioremediation
I want to develop a biological engineering tool designed to enhance the natural process of phytoremediation, where plants are used to extract or break down soil contaminants. The core idea is to create specially engineered soil bacteria that act as biological boosters for ordinary plants. This would enable plants to clean contaminated environments far more effectively. The tool is inspired by natural systems like the plant pathogen Agrobacterium tumefaciens, a microbe that hijacks plant development to create tumors. However, here I would redesign this concept for a beneficial purpose, that involves this: instead of causing disease in the plant, the engineered bacteria would send beneficial signals to the plant, first by these bacteria being able to express morphogens and then instructing the plant to grow a more extensive root system, in addition to further activate internal contaminants uptake and potencial degradation pathways.
Homework Questions Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy? Polymerase has a very low error rate of approximately 10^{-4} to 10^{-6} errors per nucleotide without proofreading. Thus, comparing to the human genome length of approximately 3 billion base pair, it represents a minimal part. Thanks to other molecular features, errors produced by the sythesis machinery can be corrected by mechanisms applied like read proof, including the polymerase’s intrinsic proofreading domain, post-replication mismatch repair (MMR) pathways involving proteins like MutS and MutL that scan and excise mismatches, and DNA damage response mechanisms such as base excision repair (BER) and nucleotide excision repair (NER).
Homework Questions Part A. Conceptual Questions Why humans eat beef but do not become a cow, eat fish but do not become fish? Because digestion breaks proteins into individual amino acids, so it means it destroys the cow’s structure and information. We absorb the amino acids and then our ribosomes rebuild human proteins based on human DNA. To make it clear, biological identity comes from genetic information, not raw material.
Part A. SOD1 Binder Peptide Design Part 1 Design short peptides that bind mutant SOD1
Which are worth advancing for therapy
Human SOD1 sequence
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ SOD1 A5V sequence
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ Binder 1: WHYPAVAVALGX Perplexity: 8.190170
Binder 2: KRYYVVGVRHKE Perplexity: 31.759185
DNA Assembly Part 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix key components include:
Phusion DNA Polymerase: This is a fusion protein comprising a traditional Pfu-like DNA polymerase and a processivity-enhancing domain. Its purpose is to synthesize new DNA strands with exceptionally high fidelity (due to its inherent 3’→5’ proofreading exonuclease activity) and high speed.
Part 1: Intracellular Artificial Neural Networks What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Cell Free Artwork Experimental Loop I made this fish-like shape at the second quadrant (on the right side of the plate)
I liked that this project brings our creative side, and we use fluorescent protein expression to reflect artistry.
For next collaborative project there could be done in modified bacteria grown in different growth media (so as to see different color expression)
Subsections of Homework
Week 1 HW: Principles and Practices
Biological Engineering application
Bioremediation with Developmental control
General purposal: Engineered bacteria that sculpt the rhizosphere for enhanced bioremediation
I want to develop a biological engineering tool designed to enhance the natural process of phytoremediation, where plants are used to extract or break down soil contaminants. The core idea is to create specially engineered soil bacteria that act as biological boosters for ordinary plants. This would enable plants to clean contaminated environments far more effectively. The tool is inspired by natural systems like the plant pathogen Agrobacterium tumefaciens, a microbe that hijacks plant development to create tumors. However, here I would redesign this concept for a beneficial purpose, that involves this: instead of causing disease in the plant, the engineered bacteria would send beneficial signals to the plant, first by these bacteria being able to express morphogens and then instructing the plant to grow a more extensive root system, in addition to further activate internal contaminants uptake and potencial degradation pathways.
This application could be relevant for addressing widespread environmental crises, such as soil contamination from illegal mining, a severe problem in countries like Ecuador, where heavy metals and toxins leach into natural ecosystems. Regarding to this, it is undoubted that current solutions often rely on specialized accumulator plants or pollutant-degrading microbes, but these can be slow, inefficient or ecologically disruptive. And so, the develoment of this tool could offer a more adaptive and powerful solution by pairing robust, fast-growing native plants with these engineered bacterial partners directly at the contamination site.
The system would work through a simple sense-and-respond logic where the engineered bacteria would be added to the soil where they naturally associate with plant roots. They would be designed to detect a specific contaminant, like a toxic metal. And upon detection, the bacteria would respond by producing and releasing natural plant growth hormones (morphogens) that stimulate the plant to rapidly produce a dense network of new roots exactly where the pollution is concentrated. This is key because it gives the plant a much greater physical capacity to absorb contaminants. Moreover, the bacteria will simultaneously provide the plant with key enzymes or co-factors for effectively inducing in the plant with new biochemical pathways to break down or neutralize toxins internally, even if the plant lacks these abilities naturally.
Governance/policy goals
Ensure environmental safety and containment
All applications of this development must occur within physically contained environments, like sealed soil plots, that can be fully removed after a trial.
The engineered bacteria must be transient and ecologically contained. This requires built-in constraints to prevent persistence or gene transfer after the remediation function is complete.
Stewardship and ecological benefit
The tool must demonstrably provide a net ecological benefit by restoring ecosystem services without causing unintended harm to non-target organisms or soil health.
Long-term monitoring of remediation sites is a crucial component of deployment.
Prevent technological exploitation and ensure local justice
The benefits of this technology must be accessible to the communities most affected by contamination, such as those near illegal mining sites in Ecuador.
Establish a local oversight council, composed of community representatives, so that they have the authority to review all research plans, monitor progress, and pause or stop the project if it violates agreed-upon ethical or cultural standards.
Foster transparency and build public trust
Create a public registry and impact dashboard that shows real data, including all field trial locations, the specific pollutants targeted, the plant-bacteria pairs used, and key environmental health metrics (soil toxin levels, native plant recovery). This will allow for independent verification of safety and benefit claims.
The core genetic safety features will be published as open-source patents or licenses.
Governance actions
Governance Action 1
Create a physical and procedural “airlock” between lab research and open-environment deployment for ensuring safety and enabling reversal.
This would involve that all field testing must progress through locked phases:
Contained Modules: sealed, soil-filled units that can be excavated intact
Fenced & Monitored Plots: With impermeable barriers and leachate capture
Open Pilot Sites
Governance Action 2
Open-Source “Kill Switch” Development Grand Challenge
Launch a prize for teams to develop and validate novel, stable “kill switches” or dependency circuits for environmental bacteria.
This would solve the core biocontainment problem through competitive, collaborative science, in order to make the safest designs freely available.
Risks of Failure: Researchers may engineer solutions that work perfectly in the lab but fail in complex field conditions. The “best” design might be too metabolically active for the bacteria to function effectively as a remediation booster.
Provide funding, training, and simple standardized kits for metagenomic sampling or pollutant testing, to establish community-run environmental monitoring stations around proposed and active remediation sites. Data will be managed on a local server and feeds into the public dashboard.
This action assumes communities have the interest and capacity to manage this technical role; assumes data collected will be scientifically robust enough for decision-making.
Evaluation of governance actions
Scored from 1-3 with, 1 as the best
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
2
1
3
• By helping respond
1
2
1
Foster Lab Safety
• By preventing incident
3
1
3
• By helping respond
3
2
3
Protect the environment
• By preventing incidents
1
2
2
• By helping respond
1
1
1
Other considerations
• Technical Feasibility
1
2
1
• Cost and Accessibility
3
2
2
• Scalability and Adaptability
2
1
1
Reflection
For a project aiming for ethical deployment of this engineered application in a context like Ecuador, Action 3 is likely the most critical starting point, as it builds the necessary social foundation. However, a robust governance strategy must pursue Action 2 to solve the core technical risk, while using a simplified version of Action 1 to provide a structured testing framework.
Week 2 HW: DNA read, write, and edit
Homework Questions
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Polymerase has a very low error rate of approximately 10^{-4} to 10^{-6} errors per nucleotide without proofreading. Thus, comparing to the human genome length of approximately 3 billion base pair, it represents a minimal part. Thanks to other molecular features, errors produced by the sythesis machinery can be corrected by mechanisms applied like read proof, including the polymerase’s intrinsic proofreading domain, post-replication mismatch repair (MMR) pathways involving proteins like MutS and MutL that scan and excise mismatches, and DNA damage response mechanisms such as base excision repair (BER) and nucleotide excision repair (NER).
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein is approximately 345 amino acids long (based on an average coding sequence of ~1,036 bp, divided by 3 nucleotides per codon). Given the degeneracy of the genetic code, where most of the 20 standard amino acids are encoded by 2–6 synonymous codons (with exceptions like methionine and tryptophan, each with 1 codon), the theoretical number of distinct DNA sequences encoding such a protein is enormous—typically on the order of 10^{100} to 10^{200} or more, calculated as the product of the number of codon options for each amino acid position. In practice, however, not all sequences function effectively due to factors such as codon usage bias (where certain codons are preferred for efficient translation in specific organisms, affecting tRNA availability and ribosomal speed), mRNA secondary structure formation that can impede translation initiation or elongation, regulatory elements embedded in the coding sequence (e.g., splicing signals, miRNA binding sites, or transcription factor motifs), GC content imbalances leading to replication or expression issues, and potential toxicity from repetitive sequences or unintended open reading frames.
What’s the most commonly used method for oligo synthesis currently?
The most commonly used method for oligonucleotide (oligo) synthesis is solid-phase phosphoramidite chemistry, often performed on microarray chips for high-throughput, multiplex production. This enables the parallel synthesis of up to 1 million oligos per chip at significantly reduced cost
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Synthesizing oligos longer than 200 nucleotides via direct chemical methods is challenging due to cumulative errors from imperfect coupling efficiency (typically 98–99% per nucleotide addition), leading to exponential yield decay and increased heterogeneity in the product pool. Side reactions, such as depurination or incomplete deprotection, further accumulate, resulting in truncated or mutated sequences that require extensive purification.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Direct oligo synthesis cannot produce a 2,000 bp gene because current chemical methods are limited to ~200–300 nt maximum lengths with acceptable yield and fidelity; beyond this, error rates and incomplete extensions make the process infeasible. Instead, longer genes are assembled from shorter oligos using techniques like PCR amplification, ligation, or enzymatic assembly (like Gibson assembly)
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids required by all animals, which cannot be synthesized endogenously and must be obtained through diet, are arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. Regarding “Lysine Contingency,” while lysine auxotrophy is a clever single-point failure mechanism, relying on just one could be vulnerable to evolutionary escape or environmental supplementation. A more robust “contingency” might involve engineering dependencies on multiple essentials (e.g., via codon recoding for serine and leucine as in the slides) or all 10, enhancing safety in applications like industrial microbes or gene drives, as escape risks are minimized when multiple biosynthetic pathways are disrupted.
What code would you suggest for AA:AA interactions?
I suggest a code analogous to basepairing but based on complementary physicochemical properties of amino acid side chains. This could be a modular interaction code categorizing the 20 amino acids into groups like charged, hydrophobic, polar, and special. Pairings would follow rules like positive-negative for ionic bonds or hydrophobic-hydrophobic for van der Waals, enabling predictable design of interfaces (e.g., coiled-coils or beta-sheets).
Homework Week 2
Part 1: Benchling & In-silico Gel Art
resembles a cactus figure
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
No access to lab in person
Part 3: DNA Design Challenge
3.1. Choose your protein
Nitrosomonas marina is a marine ammonia-oxidizing bacterium, and its AmoA (ammonia monooxygenase subunit A) is the catalytic core of the ammonia monooxygenase complex that initiates nitrification by oxidizing ammonia (NH₃) to hydroxylamine. Studying the AmoA amino acid sequence from N. marina is relevant because it is a well-annotated, reference AmoA that is widely used for comparative and phylogenetic analyses of ammonia-oxidizing bacteria. Its sequence provides a reliable benchmark for identifying conserved functional motifs, distinguishing AmoA from closely related PmoA proteins, and comparing canonical aerobic ammonia oxidation with putative alternative or iron-coupled ammonium oxidation pathways in other bacterial systems.
reverse translation of sample sequence to a 447 base sequence of most likely codons.
tatccgattaactttgtgctgccgagcaccatgattccgggcgcgctgatgctggatacc
attatgctgctgaccggcaactggctgattaccgcgctgctgggcggcggcttttggggc
ctgtttttttatccgggcaactggccgatttttggcccgacccatctgccggtggtggtg
gaaggcgtgctgctgagcgtggcggattataccggctttctgtatgtgcgcaccggcacc
ccggaatatgtgcgcctgattgaacagggcagcctgcgcacctttggcggccataccacc
gtgattgcggcgttttttagcgcgtttgtgagcatgctgatgttttgcgtgtggtggtat
tttggcaaaatttattgcaccgcgttttattatgtgcgcggcgaacgcggccgcattagc
cagaaacatgatgtgaccgcgtttggc
3.3 Codon optimization
Codon optimization is necessary because the genetic code is degenerate, meaning most amino acids are encoded by multiple codons and organisms exhibit significant variation in their synonymous codon usage preferences due to evolutionary pressures related to translational efficiency. Expressing a heterologous gene like Nitrosomonas marina amoA in a foreign host without optimization can lead to poor expression due to several molecular constraints:
the presence of codons that are rare in the host’s genome, which correlate with low abundance of their corresponding cognate tRNAs, leading to ribosomal stalling, translation attenuation, and premature termination
the formation of unfavorable mRNA secondary structures that impede ribosome binding and translocation; and
Extreme GC content that affects transcriptional efficiency and mRNA stability.
For AmoA protein, I incline for optimizing the sequence for expression in Escherichia coli. This choice is justified by the organism’s well-characterized genetics, rapid growth kinetics, high cell density cultivation, and the extensive availability of compatible expression vectors and purification tags. Furthermore, E. coli possesses a highly annotated codon usage database so this allows for precise adjustment of codon frequencies to match its endogenous tRNA pool.
The production of a recombinant protein such as AmoA from its corresponding DNA sequence can be achieved through two primary technological platforms: cell-dependent (in vivo) expression systems and cell-free (in vitro) expression systems.
In a typical bacterial expression system, the codon-optimized gene is first cloned into a plasmid vector under the control of a strong inducible promoter. This recombinant plasmid is then introduced into a host bacterium, most commonly Escherichia coli, through a process called transformation. The bacteria are cultivated in nutrient medium within shake flasks or bioreactors until an optimal cell density is reached, at which point an inducer molecule, like IPTG, is added to initiate transcription of the gene by host RNA polymerase. The resulting mRNA transcripts are subsequently bound by bacterial ribosomes, which, with the assistance of transfer RNAs charged with amino acids, catalyze the polymerization of the polypeptide chain according to the codon sequence. Following an appropriate expression period, the bacteria are harvested by centrifugation and lysed to release the intracellular contents, after which the target protein is purified using techniques such acell-free protein synthesis offers a rapid and open in vitro platform for protein production. In this approach, crude cellular extracts are prepared from cultured bacteria (or other organisms) by mechanical lysis and centrifugation to remove cell debris and genomic DNA, retaining the essential macromolecular machinery including ribosomes, tRNA molecules, aminoacyl-tRNA synthetases, and translation factors. A reaction mixture is assembled by combining this extract with the DNA template encoding AmoA, an energy regeneration system (typically containing ATP, GTP, and phosphoenolpyruvate or creatine phosphate), amino acids, and necessary cofactors. Transcription and translation proceed simultaneously in the tube, with the endogenous RNA polymerase transcribing the gene and the ribosomes immediately translating the nascent mRNA. This system offers distinct advantages for challenging proteins like membrane enzymes, as it allows for the direct supplementation of detergents, lipids, or chaperones to facilitate proper folding, and eliminates concerns regarding cellular toxicity or inclusion body formation. Additionally, cell-free reactions can be performed in small volumes for high-throughput screening or scaled up in bioreactor configurations to increase protein yields affinity chromatography, leveraging genetically encoded affinity tags fused to the protein of interest.
On the other hand, cell-free protein synthesis is a rapid and open in vitro platform for protein production. In this approach, crude cellular extracts are prepared from cultured bacteria by mechanical lysis and centrifugation to remove cell debris and genomic DNA, retaining the essential macromolecular machinery including ribosomes, tRNA molecules, aminoacyl-tRNA synthetases, and translation factors. A reaction mixture is assembled by combining this extract with the DNA template encoding AmoA, an energy regeneration system (typically containing ATP and others), amino acids, and necessary cofactors. Transcription and translation proceed simultaneously in the tube, with the endogenous RNA polymerase transcribing the gene and the ribosomes immediately translating the nascent mRNA. This system offers distinct advantages for challenging proteins like membrane enzymes, as it allows for the direct supplementation of detergents, lipids, or chaperones to facilitate proper folding, and eliminates concerns regarding cellular toxicity or inclusion body formation. Additionally, cell-free reactions can be performed in small volumes for high-throughput screening or scaled up in bioreactor configurations to increase protein yield
(i) What DNA would you want to sequence (e.g., read) and why?
To investigate the intrinsic control of neuron formation, I would sequence the FOXG1 gene, including its coding regions, regulatory promoter elements, and conserved non-coding sequences. FOXG1 is a critical transcription factor that acts as a master regulator of telencephalic development . It balances the proliferation of neural progenitors with their differentiation into neurons . Importantly, mutations, copy number variations, or deletions in FOXG1 lead to FOXG1 syndrome, a severe neurodevelopmental disorder characterized by impaired brain development and structural abnormalities . Sequencing this gene can reveal variants that disrupt its regulatory function, thereby affecting neuronal formation and transition.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Illumina sequencing due to its high accuracy and throughput, which is ideal for detecting single nucleotide variants and small insertions/deltions, indels, within the FOXG1 gene across many samples.
Generation: This is a second-generation sequencing (next-generation sequencing) technology. It relies on massively parallel sequencing of clonally amplified DNA fragments .
Input: The input is high-quality genomic DNA (gDNA) isolated from the sample of interest (e.g., neural cells or tissue) .
Input Preparation: Essential steps include: 1) Fragmentation of gDNA into smaller pieces (200-500 bp), 2) Adapter ligation, where platform-specific oligonucleotides (P5 and P7) are attached to fragment ends, and 3) Library amplification by PCR to enrich for fragments with adapters .
Essential Steps of Technology/Base Calling: 1) Clonal amplification: Fragments are amplified on a flow cell via bridge PCR to form distinct clonal clusters . 2) Sequencing by Synthesis (SBS): Fluorescently labeled, terminator-bound nucleotides are incorporated one at a time. After each cycle, the flow cell is imaged to detect the emitted fluorescence from each cluster, identifying the base . The sequencing instrument’s software performs base calling by converting these light signals into nucleotide sequences with quality scores .
Output: The primary output is FASTQ files containing millions of high-quality short reads (e.g., 150 bp paired-end), along with quality scores for each base call
I would also use Nanopore sequencing to complement Illumina data, specifically to resolve complex structural variants, copy number variations, and haplotypes in the FOXG1 region that are difficult to detect with short reads.
Generation: This is a third-generation sequencing technology. It sequences single DNA molecules in real time without requiring amplification .
Input: The input is high-molecular-weight (HMW) genomic DNA, which is essential for constructing long reads .
Input Preparation: Steps involve: 1) Optional fragmentation if a specific size is desired, though shearing is often minimized to preserve long fragments. 2) End-prep to repair DNA ends and add a poly-A tail. 3) Adapter ligation, where sequencing adapters (containing a motor protein) are attached to the template .
Essential Steps of Technology/Base Calling: 1) The prepared library is loaded onto a flow cell containing thousands of protein nanopores embedded in a membrane . 2) A motor protein guides a single DNA strand through the nanopore. 3) As the DNA passes through, it disrupts an ionic current in a characteristic way for each nucleotide. 4) Base calling is performed in real-time by sophisticated algorithms (like recurrent neural networks) that interpret these raw electrical current changes to determine the DNA sequence .
Output: The output is FASTQ files containing ultra-long reads (often >10 kb, capable of spanning entire repetitive regions)
5.2 DNA Write
Questions
(i) What DNA would you want to synthesize (e.g., write) and why?
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Answer
To investigate the regulatory function of FOXG1 in neuronal development, I would synthesize a compact genetic circuit consisting of the FOXG1 gene under the control of a neural-specific inducible promoter, along with a fluorescent reporter like GFP, linked via a self-cleaving peptide sequence to enable simultaneous visualization of FOXG1-expressing cells. This synthetic construct would be designed to test the hypothesis that precise FOXG1 dosage regulates the balance between neural progenitor proliferation and neuronal differentiation, as FOXG1 haploinsufficiency causes FOXG1 syndrome—a severe neurodevelopmental disorder characterized by impaired telencephalic development . By delivering this circuit into induced pluripotent stem cell-derived neural progenitors and modulating FOXG1 expression levels, one could observe downstream effects on neuronal fate specification and maturation, providing a platform to screen for therapeutic modulators of FOXG1 expression.
For DNA synthesis, I would employ enzymatic DNA synthesis technology rather than traditional chemical synthesis. Enzymatic synthesis, utilizing engineered terminal deoxynucleotidyl transferase (TdT) enzymes, offers superior performance for constructing this genetic circuit due to several advantages . The essential steps of this method include: (1) oligonucleotide design, where the target sequence is computationally designed and codon-optimized for expression in human cells; (2) enzymatic synthesis, where TdT enzymes incorporate modified nucleotides base-by-base onto a growing DNA strand under aqueous conditions, achieving coupling efficiencies above 99.7% ; (3) assembly, where overlapping oligonucleotides are assembled into the full-length gene construct using PCR-based or enzymatic assembly methods ; and (4) sequence verification through Sanger or next-generation sequencing to confirm 100% accuracy before cloning into an expression vector . This approach eliminates the need for a natural DNA template, enabling complete design freedom for incorporating the neural-specific promoter and reporter elements.
The primary limitation of enzymatic synthesis lies in its current commercial availability and throughput compared to established chemical synthesis platforms . While enzymatic methods can produce longer contiguous sequences (exceeding 700 bases) with lower error rates and without toxic waste generation, the technology is still emerging and may not yet match the overall yield of chemical synthesis for very large-scale projects . Additionally, secondary structures in complex sequences, such as the GC-rich regulatory elements in neural promoters, can interfere with enzymatic activity, requiring careful sequence design and validation.
5.3 DNA Edit
Questions
(i) What DNA would you want to edit and why?
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Answer
I would like to investigate the therapeutic potential of modulating FOXG1 expression in neurodevelopmental disorders, therefore I would edit the endogenous FOXG1 locus in human induced pluripotent stem cells (iPSCs) to correct pathogenic mutations associated with FOXG1 syndrome . FOXG1 syndrome results from heterozygous mutations causing haploinsufficiency, leading to severe brain structural abnormalities including corpus callosum agenesis, hippocampal malformation, and myelination deficits . Correcting the mutated allele to wild-type sequence in patient-derived iPSCs would restore normal FOXG1 dosage and allow subsequent differentiation into neural progenitors and neurons, providing a platform to study whether genetic correction rescues the cellular and molecular phenotypes observed in FOXG1-deficient neural development . This approach has direct relevance to regenerative medicine, as it could establish proof-of-concept for autologous cell replacement therapies and disease modeling.
I would employ the CRISPR/Cas9 system delivered via adeno-associated virus vectors to perform these edits . The technology edits DNA by introducing a targeted double-strand break at the FOXG1 locus, which is then repaired through homology-directed repair using an exogenous donor DNA template containing the corrected sequence .
The essential steps include:
design and synthesis of variant-specific single-guide RNAs (sgRNAs) complementary to the target mutation site and a donor DNA template encoding the wild-type FOXG1 sequence ; (
cloning of sgRNA and donor template into AAV vectors (with AAV9 preferred for neuronal applications)
delivery of AAV-CRISPR components into patient-derived iPSCs or fibroblasts
validation of successful editing through next-generation sequencing of the target locus. The input materials include sgRNAs, Cas9 endonuclease (delivered as protein or encoded in the vector), donor DNA template, and the target cells for editing .
Regarding to limitations, this editing approach include efficiency and precision challenges. Homology-directed repair in iPSCs and neurons is relatively inefficient, with reported correction rates of 20-35% in successfully transfected cells . Additionally, off-target activity remains a concern despite careful sgRNA design, as Cas9 can cleave at genomic sites with sequence similarity to the target . Delivery efficiency varies by cell type and AAV serotype, with AAV9 showing optimal transduction in fibroblasts and iPSC-derived neurons but lower efficiency in undifferentiated iPSCs . Furthermore, the risk of unintended insertions or deletions at the target site from non-homologous end joining competing with homology-directed repair can introduce additional mutations . These limitations necessitate rigorous validation through deep sequencing and clonal selection to ensure precise correction without off-target effects before therapeutic application.
Week 3 HW: Lab Automation
Assignment: Python Script for Opentrons Artwork
Python file to run on an Opentrons liquid handling robot
from opentrons import types
import string
metadata = {
'protocolName': '{YOUR NAME} - Opentrons Art - HTGAA',
'author': 'HTGAA',
'source': 'HTGAA 2026',
'apiLevel': '2.20'
}
Z_VALUE_AGAR = 2.0
POINT_SIZE = 0.75
venus_points = [(-3.3,23.1), (-1.1,23.1), (1.1,23.1), (3.3,23.1), (5.5,23.1), (7.7,23.1), (9.9,23.1), (12.1,23.1), (-3.3,20.9), (-1.1,20.9), (-7.7,18.7), (-5.5,18.7), (16.5,18.7), (-16.5,16.5), (-14.3,16.5), (-12.1,16.5), (-9.9,16.5), (-7.7,16.5), (18.7,16.5), (-16.5,14.3), (20.9,14.3), (-23.1,12.1), (-20.9,12.1), (-18.7,12.1), (23.1,12.1), (-23.1,9.9), (25.3,9.9), (-25.3,7.7), (-25.3,5.5), (27.5,5.5), (-25.3,3.3), (29.7,3.3), (-25.3,1.1), (29.7,1.1), (-25.3,-1.1), (29.7,-1.1), (-25.3,-3.3), (29.7,-3.3), (-23.1,-5.5), (29.7,-5.5), (-20.9,-7.7), (29.7,-7.7), (-18.7,-9.9), (29.7,-9.9), (-16.5,-12.1), (-14.3,-12.1), (27.5,-12.1), (-12.1,-14.3), (9.9,-14.3), (25.3,-14.3), (-9.9,-16.5), (-7.7,-16.5), (7.7,-16.5), (12.1,-16.5), (20.9,-16.5), (23.1,-16.5), (-5.5,-18.7), (14.3,-18.7), (16.5,-18.7), (18.7,-18.7), (-3.3,-20.9), (-1.1,-20.9), (1.1,-20.9), (3.3,-20.9), (5.5,-20.9), (7.7,-20.9), (-5.5,-23.1), (3.3,-23.1), (-5.5,-25.3), (-3.3,-25.3), (-1.1,-25.3), (1.1,-25.3), (3.3,-25.3), (5.5,-25.3)]
mjuniper_points = [(1.1,20.9), (3.3,20.9), (5.5,20.9), (7.7,20.9), (9.9,20.9), (12.1,20.9), (-3.3,18.7), (-1.1,18.7), (14.3,18.7), (-5.5,16.5), (16.5,16.5), (-14.3,14.3), (-12.1,14.3), (-9.9,14.3), (-7.7,14.3), (18.7,14.3), (-16.5,12.1), (20.9,12.1), (-20.9,9.9), (-7.7,9.9), (-5.5,9.9), (3.3,9.9), (5.5,9.9), (23.1,9.9), (-23.1,7.7), (-9.9,7.7), (-3.3,7.7), (1.1,7.7), (7.7,7.7), (25.3,7.7), (-23.1,5.5), (18.7,5.5), (20.9,5.5), (25.3,5.5), (-23.1,3.3), (-14.3,3.3), (-12.1,3.3), (14.3,3.3), (18.7,3.3), (20.9,3.3), (27.5,3.3), (-23.1,1.1), (-14.3,1.1), (-12.1,1.1), (14.3,1.1), (18.7,1.1), (20.9,1.1), (23.1,1.1), (27.5,1.1), (-23.1,-1.1), (12.1,-1.1), (18.7,-1.1), (20.9,-1.1), (23.1,-1.1), (27.5,-1.1), (-23.1,-3.3), (12.1,-3.3), (16.5,-3.3), (18.7,-3.3), (20.9,-3.3), (23.1,-3.3), (27.5,-3.3), (-20.9,-5.5), (5.5,-5.5), (12.1,-5.5), (16.5,-5.5), (18.7,-5.5), (20.9,-5.5), (23.1,-5.5), (27.5,-5.5), (3.3,-7.7), (12.1,-7.7), (16.5,-7.7), (18.7,-7.7), (20.9,-7.7), (23.1,-7.7), (27.5,-7.7), (-16.5,-9.9), (1.1,-9.9), (12.1,-9.9), (16.5,-9.9), (18.7,-9.9), (20.9,-9.9), (27.5,-9.9), (-12.1,-12.1), (9.9,-12.1), (12.1,-12.1), (25.3,-12.1), (-9.9,-14.3), (-7.7,-14.3), (7.7,-14.3), (12.1,-14.3), (14.3,-14.3), (20.9,-14.3), (23.1,-14.3), (-5.5,-16.5), (-3.3,-16.5), (5.5,-16.5), (14.3,-16.5), (16.5,-16.5), (18.7,-16.5), (-3.3,-18.7), (-1.1,-18.7), (1.1,-18.7), (3.3,-18.7), (5.5,-18.7), (-5.5,-20.9), (-3.3,-23.1), (-1.1,-23.1), (1.1,-23.1), (5.5,-23.1), (7.7,-23.1)]
sfgfp_points = [(14.3,20.9), (16.5,20.9), (-14.3,12.1), (-18.7,9.9), (-18.7,-7.7)]
mturquoise2_points = [(-16.5,9.9), (-16.5,3.3), (-16.5,1.1), (-7.7,-7.7), (-5.5,-9.9), (-3.3,-9.9), (-1.1,-9.9)]
point_name_pairing = [("venus", venus_points),("mjuniper", mjuniper_points),("sfgfp", sfgfp_points),("mturquoise2", mturquoise2_points)]
# Robot deck setup constants
TIP_RACK_DECK_SLOT = 9
COLORS_DECK_SLOT = 6
AGAR_DECK_SLOT = 5
PIPETTE_STARTING_TIP_WELL = 'A1'
# Place the PCR tubes in this order
well_colors = {
'A1': 'sfGFP',
'A2': 'mRFP1',
'A3': 'mKO2',
'A4': 'Venus',
'A5': 'mKate2_TF',
'A6': 'Azurite',
'A7': 'mCerulean3',
'A8': 'mClover3',
'A9': 'mJuniper',
'A10': 'mTurquoise2',
'A11': 'mBanana',
'A12': 'mPlum',
'B1': 'Electra2',
'B2': 'mWasabi',
'B3': 'mScarlet_I',
'B4': 'mPapaya',
'B5': 'eqFP578',
'B6': 'tdTomato',
'B7': 'DsRed',
'B8': 'mKate2',
'B9': 'EGFP',
'B10': 'mRuby2',
'B11': 'TagBFP',
'B12': 'mChartreuse_TF',
'C1': 'mLychee_TF',
'C2': 'mTagBFP2',
'C3': 'mEGFP',
'C4': 'mNeonGreen',
'C5': 'mAzamiGreen',
'C6': 'mWatermelon',
'C7': 'avGFP',
'C8': 'mCitrine',
'C9': 'mVenus',
'C10': 'mCherry',
'C11': 'mHoneydew',
'C12': 'TagRFP',
'D1': 'mTFP1',
'D2': 'Ultramarine',
'D3': 'ZsGreen1',
'D4': 'mMiCy',
'D5': 'mStayGold2',
'D6': 'PA_GFP'
}
volume_used = {
'venus': 0,
'mjuniper': 0,
'sfgfp': 0,
'mturquoise2': 0
}
def update_volume_remaining(current_color, quantity_to_aspirate):
rows = string.ascii_uppercase
for well, color in list(well_colors.items()):
if color == current_color:
if (volume_used[current_color] + quantity_to_aspirate) > 250:
# Move to next well horizontally by advancing row letter, keeping column number
row = well[0]
col = well[1:]
# Find next row letter
next_row = rows[rows.index(row) + 1]
next_well = f"{next_row}{col}"
del well_colors[well]
well_colors[next_well] = current_color
volume_used[current_color] = quantity_to_aspirate
else:
volume_used[current_color] += quantity_to_aspirate
break
def run(protocol):
# Load labware, modules and pipettes
protocol.home()
# Tips
tips_20ul = protocol.load_labware('opentrons_96_tiprack_20ul', TIP_RACK_DECK_SLOT, 'Opentrons 20uL Tips')
# Pipettes
pipette_20ul = protocol.load_instrument("p20_single_gen2", "right", [tips_20ul])
# PCR Plate
temperature_plate = protocol.load_labware('opentrons_96_aluminumblock_generic_pcr_strip_200ul', 6)
# Agar Plate
agar_plate = protocol.load_labware('htgaa_agar_plate', AGAR_DECK_SLOT, 'Agar Plate')
agar_plate.set_offset(x=0.00, y=0.00, z=Z_VALUE_AGAR)
# Get the top-center of the plate, make sure the plate was calibrated before running this
center_location = agar_plate['A1'].top()
pipette_20ul.starting_tip = tips_20ul.well(PIPETTE_STARTING_TIP_WELL)
# Helper function (dispensing)
def dispense_and_jog(pipette, volume, location):
assert(isinstance(volume, (int, float)))
# Go above the location
above_location = location.move(types.Point(z=location.point.z + 2))
pipette.move_to(above_location)
# Go downwards and dispense
pipette.dispense(volume, location)
# Go upwards to avoid smearing
pipette.move_to(above_location)
# Helper function (color location)
def location_of_color(color_string):
for well,color in well_colors.items():
if color.lower() == color_string.lower():
return temperature_plate[well]
raise ValueError(f"No well found with color {color_string}")
# Print pattern by iterating over lists
for i, (current_color, point_list) in enumerate(point_name_pairing):
# Skip the rest of the loop if the list is empty
if not point_list:
continue
# Get the tip for this run, set the bacteria color, and the aspirate bacteria of choice
pipette_20ul.pick_up_tip()
max_aspirate = int(18 // POINT_SIZE) * POINT_SIZE
quantity_to_aspirate = min(len(point_list)*POINT_SIZE, max_aspirate)
update_volume_remaining(current_color, quantity_to_aspirate)
pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color))
# Iterate over the current points list and dispense them, refilling along the way
for i in range(len(point_list)):
x, y = point_list[i]
adjusted_location = center_location.move(types.Point(x, y))
dispense_and_jog(pipette_20ul, POINT_SIZE, adjusted_location)
if pipette_20ul.current_volume == 0 and len(point_list[i+1:]) > 0:
quantity_to_aspirate = min(len(point_list[i:])*POINT_SIZE, max_aspirate)
update_volume_remaining(current_color, quantity_to_aspirate)
pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color))
# Drop tip between each color
pipette_20ul.drop_tip()
Post-Lab Questions
Use of automation in final project
Project: A Cell-Free Paper-Based Assay for Rapid Population Genotyping
This project will utilize cloud laboratory automation to streamline the design and optimization of toehold switch RNA sensors. The core idea is to move from a manual, low-throughput testing process to an automated, high-throughput workflow that can rapidly evaluate hundreds of sensor variants against multiple target conditions. This approach will accelerate the identification of highly specific sensors that can distinguish between populations based on single nucleotide polymorphisms (SNPs).
The automated workflow will systematically test sensor variants against target and non-target sequences to identify the most specific and sensitive candidates for field deployment. By automating the iterative design-build-test cycle, we can rapidly screen hundreds of sensor designs that would be impractical to test manually.
Use Ginkgo’s automated DNA design tools to generate a library of toehold switch variants targeting a population-specific SNP in humpback whale mitochondrial DNA
Each variant will incorporate sequence variations around the SNP to optimize discrimination
Expected Outcomes:
The automated workflow will deliver a validated set of 3-5 highly specific toehold sensors ready for field deployment. Ultimately we would be making advanced genetic analysis accessible for conservation applications in remote locations.
Summary of paper
Paper DOI: 10.1016/j.mcpro.2024.100790
Fully Automated Workflow for Integrated Sample Digestion and Evotip Loading Enabling High-Throughput Clinical Proteomics
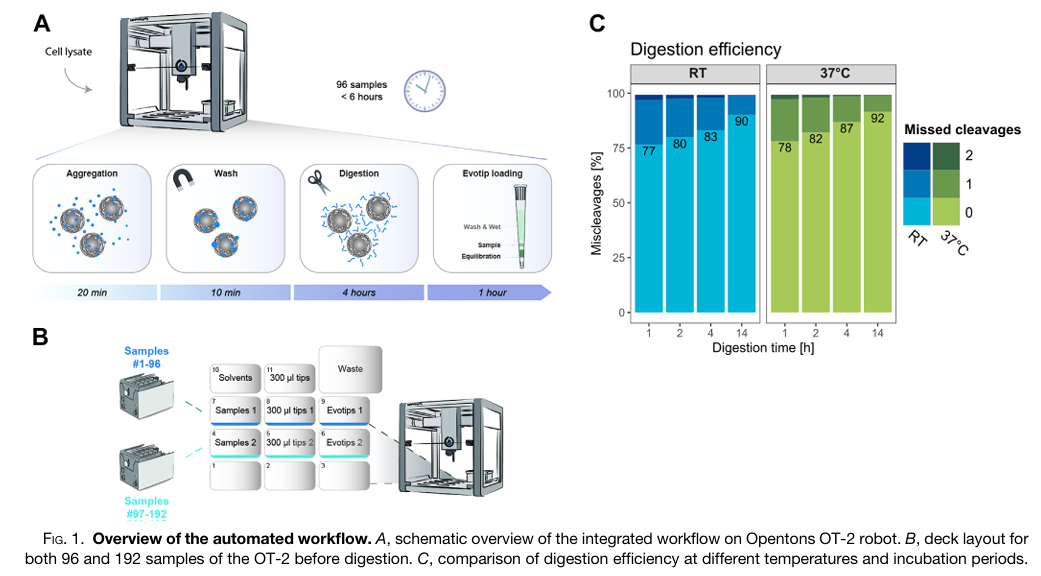
The study aimed to address a major bottleneck in clinical proteomics, meaning the manual, time-consuming, and often variable preparation of samples for mass spectrometry analysis. The authors sought to develop and validate a fully automated, high-throughput workflow using the affordable and flexible Opentrons OT-2 liquid handling robot. Their primary goal was to create an end-to-end process that could handle hundreds of samples simultaneously, from protein digestion to final loading onto Evotips for LC-MS/MS analysis, with minimal human intervention. They also aimed to expand this automated workflow to include phosphopeptide enrichment, enabling large-scale studies of cellular signaling.
To achieve this, the researchers designed a protocol on the OT-2 that integrates magnetic bead-based protein aggregation capture (PAC) digestion with direct loading of the resulting peptides onto Evotips. This method replaces traditional centrifugation steps with positive pressure, allowing the robot to process up to 192 samples in parallel within approximately six hours. They rigorously tested the system’s performance using HeLa cell lysates and human plasma samples, analyzing the results with high-speed Orbitrap mass spectrometers. Furthermore, they adapted the protocol to incorporate an automated step for enriching phosphorylated peptides using magnetic Ti-IMAC beads, demonstrating its utility in a time-course study of cellular response to drug treatment.
The automated workflow proved to be highly robust and reproducible, consistently identifying approximately 8,000 protein groups and 130,000 peptides from just 1 µg of HeLa protein digest across 192 samples. In a pilot clinical study with plasma samples from 48 metastatic melanoma patients, the robot-enabled preparation allowed for the quantification of over 600 protein groups, revealing potential biomarker candidates that differentiated responders from non-responders to immune therapy. The integrated phosphoproteomics workflow also successfully identified over 8,000 phosphorylation sites from as few as 40,000 cells and accurately mapped dynamic signaling changes in response to anisomycin treatment. The use of the Opentrons OT-2 robot was central to this research, dramatically increasing throughput and reproducibility while reducing reagent costs and hands-on time, thereby demonstrating its potential to unlock large-scale, clinically-relevant proteomic and phosphoproteomic studies.
Final Project ideas
Week 4 HW: Protein Design Part I
Homework Questions
Part A. Conceptual Questions
Why humans eat beef but do not become a cow, eat fish but do not become fish?
Because digestion breaks proteins into individual amino acids, so it means it destroys the cow’s structure and information. We absorb the amino acids and then our ribosomes rebuild human proteins based on human DNA. To make it clear, biological identity comes from genetic information, not raw material.
Why there are only 20 natural amino acids?
The standard set of 20 amino acids encoded by the nearly universal genetic code reflects evolutionary optimization rather than chemical inevitability. These amino acids provide a chemically diversity, varying in size, charge, hydrophobicity, aromaticity, and hydrogen-bonding potential. This set is sufficient to support stable folding, catalysis, and structural complexity while maintaining translational fidelity.
Can you make other non-natural amino acids? Design some new amino acids.
Yes! non-natural amino acids can be designed by modifying some side chains while preserving the α-amino and α-carboxylate backbone.
Synthetic biology approaches enable site-specific incorporation via engineered tRNA/synthetase pairs, so this way expands the genetic code and enabling novel materials or catalytic functions.
Where did amino acids come from before enzymes that make them, and before life started?
Before enzymatic biosynthesis, amino acids likely formed through abiotic chemical processes. The Miller–Urey experiment demonstrated that electrical discharge in reducing gas mixtures can produce amino acids. Additional pathways include synthesis in hydrothermal vent systems, Strecker-type reactions in aqueous environments, and extraterrestrial delivery via carbonaceous meteorites.
If you make an alpha-helix using D-amino acids, what handedness (right or left) would you expect?
Proteins composed of L-amino acids form predominantly right-handed α-helices due to stereochemical constraints of the backbone. If one constructed a polypeptide exclusively from D-amino acids, the mirror-image geometry would be favored, yielding predominantly left-handed α-helices. This inversion follows directly from chirality at the α-carbon and the allowed φ/ψ torsion angles.
Can you discover additional helices in proteins?
Beyond the α-helix, proteins contain other helical conformations such as the 3₁₀ helix and π-helix. Novel helices can be identified computationally by analyzing backbone dihedral angle distributions in high-resolution structural databases (e.g., Protein Data Bank) or experimentally via X-ray crystallography and cryo-electron microscopy. De novo protein design and foldamer chemistry further enable exploration of alternative helical geometries, including those based on non-canonical backbones.
Why most molecular helices are right-handed?
The predominance of right-handed helices in biological systems arises from the homochirality of L-amino acids. Given uniform chirality, steric constraints and favorable hydrogen-bond geometry bias the α-helix toward right-handedness. In a mirror-symmetric world of D-amino acids, left-handed helices would dominate. Thus, helix handedness reflects fundamental stereochemical asymmetry in biomolecular building blocks.
Why do beta-sheets tend to aggregate?
What is the driving force for b-sheet aggregation?
β-sheets possess extended backbone conformations that maximize inter-strand hydrogen bonding. When partially unfolded polypeptides expose β-prone segments, they can associate intermolecularly via backbone hydrogen bonds, forming extended β-sheet assemblies. Side-chain complementarity and hydrophobic interactions further stabilize aggregation.
Driving force - The primary driving force is the formation of extensive intermolecular hydrogen-bond networks along the peptide backbone, combined with hydrophobic collapse and favorable packing of side chains. The resulting cross-β architecture is thermodynamically stable and often kinetically persistent.
Why many amyloid diseases form b-sheet?
Can you use amyloid b-sheets as materials?
Amyloid fibrils share a common cross-β structural motif, in which β-strands align perpendicular to the fibril axis. This structure maximizes backbone hydrogen bonding and produces highly stable, self-templating aggregates. In diseases such as Alzheimer’s disease and Parkinson’s disease, normally soluble proteins misfold and adopt β-rich conformations that nucleate fibril formation.
Amyloid β-sheets as materials: Due to their mechanical strength, nanoscale order, and self-assembly, amyloid fibrils are being explored as biomaterials for nanowires, hydrogels, and tissue scaffolds. Their stability and tunable assembly make them promising in bioengineering applications.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins.
Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions.
1. Briefly describe the protein you selected and why you selected it.
Carboxyltransferase subunit of Mycobacterium tuberculosis acetyl-CoA carboxylase
I chose this protein because of its central role in building the unique and protective cell wall of M. tuberculosis. ACC’s job is to convert acetyl-CoA to malonyl-CoA. In M. tuberculosis , this malonyl-CoA is the fundamental building block for synthesizing the long fatty acid chains needed for its cell wall, including the very long mycolic acids.
Component of a biotin-dependent acyl-CoA carboxylase complex. This subunit transfers the CO2 from carboxybiotin to the CoA ester substrate. When associated with the alpha3 subunit AccA3, is involved in the carboxylation of acetyl-CoA and propionyl-CoA, with a preference for acetyl-CoA
Information taken from UniProt
2. Identify the amino acid sequence of your protein.
The length of the protein is: 493 aminoacids.
The most common amino acid is: A, which appears 61 times.
Sequence length for P9WQH5 (Acetyl-CoA carboxylase): 473 amino acids
Over 245 homologous sequences were identified across different Mycobacterium species and strains
This protein belongs to the AccD/PCCB family. This family comprises enzymes involved in fatty acid and amino acid metabolism. They function as carboxyltransferases, which are part of larger enzyme complexes that catalyze the carboxylation of various CoA ester substrates.
3. Identify the structure page of your protein in RCSB
Are there any other molecules in the solved structure apart from protein?
Does your protein belong to any structure classification family?
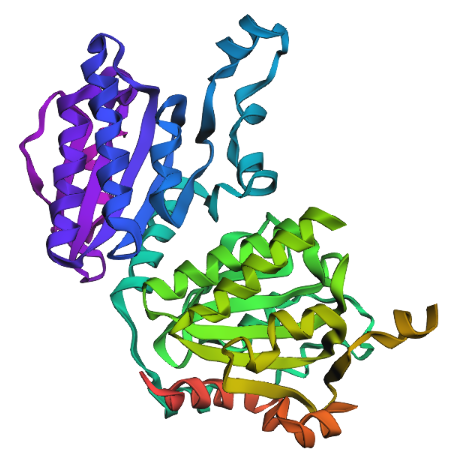
RCSB 3D Structure
Released: 2013-07-03
Resolution: 1.95 Å –> Good! :) Indicates high structural quality.
The structure contains Mg²⁺ and ATP in addition to the protein.
Structural classification: Ligase
Open the structure of your protein in any 3D molecule visualization software:
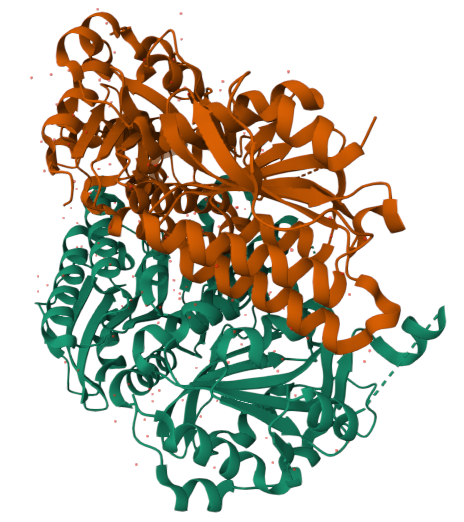
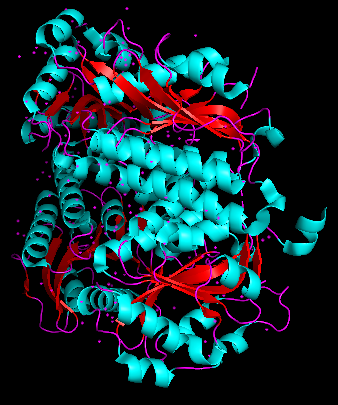
Cartoon
Colored by secondary structue
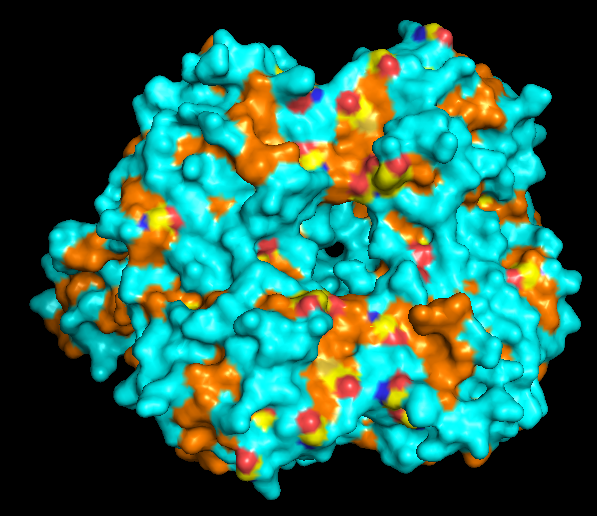
The protein is predominantly alpha-helical with only a few beta-sheets region. This is evidenced by the cyan sheets present.
When colored by residue type, hydrophobic residues appear to cluster in the interior of the protein (orange), while polar and charged residues are more solvent-exposed (cyan). This is consistent with proper folding of a soluble enzyme.
Visualization of the molecular surface reveals a compact, globular structure with noticeable surface depressions and grooves. One elongated cleft with visible depth can be observed, which may correspond to the substrate-binding site. While no large enclosed cavities are present, the surface topology suggests the presence of shallow binding pockets typical of enzymatic proteins.
Part C. Using ML-Based Protein Design Tools
Protein Language modeling
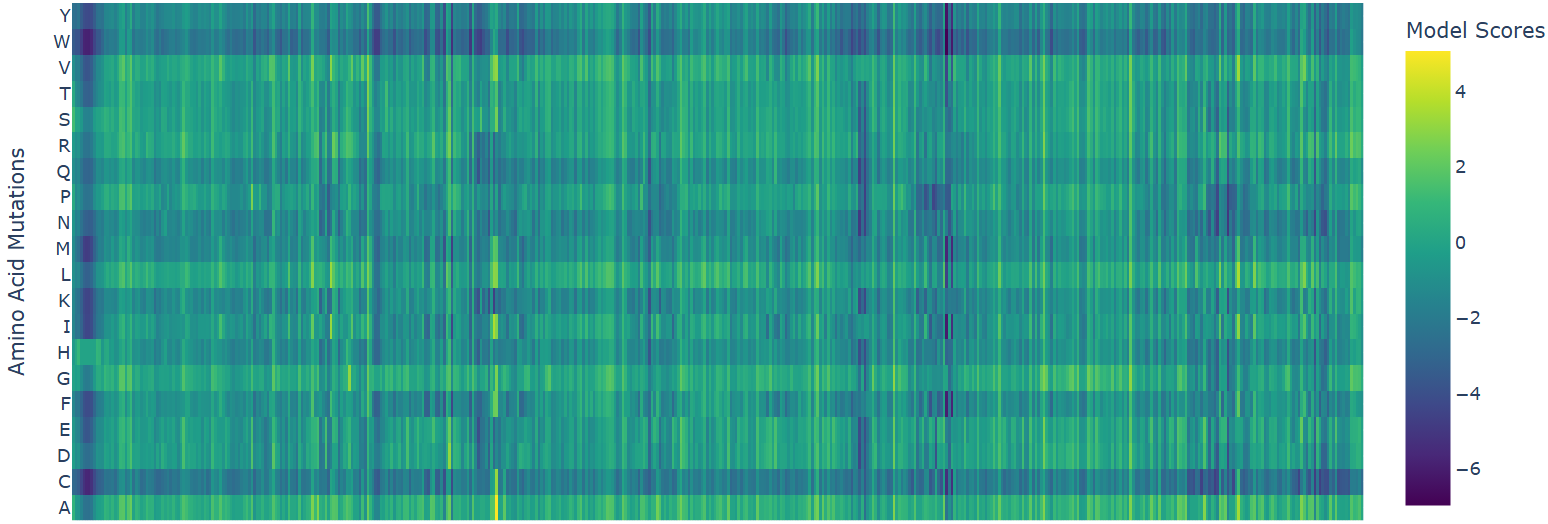
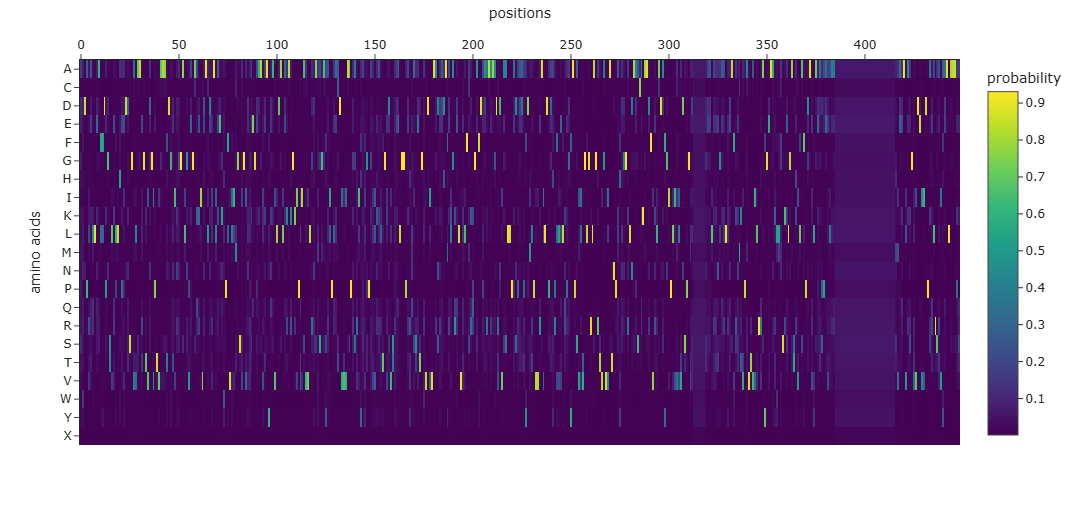
1. deep mutational scan
Heatmap
W (Tryptophan) shows interesting patterns - mutations to certain residues like S, R, Q have higher scores
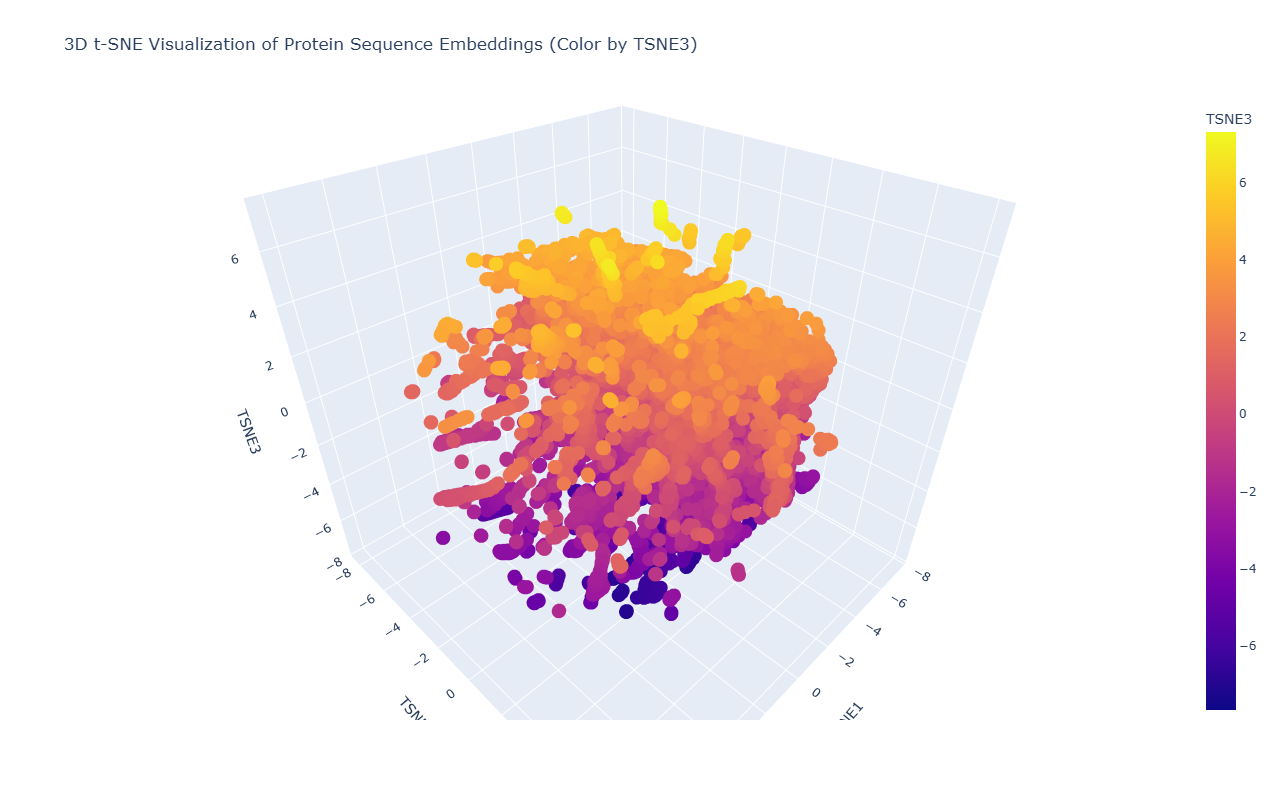
2. Latent Space Analysis
Due to the high density and overlap of points in the t-SNE visualization, it is difficult to precisely locate the position of acetyl-CoA carboxylase from Mycobacterium tuberculosis. However, based on its biological function as a key enzyme in fatty acid biosynthesis, it would be expected to cluster with other metabolic enzymes, particularly carboxylases and proteins involved in lipid metabolism.
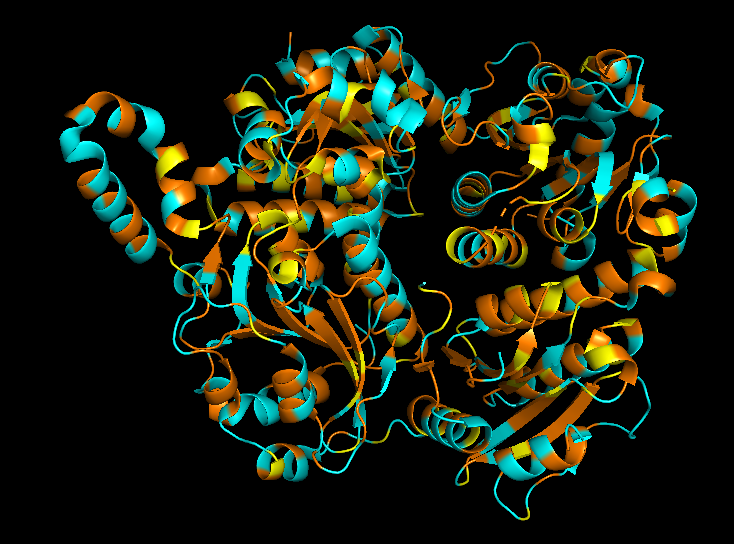
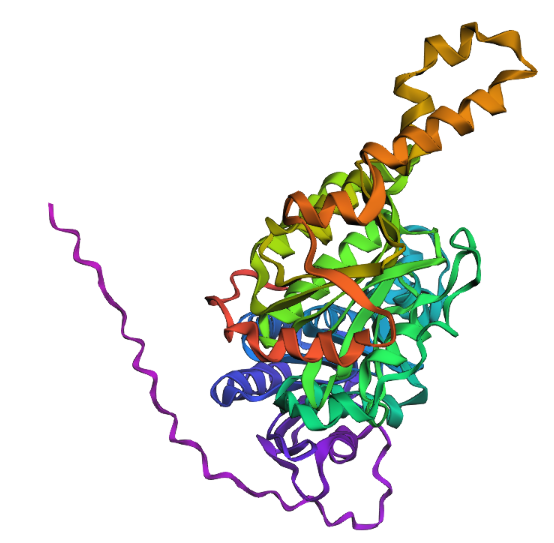
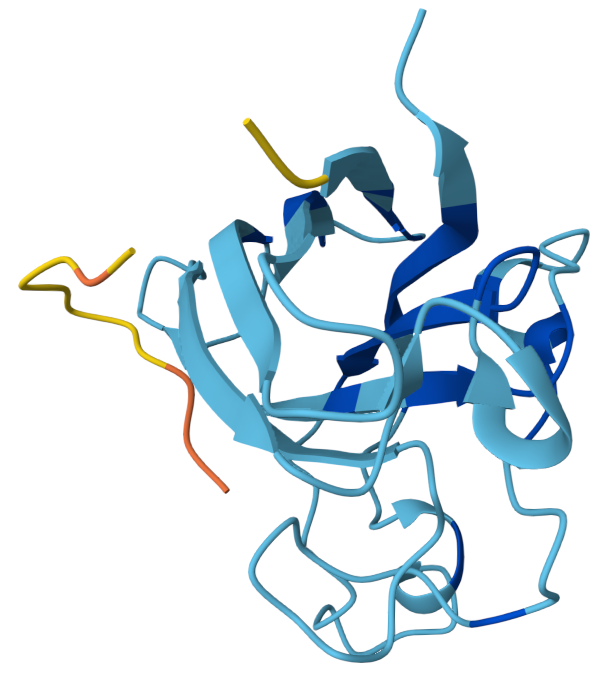
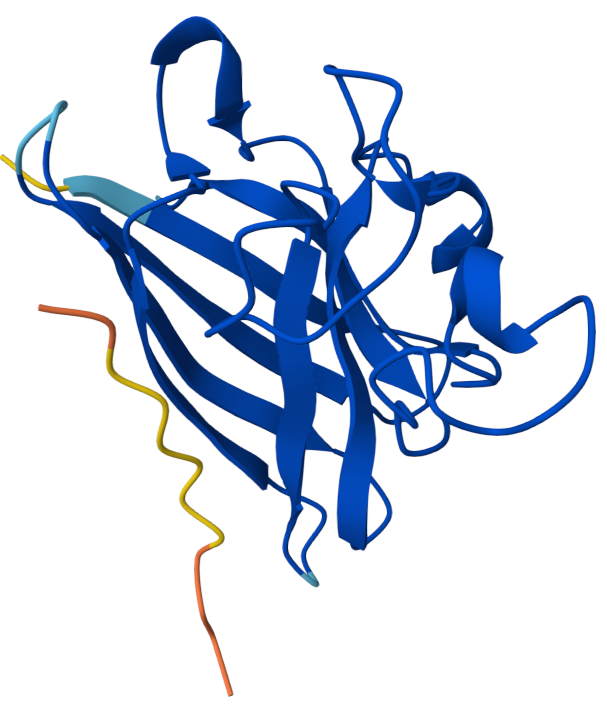
3. Protein folding
Observation: The overall fold and general domain organization are somewhat preserved, indicating that the model captures key structural features of the protein. However, the ESMFold prediction appears less compact and shows greater deviation in the arrangement of secondary structure elements compared to the reference structure.
Many positions in the protein show low specificity, where no single amino acid strongly dominates. This suggests that these regions are more tolerant to mutations and may correspond to flexible or less critical structural regions.
The protein appears to contain both highly conserved regions and more variable segments, reflecting a balance between structural stability and sequence adaptability.
New Sequence: AWDPLELLEAFFDPGSVRLLHPPDDSGVLAARGTVDGIPTVAAATDGTINGGALGPAGAQKIVDALDEALAEGWPFVLIMGSVGVLRAEGEAAKKAYQAVLDALEKAKGKIPLIAVVLRNALSGAYRIPLACDVVVMAPEAVLAEIGPGVLKALTGVDVSWEELGGPERQASETGAVDIVAADRADAFALARELVRLFAAPGAFDAAAAAAADVDVASLLPASPEDPYDMLPVVRALLDPDAPLLVLQPRYAPSVLVGLGRLAGRTVGFIATNPAHDGGRLNAASCAKAARFVELADAFGIPLVVVVDSPGTLXXXXXXGAALAEAGARLRAAFAACKVPAVTLIVRRAYGEAAELLASKSLGATKVLAFPDAVLAEASPEVLAAXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXMVEEAVKEGRVDEIIDPAKARSAIAAALAAAPA
Protein Generation
this new sequence is already an engineered version that prospectively aligns with the original one.
Part D. Group Brainstorm on Bacteriophage Engineering
Review the Bacteriophage Final Project Goals:
Increased stability (easiest)
Main Goal: Increase stability of the MS2 L lysis protein through computational design to enhance phage therapeutic efficacy, while maintaining/improving its lytic function.
Identification of Stability-Limiting Regions:The MS2 L protein has a highly basic, unstructured N-terminal domain (Domain 1, residues 1-36) that confers DnaJ dependence and may contribute to proteolytic instability. The essential C-terminal domain (residues 37-75) contains the critical LS motif (Leu48-Ser49) that mediates target interaction but may have suboptimal stability.
Rational Stabilization Without Function Loss: Mutations that increase stability must not disrupt:
The essential LS motif interaction interface
Membrane insertion capability
Oligomerization potential (≥10 monomers)
The regulatory timing mechanism
Balancing Stability with Controllable Lysis Timing: The N-terminal domain acts as a regulatory “damping” feature—complete stabilization/removal causes premature lysis (~20 min earlier), which may reduce phage progeny production. Optimal therapeutic phages require balanced lysis timing.
The computational enhancement of MS2 L lysis protein stability will be executed through a pipeline integrating sequence analysis, structure prediction, in silico mutagenesis, interaction validation, and candidate ranking.
In Phase 1, sequence analysis begins with BLASTp to identify L protein homologs across Leviviridae, followed by Clustal-Omega multiple sequence alignment anchored by the conserved LS motif identified in previous mutational studies.
Phase 2 employs structure prediction using ESMFold and ESM3, which leverage evolutionary data to model L’s conformation, complemented by Boltz-1 for generating conformational ensembles that capture the protein’s behavior in membrane environments.
AlphaFold-Multimer predicts oligomerization interfaces based on evidence that L forms high-order complexes of at least ten monomers, while FoldSeek searches for structural homologs with known stabilizing features that could inform design.
Phase 3 conducts in silico mutagenesis using ESM2 zero-shot prediction to score missense variants for evolutionary fitness, ProteinMPNN for sequence redesign of variable regions while fixing the essential LS motif, EvolvePro for directed evolution simulation, and Rosetta ddG_monomer for quantitative ΔΔG stability calculations. T
Phase 4 validates interaction interfaces using BindCraft to predict preservation of L-target binding, AlphaFold3 for L-DnaJ docking to verify retention of regulatory chaperone interaction, and molecular docking with HADDOCK to ensure stabilizing mutations do not disrupt essential contacts with the unknown lytic target.
Phase 5 integrates all outputs through a custom Python pipeline that scores each candidate mutation on stability change, evolutionary conservation, predicted function retention and DnaJ binding preservation, generating a weighted ranking of the top five to ten stabilizing mutations for experimental testing.
The known binder FLYRWLPSRRGG contains several hydrophobic and positively charged residues, which are commonly observed in the generated peptides as well, suggesting that PepMLM captures sequence patterns associated with binding.
Part 2: Evaluate Binders with AlphaFold3
Binder 1: WHYPAVAVALGX ipTM: 0.34
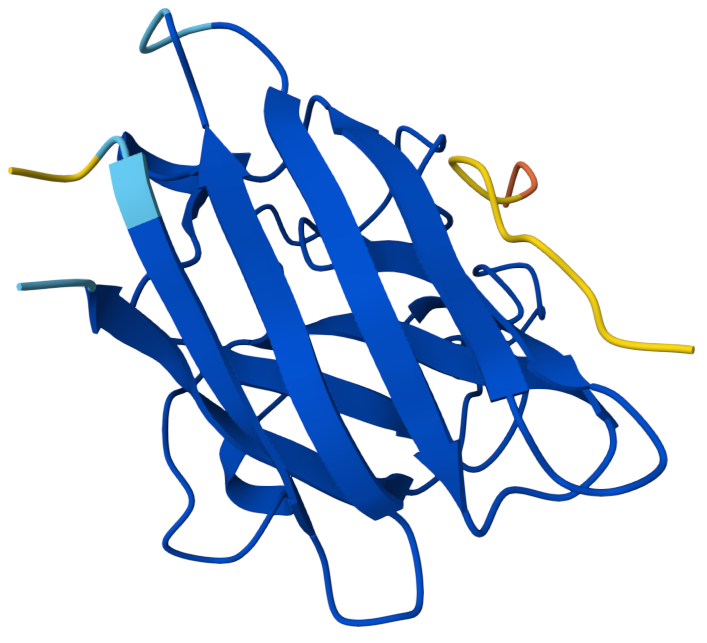
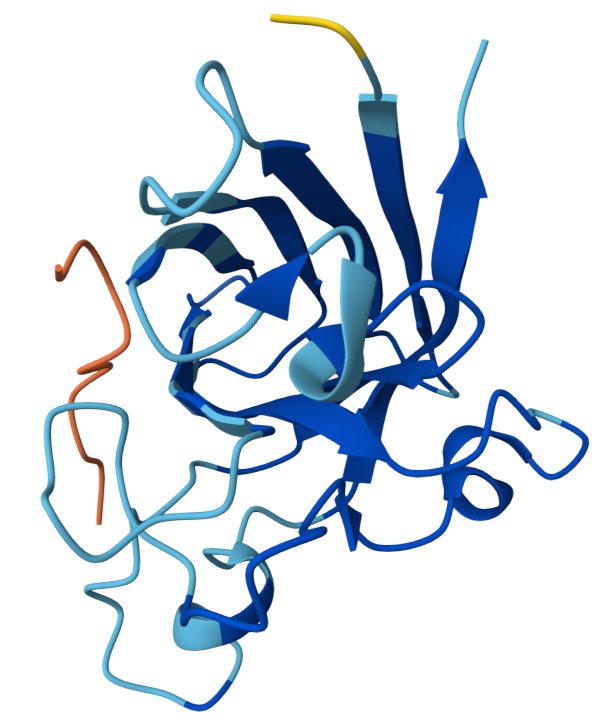
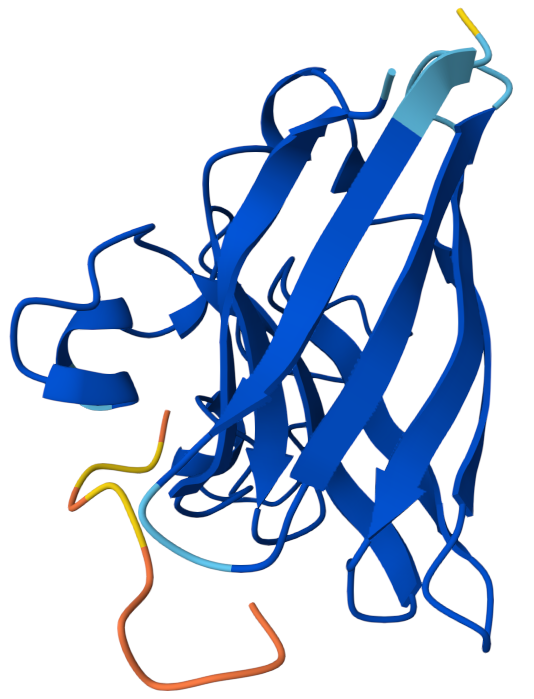
The peptide appears mostly surface-adjacent but not tightly bound to SOD1. The peptide does not appear to bind near the N-terminal region where the A4V mutation occurs. The peptide approaches the surface of the β-barrel but does not appear to form a strong interface. The peptide does not appear to approach the dimer interface in this model. The predicted complex has an ipTM score of 0.34, which indicates weak confidence in the protein–peptide interaction.
Binder 2: KRYYVVGVRHKE ipTM: 0.45
ipTM score of 0.45 indicates weak to moderate confidence in the predicted interaction. The peptide appears positioned near the outer surface of the SOD1 structure but does not form a tightly packed interface with the protein. It does not localize close to the N-terminal region where the A4V mutation occurs, which is located on a flexible loop extending from the protein. Instead, the peptide lies adjacent to the β-barrel surface, suggesting a potential but weak surface interaction rather than deep binding. The peptide appears surface-bound and largely exposed, rather than buried within a pocket.
Binder 3: HHYYVAAVELGX ipTM:0.41
This has an ipTM score of 0.41, means low confidence in the protein–peptide interaction. In the model, the peptide is positioned along the side of the β-barrel structure of SOD1 but remains partially separated from the protein surface. It does not appear to interact near the N-terminal region containing the A4V mutation. Instead, the peptide lies adjacent to the barrel-shaped core of the protein and does not appear to insert into any pocket or groove. The peptide appears loosely associated with the protein surface rather than buried, suggesting weak or transient binding.
Binder 4: WRYPPVAARHKX ipTM: 0.37
The ipTM score suggests weak confidence in the predicted interaction. The peptide is positioned near the surface loops of the SOD1 structure but does not appear to form a stable interface with the protein. It does not localize near the N-terminus where the A4V mutation is located, and instead lies along flexible surface regions away from the mutation site. The peptide approaches the outer surface of the β-barrel region, but it remains mostly exposed and does not appear to penetrate into a binding pocket. Overall, the peptide appears surface-bound and partially detached, indicating weak predicted binding.
Known Binder: FLYRWLPSRRGG iPTM: 0.38
Although the peptide FLYRWLPSRRGG is experimentally known to bind SOD1, the AlphaFold prediction still yields a relatively low ipTM score and a loosely associated binding pose. This highlights that AlphaFold is not specifically optimized for predicting short peptide–protein interactions, and therefore may underestimate the stability of such complexes.
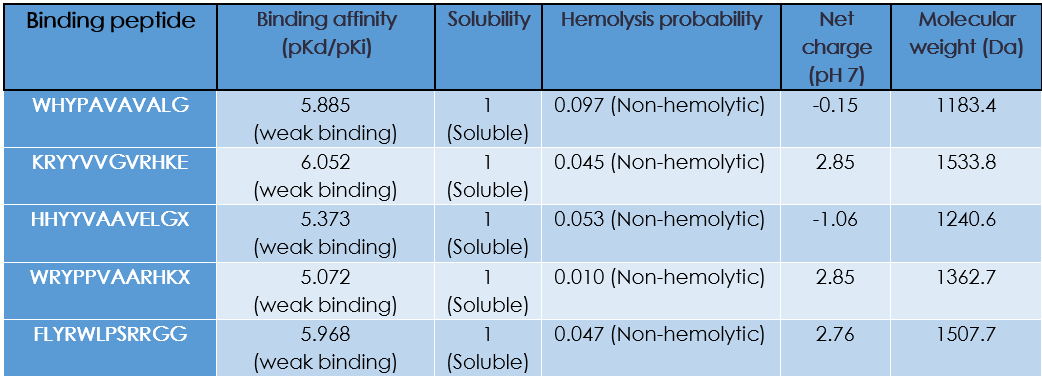
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Do peptides with higher ipTM also show stronger predicted affinity?
Yes!
Are strong binders hemolytic or poorly soluble?
They are non-hemolytic and soluble
Part 4: Generate Optimized Peptides with moPPIt
Part B: BRD4 Drug Discovery Platform Tutorial
Part C: Final Project: L-Protein Mutants
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
DNA Assembly Part
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix key components include:
Phusion DNA Polymerase: This is a fusion protein comprising a traditional Pfu-like DNA polymerase and a processivity-enhancing domain. Its purpose is to synthesize new DNA strands with exceptionally high fidelity (due to its inherent 3’→5’ proofreading exonuclease activity) and high speed.
Deoxynucleotide Triphosphates (dNTPs): These are the blocks that the polymerase uses as substrates to construct the new complementary DNA strands.
Reaction Buffer: The buffer maintains the optimal pH for enzyme activity. The magnesium ions in it act as an essential cofactor for polymerase function and influence primer annealing by stabilizing the interaction between the primer and the DNA template.
Stabilizers and Enhancers: These proprietary compounds (sugars or detergents) help maintain enzyme stability during thermal cycling and can aid in amplifying difficult templates by reducing secondary structures or preventing non-specific primer binding.
2. What are some factors that determine primer annealing temperature during PCR?
Melting Temperature: It is the temperature at which half of the DNA duplex dissociates. It is calculated based on the primer’s nucleotide composition, length, and concentration.
Primer Length and GC Content: Longer primers and those with a higher proportion of guanine (G) and cytosine (C) bases (which form three hydrogen bonds) have higher Tm values, thus generally requiring higher annealing temperatures.
Salt Concentration: The ionic strength of the reaction buffer significantly impacts Tm. Higher salt concentrations shield the negative charges of the DNA backbone, stabilizing the duplex and raising the Tm.
Primer-Template Complementarity: Perfectly matched primers will anneal at a higher, more predictable temperature. Mismatches, especially at the 3’ end, will destabilize binding and necessitate a lower Ta, though this is often avoided to maintain specificity.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Protocol Comparison
Source: PCR amplifies a specific sequence from a small amount of template DNA (e.g., genomic DNA, plasmid, or cDNA), exponentially increasing its copy number. Restriction digestion starts with a large, purified DNA molecule (like a plasmid or a PCR product) and cuts it at specific recognition sites, producing a finite number of fragments without amplification.
Mechanism: PCR uses thermal cycling and a DNA polymerase to synthesize new DNA. Restriction digestion is an incubation at a constant temperature where endonucleases hydrolyze the phosphodiester backbone at specific palindromic sequences.
Product: PCR products are defined by the primers used, resulting in fragments with specific, known ends (which can be blunt or, if designed with overhangs, effectively sticky after processing). Restriction digestion yields fragments whose ends are dictated by the location and type of restriction site (blunt or sticky overhangs).
Preferential Usage
PCR is preferable when: The goal is to amplify a gene from a genome or cDNA for cloning, to introduce specific mutations or sequences (like restriction sites or tags) via the primers, or when the starting template is scarce. It is also the method of choice for diagnostic screening (colony PCR).
Restriction Digest is preferable when: The goal is to prepare a vector (plasmid backbone) for cloning by creating compatible ends, to sub-clone a fragment from an existing plasmid, to analyze the size or orientation of an insert (diagnostic digest), or when the source DNA is already abundant and contains the fragment of interest within convenient restriction sites.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Ensure that the ends of your linear fragments share complementary sequences. Typically, overlaps of 20–40 base pairs (bp) are designed. The PCR primers used to amplify an insert must have a 5’ extension that is complementary to the end of the adjacent fragment (e.g., the vector). Therefore, the PCR product itself will have these overhangs built into its ends.
The DNA fragments must have clean, blunt ends or ends compatible with the assembly mechanism. Gibson Assembly relies on the 3’→5’ exonuclease activity of the T5 exonuclease to chew back one strand, creating single-stranded 3’ overhangs that allow complementary overlaps to anneal. Therefore, your fragments must be free of damaged ends or unusual secondary structures at the termini.
Following PCR or restriction digest, the DNA must be purified (via gel extraction or column cleanup) to remove enzymes, primers, dNTPs, and buffer components. Residual polymerase or restriction enzymes could interfere with the Gibson Assembly master mix’s enzymes (exonuclease, polymerase, ligase).
While not part of the physical prep, in silico validation is crucial. Use software to confirm that the designed overlaps are unique and have a suitable $T_m$ (~50°C, the Gibson reaction temperature) to ensure proper annealing during the 50°C incubation step.
5. How does the plasmid DNA enter the E. coli cells during transformation?
Chemical Transformation (Heat Shock): Cells are treated with ice-cold $CaCl_2$ (or other divalent cations), which makes the cell membrane more permeable by neutralizing the repulsion between the negatively charged DNA backbone and the negatively charged lipopolysaccharides on the cell surface. The cations are thought to create patches of positive charge and induce structural changes in the membrane. A brief heat shock (usually 42°C) creates a thermal gradient that is believed to either create pores in the membrane or trigger an influx of buffer into the periplasm, effectively dragging the DNA associated with the membrane into the cell.
Electroporation: This method uses a high-voltage electrical pulse to temporarily destabilize the phospholipid bilayer. The electrical current creates transient pores (electropores) in the cell membrane through which the negatively charged DNA is driven by the electrical potential. Once the pulse subsides, the membrane pores reseal, trapping the plasmid inside.
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a powerful, seamless DNA cloning method that utilizes Type IIS restriction enzymes to digest and ligate DNA fragments in a one-tube reaction. Unlike traditional enzymes, Type IIS enzymes (such as BsaI or BsmBI) cut outside their recognition sequence, generating short, unique, single-stranded overhangs of 4 base pairs. Because the recognition site is removed from the final assembly junction, the reaction can be programmed to cut, ligate, and re-cut until the correct, seamless product is formed.
In a typical protocol, DNA fragments (PCR products or vectors) are designed with flanking Type IIS sites that generate complementary overhangs. These fragments, along with the destination vector, are mixed in a single tube with the Type IIS enzyme and T4 DNA Ligase. The reaction is incubated in a thermal cycler, alternating between the optimal temperature for the enzyme (e.g., 37°C) and for the ligase (e.g., 16°C). During the digestion phase, fragments are released with specific sticky ends. If they are designed to ligate together correctly, they form a stable assembly that lacks the original enzyme recognition sites. Incorrect assemblies or uncut fragments retain the recognition sites and are subject to re-digestion. This “cut-ligate” cycling drives the reaction towards the desired final construct with very high efficiency, making it ideal for modular cloning (MoClo) and assembling multiple parts simultaneously.
Asimov Kernel Part
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Part 1: Intracellular Artificial Neural Networks
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Week 9 HW:
lol
Week 10 HW: imagin and measurement
Week 11 HW: Building Genomes
Cell Free Artwork Experimental Loop
I made this fish-like shape at the second quadrant (on the right side of the plate)
I liked that this project brings our creative side, and we use fluorescent protein expression to reflect artistry.
For next collaborative project there could be done in modified bacteria grown in different growth media (so as to see different color expression)
Cell Free Composition Master mix
main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix
PEP‑NTP starts with fully charged NTPs so this means translation begins instantly.
NMP‑Ribose‑Glucose: cells must first convert NMP → NDP → NTP, which takes time.
PEP-NTP: NTPs are expensive, needs pH buffers
NMP‑Ribose‑Glucose runs longer because glucose/ribose are abundant, cheap and slowly metabolized.