Week 4 HW: Protein Design Part I
Homework Questions
Part A. Conceptual Questions
- Why humans eat beef but do not become a cow, eat fish but do not become fish?
Because digestion breaks proteins into individual amino acids, so it means it destroys the cow’s structure and information. We absorb the amino acids and then our ribosomes rebuild human proteins based on human DNA. To make it clear, biological identity comes from genetic information, not raw material.
- Why there are only 20 natural amino acids?
The standard set of 20 amino acids encoded by the nearly universal genetic code reflects evolutionary optimization rather than chemical inevitability. These amino acids provide a chemically diversity, varying in size, charge, hydrophobicity, aromaticity, and hydrogen-bonding potential. This set is sufficient to support stable folding, catalysis, and structural complexity while maintaining translational fidelity.
- Can you make other non-natural amino acids? Design some new amino acids.
Yes! non-natural amino acids can be designed by modifying some side chains while preserving the α-amino and α-carboxylate backbone. Synthetic biology approaches enable site-specific incorporation via engineered tRNA/synthetase pairs, so this way expands the genetic code and enabling novel materials or catalytic functions.
- Where did amino acids come from before enzymes that make them, and before life started?
Before enzymatic biosynthesis, amino acids likely formed through abiotic chemical processes. The Miller–Urey experiment demonstrated that electrical discharge in reducing gas mixtures can produce amino acids. Additional pathways include synthesis in hydrothermal vent systems, Strecker-type reactions in aqueous environments, and extraterrestrial delivery via carbonaceous meteorites.
- If you make an alpha-helix using D-amino acids, what handedness (right or left) would you expect?
Proteins composed of L-amino acids form predominantly right-handed α-helices due to stereochemical constraints of the backbone. If one constructed a polypeptide exclusively from D-amino acids, the mirror-image geometry would be favored, yielding predominantly left-handed α-helices. This inversion follows directly from chirality at the α-carbon and the allowed φ/ψ torsion angles.
- Can you discover additional helices in proteins?
Beyond the α-helix, proteins contain other helical conformations such as the 3₁₀ helix and π-helix. Novel helices can be identified computationally by analyzing backbone dihedral angle distributions in high-resolution structural databases (e.g., Protein Data Bank) or experimentally via X-ray crystallography and cryo-electron microscopy. De novo protein design and foldamer chemistry further enable exploration of alternative helical geometries, including those based on non-canonical backbones.
- Why most molecular helices are right-handed?
The predominance of right-handed helices in biological systems arises from the homochirality of L-amino acids. Given uniform chirality, steric constraints and favorable hydrogen-bond geometry bias the α-helix toward right-handedness. In a mirror-symmetric world of D-amino acids, left-handed helices would dominate. Thus, helix handedness reflects fundamental stereochemical asymmetry in biomolecular building blocks.
- Why do beta-sheets tend to aggregate? What is the driving force for b-sheet aggregation?
β-sheets possess extended backbone conformations that maximize inter-strand hydrogen bonding. When partially unfolded polypeptides expose β-prone segments, they can associate intermolecularly via backbone hydrogen bonds, forming extended β-sheet assemblies. Side-chain complementarity and hydrophobic interactions further stabilize aggregation.
Driving force - The primary driving force is the formation of extensive intermolecular hydrogen-bond networks along the peptide backbone, combined with hydrophobic collapse and favorable packing of side chains. The resulting cross-β architecture is thermodynamically stable and often kinetically persistent.
- Why many amyloid diseases form b-sheet? Can you use amyloid b-sheets as materials?
Amyloid fibrils share a common cross-β structural motif, in which β-strands align perpendicular to the fibril axis. This structure maximizes backbone hydrogen bonding and produces highly stable, self-templating aggregates. In diseases such as Alzheimer’s disease and Parkinson’s disease, normally soluble proteins misfold and adopt β-rich conformations that nucleate fibril formation.
Amyloid β-sheets as materials: Due to their mechanical strength, nanoscale order, and self-assembly, amyloid fibrils are being explored as biomaterials for nanowires, hydrogels, and tissue scaffolds. Their stability and tunable assembly make them promising in bioengineering applications.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions.
1. Briefly describe the protein you selected and why you selected it.
Carboxyltransferase subunit of Mycobacterium tuberculosis acetyl-CoA carboxylase
Amino acids: 494 Organism: Mycobacterium tuberculosis
I chose this protein because of its central role in building the unique and protective cell wall of M. tuberculosis. ACC’s job is to convert acetyl-CoA to malonyl-CoA. In M. tuberculosis , this malonyl-CoA is the fundamental building block for synthesizing the long fatty acid chains needed for its cell wall, including the very long mycolic acids.
**Sequence:**
MGSSHHHHHHSSGLVPRGSHMTIMAPEAVGESLDPRDPLLRLSNFFDDGSVELLHERDRSGVLAAAGTVNGVRTIAFCTDGTVMGGAMGVEGCTHIVNAYDTAIEDQSPIVGIWHSGGARLAEGVRALHAVGQVFEAMIRASGYIPQISVVVGFAAGGAAYGPALTDVVVMAPESRVFVTGPDVVRSVTGEDVDMASLGGPETHHKKSGVCHIVADDELDAYDRGRRLVGLFCQQGHFDRSKAEAGDTDIHALLPESSRRAYDVRPIVTAILDADTPFDEFQANWAPSMVVGLGRLSGRTVGVLANNPLRLGGCLNSESAEKAARFVRLCDAFGIPLVVVVDVPGYLPGVDQEWGGVVRRGAKLLHAFGECTVPRVTLVTRKTYGGAYIAMNSRSLNATKVFAWPDAEVAVMGAKAAVGILHKKKLAAAPEHEREALHDQLAAEHERIAGGVDSALDIGVVDEKIDPAHTRSKLTEALAQAPARRGRHKNIPL
Component of a biotin-dependent acyl-CoA carboxylase complex. This subunit transfers the CO2 from carboxybiotin to the CoA ester substrate. When associated with the alpha3 subunit AccA3, is involved in the carboxylation of acetyl-CoA and propionyl-CoA, with a preference for acetyl-CoA
Information taken from UniProt
2. Identify the amino acid sequence of your protein.
The length of the protein is: 493 aminoacids.
The most common amino acid is: A, which appears 61 times.
Sequence length for P9WQH5 (Acetyl-CoA carboxylase): 473 amino acids
Amino Acid Frequencies: A: 61 (12.90%) V: 52 (10.99%) G: 50 (10.57%) L: 38 (8.03%) R: 33 (6.98%) D: 32 (6.77%) E: 27 (5.71%) P: 23 (4.86%) S: 23 (4.86%) T: 22 (4.65%) I: 20 (4.23%) H: 16 (3.38%) F: 14 (2.96%) K: 14 (2.96%) M: 10 (2.11%) N: 10 (2.11%) Q: 9 (1.90%) Y: 8 (1.69%) C: 7 (1.48%) W: 4 (0.85%)
https://web.expasy.org/protparam/ Number of amino acids: 493 Most frequent amino acid: Ala (A) 61 (count) 12.4%
Over 245 homologous sequences were identified across different Mycobacterium species and strains
This protein belongs to the AccD/PCCB family. This family comprises enzymes involved in fatty acid and amino acid metabolism. They function as carboxyltransferases, which are part of larger enzyme complexes that catalyze the carboxylation of various CoA ester substrates.
3. Identify the structure page of your protein in RCSB
Are there any other molecules in the solved structure apart from protein? Does your protein belong to any structure classification family?
RCSB 3D Structure
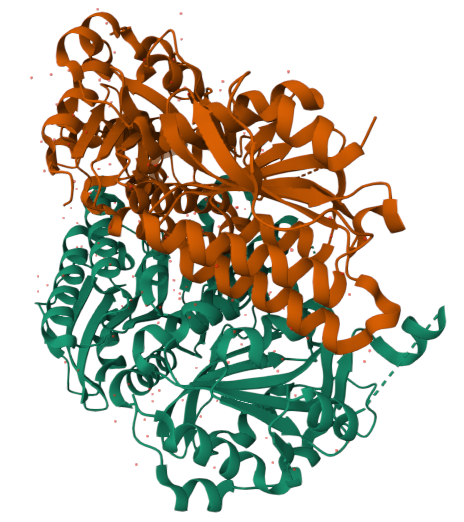
Released: 2013-07-03
Resolution: 1.95 Å –> Good! :) Indicates high structural quality.
The structure contains Mg²⁺ and ATP in addition to the protein.
Structural classification: Ligase
- Open the structure of your protein in any 3D molecule visualization software:
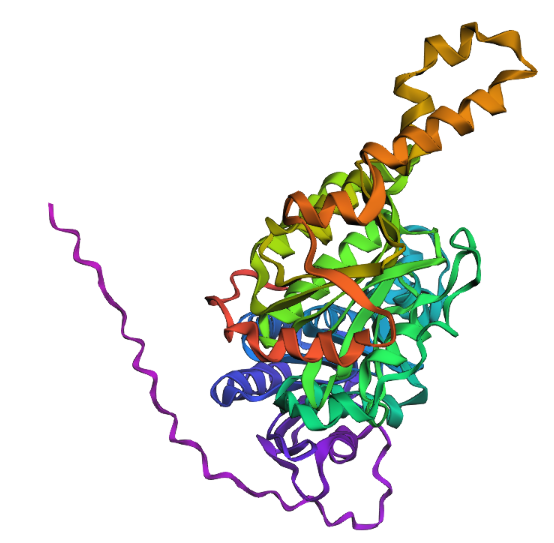
Cartoon
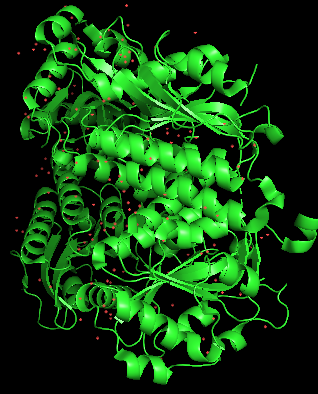
Colored by secondary structue
The protein is predominantly alpha-helical with only a few beta-sheets region. This is evidenced by the cyan sheets present.
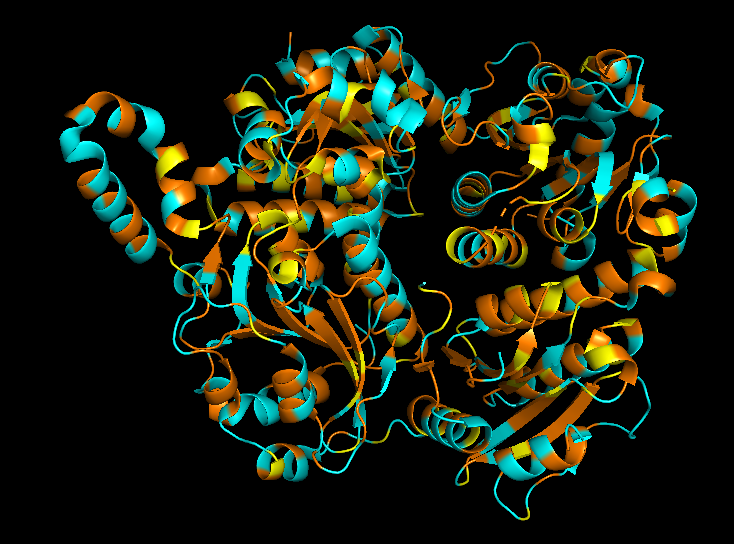
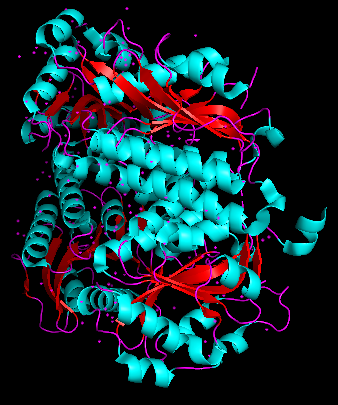
When colored by residue type, hydrophobic residues appear to cluster in the interior of the protein (orange), while polar and charged residues are more solvent-exposed (cyan). This is consistent with proper folding of a soluble enzyme.
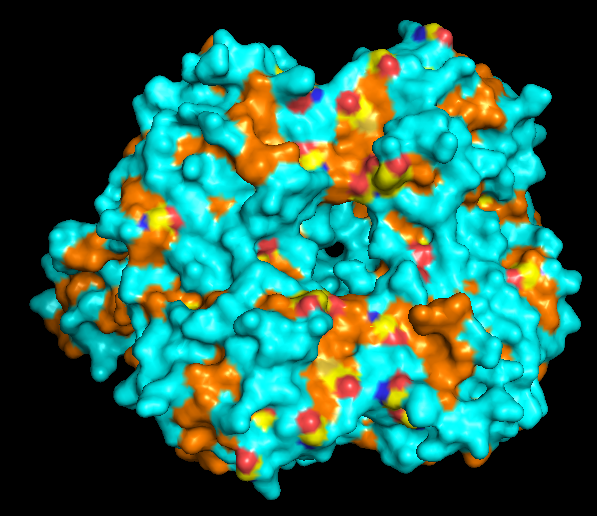
Visualization of the molecular surface reveals a compact, globular structure with noticeable surface depressions and grooves. One elongated cleft with visible depth can be observed, which may correspond to the substrate-binding site. While no large enclosed cavities are present, the surface topology suggests the presence of shallow binding pockets typical of enzymatic proteins.
Part C. Using ML-Based Protein Design Tools
Protein Language modeling
1. deep mutational scan
Heatmap
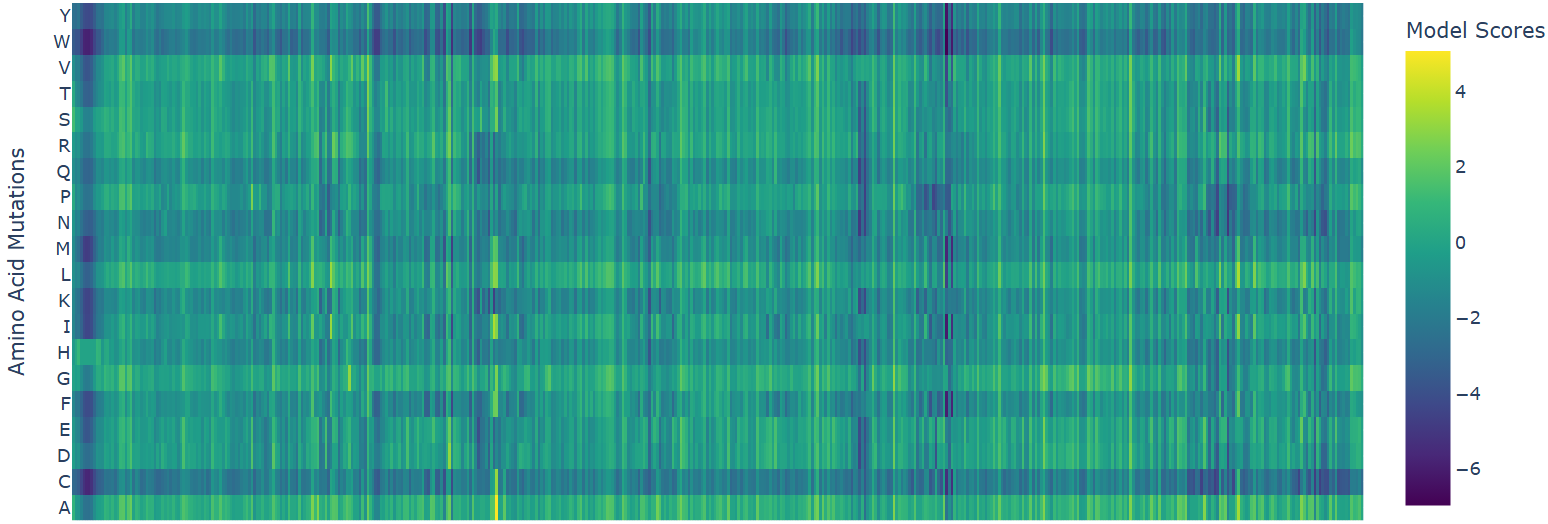
- W (Tryptophan) shows interesting patterns - mutations to certain residues like S, R, Q have higher scores
2. Latent Space Analysis
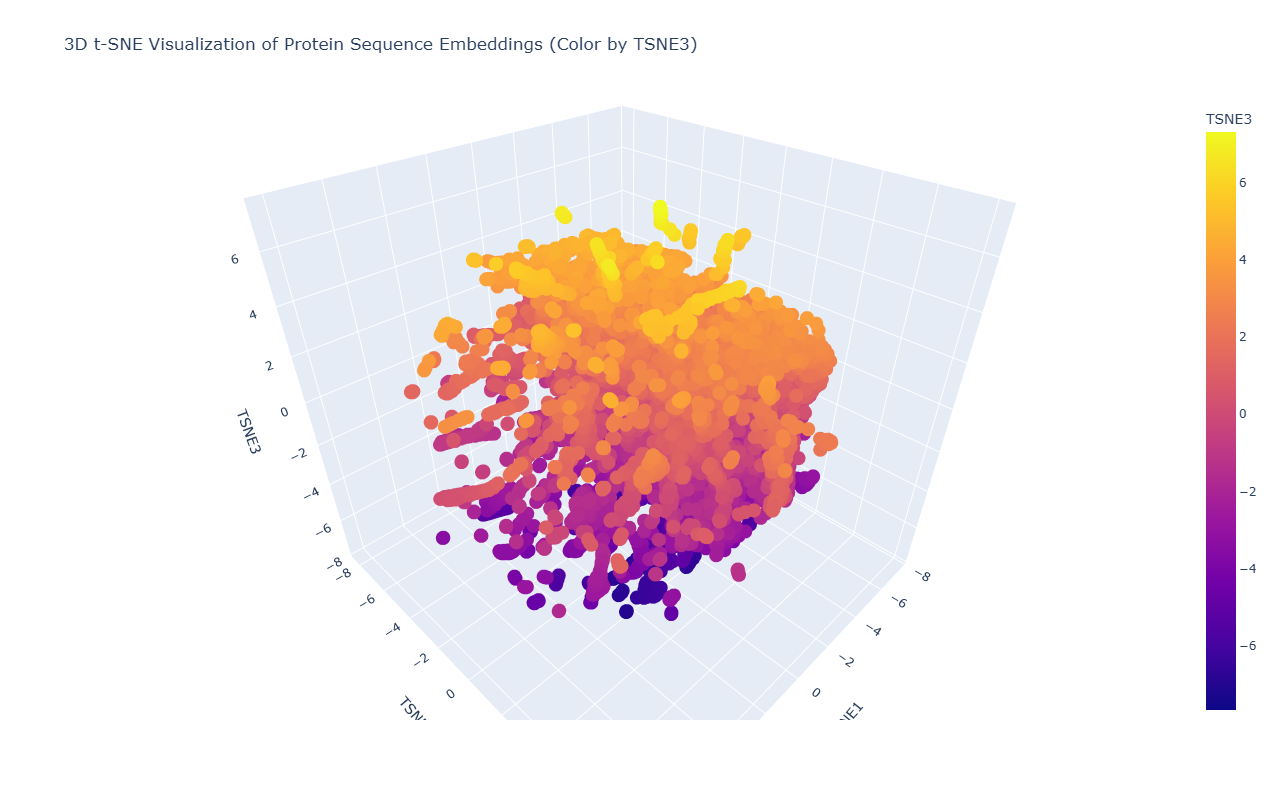
Due to the high density and overlap of points in the t-SNE visualization, it is difficult to precisely locate the position of acetyl-CoA carboxylase from Mycobacterium tuberculosis. However, based on its biological function as a key enzyme in fatty acid biosynthesis, it would be expected to cluster with other metabolic enzymes, particularly carboxylases and proteins involved in lipid metabolism.
3. Protein folding
Observation: The overall fold and general domain organization are somewhat preserved, indicating that the model captures key structural features of the protein. However, the ESMFold prediction appears less compact and shows greater deviation in the arrangement of secondary structure elements compared to the reference structure.

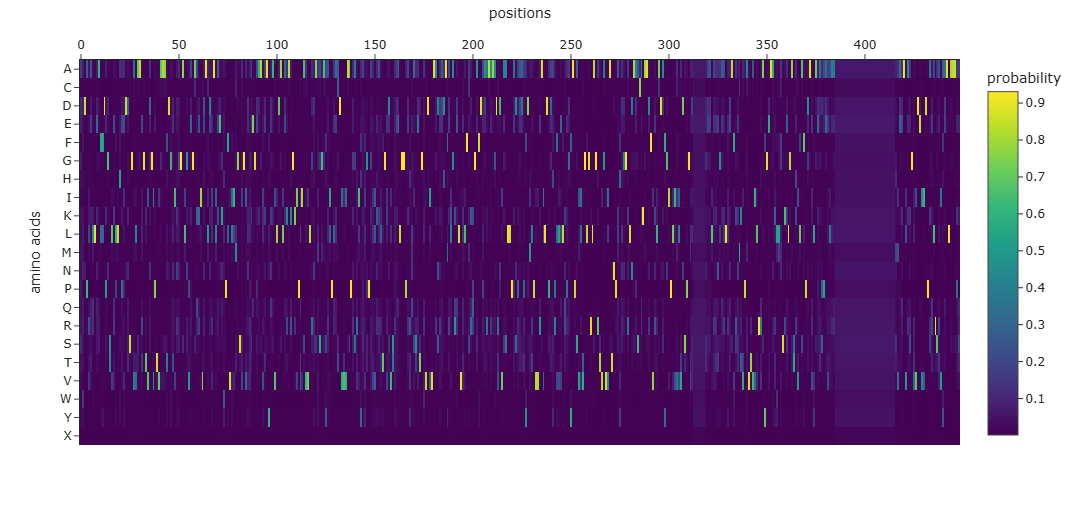
Many positions in the protein show low specificity, where no single amino acid strongly dominates. This suggests that these regions are more tolerant to mutations and may correspond to flexible or less critical structural regions.
The protein appears to contain both highly conserved regions and more variable segments, reflecting a balance between structural stability and sequence adaptability.
New Sequence: AWDPLELLEAFFDPGSVRLLHPPDDSGVLAARGTVDGIPTVAAATDGTINGGALGPAGAQKIVDALDEALAEGWPFVLIMGSVGVLRAEGEAAKKAYQAVLDALEKAKGKIPLIAVVLRNALSGAYRIPLACDVVVMAPEAVLAEIGPGVLKALTGVDVSWEELGGPERQASETGAVDIVAADRADAFALARELVRLFAAPGAFDAAAAAAADVDVASLLPASPEDPYDMLPVVRALLDPDAPLLVLQPRYAPSVLVGLGRLAGRTVGFIATNPAHDGGRLNAASCAKAARFVELADAFGIPLVVVVDSPGTLXXXXXXGAALAEAGARLRAAFAACKVPAVTLIVRRAYGEAAELLASKSLGATKVLAFPDAVLAEASPEVLAAXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXMVEEAVKEGRVDEIIDPAKARSAIAAALAAAPA
- Protein Generation
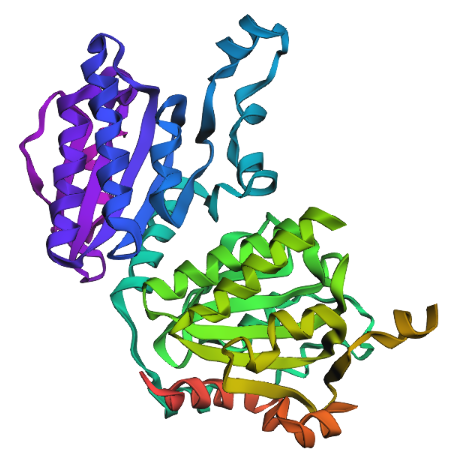
this new sequence is already an engineered version that prospectively aligns with the original one.
Part D. Group Brainstorm on Bacteriophage Engineering
Review the Bacteriophage Final Project Goals:
Increased stability (easiest)
Main Goal: Increase stability of the MS2 L lysis protein through computational design to enhance phage therapeutic efficacy, while maintaining/improving its lytic function.
Identification of Stability-Limiting Regions:The MS2 L protein has a highly basic, unstructured N-terminal domain (Domain 1, residues 1-36) that confers DnaJ dependence and may contribute to proteolytic instability. The essential C-terminal domain (residues 37-75) contains the critical LS motif (Leu48-Ser49) that mediates target interaction but may have suboptimal stability.
Rational Stabilization Without Function Loss: Mutations that increase stability must not disrupt:
The essential LS motif interaction interface
Membrane insertion capability
Oligomerization potential (≥10 monomers)
The regulatory timing mechanism
Balancing Stability with Controllable Lysis Timing: The N-terminal domain acts as a regulatory “damping” feature—complete stabilization/removal causes premature lysis (~20 min earlier), which may reduce phage progeny production. Optimal therapeutic phages require balanced lysis timing.
The computational enhancement of MS2 L lysis protein stability will be executed through a pipeline integrating sequence analysis, structure prediction, in silico mutagenesis, interaction validation, and candidate ranking.
In Phase 1, sequence analysis begins with BLASTp to identify L protein homologs across Leviviridae, followed by Clustal-Omega multiple sequence alignment anchored by the conserved LS motif identified in previous mutational studies.
Phase 2 employs structure prediction using ESMFold and ESM3, which leverage evolutionary data to model L’s conformation, complemented by Boltz-1 for generating conformational ensembles that capture the protein’s behavior in membrane environments.
AlphaFold-Multimer predicts oligomerization interfaces based on evidence that L forms high-order complexes of at least ten monomers, while FoldSeek searches for structural homologs with known stabilizing features that could inform design.
Phase 3 conducts in silico mutagenesis using ESM2 zero-shot prediction to score missense variants for evolutionary fitness, ProteinMPNN for sequence redesign of variable regions while fixing the essential LS motif, EvolvePro for directed evolution simulation, and Rosetta ddG_monomer for quantitative ΔΔG stability calculations. T
Phase 4 validates interaction interfaces using BindCraft to predict preservation of L-target binding, AlphaFold3 for L-DnaJ docking to verify retention of regulatory chaperone interaction, and molecular docking with HADDOCK to ensure stabilizing mutations do not disrupt essential contacts with the unknown lytic target.
Phase 5 integrates all outputs through a custom Python pipeline that scores each candidate mutation on stability change, evolutionary conservation, predicted function retention and DnaJ binding preservation, generating a weighted ranking of the top five to ten stabilizing mutations for experimental testing.