Week 5 HW: Protein Design Part II
Part A. SOD1 Binder Peptide Design
Part 1
Design short peptides that bind mutant SOD1
Which are worth advancing for therapy
Human SOD1 sequence
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
SOD1 A5V sequence
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Binder 1: WHYPAVAVALGX Perplexity: 8.190170
Binder 2: KRYYVVGVRHKE Perplexity: 31.759185
Binder 3: HHYYVAAVELGX Perplexity: 18.370893
Binder 4: WRYPPVAARHKX Perplexity: 12.978095
FLYRWLPSRRGG (known binder)
The known binder FLYRWLPSRRGG contains several hydrophobic and positively charged residues, which are commonly observed in the generated peptides as well, suggesting that PepMLM captures sequence patterns associated with binding.
Part 2: Evaluate Binders with AlphaFold3
Binder 1: WHYPAVAVALGX ipTM: 0.34
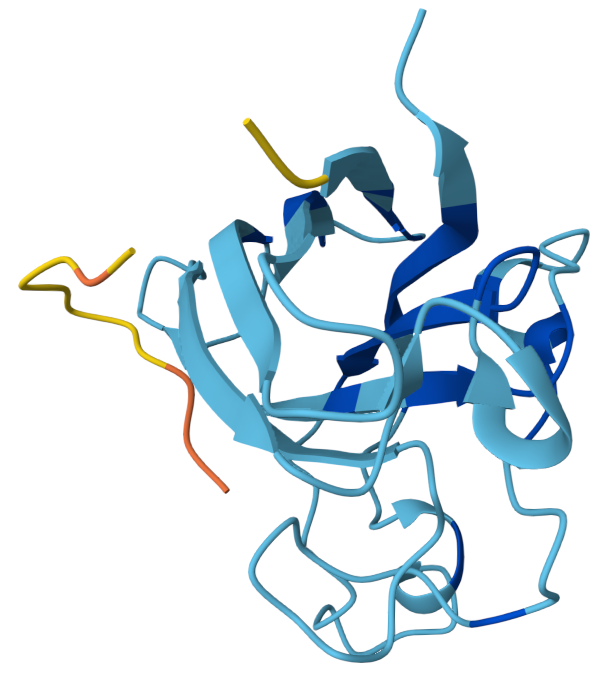
The peptide appears mostly surface-adjacent but not tightly bound to SOD1. The peptide does not appear to bind near the N-terminal region where the A4V mutation occurs. The peptide approaches the surface of the β-barrel but does not appear to form a strong interface. The peptide does not appear to approach the dimer interface in this model. The predicted complex has an ipTM score of 0.34, which indicates weak confidence in the protein–peptide interaction.
Binder 2: KRYYVVGVRHKE ipTM: 0.45
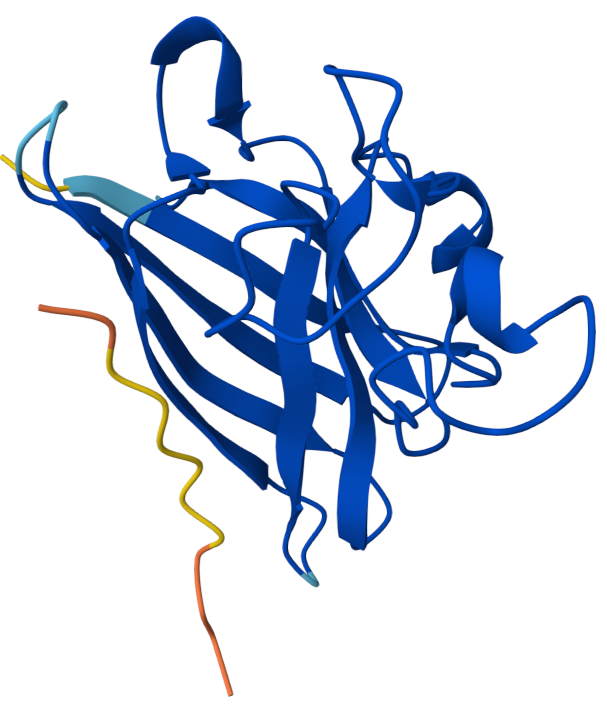
ipTM score of 0.45 indicates weak to moderate confidence in the predicted interaction. The peptide appears positioned near the outer surface of the SOD1 structure but does not form a tightly packed interface with the protein. It does not localize close to the N-terminal region where the A4V mutation occurs, which is located on a flexible loop extending from the protein. Instead, the peptide lies adjacent to the β-barrel surface, suggesting a potential but weak surface interaction rather than deep binding. The peptide appears surface-bound and largely exposed, rather than buried within a pocket.
Binder 3: HHYYVAAVELGX ipTM:0.41
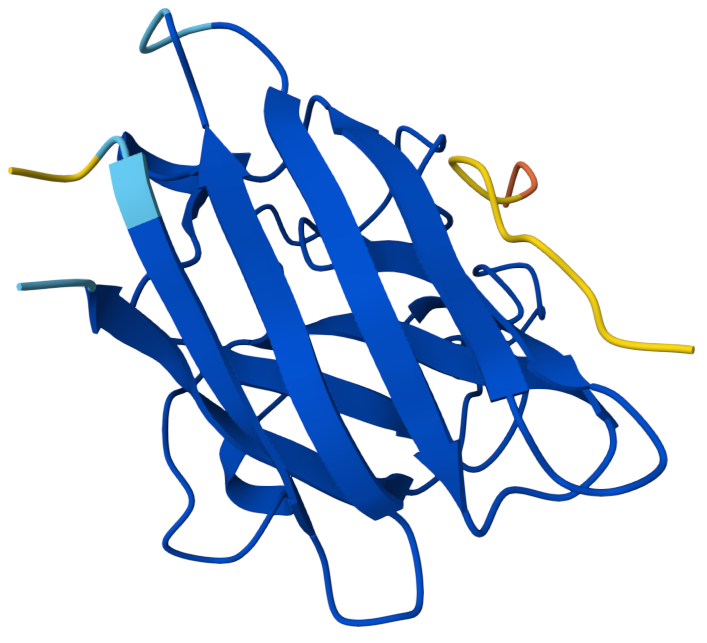
This has an ipTM score of 0.41, means low confidence in the protein–peptide interaction. In the model, the peptide is positioned along the side of the β-barrel structure of SOD1 but remains partially separated from the protein surface. It does not appear to interact near the N-terminal region containing the A4V mutation. Instead, the peptide lies adjacent to the barrel-shaped core of the protein and does not appear to insert into any pocket or groove. The peptide appears loosely associated with the protein surface rather than buried, suggesting weak or transient binding.
Binder 4: WRYPPVAARHKX ipTM: 0.37
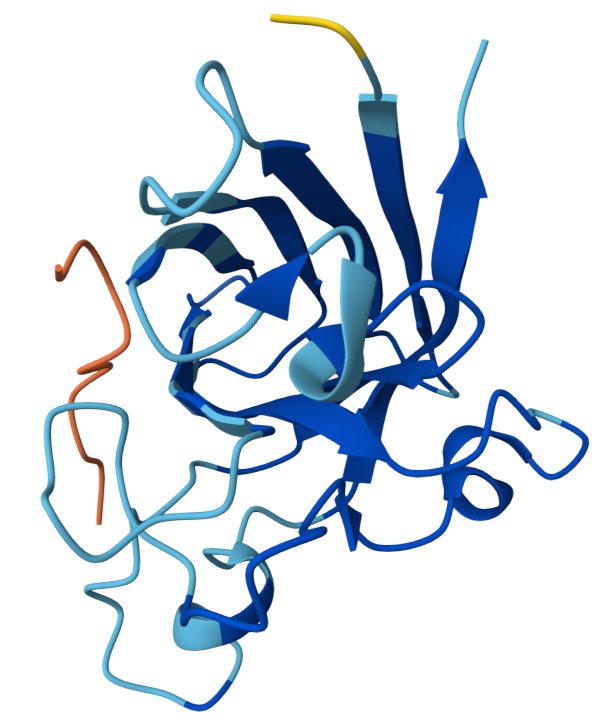
The ipTM score suggests weak confidence in the predicted interaction. The peptide is positioned near the surface loops of the SOD1 structure but does not appear to form a stable interface with the protein. It does not localize near the N-terminus where the A4V mutation is located, and instead lies along flexible surface regions away from the mutation site. The peptide approaches the outer surface of the β-barrel region, but it remains mostly exposed and does not appear to penetrate into a binding pocket. Overall, the peptide appears surface-bound and partially detached, indicating weak predicted binding.
Known Binder: FLYRWLPSRRGG iPTM: 0.38
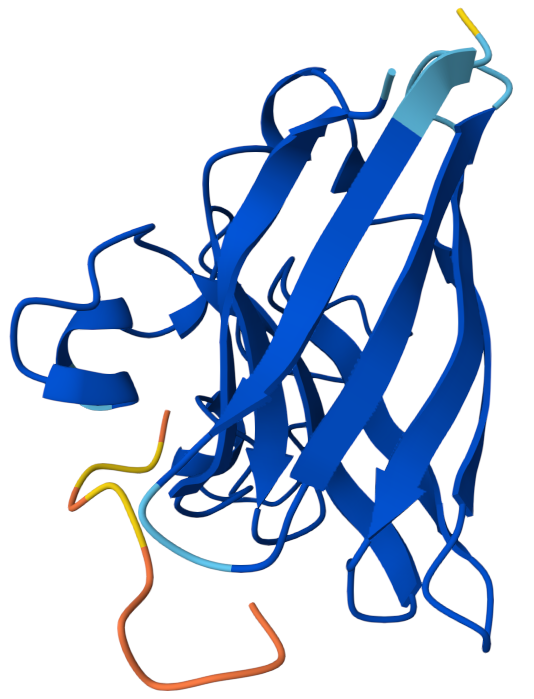
Although the peptide FLYRWLPSRRGG is experimentally known to bind SOD1, the AlphaFold prediction still yields a relatively low ipTM score and a loosely associated binding pose. This highlights that AlphaFold is not specifically optimized for predicting short peptide–protein interactions, and therefore may underestimate the stability of such complexes.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
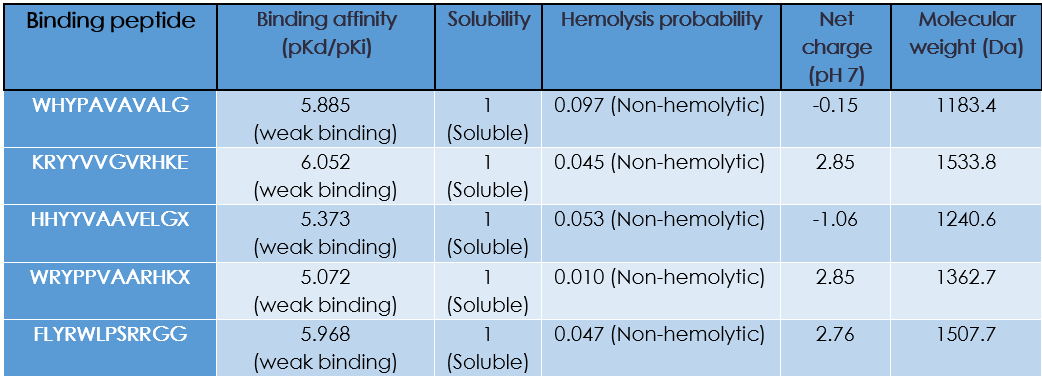
Do peptides with higher ipTM also show stronger predicted affinity?
Yes!
Are strong binders hemolytic or poorly soluble?
They are non-hemolytic and soluble