Describe a biological engineering application or tool you want to develop and why.
Virus Hunting
The usage of virus hunting to discover viruses in animal populations that might become a pandemic and exploit it as a gene therapy tool.
first of all the viruses are isolated from hosts of interest, then sequencing their genome, then characterize the virus.
Following steps will be:
Developing arrays for the virus detection providing a faster and cheaper way.
Exploiting the virus replication machinery to deliver compounds / biopharmaceuticals to humans or animals.
Disocvering potential pandemic pathogens early will prevent its outbreak and prepare us well.
Describe one or more governance policy goals related to ensuring this application contributes to an ethical future & prevents harm.
Biosafety and biosecurity aims to prevent loss, theft and misuse of highconsequence material. This can be done by providing and implementing risk control measures that address the risks associated with conducting high-consequence research and working
with high-consequence material, including other biosecurity-relevant material.
The intrinsic risks of working with biological agents are not only of a biosafety nature, such as exposure or unintentional release, but also of biosecurity, which includes the theft, misuse, or intended release of biological material.
Describe at least three different potential governance actions by considering the purpose, design, assumptions, and risks of failures & “success”
Development of a board to organize and authorize the suitable scientist for conducting virus hunting
Purpose: The aim is to allow only trained professionals to conduct such procedures
Design: Every country will have a trusted board that will allow and oversee the virus hunting procedures and these boards will be under the supervision of a central board that will get periodic reports
Assumptions: Incorrect selection of personnel might lead to inproper viral isolation and process organization leading to its outbreak
Risks of Failures & Success: This action might fall if not properly implemented
Development of an agreed upon method of biological materials disposal
Purpose: The aim is to control and oversee disposal methods to prevent any outbreaks
Design: Professionals will be further trained
Assumptions: Ignoring the right protocol for disposal may lead to an outbreak
Risks of Failures & Success: not providing the right training and control
Providing enough funds to conduct the required procedures in the countries of interest
Purpose: this action aim to fund labs at developing countries of interest
Design: The organization will provide the fund and supervise its implementation to buy the right equipment and tools
Assumption: Corruption or not providing the fund will hinder the virus hunting procedures in that country
Risk of Failures & Success: Not providing enough funds will stop the required procedures
Score each of your governance actions against your rubric of policy goals.
Does the option:
Authorizing Board
Biological Materials Disposal
Funds
Enhance Biosecurity
• By preventing incidents
1
2
3
• By helping respond
1
2
3
Foster Lab Safety
• By preventing incident
2
1
3
• By helping respond
1
2
3
Protect the environment
• By preventing incidents
2
1
3
• By helping respond
1
2
3
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
1
• Feasibility?
1
2
3
• Not impede research
2
1
3
• Promote constructive applications
3
2
1
Based on scores, describe which governance option or combination of options, you would prioritize, and why.
Based on the scores:
I would prioritize the formation of the board because it is the base upon which every other step will follow.
I would prioritize as well providing enough funds especially for developing countries in which many have the knowledgeable scientists but not enouhg funds for buying the necessary equipment.
As part of your final project, design one or more strategies to ensure that your project, and what it enables, contributes to growing an ethical biological future.
Justice
Equitable Access to All Populations
The platform is intentionally designed toward a low-cost system rather than instrument-dependent workflows, so that the end product is deployable in under
resourced clinics in low and middle income regions where AD diagnostic infrastructure is nearly absent. Equitable access partnerships and open licensing models will be established from the outset, not as an afterthought, to prevent the technology from benefiting high-income populations exclusively.
Assignment (Week 2 Lecture Prep)
Homework Questions from Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate of DNA polymerase is 1:10^6, meaning that it makes 1 mistake per 1000,000 bases
The human genome contains approximately 3.2 billion base pairs. If polymerase copied it at its raw error rate, each replication would produce:
3,200,000,000 ÷ 1000,000 = 3,200 errors per cell division which is not good
Biology deal with that discrepancy through three-layer error correction system; nucleotide selectivety, proofreading and mismatch repair (MutS repair system)
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The genetic code uses 3-base codons (triplets) to specify each amino acid. There are 4 DNA bases (A, T, G, C), giving 4³ = 64 possible codons. Therefore, there are more codons (64) than amino acids (20). So most amino acids can be coded by 2–6 different codons, averaging about 3. A typical human protein is ~375 amino acids long. If each position has ~3 coding options: 3³⁷⁵ ≈ 10¹⁷⁹ possible DNA sequences for the same protein. So the number of DNA sequences that could theoretically encode the same protein is astronomically larger than the number of atoms in existence.
There are a variety of reasons that all of these different codes don’t work to code for the protein of interest;
Codon Usage Bias: Different organisms prefer certain synonymous codons over others, because the matching tRNA molecules are more or less abundant
mRNA Secondary Structure: The mRNA sequence folds back on itself into hairpins, loops, and stem structures based on its nucleotide sequence.
Splicing Signals are Embedded in the Coding Sequence
CpG Dinucleotides and Methylation
Cryptic Regulatory Elements
Translation Speed and Protein Folding
Homework Questions from Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
Solid-phase Phosphoramidite method by Caruthers
Why is it difficult to make oligos longer than 200nt via direct synthesis?
The fundamental problem is compounding error. Each coupling step is ~99% efficient, meaning ~1% of strands fail at every base addition. Over hundreds of cycles this multiplies catastrophically
Why can’t you make a 2000bp gene via direct oligo synthesis?
Due to compounding error, at 99% coupling efficiency, a 2000-mer would yield:
0.99²⁰⁰⁰ ≈ 0.000002% perfect product
Therefore, the solution, will be to assemble a 2000bp gene from many short oligos (~40–60mers) rather than attempting to synthesize it as one continuous chain.
The Lysine Contingency (a fictional genetic failsafe from the Jurassic Park franchise) was a genetic alteration performed in the dinosaur genome. The modification knocked out the ability of the dinosaurs to produce the amino acid lysine. This forced the dinosaurs to depend on lysine supplements provided by the park’s veterinary staff. Since all mammals can synthesize Lysine, the lysine contingency is useless, since dinosaurs can simple prey on other animals to have lysine in their diet
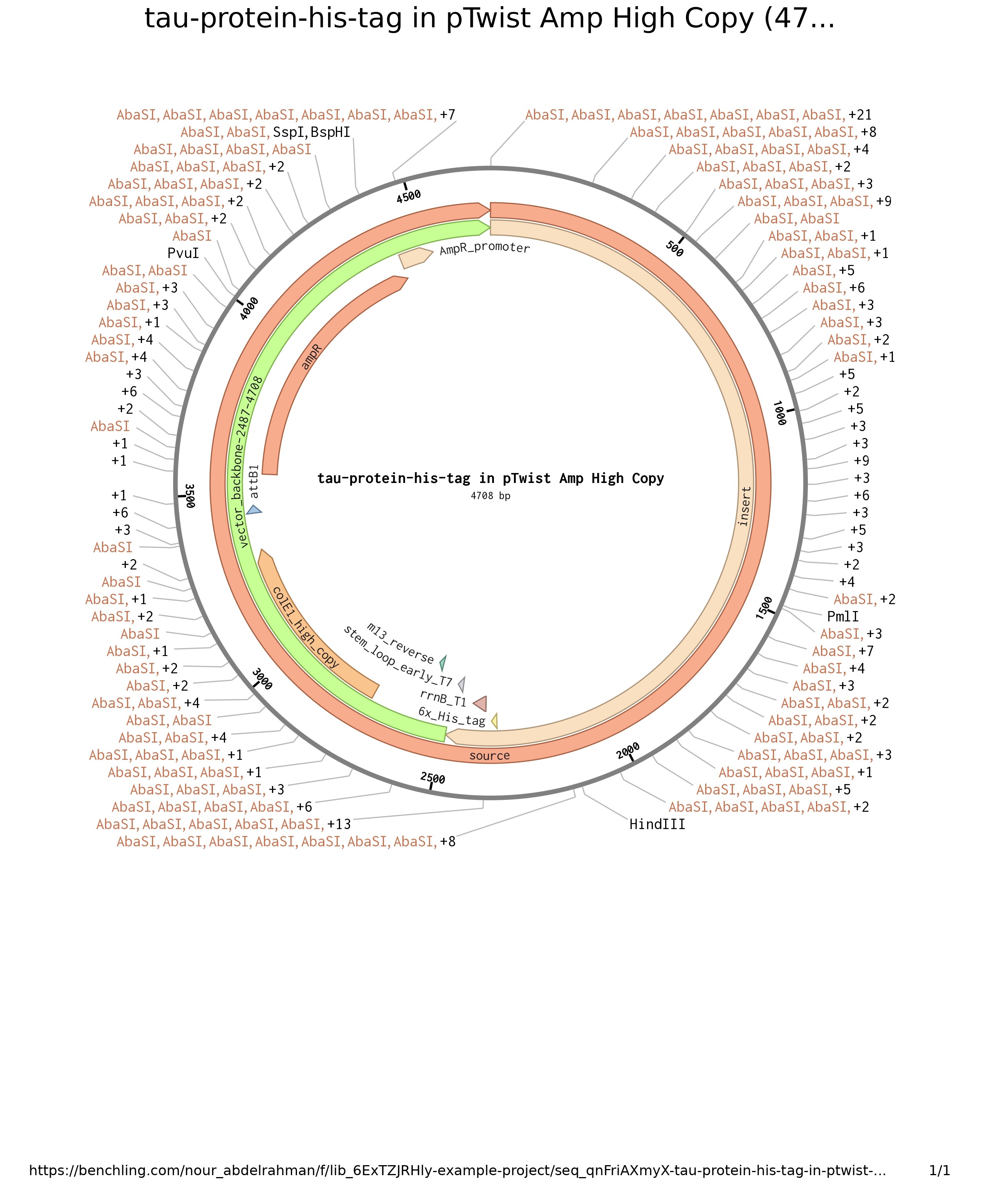
Week 2 HW: DNA Read, Write and Edit
Part 0: Attend or watch all lecture and recitation videos. [DONE]
Part 1: Benchling & In-silico Gel Art
Make a free account at benchling.com
Import the Lambda DNA.
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
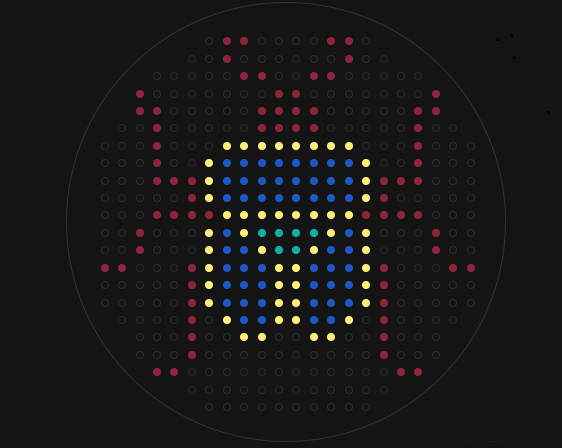
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
I imagine the pattern as a hand making number one
Part 3: DNA Design Challenge
Choose your protein.
I chose tau protein that it’s hyperphosphorylation is involved in Alzheimer’s disease progression
Codon optimization is conducted to increase the efficiency of expression. For example, although each amino acid has more than one codon, their efficiency varies, therefore, the optimization aims to choose the most efficient codons for increased translation efficiency and stable mRNA structure.
I chose E.coli to optimize the codon for, because it of its fast replication and versatile applications
What DNA would you want to sequence (e.g., read) and why?
The gene I’m interested in is the APP (Amyloid Beta Precursor Protein) Gene, this gene is involved in Alzheimer disease. I chose this gene because I’m interested in using synthetic biology to understand neurodegenerative disorders, especially Alzheimer’s disease.
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I chose PacBio sequencing technology, it is a third-generation sequencing technology, that have the ability to produce long and higly accurate DNA reads. It is based on single molecule real-time (SMRT) sequencing principle.
for preparing the input, the DNA is prepared into the SMRTbell library by ligating hairpin adapters to double-stranded DNA on both ends, forming a circular template. Primers and polymerases are added to this library, which is loaded onto the sequencing instrument that contains the SMRT Cell and ZMWs. A single template DNA is immobilized in each ZMW. As the polymerase adds fluorescently labeled nucleotides into the growing DNA strand, light is emitted. This light emission is measured in real time and these signals are converted into nucleotide sequences.
the first step is library costruction which involves several steps to prepare DNA for sequencing:
DNA is cleaved into fragments of the desired size and it undergoes end repair.
Then, adaptors with hairpin structures are ligated to both ends of the DNA fragments which creates single-stranded circular structures called SMRTbell templates.
Finally, the templates are purified and loaded onto the PacBio sequencing instrument.
the output is fluorescent signals that are translated into base sequences then alignment and assembly are conducted
DNA Write
What DNA would you want to synthesize (e.g., write) and why?
I would synthesize genetic circuit that sense the presence of high amount of hyperphosphorylation in the brain for example.
What technology or technologies would you use to perform this DNA synthesis and why?
I would choose oxford nanopore for synthesizing the genetic circuit.
DNA Edit
What DNA would you want to edit and why?
I would want to edit a gene that have a disease-causing mutation.
What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-Cas to edit the gene, the main steps involves designing gRNA that will guide the Cas9 to cut the specific site
Week 3 HW: Lab Automation
Python Script for Opentrons Artwork
I chose to make the Egyptian Beetle
The Egyptian scarab beetle Scarabaeus sacer is a dung beetle native to the Mediterranean region and North Africa. Ancient Egyptians considered it one of the most sacred animals, associating it with Khepri, the god of the rising sun, because the beetle’s behavior of rolling dung balls across the ground resembled the sun being pushed across the sky. Scarab amulets were among the most widely produced objects in ancient Egypt, worn by both the living and the dead as protective charms, and heart scarabs were placed on mummies to protect the deceased during judgment in the afterlife. The beetle lays its eggs inside dung balls, and when the larvae hatch and emerge from the ground, Egyptians interpreted this as spontaneous creation, reinforcing the scarab’s association with regeneration and eternal life. Today the scarab remains one of the most recognizable symbols of ancient Egyptian civilization, widely reproduced in art, jewelry, and archaeology.
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots
This paper presents AssemblyTron, an open-source Python-based automation framework designed to facilitate DNA assembly workflows using the Opentrons OT-2 robotic platform. It integrates outputs from DNA design software (specifically j5) with robotic liquid handling protocols to automate multiple DNA assembly methods, including Golden Gate, AQUA, IVA, and site-directed mutagenesis. The system effectively automates critical steps such as PCR setup, enzyme treatments, assembly reactions, and transformations, thereby reducing manual labor, human error, costs, and training time.
The novel biological application demonstrated is the automated, high-fidelity construction of complex multigene DNA constructs and mutants, exemplified by assembling chromoprotein expression plasmids and performing site-directed mutagenesis with accuracy comparable to manual methods. These capabilities significantly accelerate synthetic biology research, enabling rapid and reliable genetic engineering workflows with minimal manual intervention.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more.
A cloud laboratory can be used to perform repetitive steps saving time and energy and increasing reproducibility and accuracy. For the final project (CRISPR-Cas13a based biosensor for Alzheimer’s blood biomarker detection), the following can be used:
Echo525: for transfering the small volume of gRNA and produced miRNA into the wells.
ATC: thermal cycler for DNA constructs amplification
Multiflow: transfer gRNA and produced miRNA different combinations to 96-well
Inheco: incubate the plate at 37°C for the CRISPR to perform its cleavage activity
Spark: measure the fluorescence upon binding of the cas13a protein, miRNA and the target biomarker giving insights into the best quantities of DNA parts and designs.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
because after eating the food is processed / digested to eventually its initial constituents as amino acids, fatty acids, vitamins, etc then the essential building blocks (nucleotides) that goes in building the human body
Why are there only 20 natural amino acids?
According to Doig, there are reasons for the selection of every amino acid making them a near ideal group. The factors taken into account included each amino acid’s component atoms, functional groups and biosynthetic cost.
Forming soluble, stable protein structures with close‐packed cores and ordered binding pockets needed the variety of amino acids we see today. Multiple hydrophobic proteins are needed.
Stephen Freeland and his team investigated size, charge and hydrophobicity parameters of proteins to show that the amino acids adopted by biology were not chosen randomly.
Can you make other non-natural amino acids? Design some new amino acids.
Yes, non-natural amino acids also known as non-canonical or non-proteinogenic amino acids can be synthesized chemically or produced naturally as secondary metabolites in several organisms, such as bacteria, fungi, plants, or marine organisms.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids were created through purely natural chemical and physical processes driven by energy. These building blocks formed spontaneously in two primary ways: through atmospheric synthesis on early Earth, and through delivery by space rocks.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
If α-helix was made using D-amino acids, the mirror image of natural amino acids, a left-handed helix is expected instead of the normal right-handed one.
Can you discover additional helices in proteins?
Yes, Proteins contain five helical structures, each defined by distinct hydrogen bonding patterns. The α-helix (i→i+4) is the most abundant, the 3₁₀ helix (i→i+3) is tighter and found at helix termini, and the π-helix (i→i+5) is wider, occurring in ~15% of protein structures almost exclusively near active sites. The PPII helix is left-handed with no intrachain hydrogen bonds yet critically mediates cell signaling through SH3 domain interactions, while the collagen triple helix consists of three intertwined PPII strands stabilized by interchain hydrogen bonds forming the structural basis of connective tissue.
Why are most molecular helices right-handed?
Proteins are almost exclusively built from L-amino acids, and this stereochemical bias of nature is directly reflected at the secondary structure level, where right-handed helices are strongly preferred over left-handed helices.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets inherently aggregate because their edge strands possess exposed, unsatisfied hydrogen bonding potentials and sticky hydrophobic patches, making them chemically predisposed to self-assembly. This process often associated with amyloid fiber formation is driven by a combination of thermodynamics, specific atomic interactions, and conformational changes.
The driving forces for aggregation are:
The Hydrophobic Effect: In an aqueous environment, the primary thermodynamic driver for protein aggregation is the minimization of free surface energy. When unstructured proteins misfold into β-sheets, hydrophobic side chains that are normally buried inside a properly folded protein become exposed to the solvent. To minimize unfavorable contact with water, these exposed hydrophobic regions clump together with other β-strands, squeezing out water molecules and increasing the overall entropy of the surrounding solvent.
Intermolecular Hydrogen Bonding: In a single β-sheet, the backbone C=O and N-H groups are locked into hydrogen bonds with adjacent strands. However, the outer edge strands of a β-sheet have exposed, unsatisfied hydrogen bonding groups. These exposed groups eagerly accept or donate hydrogen bonds to other β-sheet edges, leading to a highly stable, repeating “cross-β” architecture.
Steric Interdigitation: β-sheets often have amino acid side chains that alternate pointing “up” and “down” from the sheet. When two sheets stack face-to-face, these interlocking side chains fit into one another like teeth on a zipper (a phenomenon often called “steric zipper” or knob-into-hole packing). This precise, stable alignment is stabilized by powerful van der Waals forces and can sometimes involve aromatic stacking.
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Protein aggregation into highly ordered, regularly repeated cross-β sheet structures called amyloid fibrils is closely associated with human disorders
The reason β-sheets dominate is thermodynamic and structural. Formation of the β-sheet conformation from native protein structures can be induced by high protein concentrations, metal binding, acidic pH, amino acid mutation, and interaction with lipid membranes. All amyloid proteins exhibit a characteristic non-native β-sheet state and aggregate spontaneously into extended fibrils that precipitate out of solution.
The driving force is hydrogen bonding. The formation of ordered arrays of hydrogen bonds drives the formation of β-sheets within disordered oligomeric aggregates. Initially individual β-sheets form with random orientations, which subsequently align into protofilaments as their lengths increase.
Yes, Amyloids are protein-based biomaterials composed of fibrils with cross-β cores. Previously only associated with degenerative diseases, amyloids remain active and functional both in vivo and in vitro conditions, enabling a variety of applications in medicine, nanotechnology, and biotechnology.
Design a β-sheet motif that forms a well-ordered structure.
Since β-sheets are inherently prone to runaway aggregation, designing a β-sheet that folds into a well-ordered, non-aggregating structure is quite challenging.
For the design to simultaneously promote folding and prevent uncontrolled self-association, there are six key design principles:
Strand Sequence: Alternating Hydrophobic/Polar Pattern creating one hydrophobic face (the core) and one hydrophilic face (the solvent-exposed surface).
Turn/Loop Design: The Critical Nucleation Point: The turn connecting two β-strands (forming a β-hairpin) is the nucleation site for folding, a poorly designed turn means the sheet never forms.
Edge Strand Protection: Preventing Aggregation: Exposed β-strand edges have free hydrogen bond donors and acceptors that readily recruit additional strands, causing aggregation into amyloid-like fibrils.
Capping Motifs: Stabilizing the Termini using N-cap and C-cap residues.
Part B: Protein Analysis and Visualization
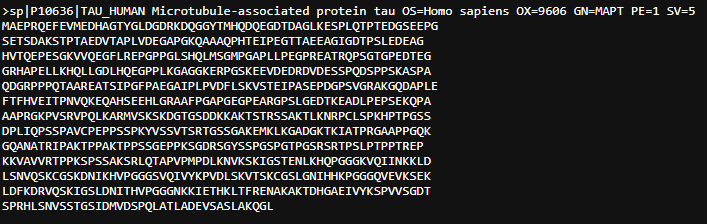
Briefly describe the protein you selected and why you selected it.
I select tau protein. It is a microtubule-associated protein that promotes microtubule assembly and stability, and might be involved in the establishment and maintenance of neuronal polarity. In neurodegeneration, this protein becomes hyperphosphorylated, detaches from microtubules, and aggregates into toxic, insoluble neurofibrillary tangles (NFTs). Since I’m interested in using synthetic biology to understand more neurodegenerative disorders, this protein is of interest.
Another protein is Amyloid beta precursor like protein 1.
It may play a role in postsynaptic function. The C-terminal gamma-secretase processed fragment, ALID1, activates transcription activation through APBB1 (Fe65) binding. Couples to JIP signal transduction through C-terminal binding. May interact with cellular G-protein signaling pathways. Can regulate neurite outgrowth through binding to components of the extracellular matrix such as heparin and collagen I. The gamma-CTF peptide, C30, is a potent enhancer of neuronal apoptosis.
Identify the amino acid sequence of your protein.
Tau Protein
Sequence Length: 758 amino acids
The most common amino acid is: Proline, which appears 93 times.
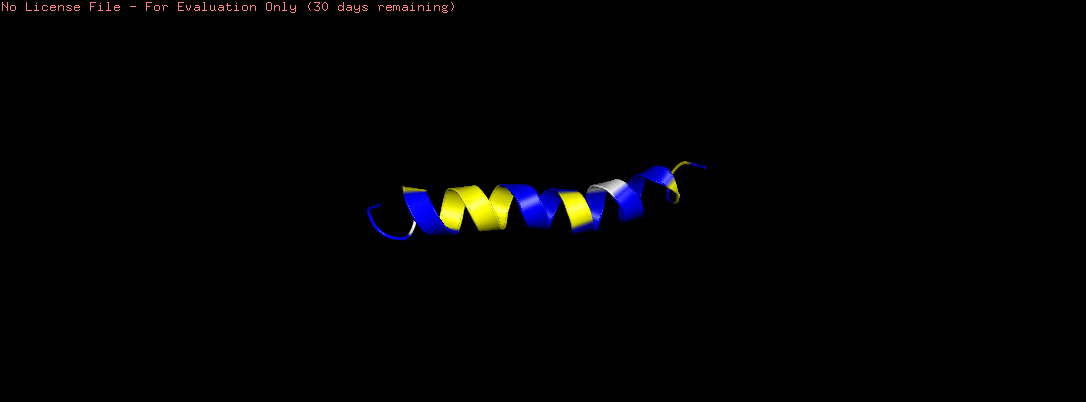
Open the structure of your protein in any 3D molecule visualization software
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”
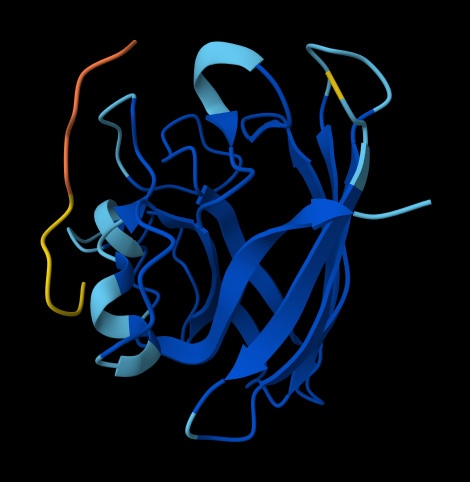
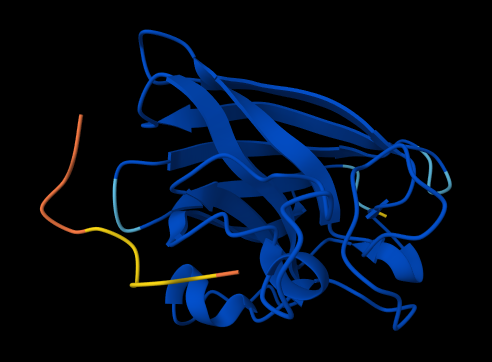
the protein that will be visualized is APP (Amyloid-beta precursor protein)
Cartoon
Ribbon
Ball and Stick
Color the protein by secondary structure. Does it have more helices or sheets?
The helix is colored in cyan and the sheet in magenta, from the image the helices are more
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The hydrophobic residues (ALA, VAL, LEU, ILE, MET, PHE, TRP, PRO) are colored in yellow
The hydrophilic residues (SER, THR, ASN, GLN, TYR, CYS, LYS, ARG, HIS, ASP, GLU) are colored in blue
GLY (neutral) is colored in white
From the image the hydrophilic residues are grouped together and they are more then the hydrophobic ones
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
No, it doesn’t have any holes
Part C: Using ML-Based Protein Design Tools
I chose the Amyloid Beta-Peptide like protein
C1. Protein Language Modeling
Deep Mutational Scans
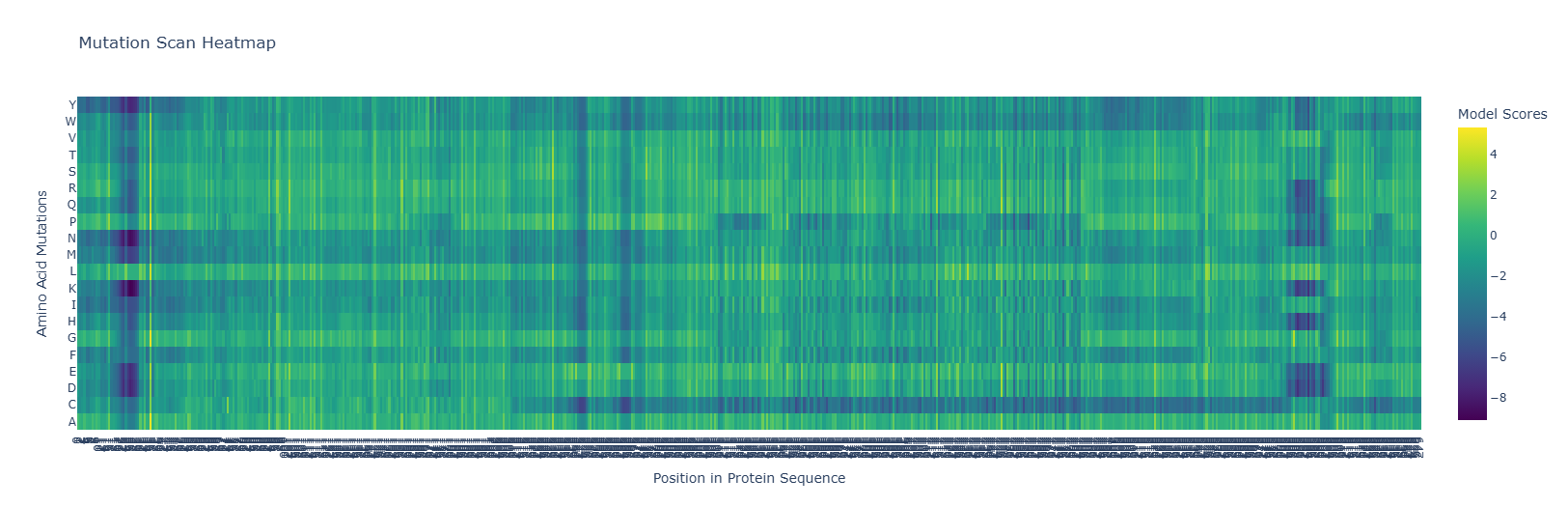
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Fasta File
1AMC_1|Chain A|AMYLOID BETA-PEPTIDE|Homo sapiens (9606) old
DAEFRHDSGYEVHHQKLVFFAEDVGSNK old
The heatmap shows hotspots for mutations that are beneficial or detrimental to the function of the protein.
Notice the dark blue regions, where the LLR values are negative, indicating that mutations that are likely detrimental to function, and lighter yellow regions where the LLR values are positive, indicating mutations that are likely beneficial to the function of the protein. Also, note how there are dark bands running vertically indicating regions which are likely evolutionarily conserved, and brighter bands running vertically indicating regions of the protein which may in fact be preferable over the wild-type sequence. Note also, for some regions of the protein, there are amino acid mutations which are likely to be detrimental to functioning for entire regions of the protein, indicated by dark bands running horizontally along most of the protein. Similarly, we see brighter bands of yellow running horizontally, indicating almost any residue mutated to that amino acid would be preferential to the wild type.
for example replacing by is prefereable. mutations at as well are favourable
Latent Space Analysis
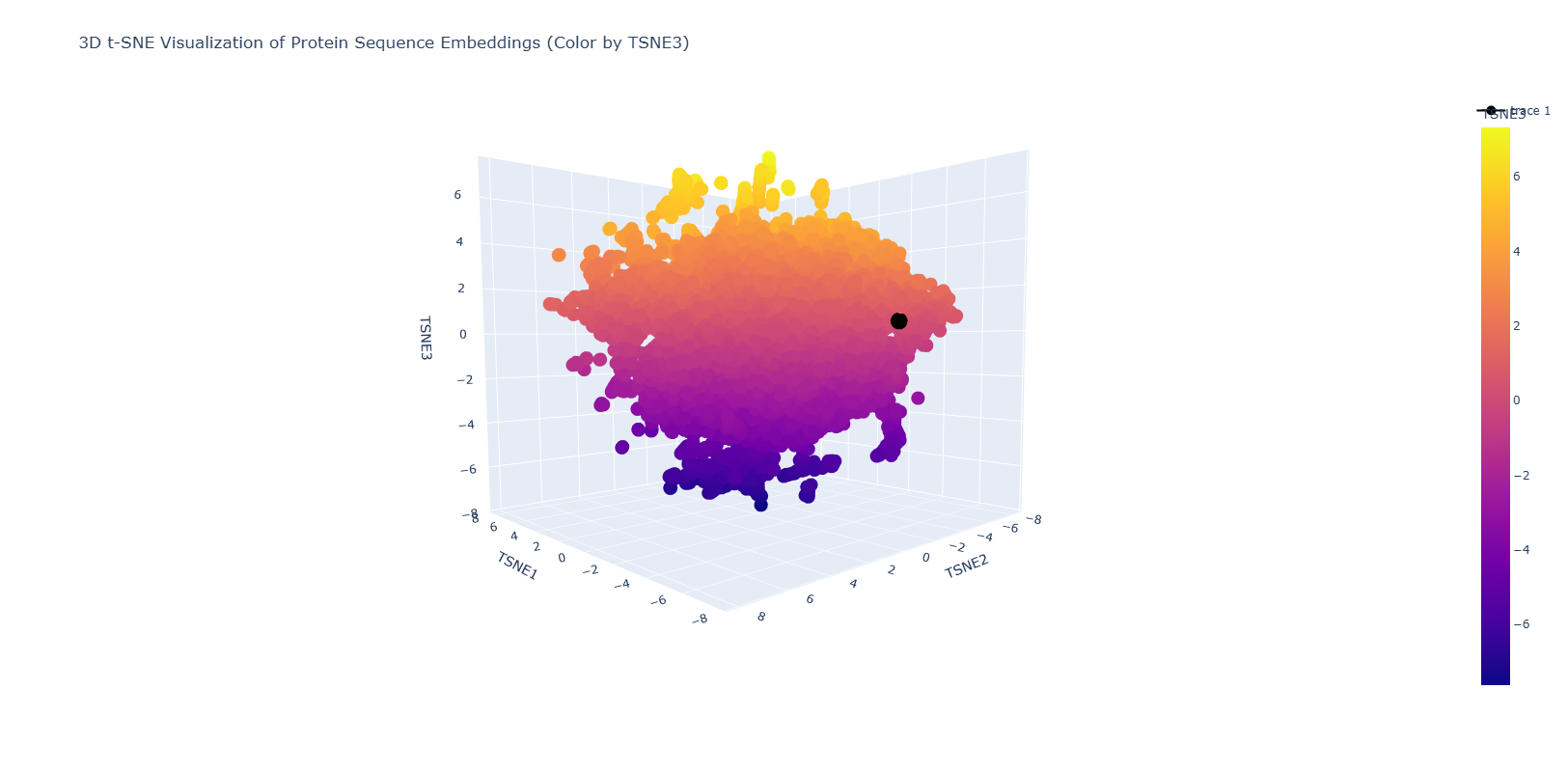
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
No, they don’t match it
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Mutation done: replacing position 22 (L) by K
Mutation: replacing positions 20 to 29 LPLLLPLLLL with NNNNNNNNNN
Part D: Group Brainstorm on Bacteriophage Engineering
1. Find a group of ~3–4 students
2. Read through the Phage Reading material listed under “Reading & Resources” below.
3. Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
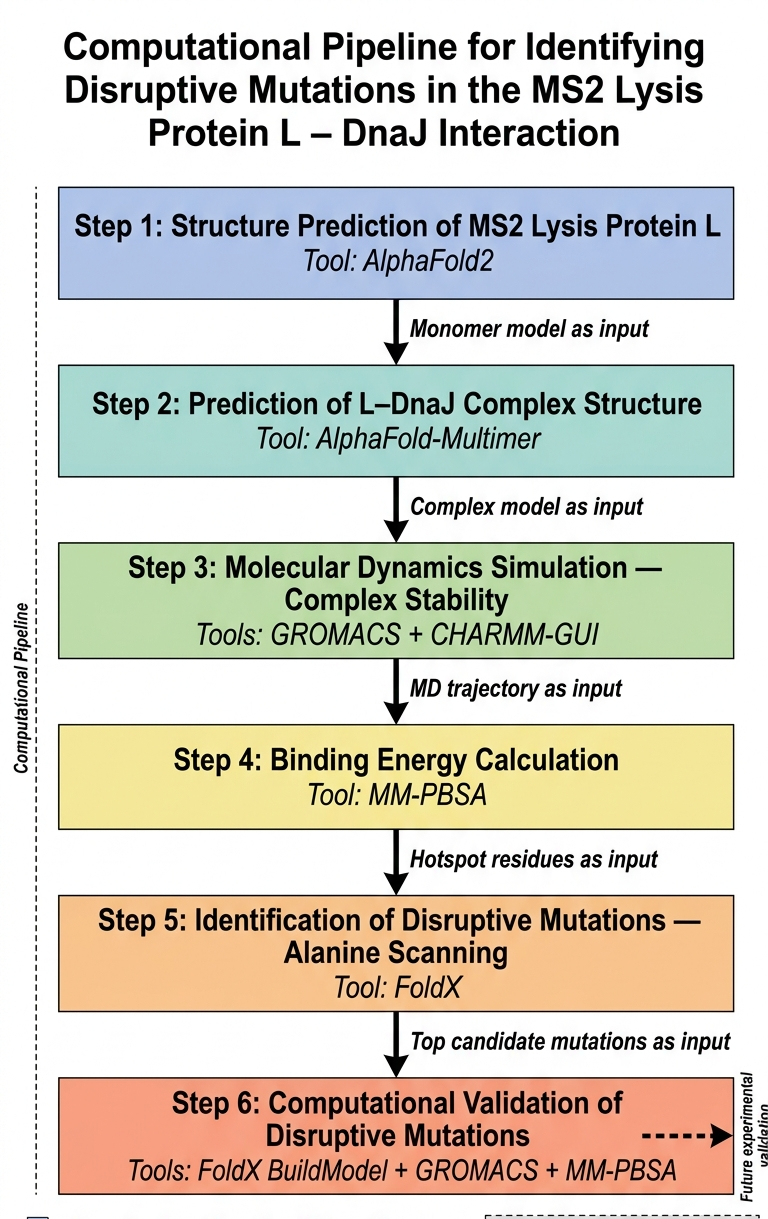
The main goal that I think can be adressed computationally is disrupting the lysis protein interaction with E. coli chaperone DnaJ
The proposed approache is predicting the 3D structure of the complex and then figuring out the genes responsible for the interaction between the lysis protein and the DnaJ and then introdcing mutations to these genes for the aim of disrupting the complex binding
The tools that might help solve the chosen sub-problem are:
ESMFold or AlphaFold2 for 3D structure prediction of L lysis protein
AlphaFold-Multimer to predict how L and DnaJ physically interact
test the complex stability (to validates whether the AlphaFold-Multimer prediction is physically reasonable)
Navigate to the AlphaFold Server: alphafoldserver.com
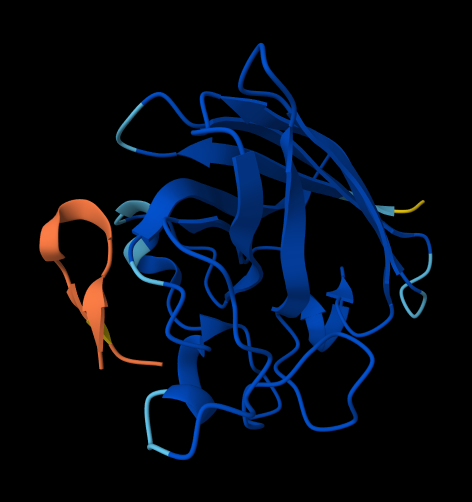
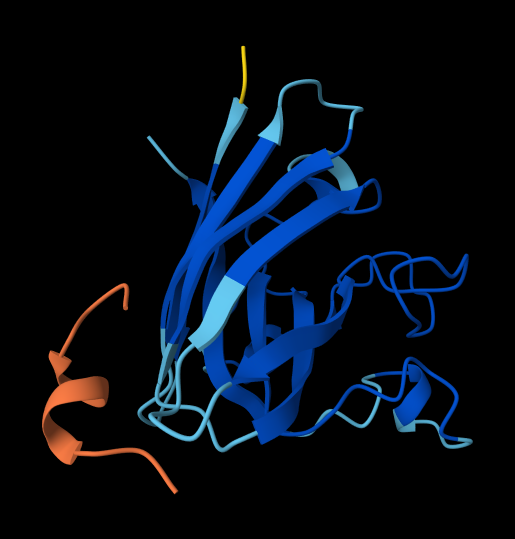
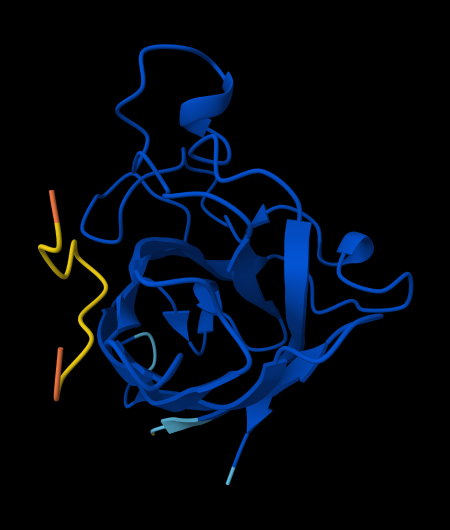
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
ipTM measures the accuracy of the predicted relative positions of the subunits within the complex. Values higher than 0.8 represent confident high-quality predictions, while values below 0.6 suggest likely a failed prediction. ipTM values between 0.6 and 0.8 are a gray zone where predictions could be correct or incorrect.
WHYGAAGARLKE
ipTM = 0.3
since the value is below 0.6, it suggest likely a failed prediction.
WHYPAAVAEWGK
ipTM = 0.28
since the value is below 0.6, it suggest likely a failed prediction.
WRSPATAVAHKK
ipTM = 0.6
the value is 0.6 and it is highest value obtained suggesting it could be correct or incorrect
WLYYPAALEHGE
ipTM = 0.32
since the value is below 0.6, it suggest likely a failed prediction.
FLYRWLPSRRGG
ipTM = 0.37
since the value is below 0.6, it suggest likely a failed prediction.
Pt 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
moPPit peptides
first peptide generated with moPPit
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
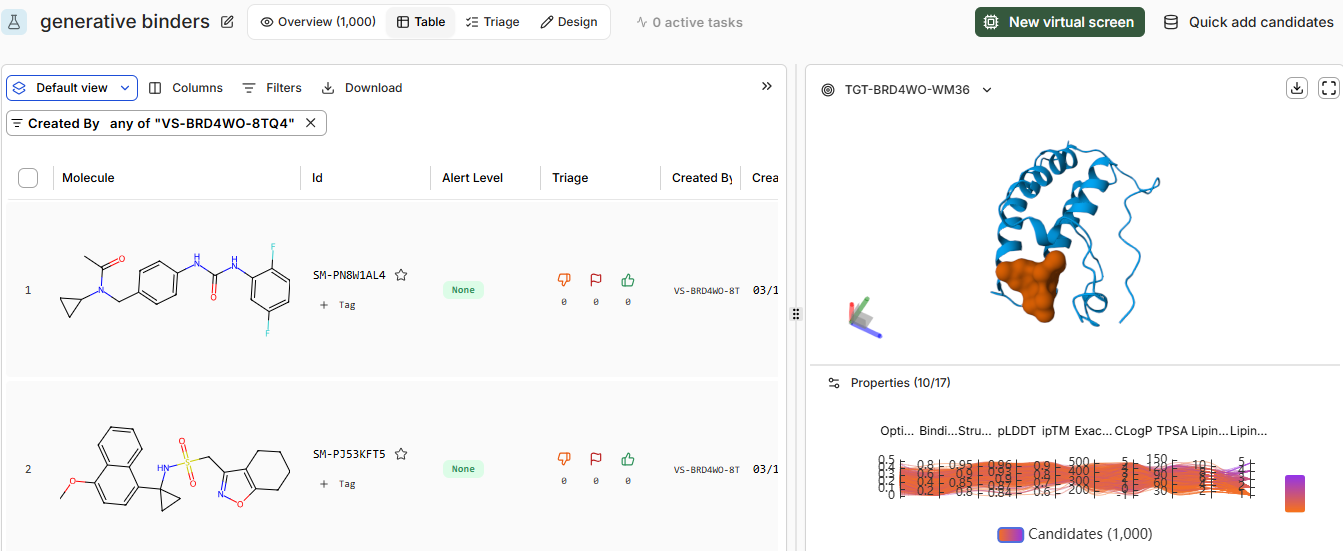
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
Pt 0: Sign-up to Boltz Lab
Pt 1: Structural Predictions in the Sandbox
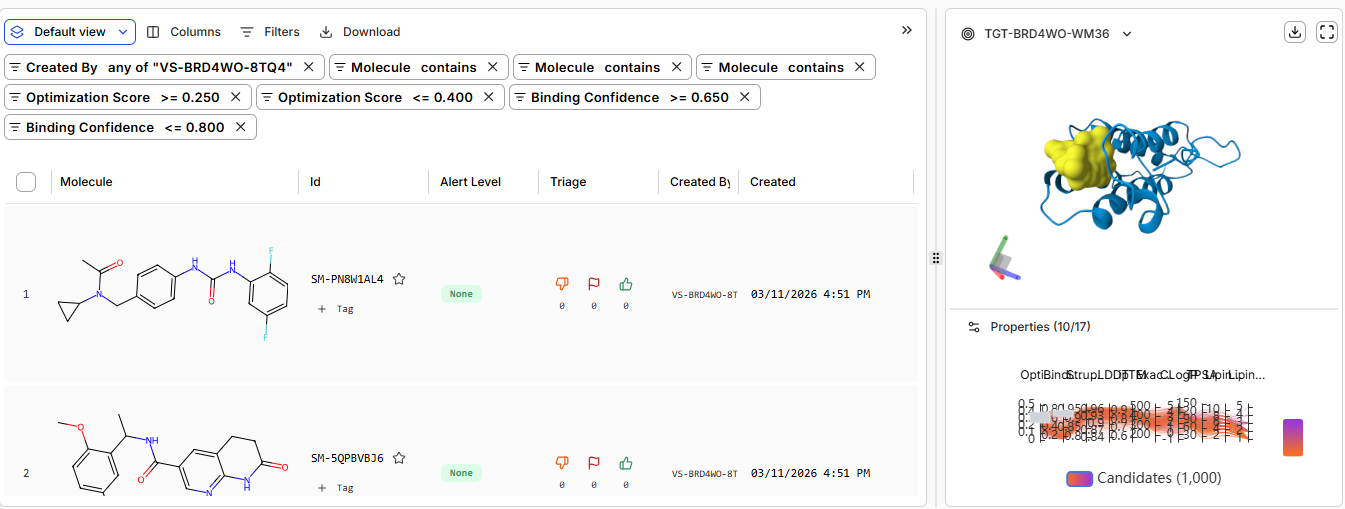
Cmpound
Binding Confidence
Optimization Score
Structure Confidence
Hit
0.45
0.23
0.98
Lead
0.75
0.26
0.98
JQ1
0.96
0.44
0.99
Discussion Questions
Does Binding Confidence increase as you move from hit to clinical candidate? What would
you expect, and why might it deviate?
Binding confidence which means how confidently the ligand is placed in the binding site is higher when JQ1 was chosen as the ligand and lower in hit
Inspect the predicted binding pose for JQ1. Can you identify potential key binding
interactions.
Compare the Optimization Scores. How do the scores compare for JQ1 vs the Lead.
Optimization score for JQ1 is 0.44 while 0.26 for Lead, indicating higher tight binding with JQ1\
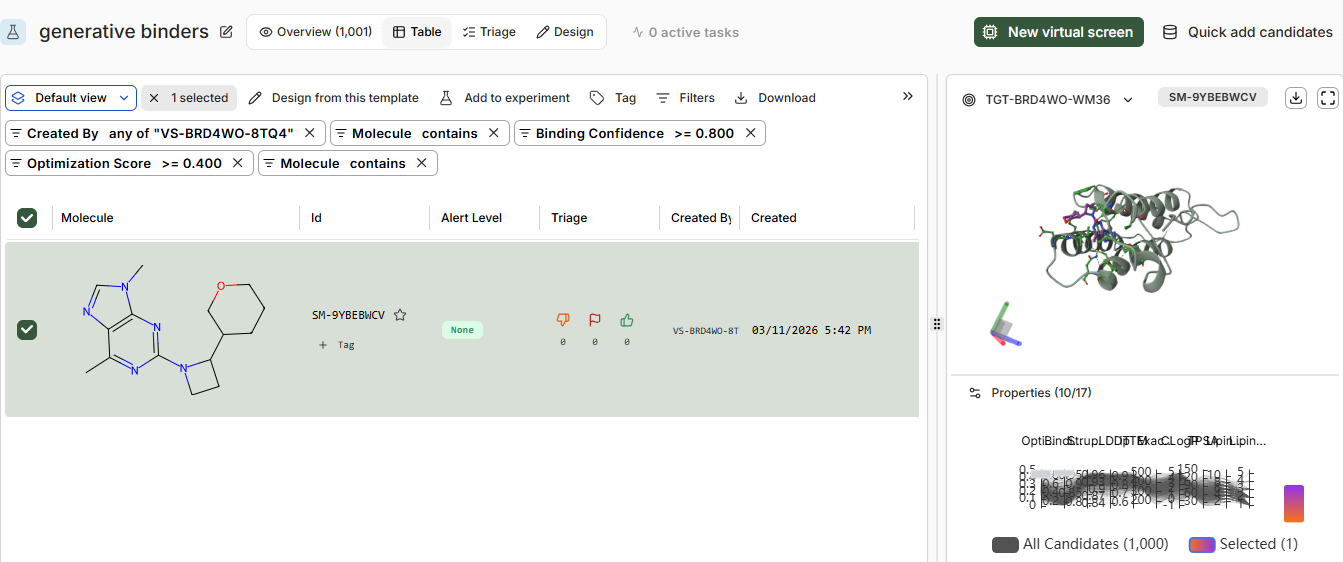
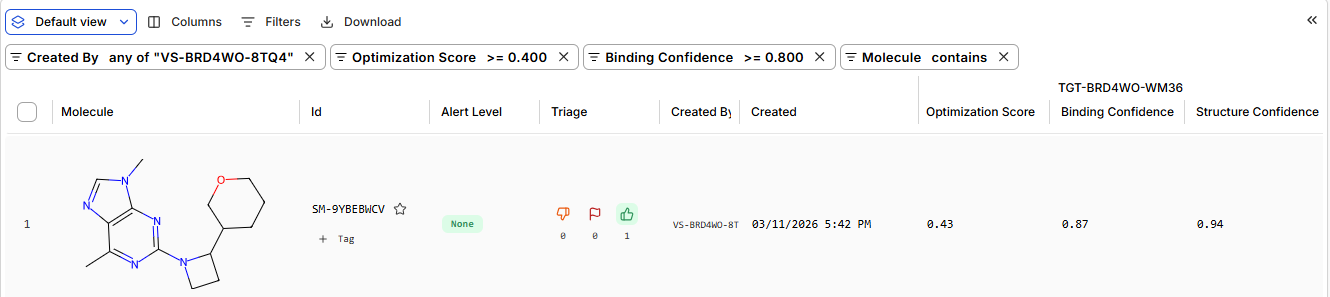
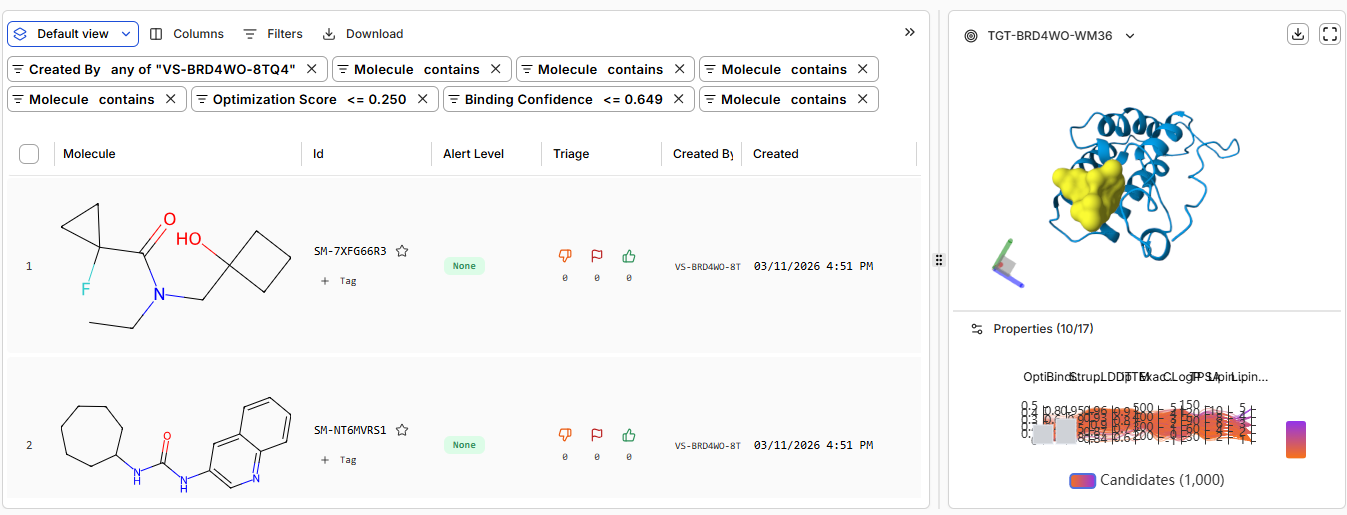
Pt 2: Setting Up a BRD4 Design Project
the predicted structure from boltz vs from RCSB
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The components of the Phusion High-Fidelity PCR Master Mix are the following:
Phusion DNA Polymerase, incorporates nucleotides to “fill in” the gaps in the annealed DNA fragments. it is a hot-start, proofreading PCR enzyme, enabling generation of PCR amplicons with high sequence accuracy, sensitivity, and specificity. Phusion DNA Polymerase is a thermostable polymerase that possesses 5´→ 3´ polymerase activity, 3´→ 5´ exonuclease activity and will generate blunt-ended products.
nucleotides: building blocks for new DNA strands during amplification.
Buffer: it provides the optimal pH, ionic strength, and Mg²⁺ concentration (1.5 mM final) required for Phusion DNA polymerase to bind primers, extend DNA efficiently, and maintain its high fidelity
What are some factors that determine primer annealing temperature during PCR?
The specific primer annealing temperature depends on specific length and sequence of the primers.
It depends also on melting temperature of the primers and therefore GC content
TM = 4(G + C) + 2(A+T)
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Restriction Enzyme Digest
It is a process in which DNA is cut at specific sites, dictated by the surrounding DNA sequence.
It is accomplished by incubation of the target DNA molecule with restriction enzymes - enzymes that recognize and bind specific DNA sequences and cleave at specific nucleotides either within the recognition sequence or outside of the recognition sequence.
Restriction digestion can result in the production of blunt ends (ends of a DNA molecule that end with a base pair) or sticky ends
Restriction digestion is usually used to prepare a DNA fragment for subsequence molecular cloning
The results of a restriction digestion can be evaluated by gel electrophoresis, in which the products of the digestion are separated by molecule length
The components of a typical restriction digestion reaction include the DNA template, the restriction enzyme of choice, a buffer and sometimes BSA protein. The reaction is incubated at a specific temperature required for optimal activity of the restriction enzyme and terminated by heat.
reaction mixing, incubation at specific temperatures and time
PCR
is a method for amplifying DNA. millions of copies of a DNA sequence can be generated from a single copy or just a few copies of DNA
PCR protocols consist of assembling a PCR reaction mix containing Taq polymerase (a thermostable DNA polymerase that can withstand the high temperatures required for thermal cycling), primers (short DNA sequences that define the target region for amplification), deoxynucleotide triphosphates (dNTPs, the building blocks of DNA) and MgCl2 (Taq polymerase co-factor) in a buffered solution. The reaction mixture undergoes three basic thermal cycling steps including (1) denaturation (usually at 95°C, (2) annealing (usually lowest primer melting temperature - 5°C, (3) extension (usually at 72°C).
components mixing , 2. denaturation step where the double stranded DNA denatures into single strands, annealing for primers annealing then extension where DNA polymerase extends the DNA creating million copies of required DNA fragment
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
after generating dna fragments by pcr, run agarose gel to check for size and yield
How does the plasmid DNA enter the E. coli cells during transformation?
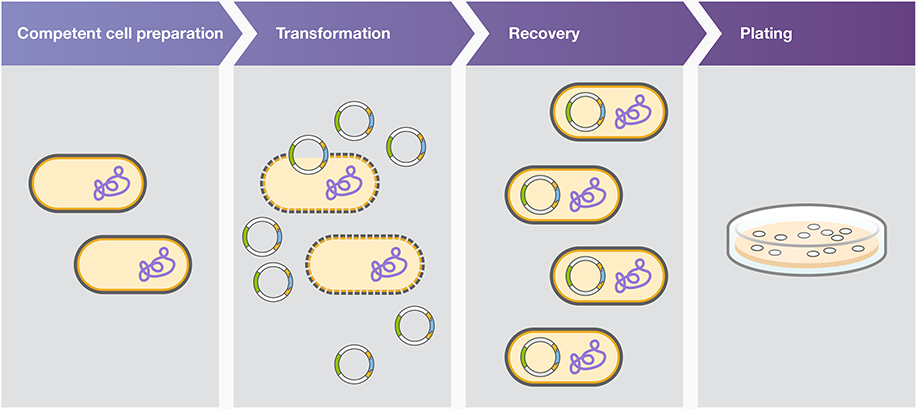
Heat shock transformation is also known as chemical transformation and calcium chloride transformation. This method involves subjecting the cells to a sudden increase in temperature, often achieved by briefly immersing them in hot water or placing them in a heating block, followed by a rapid decrease in temperature through incubation on ice. The heat shock causes the cell membrane to become more permeable, facilitating the uptake of exogenous DNA.
Plasmid uptake by chemically competent cells is facilitated by heat shock, and plasmid uptake by electrocompetent cells is facilitated by electroporation.
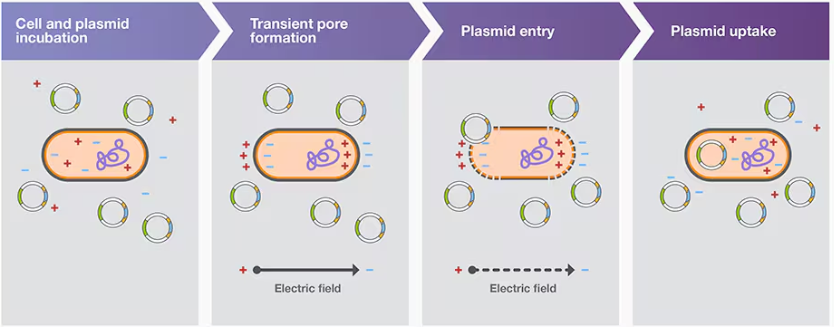
Electroporation transformation
Bacterial transformation aided by electroporation is called electroporation transformation; electroporation involves using an electroporator to subject competent cells and the plasmid carrying DNA construct to a brief pulse of a high-voltage electric field (Figure 3B). This treatment induces transient pores in cell membranes, which permits plasmid entry into the cells
One of the main issues with electroporation is arcing, or electric discharge, which may lower cell viability and transformation efficiency. Arcing often results from electroporation in conductive buffers, such as those containing MgCl2 and phosphates.
it enters through creating pores in bacterial membrane, these pores can be created by either heat shock
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
References
Week 7 HW: Genetic Circuits Pt.2
Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
The main advantage IANNs hold over traditional genetic circuits is scalability and the ability to support multilayer networks for complex decision-making. Traditional genetic circuits limitations include poor predictability and the struggle to reliably program multiple functions simultaneously due to inherent scalability limitations. On the other hand, ANNs have good predictability offering improved robustness for complex designs. Because of multiple layers and non-linear activations, neural networks can model complex, non-linear decision boundaries
Traditional genetic circuits have input/output behaviors that function as Boolean operations. They process discrete signals (ON/OFF, high/low expression) through logic gates like AND, OR, and NOT, producing binary outputs based on truth tables. Moreover, the output layer in the ANNs producing the final prediction may be binary, multi-class or a continuous value.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Application of CNNs: tumor and MSI detection in gastrointestinal cancer
Convolutional Neural Networks (CNNs) are deep learning models designed to analyze structured grid-like data such as images.
The CNNs were used as automatic tumor detector to predict MSI (Microsatellite instability) that determines if the patient with gastrointestinal cancer will respond will to immunotherapy. The authors used hematoxylin and eosin (H&E)-stained histology slides as an input
For tumor detection in gastrointestinal cancer, the authors trained a convolutional neural network with deep residual learning (resnet18)12 model to classify tumor versus normal tissue by transfer learning. Transfer learning means reusing a pre-trained neural network model on a new but related task, instead of training from scratch. For MSI detection, we trained another resnet18 model for each tumor type.
input/output behavior
Input: Tiles extracted from digitized histology slides.
Output: For each tile, a probability score indicating tumor vs. normal or MSI vs. MSS status.
Behavior: The neural network processes image features within each tile to generate these probability scores, enabling localized tissue characterization and subsequent patient-level molecular classification.
The mentioned limitations of CNN were:
Classifying ability is limited to cancer type and ethnicity in the training set. therefore, larger training cohorts are needed to boost classification performance because rare morphological variants can be learned by the network
The required tissue size. To define its lower limit, they generated ‘virtual biopsies’ and found that performance plateaued at approximately 100 tiles of 256 μm edge length, suggesting that biopsies are sufficient for MSI prediction
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Rigid fungal composites
They are created by combining fungi with lignocellulosic fibers or particles, producing materials with varying properties depending on the used finishing method (e.g., hot/cold pressing) , followed by the used substrate, as well as fungal species and strains, particularly the growth behavior and hyphal type, besides substrate nutritional profile and growth conditions
Disadvantages: their mechanical strength and moisture uptake limit their use primarily to non-weight-bearing applications, such as interior panels and acoustic absorption
Advantages: biodegradable and have demonstrated potential in architectural designs
Examples
Mycotectural Alpha (2009): Utilized G. lucidum-bound sawdust for its construction.
Hy-Fi (2014): A cluster of circular towers made from mycelium-based bricks.
MycoTree (2017): Featured mycelium-bound composite blocks in its installation.
My-Co Space (2021): Showcased elements of hemp-grown F. fomentarius on a supporting structure.
Flexible Fungal Materials
Flexible fungal materials have diverse applications, including fungal wound dressings (e.g., F. fomentarius), medical cell scaffolds, paper like materials, fungal chitin nanomaterials, filters for water treatment, and meat analogs
Disadvantages: limited availability and fragility
Advantages: sustainable, biodegradable, and customizable, and their properties depend on the fungal strain, substrate, growth regime, and post-processing techniques (e.g., drying, pigmenting, plasticizing) for enhancing (microbiological) robustness and appearance. Mycelium-based foams and leather alternatives, made from agricultural waste, are cruelty-free and more eco-friendly than traditional materials, as they generate less pollution and use less water. They are lightweight, offer good thermal and acoustic insulation, and require fewer resources to produce
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Filamentous fungi are considered as unique cell factories for protein production due to the high efficiency of protein secretion and superior capability of post-translational modifications. Therefore, they can be engineered to secrete proteins with higher efficiency.
Genetically engineered fungi have diverse applications across food, industry, medicine, and agriculture due to their eukaryotic biology and secretion capabilities.
Applications:
Food Prodcution: production of high-protein, meat-like alternatives with enhanced nutrition
Industrial Biotech: industrial enzymes where fungi serve as cell factories for secreted enzymes like glucoamylase or cellulases used in biofuels, detergents, and food processing
Pharmaceuticals: where fungi produce secondary metabolites (antibiotics, anticancer drugs)
Both bacteria and fungi have their unique properties in synthetic biology. Synthetic biology in fungi offers key advantages over bacteria, particularly for complex eukaryotic pathways, due to their eukaryotic machinery and natural industrial traits.
Bacteria is a prokaryote with simple cell wall and fast growth rate. they are versatie and easily genetically manipulated. They are extensively used in the production of antibiotics, enzymes, and biofuels.
In agriculture, bacteria serve as biofertilizers and biopesticides, enhancing soil fertility and protecting crops from pests and diseases.
In medicine, bacteria are harnessed to produce therapeutic proteins and vaccines, and they are central to the development of new antibiotics
Fungi is an eukaryote with thick chitinous cell wall and slow growth rate. They have long been utilized in biotechnology for their ability to produce a wide range of metabolites, including antibiotics, enzymes, and organic acids. Fungi grow on cheap, complex substrates like lignocellulose or waste, reducing costs compared to bacteria’s need for purified sugars.
They have applications in many sectors, for example:
Food industry: production of bread, beer, and cheese.
Agriculture: they improve agricultural crop yield and quality by enhancing plant physiology and stress tolerance.
Environmental sustainability: they play a significant role by decomposing organic matter, thus recycling nutrients in ecosystems.
Medicine: they are sources of important pharmaceuticals, such as penicillin, and are being explored for their potential in developing new drugs.
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
The main advantages of cell-free protein synthesis (CFPS) over traditional in vivo methods include
Cell-free systems do not need time-consuming cloning steps
Easy manipulation of reaction conditions
High-throughput potential
Synthesis of difficult to express proteins, such as toxic and transmembrane proteins. In addition, the absence of the cellular membrane allows the synthesis of modified proteins with statistically as well as site-specifically embedded non-canonical amino acids
CFPS is easily adaptable to the translational requirements of a particular target protein, and the synthesis conditions can be adjusted for a desired subsequent analytical setup
Novel automated high-throughput systems are being developed due to the simple handling of liquids and the easy scalability of cell-free reactions
Via the removal of the cell membranes and redundant parts of cells, CFPS has provided flexibility in directly dissecting and manipulating the Central Dogma with rapid feedback. non-native chemicals can be introduced directly into the system, allowing greater flexibility in the selection of regulating reagents
Such an open nature of the CFPS enables the first-ever programming of modular cellular mimicking processes with active transcription and translation support.
ease of use, rapid protein production, and minimal requirements for lab space, equipment, and expertise compared to traditional methods
Flexibility: The cell-free expression system can utilize various template DNAs, including PCR products, plasmid DNA, and synthetic DNA, making it suitable for expressing different types of proteins.
Two cases where cell-free expression is more beneficial than cell production.
Cell-free protein expression lets researchers incorporate unnatural labels or amino acids into targets of interest, as well as express toxic proteins
The accessibility of cell-free reactions enables optimization impossible in cells. Researchers can directly adjust pH, ionic strength, redox potential, metal ion concentrations, or temperature without considering cellular viability. Specific folding catalysts, chaperones, or cofactors can be added at precise concentrations. For disulfide-bonded proteins, the oxidation-reduction balance can be fine-tuned by adding specific ratios of reduced and oxidized glutathione. For metalloproteins, appropriate metal ions can be supplemented. This level of control over the biochemical environment enables optimization of yield and proper folding for challenging targets that fail in standard cellular environments.
Describe the main components of a cell-free expression system and explain the role of each component.
There are three fundamental components:
Cell-Free Extract: This is the heart of the system, containing the essential cellular components for protein synthesis, such as ribosomes, tRNA, amino acids, and enzymes. The source of the extract can vary, with commonly used ones including E. coli, rabbit reticulocyte, and wheat germ extracts.
DNA Template: Researchers provide the genetic information for the desired protein in the form of a DNA template. This template typically contains a promoter sequence to initiate transcription and a coding sequence for the target protein.
Energy and Cofactors: Energy sources (e.g., ATP, GTP) and cofactors (e.g., magnesium ions) are supplied to facilitate transcription and translation processes.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical in cell-free systems because protein synthesis is ATP-intensive, and there is no living metabolism in the reaction mix to continuously replenish ATP as a cell would. Without regeneration, ATP is depleted quickly, protein synthesis slows or stops, and yield drops; stable energy supply is also a major determinant of reaction duration and cost.
Current energy module engineering solutions:
ATP regeneration systems for CFPS
using phosphoenolpyruvate (PEP) with pyruvate kinase, creatine phosphate with creatine kinase, or acetyl phosphate with acetate kinase. These systems help maintain energy levels throughout the protein synthesis process, significantly improving yield and duration of the reaction.
enzymes like creatine kinase or acetate kinase that regenerate ATP from ADP using high-energy phosphate donors.
Secondary energy sources such as phosphoenolpyruvate, creatine phosphate, and acetyl phosphate can be incorporated into cell-free protein synthesis systems to enhance energy availability. These compounds serve as phosphate donors in enzymatic reactions that regenerate ATP.
Continuous-exchange cell-free protein synthesis systems: Continuous-exchange cell-free protein synthesis systems involve the continuous supply of energy substrates and removal of inhibitory byproducts during the reaction. These systems utilize specialized reaction chambers with semi-permeable membranes that allow small molecules to diffuse while retaining larger components like ribosomes and enzymes. This approach significantly extends reaction lifetimes and increases protein yields by preventing energy depletion and byproduct accumulation that typically limit batch reactions.
Secondary energy sources and cofactors
Beyond primary ATP regeneration, cell-free protein synthesis energy modules incorporate secondary energy sources and essential cofactors. These include NAD+/NADH, NADP+/NADPH, and GTP, which support various biochemical reactions during protein synthesis. Optimized ratios of these cofactors are critical for maintaining redox balance and ensuring efficient translation. Some systems also utilize glucose or maltose with appropriate enzymes to create a continuous energy supply pathway, enhancing the overall efficiency and productivity of the cell-free system.
Engineered extracts for improved energy efficiency
Specially engineered cell extracts can significantly improve the energy efficiency of cell-free protein synthesis systems. These extracts are often derived from modified organisms with enhanced metabolic pathways or reduced energy-consuming side reactions. By eliminating competing pathways that deplete energy resources and optimizing the concentration of key enzymes involved in energy metabolism, these engineered extracts can sustain protein synthesis for longer periods with higher yields. Some approaches include genetic modifications to reduce phosphatase activity or enhance glycolytic flux.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free expression systems
The E. coli based CFPS system has redefined the scale standard for protein synthesis. Its core advantage lies in the simplicity and metabolic robustness of the prokaryotic machinery, allowing for high concentration yields in batch and continuous-flow reactions. This platform is the undisputed leader in low-cost, high-throughput synthesis for projects where functional folding (e.g., disulfide bonds or glycosylation) is not a critical factor.
Scale Advantage: Capable of producing up to 2 mg/mL of protein, dramatically reducing the cost per gram—similar to Twist’s $0.003/bp DNA synthesis advantage.
Speed Metric: Protein production can be completed within 2–4 hours, allowing for parallel synthesis of hundreds of constructs in a single day.
Modification Niche: Highly adaptable for specialized labeling, such as efficient incorporation of non-natural amino acids (CFPS for Non-Natural Amino Acid Incorporation Service), due to easy depletion of natural amino acids in the lysate.
Limitation: Lacks the machinery (PDI, chaperones, microsomal membranes) for correct folding and processing of complex eukaryotic proteins, often resulting in inclusion bodies or inactive constructs.
Eukaryotic cell-free expression systems
Eukaryotic CFPS systems have specialized in overcoming the functional bottlenecks of prokaryotic expression. By retaining cell-specific endogenous elements—including ribosomes, tRNA pools, and PTM enzymes, they achieve functional integrity for complex targets, aligning with the “clinical-grade accuracy” of GenScript.
Systems based on mammalian cells (HEK293, CHO) are essential for therapeutic protein research.
Fidelity Core: The presence of microsomal membranes allows for co-translational or post-translational translocation, critical for synthesizing Cell-Free Membrane Protein Expression and functional antibodies.
Key PTM: Capable of performing initial N-glycosylation and forming correct disulfide bonds, reaching a functional correctness standard of 99% for single-chain variable fragments (scFv).
Limitation: Production yields are generally 5–10 times lower than E. coli systems, and the lysate preparation process is costly.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Design an example of a useful synthetic minimal cell as follows
According to S. Cao, L. C. da Silva, K. Landfester, Angew. Chem. Int. Ed.2022, 61, e202205266; Angew. Chem.2022, 134, e202205266.
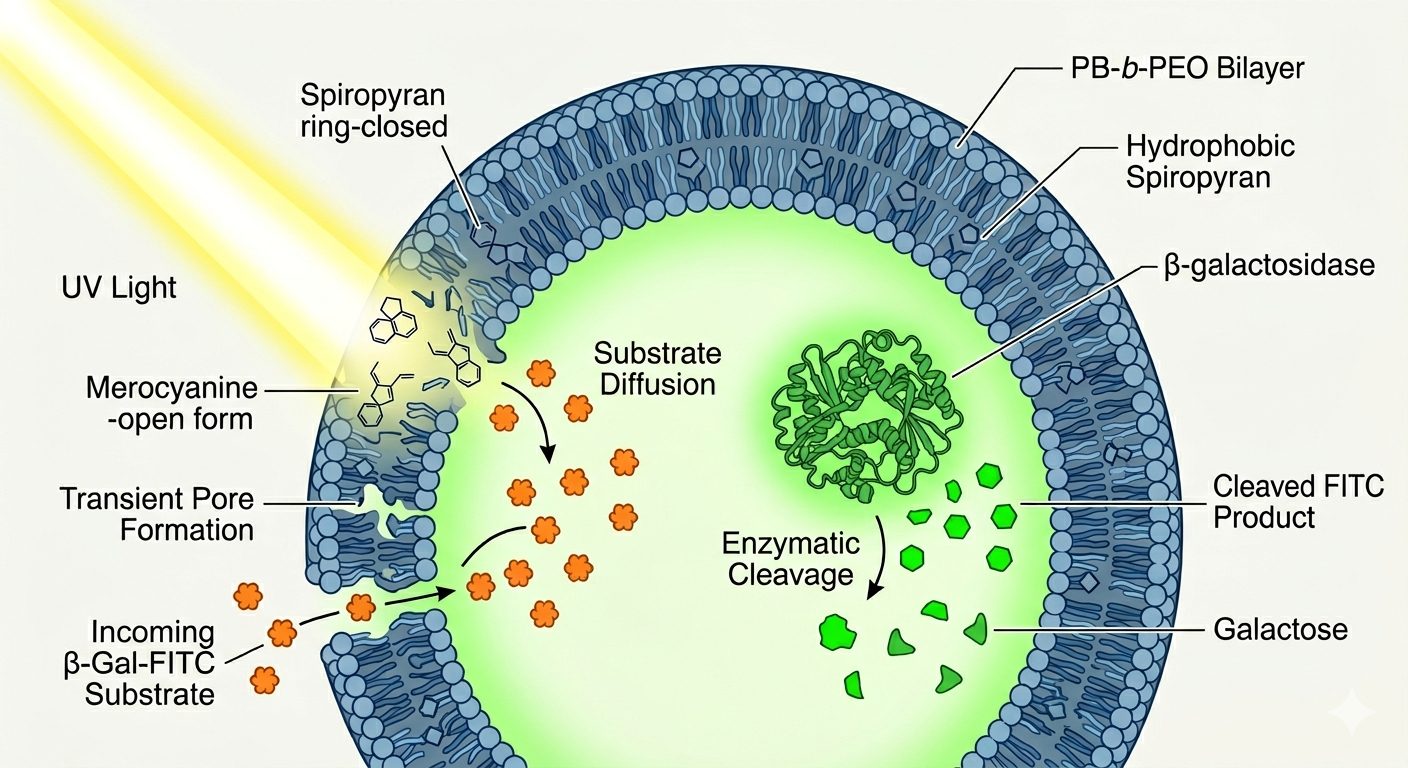
Pick a function and describe it.
The synthetic cell functions as a light-activated micro-reactor that controls internal enzymatic reactions on demand. Its input is dual: a UV light signal that opens membrane permeability, and an external substrate (β-Gal-FITC) that enters the vesicle as a result. Its output is the fluorescent molecule FITC, produced when the encapsulated enzyme β-galactosidase hydrolyzes the substrate inside the polymersome, providing a directly measurable chemical signal that confirms successful reaction activation.
Controlling enzymatic reactions solely through cell-free transcription and translation without encapsulation is difficult because the system relies on compartmentalization and membrane-based regulation of substrate access. Encapsulation provides the spatial and temporal control needed for light-activated permeability, which would be challenging to replicate with free, unencapsulated systems.
Yes, natural cells can be genetically modified to achieve light-controlled membrane transport and enzymatic activity, similar to the synthetic system, using light-sensitive proteins or channels
The desired outcome of the synthetic cell operation is to achieve precise, light-controlled regulation of membrane permeability, enabling selective transport of small hydrophilic molecules into and out of the compartments, and to facilitate dynamic internal processes such as enzymatic reactions and biomolecular coacervation, mimicking natural cellular functions with controllable activation and deactivation triggered by light stimuli.
Design all components that would need to be part of your synthetic cell
The membrane of the giant polymersomes is made of a copolymer consisting of poly(butadiene)-block-poly(ethylene oxide) (PB-b-PEO). Additionally, it incorporates spiropyran-based photo-transducers, specifically SP-C16, which are hydrophobic molecules embedded within the polymer matrix to enable light-responsive behavior.
Inside the giant polymersomes, various hydrophilic molecules and macromolecular cargoes are encapsulated. These include water-soluble molecules like FITC-labeled poly-L-lysine, fluorescent dyes, as well as larger biomolecules like bovine serum albumin and dextran. The system was also used to encapsulate enzymes like β-galactosidase and other active components for constructing light-controlled microreactors and dynamic cell-like systems
The system described is synthetic and biomimetic; it does not originate from a natural organism. Instead, it is engineered using polymeric materials and designed to mimic cellular functions in a controlled, artificial environment.
The synthetic cell communicates with the environment through light-controlled membrane permeability. By using spiropyran-based modulators, the membrane can be made permeable or impermeable to small molecules in response to light, allowing controlled exchange of signals and substrates, and enabling interactions with its surroundings.
Experimental details
To design a synthetic cell mimicking the functions described— such as light-activated membrane permeability, controlled molecular transport, and internal biochemical reactions, I will select lipids and genes that facilitate membrane stability, responsiveness to stimuli, and internal functionalities.
Lipids:
Phospholipids: Since natural membranes are lipid-based, incorporating phospholipids like phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylserine (PS) can provide a biocompatible and flexible membrane.
Cholesterol: To modulate membrane fluidity and stability.
Light-responsive lipids: Incorporate lipids conjugated with photoresponsive groups (similar to spiropyran derivatives) that can undergo structural changes upon light irradiation, affecting membrane permeability.
Synthetic amphiphiles: Custom-designed lipids with specific functional groups for stimulus-responsive behaviors.
Genes:
Enzymes for internal reactions: Genes encoding enzymes such as β-galactosidase (as used in the study) or other catalytic proteins to perform the desired biochemical transformations.
Transporter genes: For internal regulation, genes encoding for synthetic or natural transporter proteins can be introduced to enhance selectivity of transport.
Regulatory genes: To allow dynamic response or adaptation within the synthetic cell, synthetic regulatory gene circuits could be designed.
The function of the light-activated synthetic cell system can be assessed through several experimental approaches:
Permeability Testing by using fluorescently labeled small molecules outside the system and monitoring their diffusion into the system upon light activation using fluorescence microscopy or spectroscopy.
Internal Biochemical Reactions by incorporate a fluorescent substrate inside the system and then measuring the production of fluorescent products using confocal microscopy or fluorometry before and after light activation to verify enzyme activity and substrate access
Substrate Conversion and Product Release by quantifing reaction products in the external medium using spectroscopic methods or HPLC
Dynamic Response and Control by performing time-resolved studies applying light stimuli repeatedly to test the reversibility and responsiveness of permeability and internal reactions.
Write a one-sentence summary pitch sentence describing your concept.
a freeze-dried embedded biosensor in fabric that detects cortisol levels in diabetic patients
How will the idea work, in more detail? Write 3-4 sentences or more.
for the idea to work, first the sensor should be designed and synthetized, following that, it will be freeze-dried and embedded on a fabric that upon activation / hydration using sweat produce a detectable colorimetric output indicating the high level presence of the biomarker (cortisol)
What societal challenge or market need will this address?
It will mainly address patients suffering from metabolic and endocrine dieases to mental health patients and healthy individuals seeking performance and wellness optimization
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
activation is a big limitation since the sensor is acitvated by any liquid, so the specificity to be activated by sweat is a challenge that can be solved by the option of protecting the sensor from external liquids and activate it on-demand
the second limitation is the usage time and this can be addressed through replacing something in the fabric not the whole system, therefore, increasing the usage time with affecting the system.
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Long-duration space missions expose astronauts to profound physiological and psychological stressors that significantly compromise health, cognitive performance, and behavioral integrity. Diagnostic health technologies during space missions pose many challenges, including limited availability of medical personnel, tools, and equipment. Cortisol, the primary stress hormone measurable non-invasively in sweat represents a scientifically compelling real-time biomarker for simultaneously tracking physiological and psychological deterioration. Dynamic monitoring of specific biomarkers is essential in the diagnosis of fluctuating conditions. Developing autonomous, wearable biosensing platforms capable of continuous, non-invasive biomarker detection is therefore both scientifically urgent and mission-critical for safe deep-space human exploration.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
The primary molecular targets proposed are cortisol (a steroid hormone), cell-free circulating RNA (cfRNA), and the HPA axis-responsive gene transcripts, specifically FKBP5 and NR3C1 (glucocorticoid receptor gene)
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Long-duration spaceflight chronically activates the hypothalamic-pituitary-adrenal (HPA) axis through persistent psychological and physiological stressors, resulting in sustained cortisol elevation and downstream immune suppression. Elevated cortisol during spaceflight activates the HPA axis, suppressing cell-mediated immunity and triggering reactivation of latent herpesviruses. At the molecular level, variations in NR3C1 and FKBP5 alter glucocorticoid receptor sensitivity and HPA axis dynamics, while chronic stress induces persistent HPA activation and maladaptive physiological responses. Since cortisol is detectable non-invasively in saliva, and FKBP5/NR3C1 transcripts reflect real-time HPA axis status, these targets collectively enable continuous, non-invasive monitoring of both psychological stress and immune health, critical capabilities currently absent from deep-space mission medicine.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Hypothesis: freeze-dried, cell-free synthetic biology circuits embedded in a wearable fabric, activated by saliva, can simultaneously detect salivary cortisol protein and FKBP5/NR3C1 mRNA transcripts, providing a non-invasive, real-time dual-layer readout of HPA axis status in astronauts during deep-space missions.
This reasoning converges on three evidence pillars: first, freeze-dried cell-free synthetic circuits embedded in textiles are activated upon rehydration from liquids and can detect metabolites and nucleic acid signatures, with detection limits rivalling quantitative PCR. Second, significant correlations exist between free cortisol levels in saliva and blood, attributed to cortisol’s small molecular weight and lipophilicity enabling diffusion through glandular epithelial membranes. Third, since FKBP5 and NR3C1 transcripts are present in salivary cells and reflect real-time HPA axis regulatory status, their co-detection alongside cortisol protein would distinguish acute stress from chronic HPA breakdown, enabling autonomous, physician-independent health surveillance.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
The experiment proceeds in three phases. First, freeze-dried cell-free toehold switch biosensors targeting FKBP5 and NR3C1 mRNA transcripts and a cortisol-specific aptamer module will be validated in vitro using synthetic RNA and recombinant cortisol in artificial saliva, with nuclease-free buffer and non-target RNA as controls. Second, human saliva collected before and after the Trier Social Stress Test from healthy volunteers will validate biosensor performance against gold-standard ELISA and qRT-PCR. The cortisol sensor reliability will be confirmed by comparison to ELISA immunoassay across stressed and non-stressed conditions. Third, biosensors will be integrated into textile substrates and tested for colorimetric output, storage stability, and reproducibility upon deliberate saliva application, simulating operational astronaut use.
Part B: Individual Final Project
Put your chosen final project slide in the appropriate slide deck following the instructions on slide 1
Submit this Final Project selection form if you have not already.
Begin planning how you will write your final project documentation based on the guidelines
Prepare your first DNA order and put it in the “Twist (MIT)” or “Twist (Nodes)” tab of the 2026 HTGAA Ordering: DNA, Reagents, Consumables spreadsheet, as appropriate.
Week 10 HW: Imaging and Measurement
Final Project
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
In my project, I will measure the fluorescence that indicate the intensity of the biomarker present. Additionally, after the production of miRNA, I will measure their molecular weight to make sure they are right.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
I will measure it using UV Spectrophotometer for fluorescence and LC-MS for molecular weight
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Mass spectrometry will be used mainly for molecular weight determination to get the chromatogram and make sure from it that the produced peaks are correct.
Homework: Waters Part I — Molecular Weight
The calculated molecular weight based on the predicted eGFP amino acid sequence is:
Theoretical pI is 5.90
Theoretical molecular weight is 28006.60 Da
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Determine z for each adjacent pair of peaks (n, n+1) using:
the selected two charge states are (m/zn+1) = 875.4421 and (m/zn) = 903.7148
therefore, the z = 30.96 = 31
Determine the MW of the protein using the relationship between m/zn, MW and z
MW = (n x m/zn) - n
n (number of charges) = 31
MW = (31 x 903.7148) - 31 = 27,984.1588
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1
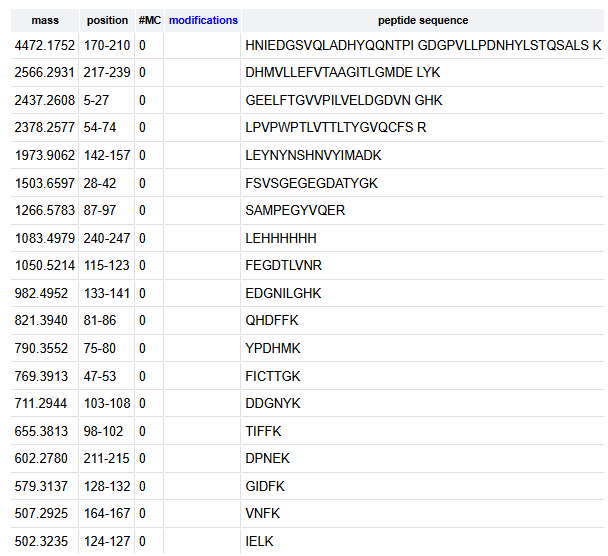
How many peptides will be generated from tryptic digestion of eGFP?
The number of peptides generated when using trypsin to perform the digest is 19 peptides
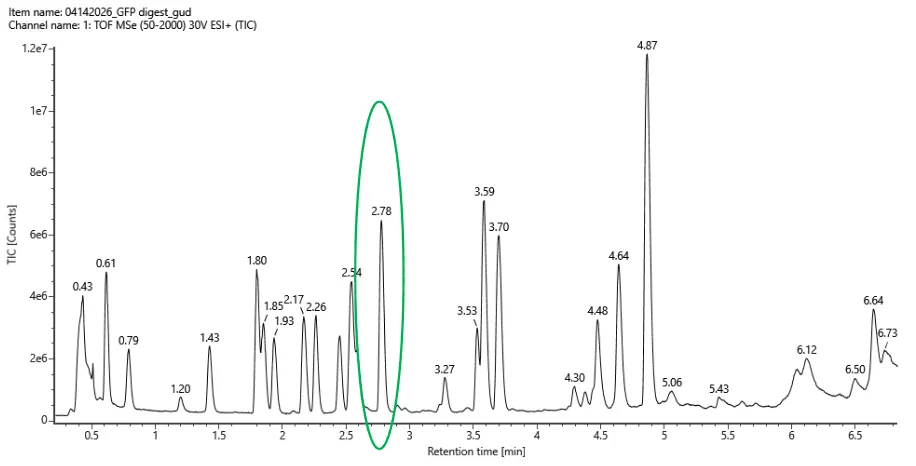
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
The number of chromatographic peaks in the eGFP peptide map between 0.5 and 6 mins are 21 labeled peaks
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
No, the number of predicted peptides are lower than the number of peptide peaks in the chromatogram.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its m/z and z.
the m/z of the peptide shown is 525.76
since the peptides are separated by app. 0.5 m/z, therefore, the charge (z) of the peptide is 2
the mass of the singly charged form of the peptide ([M+H]+)
m/z = (M + nH) / n
525.76 = (M + 2H)/2
1051.52 = M + 2 (1.00727)
M = 1049.5
therefore, M+H = 1050.51
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
What is the percentage of the sequence that is confirmed by peptide mapping?
88 %
Homework: Waters Part IV — Oligomers
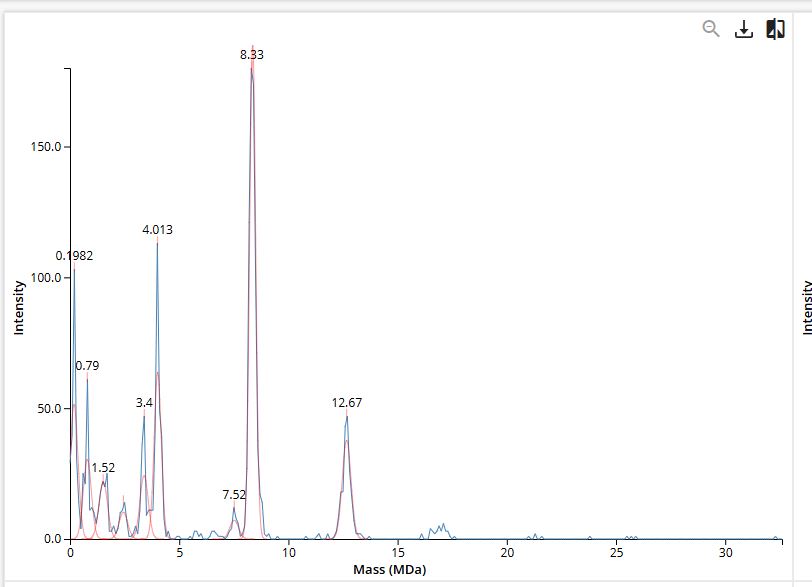
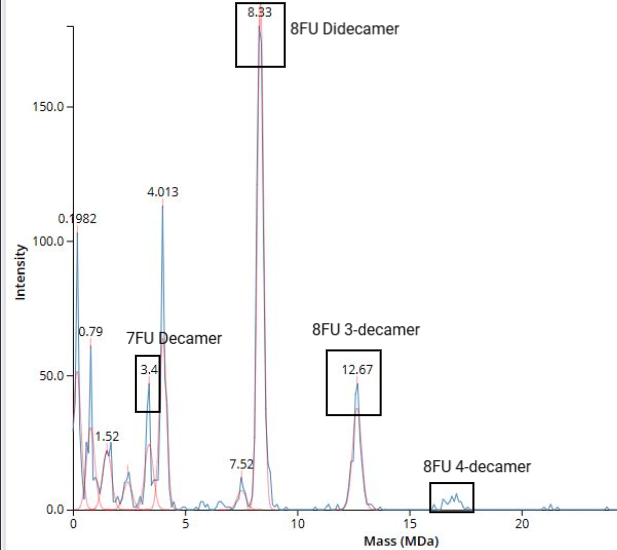
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
7FU decamer = 7FU mass x 10 = 340 kDa x 10 = 3400 kDa = 3.4 MDa
8FU didecamer = 8FU mass x 20 = 400 kDa x 20 = 8000 kDa = 8 MDa
8FU 3-decamer = 8FU mass x 30 = 400 kDa x 30 = 12000 kDa = 12 MDa
8FU 4-decamer = 8FU mass x 40 = 400 kDa x 40 = 16000 kDa = 16 MDa
Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I contributed 123 pixels to the global artwork experiment by making HTGAA letters at the bottom left. I liked the collaborative work and that it represents all of us. I think it can be better by including more colors.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition, provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
Component
Role
E. coli Lysate
• BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
As a whole-cell lysate, it provides the complete “hardware” needed for protein synthesis that was harvested during the exponential growth phase of the bacteria. It serves as the core transcription-translation machinery in cell-free protein synthesis (CFPS) systems. The lysate contains endogenous metabolic enzymes that work in tandem with added secondary energy substrates. T7 RNA Polymerase (from DE3 lysogen): Specifically transcribes T7 promoter-driven genes. BL21 Star mutation (mutation in the rne131 gene): Reduces RNase E activity, stabilizing mRNA longer for higher protein yields.
Salts/Buffer
• Potassium Glutamate
act as the primary salt to maintain the chemical and osmotic environment necessary for the molecular machinery to function.
• HEPES-KOH pH 7.5
serves as the primary buffering agent, maintaining the precise chemical environment required for delicate enzymatic reactions to occur outside the cell. It maintains pH homeostasis, in addition it does not readily cross biological membranes and has limited interaction with metal ions.
• Magnesium Glutamate
It serves as the primary source of magnesium cations. Mg ions are needed to drive the association of the 30S and 50S ribosomal subunits into the active 70S complex and it neutralize the strong negative charges of the ribosomal RNA (rRNA) phosphate backbone, allowing the complex to fold into the precise 3D shape required for translation. Glutamate anion is naturally dominant in the E. coli cytoplasm, allowing for high magnesium concentrations without the enzymatic inhibition typically caused by chloride ions.
• Potassium phosphate monobasic
It provides the essential inorganic phosphate required for metabolic pathways within the lysate to recycle ADP and GDP back into ATP and GTP. It also acts as a buffer to maintain a steady pH, preventing the metabolic byproducts of the reaction from becoming too acidic and denaturing the ribosomes or RNA polymerase. It supplies potassium cations
• Potassium phosphate dibasic
stabilize the pH of the reaction mixture by acting as a proton acceptor to prevent the pH from dropping into a range that would denature the ribosomes. Dibasic phosphate provides a ready source of inorganic phosphate required by endogenous enzymes in the E. coli lysate to re-phosphorylate ADP and GDP
Energy / Nucleotide System
• Ribose
It is needed for Nucleotide Synthesis (ATP, GTP, CTP, UTP). It is needed also for RNA Stability by providing the structural backbone for the mRNA being transcribed and the tRNA used during the translation process. Ribose can enter the Pentose Phosphate Pathway to generate NADPH, which provides reducing power and further metabolic intermediates that support the overall “health” of the lysate during prolonged incubation.
• Glucose
It is the Primary energy source via glycolysis. It is needed also for ATP Regeneration: Through the glycolytic pathway, glucose helps regenerate ATP and GTP from ADP and GDP, providing the chemical energy necessary for peptide bond formation.
• AMP, CMP, GMP, UMP
precursors that are phosphorylated to NTPs (Nucleoside Triphosphates): ATP, CTP, GTP, UTP respectively that are needed during transcription for mRNA synthesis. ATP is the main energy, it is used for Aminoacylation, which is the process of “charging” or loading each tRNA with its specific amino acid. GTP is the specific energy source for the Ribosome. It powers “translocation,” which is the physical movement of the ribosome as it slides down the mRNA and “clicks” the next amino acid into place. CTP & UTP are primarily used as building blocks for the mRNA instructions, they also help maintain the metabolic balance
• Guanine
It is the base component of GMP and GTP. During transcription, the T7 RNA polymerase incorporates guanine into the growing mRNA strand whenever it encounters a cytosine on the DNA template. it acts as a key part of the genetic code (codons), and the core of the GTP energy molecule that powers the physical movement of the protein-building machinery.
Translation Mix (Amino Acids)
• 17 Amino Acid Mix
The 17 Amino Acid Mix provides direct substrates for protein synthesis. The mixture excludes Cys, Ile, Met, Trp that are added separately as radioactive or fluorescently tagged to track or visualize the protein later.
• Tyrosine
It acts as a structural building block for the protein chain, a critical component for protein folding, and a primary tool for quantifying and labeling the synthesized product due to its unique chemical and optical properties (is responsible for the natural UV light absorbance of proteins).
• Cysteine
It is a sulfur-containing amino acid that serves as a critical structural stabilizer and a specific target for biochemical labeling. It is the only amino acid capable of forming covalent disulfide bonds (S-S) between different parts of a protein chain.
Additives
• Nicotinamide
It is a precursor for NAD+/NADH. NAD is required to facilitate the flow of electrons during the breakdown of energy sources like glucose or glutamate. It drives energy regeneration by maintaining the pool of NAD+, the system can continue the process of oxidative or substrate-level phosphorylation. This ensures a steady supply of ATP and GTP. It can act as a stabilizing agent for certain enzymes
Backfill
• Nuclease Free Water
It serves as the primary solvent to dissolve and dilute all other components. It ensures that no microbial or chemical contaminants are introduced and prevents DNA/RNA degradation by trace DNase/RNase contamination
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix. (2-3 sentences)
The 20-hr protocol was optimized by LLMs and autonomous labs with minimal human intervention, primarily limited to preparation, loading and unloading of reagents and consumables. Additionally, the 20-hr incubation protocol over six iterative steps produced sfGFP at a specific cost of $422/g, reducing CFPS specific cost by 40% and increasing protein titers by 27% relative to SOTA, while the 1-hr protocol optimized a reagent formulation that can produce 2.4 ± 0.3 g/L of protein product at the 15-µL scale ~$55/gprotein and 3.7 ± 0.2 g/L ~$36/gprotein at the 4-mL scale with oxygen supplementation. The 1-hr protocol screened 58 cell-free reagent components and their concentrations in 1,231 different reaction combinations, while the 20-hr protocol executed 480 384-well plate experimental designs comprising 29,527 unique reaction compositions.The knowledge acquisition also needs to be addressed. In the 20-hr protocol, GPT-5 was provided with access to the internet, a computer, data analysis packages, and the SOTA preprint.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Guanine is the base component of GMP and GTP. During transcription, the T7 RNA polymerase incorporates guanine into the growing mRNA strand whenever it encounters a cytosine on the DNA template.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems.
sfGFP (Superfolder GFP) is a basic (constitutively fluorescent) green fluorescent protein published in 2005, derived from Aequorea victoria. It is reported to be a very rapidly-maturing weak dimer.
Maturation Time (min)
13.6
Lifetime (ns)
3.1
mRFP1 is a basic (constitutively fluorescent) red fluorescent protein published in 2002, derived from Discosoma sp.. It is reported to be a somewhat slowly-maturing monomer with low acid sensitivity.
Maturation (min)
60.0
pKa
4.5
mKO2 is a basic (constitutively fluorescent) orange fluorescent protein published in 2008, derived from Verrillofungia concinna. It has moderate acid sensitivity.
Maturation (min)
108.0
pKa
5.5
mTurquoise2 is a basic (constitutively fluorescent) cyan fluorescent protein published in 2012, derived from Aequorea victoria. It is reported to be a rapidly-maturing monomer with very low acid sensitivity.
Maturation (min)
33.5
pKa
3.1
Lifetime (ns)
4.0
mScarlet is a basic (constitutively fluorescent) red fluorescent protein published in 2016, derived from synthetic construct. It has moderate acid sensitivity.
Maturation (min)
174
pKa
5.3
Lifetime (ns)
3.9
Electra2 is a basic (constitutively fluorescent) blue fluorescent protein published in 2022, derived from Entacmaea quadricolor.
Lifetime (ns)
3.4
Glossary of Terms
Maturation time: Time from translation to active form. Maturation is the time (in minutes) required (due to protein folding and chromophore maturation) for fluorescence to obtain a half-maximal value.
pKa is a measure of the acid sensitivity of a fluorescent protein. It is the pH at which fluorescence intensity drops to 50% of its maximum value.
Fluorescence Lifetime: The amount of time (in nanoseconds) after photon absorption that it takes the fluorophore to relax to the ground state is referred to as the fluorescence lifetime. When a population of fluorophores is excited, the lifetime is the time it takes for the number of excited molecules to decay to 1/e or 36.8% of the original population.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
protein: mScarlet
reagents: by increasing Magnesium Glutamate, the stability of the ribosomes and tRNA, which is optimal for translation, therefore, the expected effect is increase in fluorescence.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment.