Week 4 HW: Protein Design Pt. 1
Part A: Conceptual Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- Beef contains about 25% protein, therefore, per 500 gms of meat, there is 125 gm protein
- since 1 gm protein contains 6.022e+23 Da, therefore, 125 gm contains 7.528e+25 Da
- 7.528e+25 Da / 100 Da = 7.528 × 10²³ amino acids per 500 gm of meat
Ref: https://www.hopkinsmedicine.org/-/media/bariatrics/nutrition_protein_content_common_foods.pdf
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- because after eating the food is processed / digested to eventually its initial constituents as amino acids, fatty acids, vitamins, etc then the essential building blocks (nucleotides) that goes in building the human body
- Why are there only 20 natural amino acids?
- According to Doig, there are reasons for the selection of every amino acid making them a near ideal group. The factors taken into account included each amino acid’s component atoms, functional groups and biosynthetic cost. Forming soluble, stable protein structures with close‐packed cores and ordered binding pockets needed the variety of amino acids we see today. Multiple hydrophobic proteins are needed.
- Stephen Freeland and his team investigated size, charge and hydrophobicity parameters of proteins to show that the amino acids adopted by biology were not chosen randomly.
Ref: https://www.chemistryworld.com/features/why-are-there-20-amino-acids/3009378.article
- Can you make other non-natural amino acids? Design some new amino acids.
- Yes, non-natural amino acids also known as non-canonical or non-proteinogenic amino acids can be synthesized chemically or produced naturally as secondary metabolites in several organisms, such as bacteria, fungi, plants, or marine organisms.
Ref: https://pmc.ncbi.nlm.nih.gov/articles/PMC9044140/
- Where did amino acids come from before enzymes that make them, and before life started?
- Amino acids were created through purely natural chemical and physical processes driven by energy. These building blocks formed spontaneously in two primary ways: through atmospheric synthesis on early Earth, and through delivery by space rocks.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
- If α-helix was made using D-amino acids, the mirror image of natural amino acids, a left-handed helix is expected instead of the normal right-handed one.
- Can you discover additional helices in proteins?
- Yes, Proteins contain five helical structures, each defined by distinct hydrogen bonding patterns. The α-helix (i→i+4) is the most abundant, the 3₁₀ helix (i→i+3) is tighter and found at helix termini, and the π-helix (i→i+5) is wider, occurring in ~15% of protein structures almost exclusively near active sites. The PPII helix is left-handed with no intrachain hydrogen bonds yet critically mediates cell signaling through SH3 domain interactions, while the collagen triple helix consists of three intertwined PPII strands stabilized by interchain hydrogen bonds forming the structural basis of connective tissue.
- Why are most molecular helices right-handed?
- Proteins are almost exclusively built from L-amino acids, and this stereochemical bias of nature is directly reflected at the secondary structure level, where right-handed helices are strongly preferred over left-handed helices.
- Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets inherently aggregate because their edge strands possess exposed, unsatisfied hydrogen bonding potentials and sticky hydrophobic patches, making them chemically predisposed to self-assembly. This process often associated with amyloid fiber formation is driven by a combination of thermodynamics, specific atomic interactions, and conformational changes.
The driving forces for aggregation are:
The Hydrophobic Effect: In an aqueous environment, the primary thermodynamic driver for protein aggregation is the minimization of free surface energy. When unstructured proteins misfold into β-sheets, hydrophobic side chains that are normally buried inside a properly folded protein become exposed to the solvent. To minimize unfavorable contact with water, these exposed hydrophobic regions clump together with other β-strands, squeezing out water molecules and increasing the overall entropy of the surrounding solvent.
Intermolecular Hydrogen Bonding: In a single β-sheet, the backbone C=O and N-H groups are locked into hydrogen bonds with adjacent strands. However, the outer edge strands of a β-sheet have exposed, unsatisfied hydrogen bonding groups. These exposed groups eagerly accept or donate hydrogen bonds to other β-sheet edges, leading to a highly stable, repeating “cross-β” architecture.
Steric Interdigitation: β-sheets often have amino acid side chains that alternate pointing “up” and “down” from the sheet. When two sheets stack face-to-face, these interlocking side chains fit into one another like teeth on a zipper (a phenomenon often called “steric zipper” or knob-into-hole packing). This precise, stable alignment is stabilized by powerful van der Waals forces and can sometimes involve aromatic stacking.
- Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Protein aggregation into highly ordered, regularly repeated cross-β sheet structures called amyloid fibrils is closely associated with human disorders
The reason β-sheets dominate is thermodynamic and structural. Formation of the β-sheet conformation from native protein structures can be induced by high protein concentrations, metal binding, acidic pH, amino acid mutation, and interaction with lipid membranes. All amyloid proteins exhibit a characteristic non-native β-sheet state and aggregate spontaneously into extended fibrils that precipitate out of solution.
The driving force is hydrogen bonding. The formation of ordered arrays of hydrogen bonds drives the formation of β-sheets within disordered oligomeric aggregates. Initially individual β-sheets form with random orientations, which subsequently align into protofilaments as their lengths increase.
Yes, Amyloids are protein-based biomaterials composed of fibrils with cross-β cores. Previously only associated with degenerative diseases, amyloids remain active and functional both in vivo and in vitro conditions, enabling a variety of applications in medicine, nanotechnology, and biotechnology.
- Design a β-sheet motif that forms a well-ordered structure.
- Since β-sheets are inherently prone to runaway aggregation, designing a β-sheet that folds into a well-ordered, non-aggregating structure is quite challenging.
- For the design to simultaneously promote folding and prevent uncontrolled self-association, there are six key design principles:
- Strand Sequence: Alternating Hydrophobic/Polar Pattern creating one hydrophobic face (the core) and one hydrophilic face (the solvent-exposed surface).
- Turn/Loop Design: The Critical Nucleation Point: The turn connecting two β-strands (forming a β-hairpin) is the nucleation site for folding, a poorly designed turn means the sheet never forms.
- Edge Strand Protection: Preventing Aggregation: Exposed β-strand edges have free hydrogen bond donors and acceptors that readily recruit additional strands, causing aggregation into amyloid-like fibrils.
- Capping Motifs: Stabilizing the Termini using N-cap and C-cap residues.
Part B: Protein Analysis and Visualization
- Briefly describe the protein you selected and why you selected it.
- I select tau protein. It is a microtubule-associated protein that promotes microtubule assembly and stability, and might be involved in the establishment and maintenance of neuronal polarity. In neurodegeneration, this protein becomes hyperphosphorylated, detaches from microtubules, and aggregates into toxic, insoluble neurofibrillary tangles (NFTs). Since I’m interested in using synthetic biology to understand more neurodegenerative disorders, this protein is of interest.
Another protein is Amyloid beta precursor like protein 1.
- It may play a role in postsynaptic function. The C-terminal gamma-secretase processed fragment, ALID1, activates transcription activation through APBB1 (Fe65) binding. Couples to JIP signal transduction through C-terminal binding. May interact with cellular G-protein signaling pathways. Can regulate neurite outgrowth through binding to components of the extracellular matrix such as heparin and collagen I. The gamma-CTF peptide, C30, is a potent enhancer of neuronal apoptosis.
Identify the amino acid sequence of your protein.
Tau Protein
Sequence Length: 758 amino acids
The most common amino acid is: Proline, which appears 93 times.
Amino Acid Frequencies: P: 93 (12.27%) G: 82 (10.82%) S: 79 (10.42%) K: 64 (8.44%) A: 60 (7.92%) E: 59 (7.78%) T: 50 (6.60%) D: 43 (5.67%) L: 43 (5.67%) V: 41 (5.41%) Q: 33 (4.35%) R: 30 (3.96%) H: 20 (2.64%) I: 20 (2.64%) N: 13 (1.72%) M: 9 (1.19%) F: 9 (1.19%) Y: 6 (0.79%) C: 4 (0.53%)
there are 10 protein homologs
yes, it belongs to the microtubule-associated protein family
APP
Sequence length: 650 Amino Acids (fasta file: https://rest.uniprot.org/uniprotkb/P51693.fasta)
The most common amino acid is L appearing 70 times
Amino Acid Frequencies: L: 70 E: 67 P: 61 R: 57 G: 51 A: 51 Q: 46 S: 43 V: 36 D: 26 T: 23 H: 22 M: 16 I: 16 F: 14 K: 14 C: 12 Y: 11 N: 10 W: 4
250 homolgs (reference: https://www.uniprot.org/blast/uniprotkb/ncbiblast-R20260307-104740-0382-64643955-p1m/overview)
it belongs to the APP family.
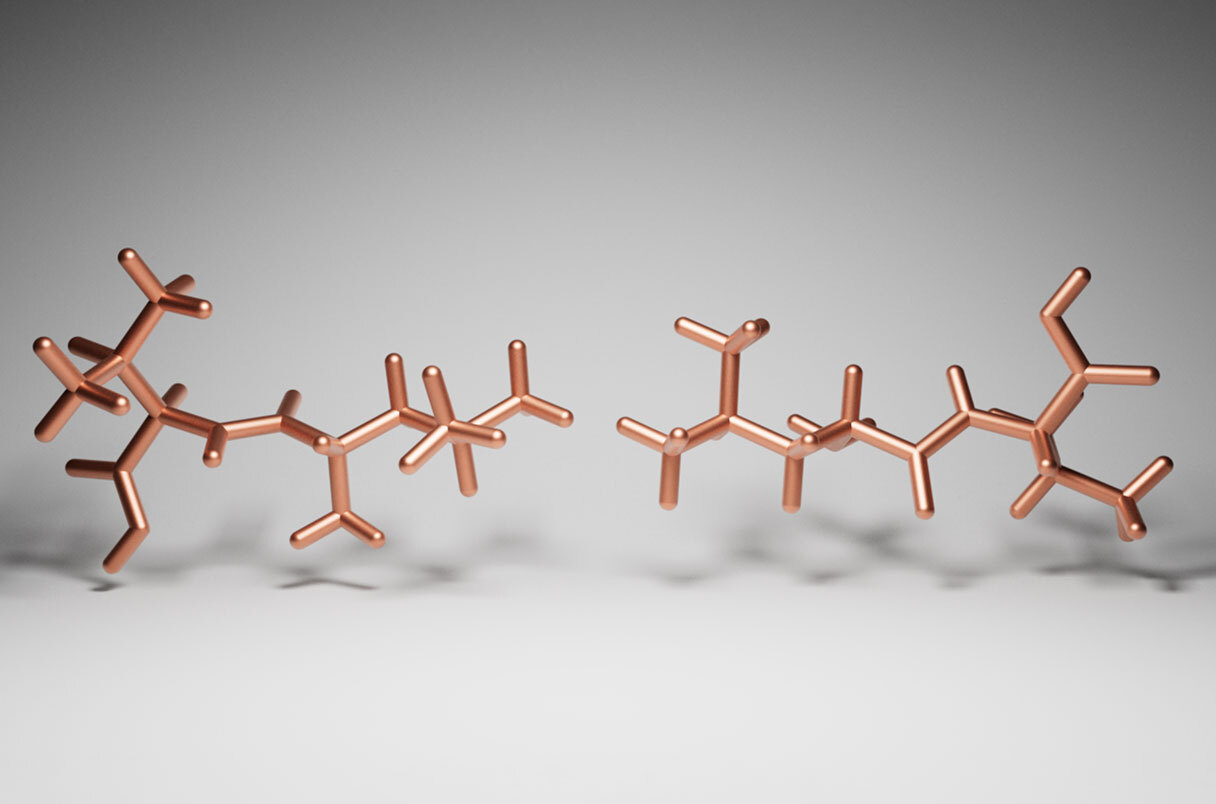
- Identify the structure page of your protein in RCSB
Tau Protein
- the structure was solved/released in 2015-07-08
- it is a high-quality NMR structure
- No, it doesn’t belong to any structure classification family
APP like Protein
- Good resoltuion. Resolution: 2.60 Å
References:
- https://www.uniprot.org/uniprotkb/P10636/entry
- https://www.ebi.ac.uk/pdbe/scop/search?t=txt;q=tau%20protein
- https://www.ebi.ac.uk/pdbe/entry/pdb/4RD9
- Open the structure of your protein in any 3D molecule visualization software
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”
the protein that will be visualized is APP (Amyloid-beta precursor protein)
Cartoon

Ribbon

Ball and Stick
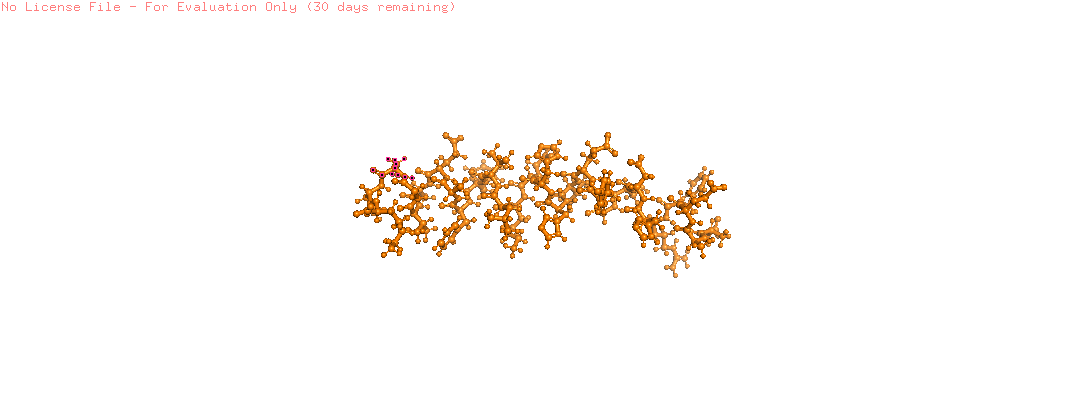
Color the protein by secondary structure. Does it have more helices or sheets?
The helix is colored in cyan and the sheet in magenta, from the image the helices are more

- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
- The hydrophobic residues (ALA, VAL, LEU, ILE, MET, PHE, TRP, PRO) are colored in yellow
- The hydrophilic residues (SER, THR, ASN, GLN, TYR, CYS, LYS, ARG, HIS, ASP, GLU) are colored in blue
- GLY (neutral) is colored in white
- From the image the hydrophilic residues are grouped together and they are more then the hydrophobic ones


- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)? No, it doesn’t have any holes
Part C: Using ML-Based Protein Design Tools
I chose the Amyloid Beta-Peptide like protein
C1. Protein Language Modeling
Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Fasta File
1AMC_1|Chain A|AMYLOID BETA-PEPTIDE|Homo sapiens (9606) old
DAEFRHDSGYEVHHQKLVFFAEDVGSNK old
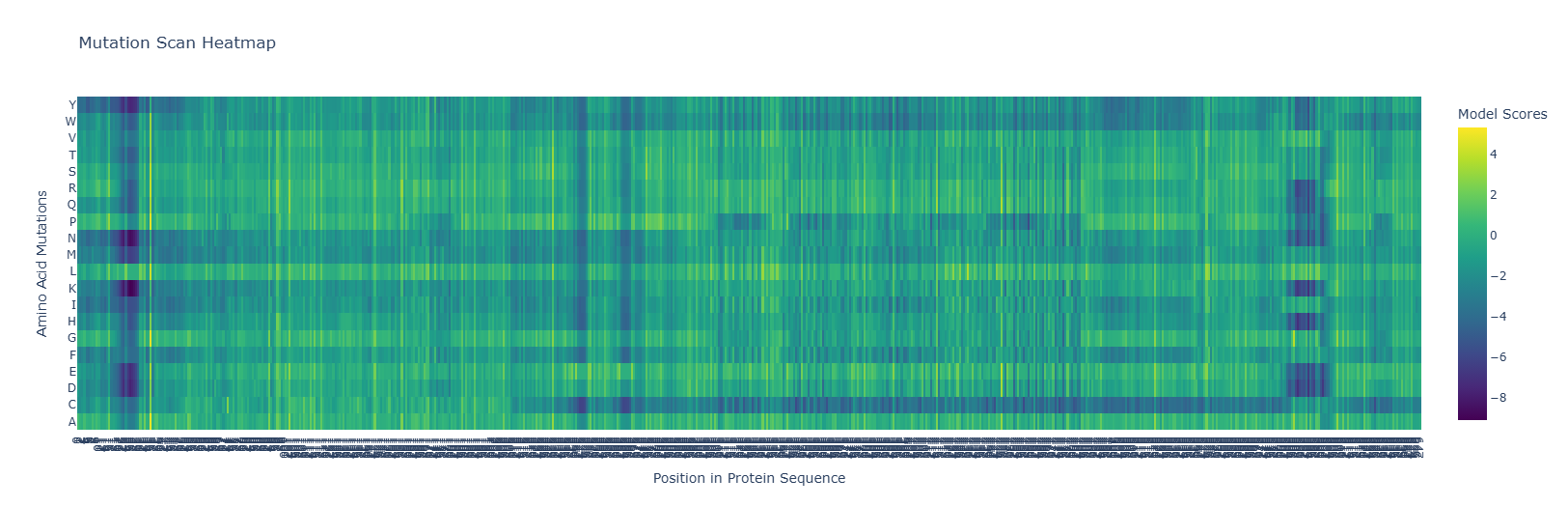
The heatmap shows hotspots for mutations that are beneficial or detrimental to the function of the protein. Notice the dark blue regions, where the LLR values are negative, indicating that mutations that are likely detrimental to function, and lighter yellow regions where the LLR values are positive, indicating mutations that are likely beneficial to the function of the protein. Also, note how there are dark bands running vertically indicating regions which are likely evolutionarily conserved, and brighter bands running vertically indicating regions of the protein which may in fact be preferable over the wild-type sequence. Note also, for some regions of the protein, there are amino acid mutations which are likely to be detrimental to functioning for entire regions of the protein, indicated by dark bands running horizontally along most of the protein. Similarly, we see brighter bands of yellow running horizontally, indicating almost any residue mutated to that amino acid would be preferential to the wild type.
for example replacing by is prefereable. mutations at as well are favourable
Latent Space Analysis
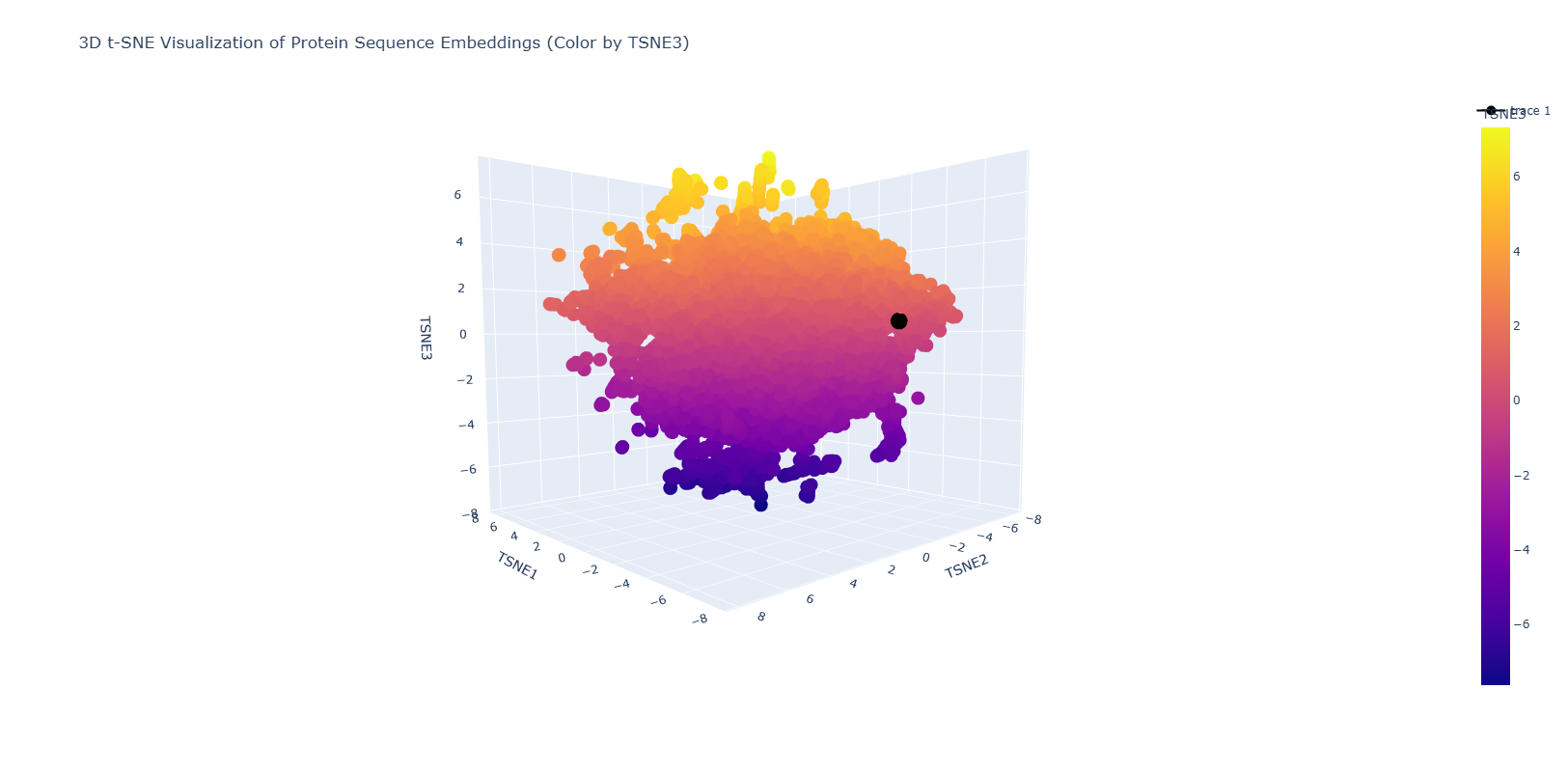
- Use the provided sequence dataset to embed proteins in reduced dimensionality.
- Analyze the different formed neighborhoods: do they approximate similar proteins?
- Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
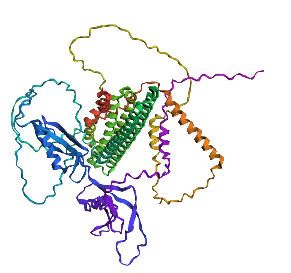
No, they don’t match it
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations? Mutation done: replacing position 22 (L) by K
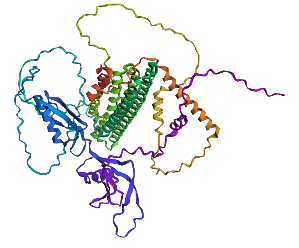
Mutation: replacing positions 20 to 29 LPLLLPLLLL with NNNNNNNNNN
Part D: Group Brainstorm on Bacteriophage Engineering
- 1. Find a group of ~3–4 students
- 2. Read through the Phage Reading material listed under “Reading & Resources” below.
- 3. Review the Bacteriophage Final Project Goals for engineering the L Protein:
- Increased stability (easiest)
- Higher titers (medium)
- Higher toxicity of lysis protein (hard)
- Brainstorm Session
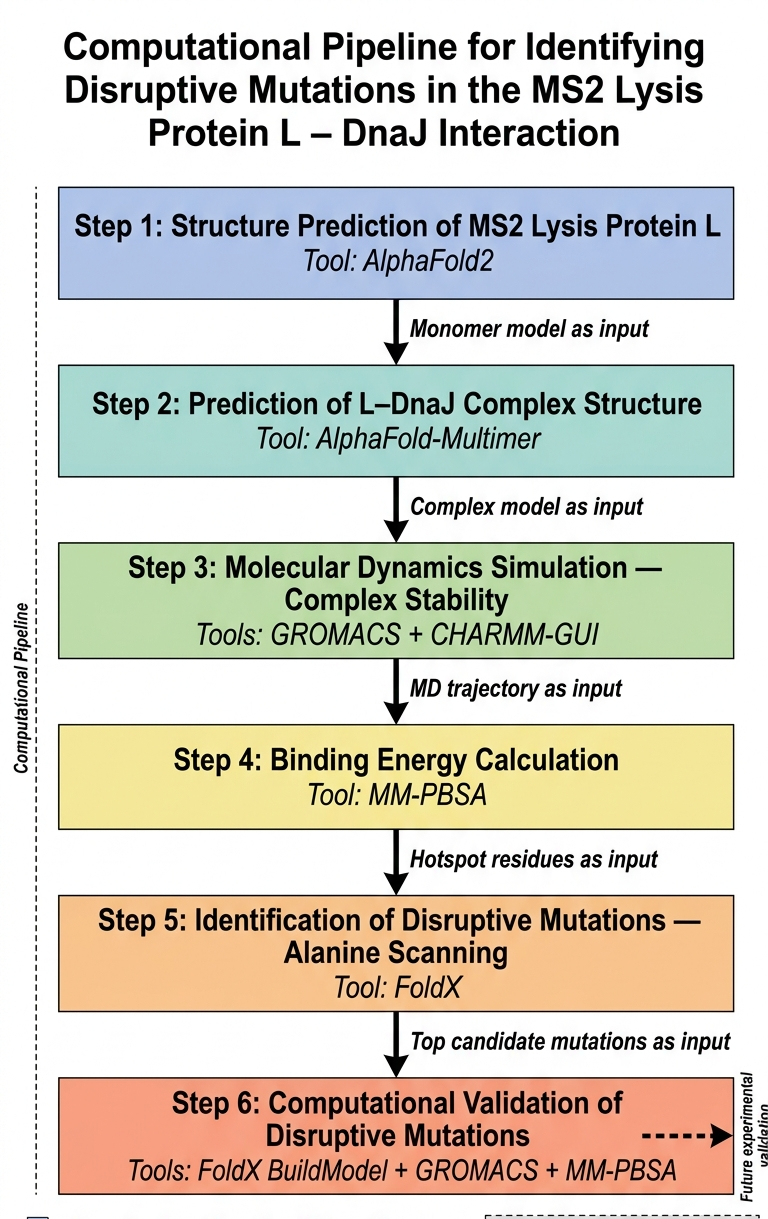
The main goal that I think can be adressed computationally is disrupting the lysis protein interaction with E. coli chaperone DnaJ
The proposed approache is predicting the 3D structure of the complex and then figuring out the genes responsible for the interaction between the lysis protein and the DnaJ and then introdcing mutations to these genes for the aim of disrupting the complex binding
The tools that might help solve the chosen sub-problem are:
- ESMFold or AlphaFold2 for 3D structure prediction of L lysis protein
- AlphaFold-Multimer to predict how L and DnaJ physically interact
- test the complex stability (to validates whether the AlphaFold-Multimer prediction is physically reasonable)
- calculate complex binding energy
- design disruptive mutations
- Introduce Mutations and Re-predict Structure
The potential pitfalls are:
- Unreliable structure prediction
The pipeline Schematic:
References