Week 5 HW: Protein Design Pt. 2
Part A: SOD1 Binder Peptide Design (From Pranam)
Pt 1: Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
- Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
- Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
- To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
- Record the perplexity scores that indicate PepMLM’s confidence in the binders.
human SOD1 sequence
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V mutation
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Generated Peptides
| Peptides | perplexity | Type |
|---|---|---|
| WHYGAAGARLKE | 10.803203 | Generated peptide No. 1 |
| WHYPAAVAEWGK | 10.861847 | Generated peptide No. 2 |
| WRSPATAVAHKK | 8.193301 | Generated peptide No. 3 |
| WLYYPAALEHGE | 14.861894 | Generated peptide No. 4 |
| FLYRWLPSRRGG | SOD1-binding peptide |
Colab Link: https://colab.research.google.com/drive/1mFeOfeeTxAycc_tvqmw2YbZpVIvX6E3E?authuser=2#scrollTo=VtfbXYndhyle
Pt 2: Evaluate Binders with AlphaFold3
- Navigate to the AlphaFold Server: alphafoldserver.com
- For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
- Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
- In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
ipTM measures the accuracy of the predicted relative positions of the subunits within the complex. Values higher than 0.8 represent confident high-quality predictions, while values below 0.6 suggest likely a failed prediction. ipTM values between 0.6 and 0.8 are a gray zone where predictions could be correct or incorrect.
WHYGAAGARLKE
ipTM = 0.3 since the value is below 0.6, it suggest likely a failed prediction.
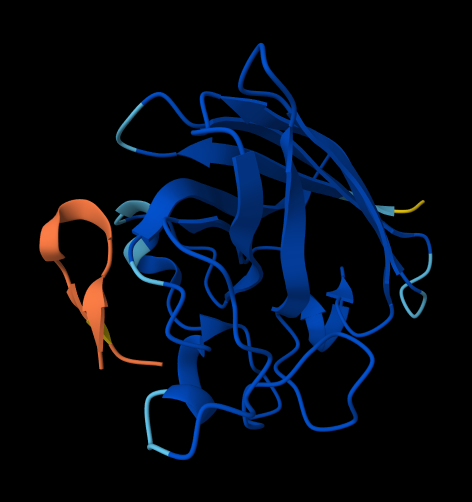
WHYPAAVAEWGK
ipTM = 0.28 since the value is below 0.6, it suggest likely a failed prediction.
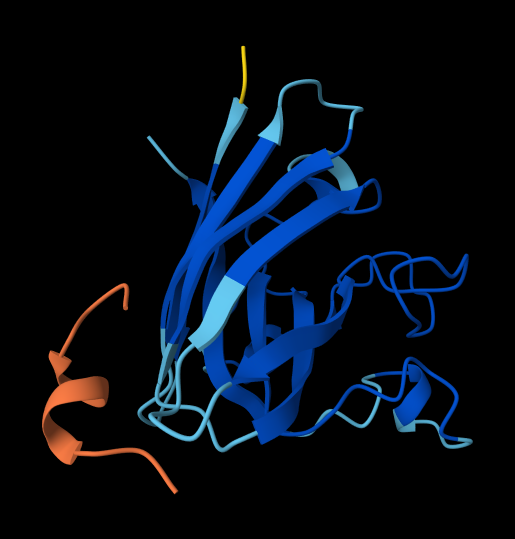
WRSPATAVAHKK
ipTM = 0.6 the value is 0.6 and it is highest value obtained suggesting it could be correct or incorrect
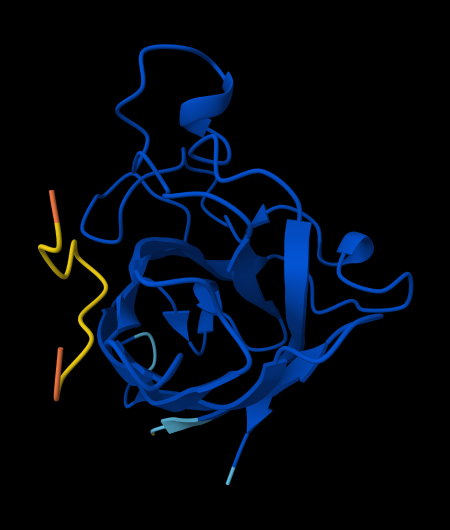
WLYYPAALEHGE
ipTM = 0.32 since the value is below 0.6, it suggest likely a failed prediction.
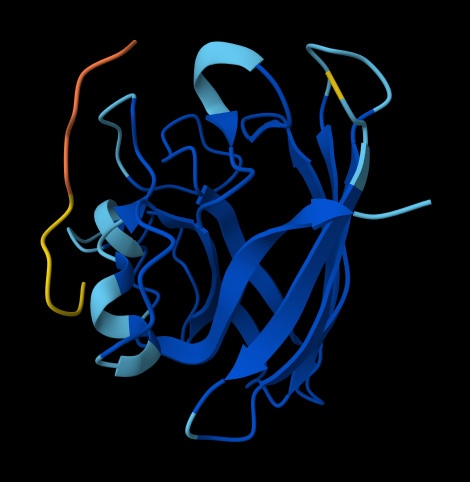
FLYRWLPSRRGG
ipTM = 0.37 since the value is below 0.6, it suggest likely a failed prediction.
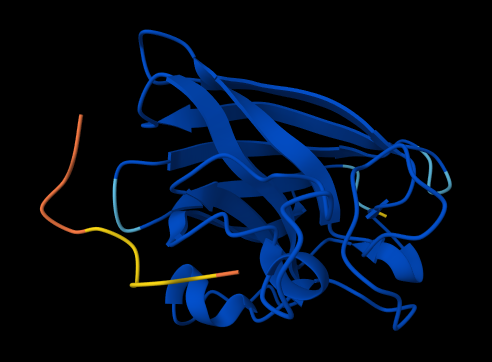
Link: https://alphafoldserver.com/fold/44913f6ed245c97c
Pt 3: Evaluate Properties of Generated Peptides in the PeptiVerse
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
WHYGAAGARLKE
| Property | Prediction | Value | Unit |
|---|---|---|---|
| Solubility | Soluble | 1.000 | Probability |
| Hemolysis | Non-hemolytic | 0.025 | Probability |
| Binding Affinity | Weak binding | 5.546 | pKd/pKi |
| Molecular weight | 1358.5 | Da | |
| Net Charge (pH 7) | 0.85 | ||
| ipTM | 0.3 |
WHYPAAVAEWGK
| Property | Prediction | Value | Unit |
|---|---|---|---|
| Solubility | Soluble | 1.000 | Probability |
| Hemolysis | Non-hemolytic | 0.023 | Probability |
| Binding Affinity | Weak binding | 5.037 | pKd/pKi |
| Molecular weight | 1414.6 | Da | |
| Net Charge (pH 7) | -0.15 | ||
| ipTM | 0.28 |
WRSPATAVAHKK
| Property | Prediction | Value | Unit |
|---|---|---|---|
| Solubility | Soluble | 1.000 | Probability |
| Hemolysis | Non-hemolytic | 0.011 | Probability |
| Binding Affinity | Weak binding | 4.520 | pKd/pKi |
| Molecular weight | 1351.6 | Da | |
| Net Charge (pH 7) | 2.85 | ||
| ipTM | 0.6 |
WLYYPAALEHGE
| Property | Prediction | Value | Unit |
|---|---|---|---|
| Solubility | Soluble | 1.000 | Probability |
| Hemolysis | Non-hemolytic | 0.037 | Probability |
| Binding Affinity | Weak binding | 5.588 | pKd/pKi |
| Molecular weight | 1448.6 | Da | |
| Net Charge (pH 7) | -2.14 | ||
| ipTM | 0.32 |
FLYRWLPSRRGG
| Property | Prediction | Value | Unit |
|---|---|---|---|
| Solubility | Soluble | 1.000 | Probability |
| Hemolysis | Non-hemolytic | 0.047 | Probability |
| Binding Affinity | Weak binding | 5.968 | pKd/pKi |
| Molecular weight | 1507.7 | Da | |
| Net Charge (pH 7) | 2.76 | ||
| ipTM | 0.37 |
Link: https://huggingface.co/spaces/ChatterjeeLab/PeptiVerse
Pt 4: Generate Optimized Peptides with moPPIt
moPPit peptides first peptide generated with moPPit
- Open the moPPit Colab linked from the HuggingFace moPPIt model card
- Make a copy and switch to a GPU runtime.
- In the notebook:
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
- After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
| Generated Peptides | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|
| HMCVNYQKKTKN | 0.9812432918697596 | 0.8333333134651184 | 6.3176960945129395 | 0.7024670839309692 |
| STDTCTGRFKQK | 0.9649077206850052 | 0.9166666865348816 | 5.785930633544922 | 0.8285709619522095 |
| KKKTYSKKGDFY | 0.9740752298384905 | 0.9166666865348816 | 5.8559699058532715 | 0.56052565574646 |
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
- Pt 0: Sign-up to Boltz Lab
- Pt 1: Structural Predictions in the Sandbox
| Cmpound | Binding Confidence | Optimization Score | Structure Confidence |
|---|---|---|---|
| Hit | 0.45 | 0.23 | 0.98 |
| Lead | 0.75 | 0.26 | 0.98 |
| JQ1 | 0.96 | 0.44 | 0.99 |
Discussion Questions
Does Binding Confidence increase as you move from hit to clinical candidate? What would you expect, and why might it deviate? Binding confidence which means how confidently the ligand is placed in the binding site is higher when JQ1 was chosen as the ligand and lower in hit
Inspect the predicted binding pose for JQ1. Can you identify potential key binding interactions.
Compare the Optimization Scores. How do the scores compare for JQ1 vs the Lead. Optimization score for JQ1 is 0.44 while 0.26 for Lead, indicating higher tight binding with JQ1\
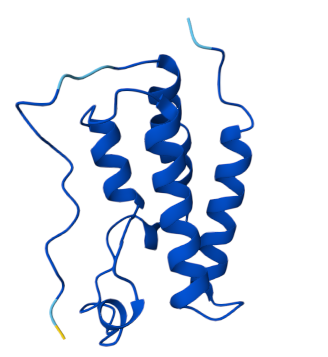
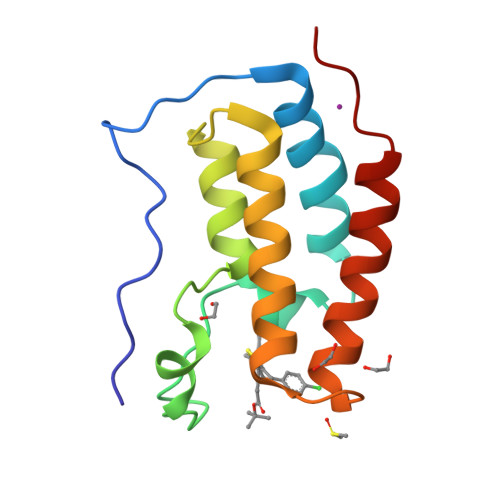
Pt 2: Setting Up a BRD4 Design Project the predicted structure from boltz vs from RCSB
Boltz
RCSB
Pocket Structure Prediction
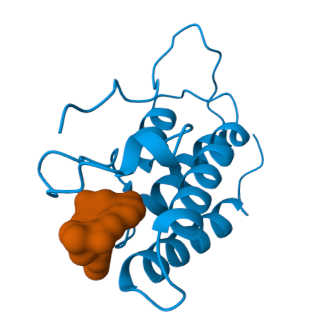
- Pt 3: Running Your Virtual Screen
Generating 1000 binders
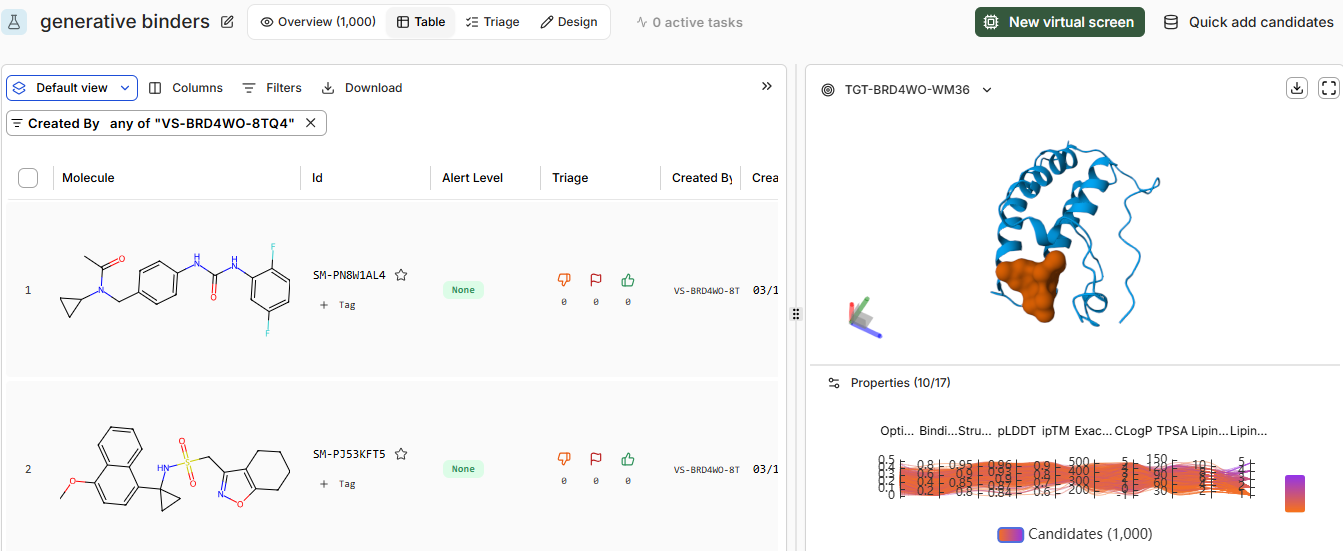
- Pt 4: Analysis and Discussion
high confidence binders only one
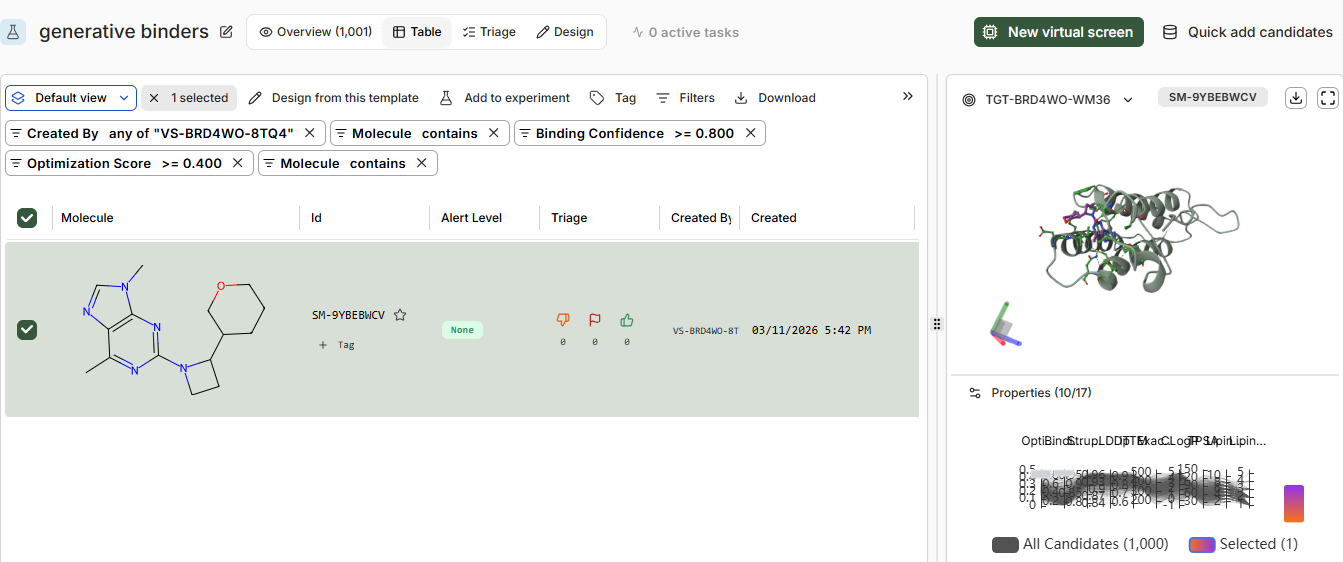
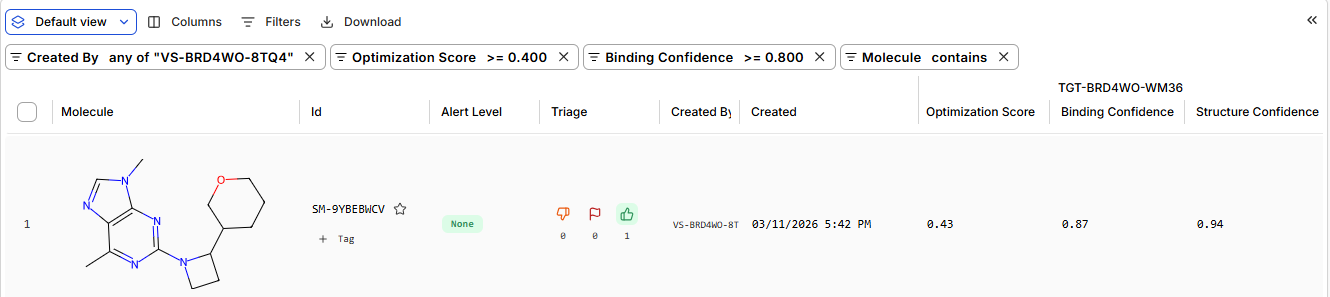
moderate confidence 23 binders
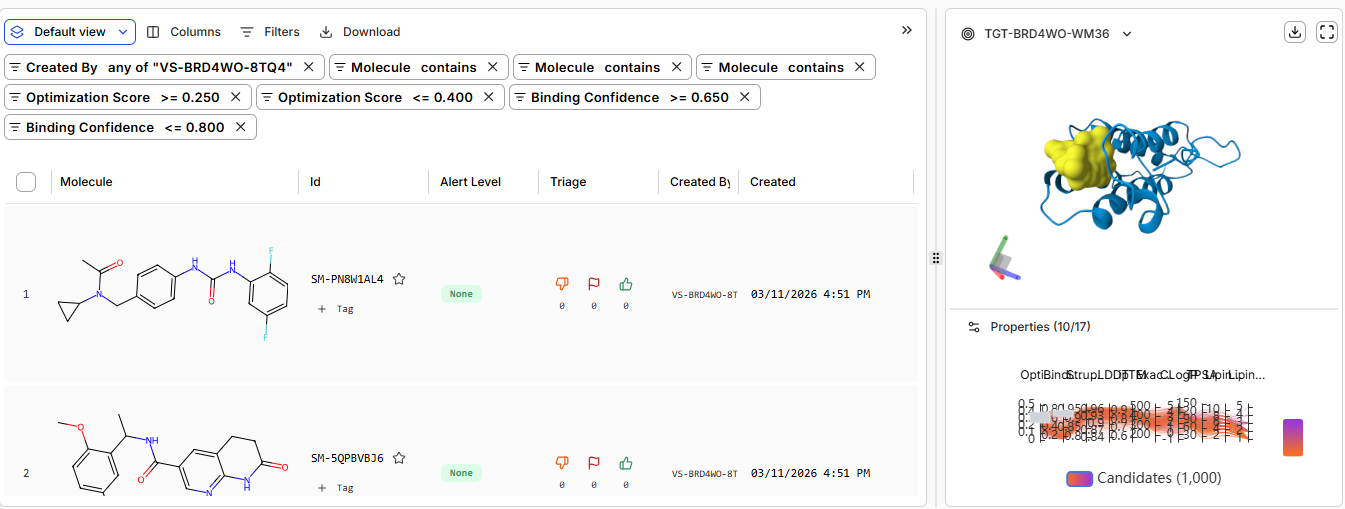
low confidence / non-binders 621 binders
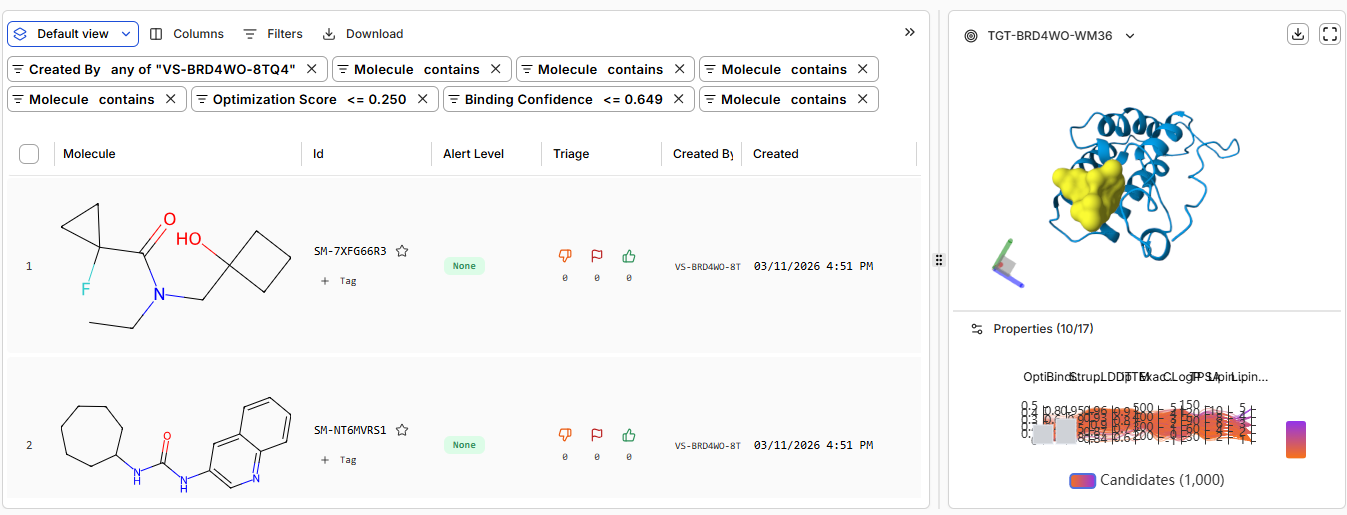
Links:
- https://lab.boltz.bio/app/nour-abdelrahman-htgaa-Uz4g/p/brd-4-workshop-f6Wt/experiments/generative-binders-k84A/virtual-screens/fa4123a5-cc15-4cc4-8b5c-287349f74144/overview
- https://www.rcsb.org/structure/3MXF
Part C: Final Project: L-Protein Mutants
L-Protein Engineering
- Run the notebook