Week 5 HW: Protein Design Part ii

𓃠 Week 5 Homework 𓃠
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
Here is the Human SOD1 sequence from Uniprot (P00441)
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Here it is again after adding the A4V mutation
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Here is the produced peptides:
| Index | Binder | Pseudo Perplexity |
|---|---|---|
| 1 | HRSYAVALRHGK | 15.152948 |
| 2 | HHSGPVAVRWKX | 11.715797 |
| 3 | WRSPAAAVEHWX | 9.166994 |
| 4 | WRYGVVGVRLWE | 14.868017 |
| 5 | FLYRWLPSRRGG | n/a |
Part 2: Evaluate Binders with AlphaFold3
Some of the peptides had X in it and Alphafold seems to reject it, i asked Gemini and it mentioned it should be swapped, Safe Options include Alanine (A) or Glycine (G) or a Rational swap where i can choose a hydrophobic Leucine (L) or Valine (V) or a hydrophilic Lysine (K) or Arginine (R) swap that depends on the pocket i am binding to, for now though i have decided to go with an Alanine (A) swap for simplicity.
Peptide 1 (HRSYAVALRHGK)
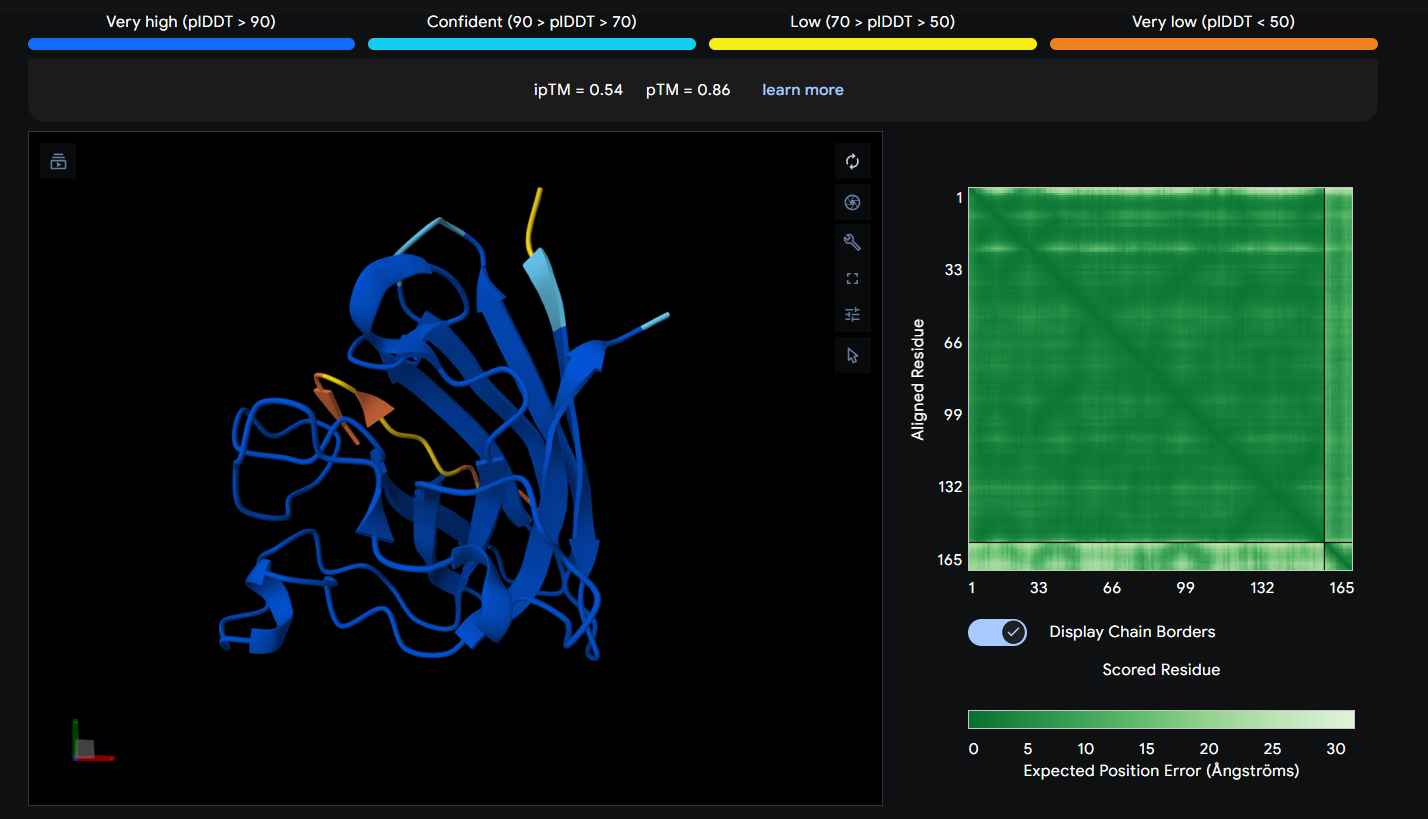
The Peptide seems to be floating really close near a side of the Dimer Interface but it is not totally sticking to it
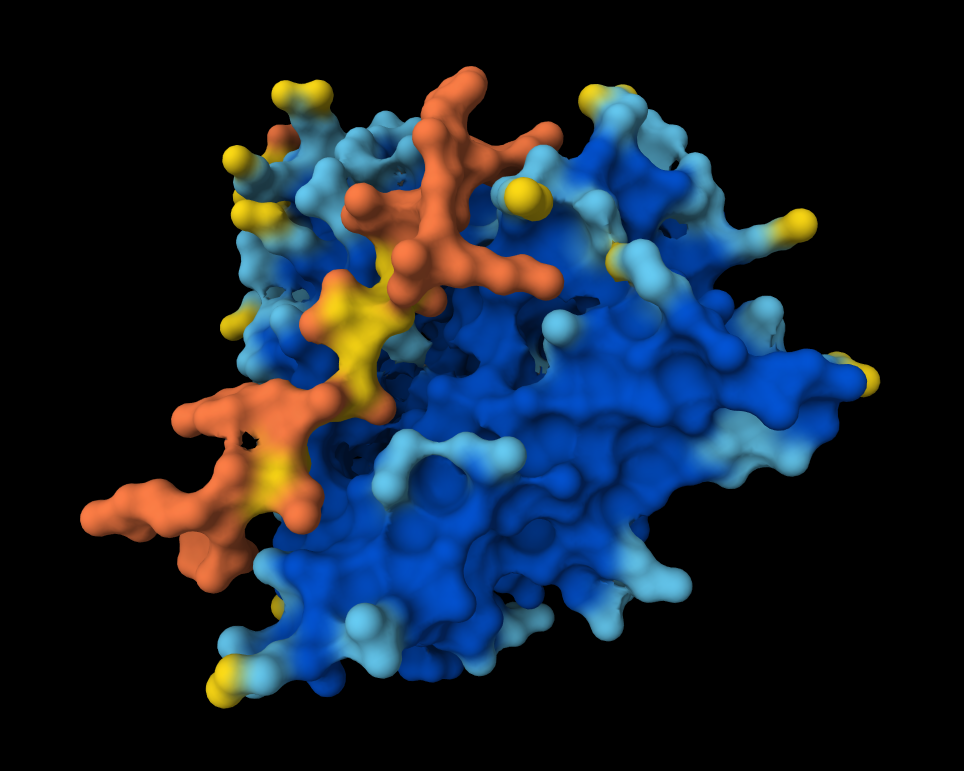
Here in this Molecular Surface View, it shows that the Peptide is floating and not touching the protein surface, yet it looks to be fitting into the protein surface groove well.
Peptide 2 (HHSGPVAVRWKX)
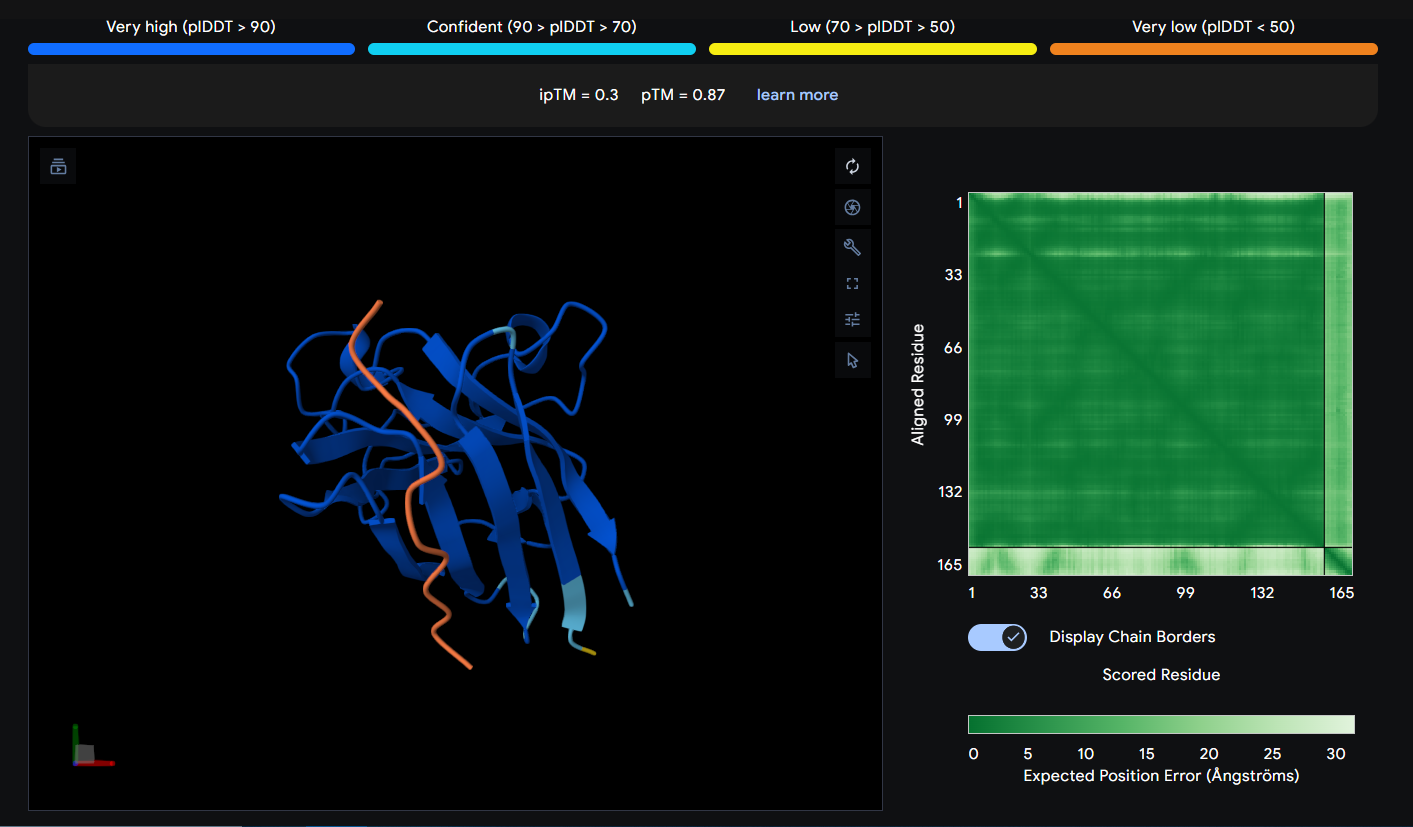
This Peptide seems to be floating near the Dimer Interface but not actually stuck to it.
Peptide 3 (WRSPAAAVEHWX)
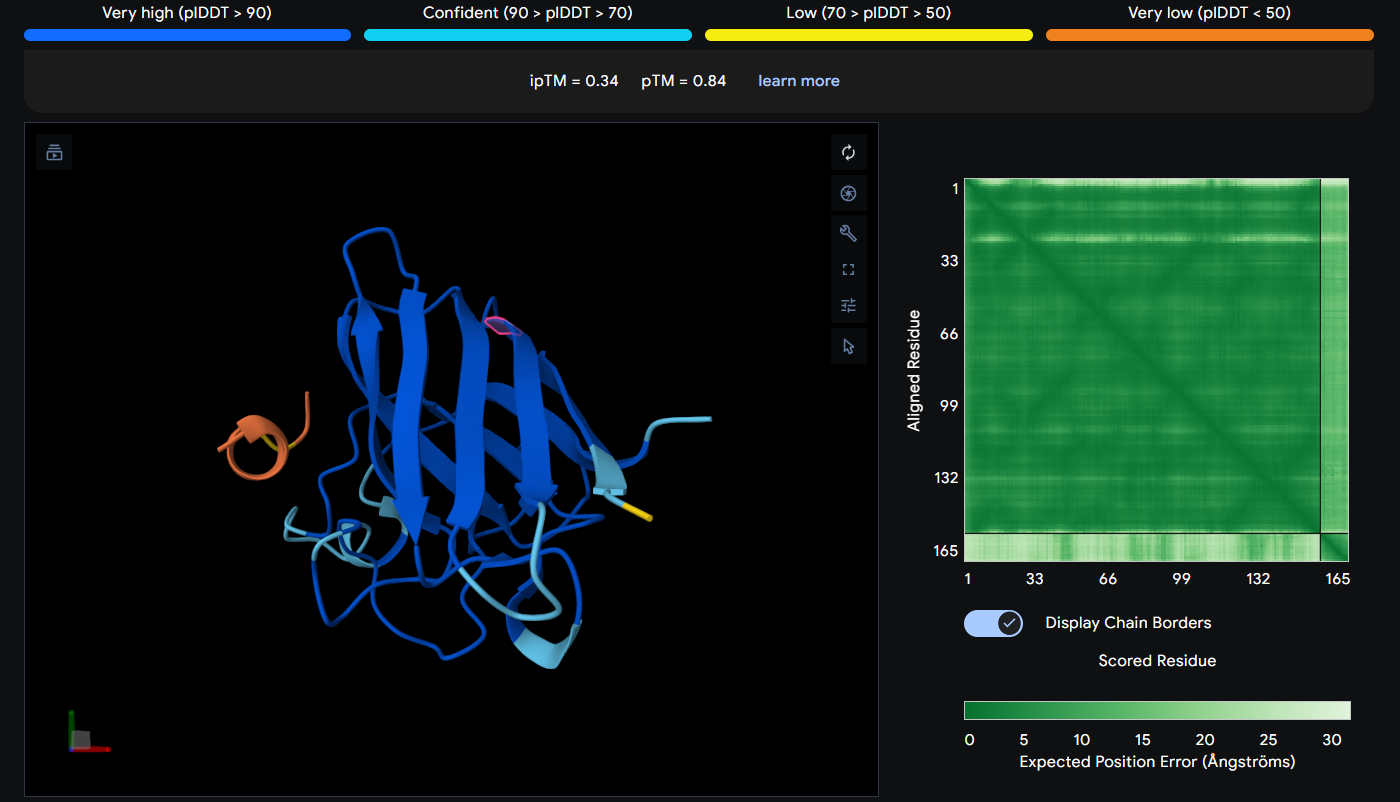
The Peptide seems to be floating away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface.
Peptide 4 (WRYGVVGVRLWE)
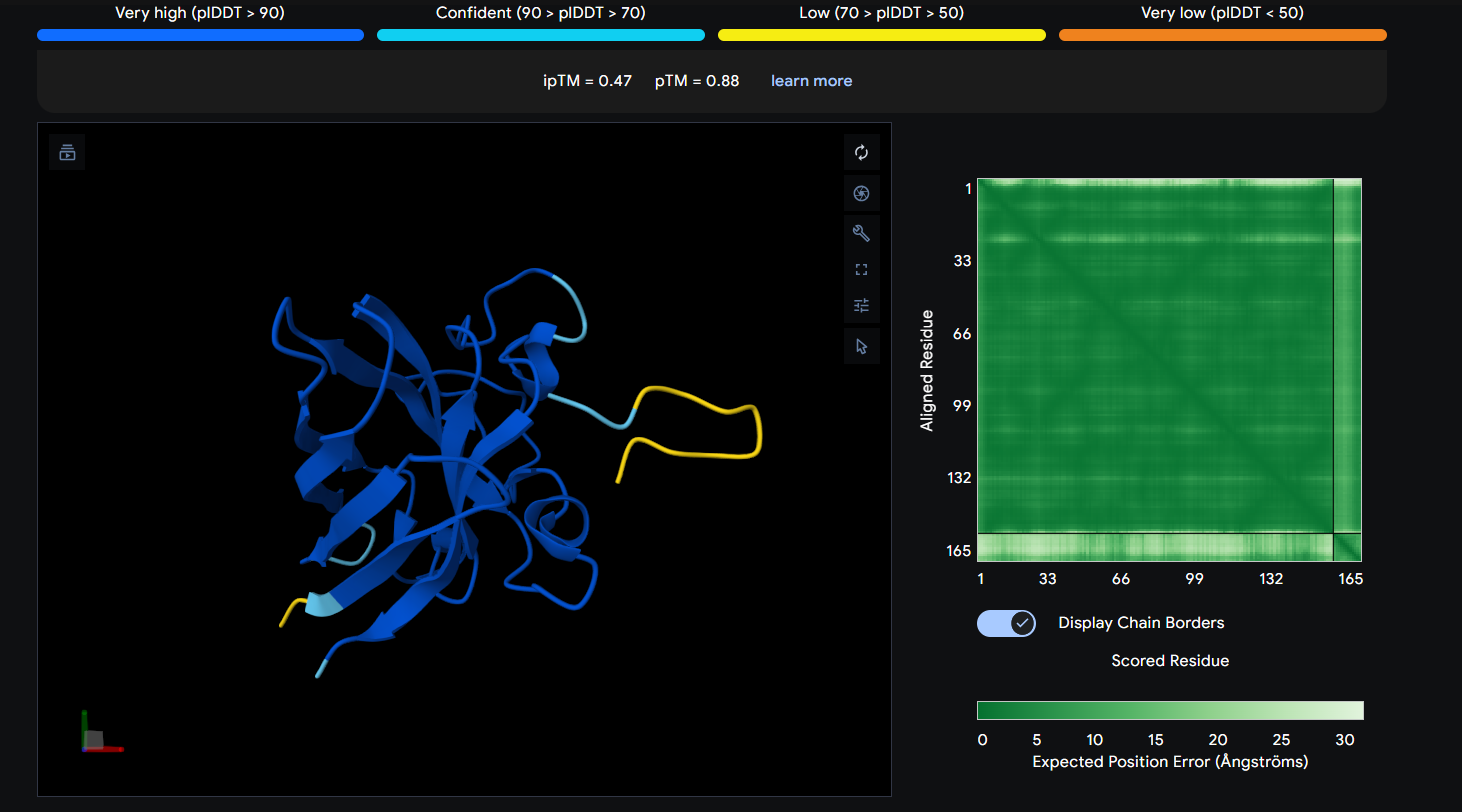
The Peptide seems to be buried into a groove into the protein, yet does not engaging with the N-terminus, the beta barrel or the Dimer Interface.
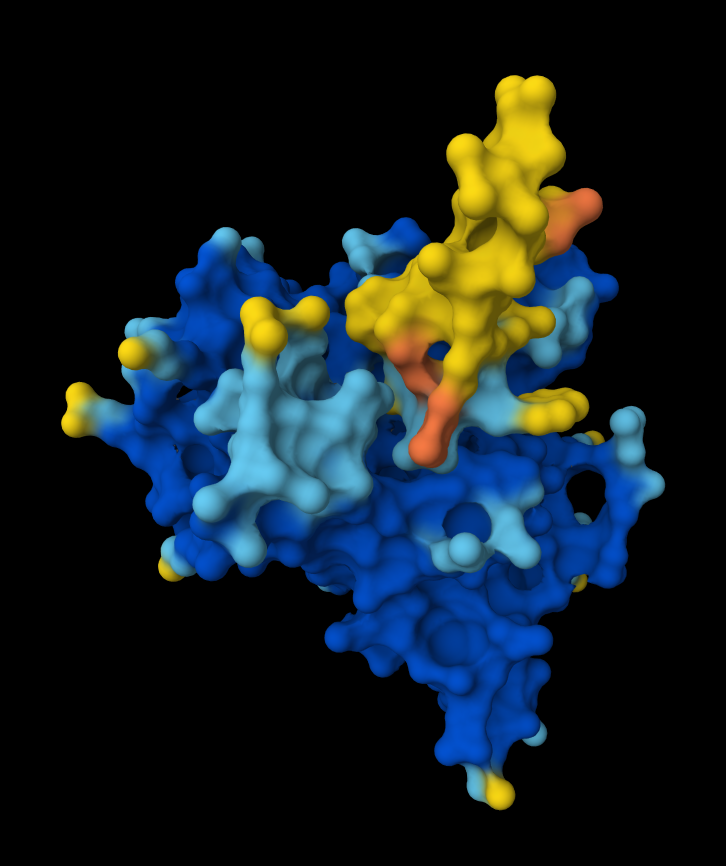
This image shows clearly how the peptide seems to be holding into a groove into the protein.
Peptide 5 (Control SOD1-binding peptide) (FLYRWLPSRRGG)
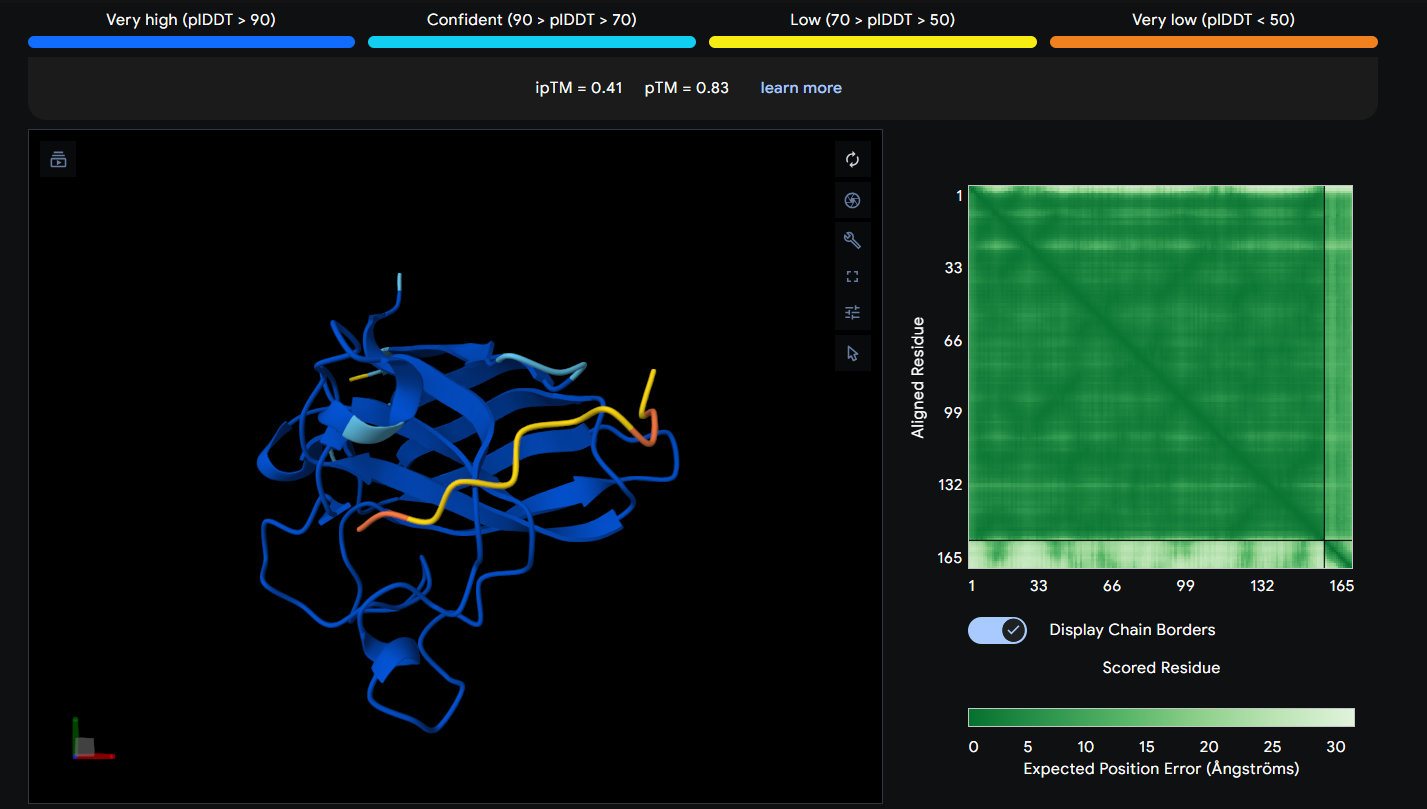
The Peptide seems to be floating away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface.
| Index | Peptide | ipTM Score |
|---|---|---|
| 1 | HRSYAVALRHGK | 0.54 |
| 2 | HHSGPVAVRWKX | 0.3 |
| 3 | WRSPAAAVEHWX | 0.34 |
| 4 | WRYGVVGVRLWE | 0.47 |
| 5 | FLYRWLPSRRGG | 0.41 |
The ipTM score tells you how confident AlphaFold is that these two chains actually interact. It ranges from 0 to 1. Generally, and as noted in AlphaFold, Scores beyond 0.8 are confident high-quality predictions) while below 0.6 are a failed prediction, in between is considered a grey zone where predictions could be correct or incorrect.
Looking at my ipTM scores, it seems like all the generated peptides fall into the failed prediction zone, with Peptide 1 almost surpassing it with a score of 0.54, Peptides 1 (0.54) & 4 (0.47) seem to have a higher score than the control Peptide 5, while Peptides 2 (0.3) & 3 (0.34) have very low scores.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Peptide 1 (HRSYAVALRHGK)
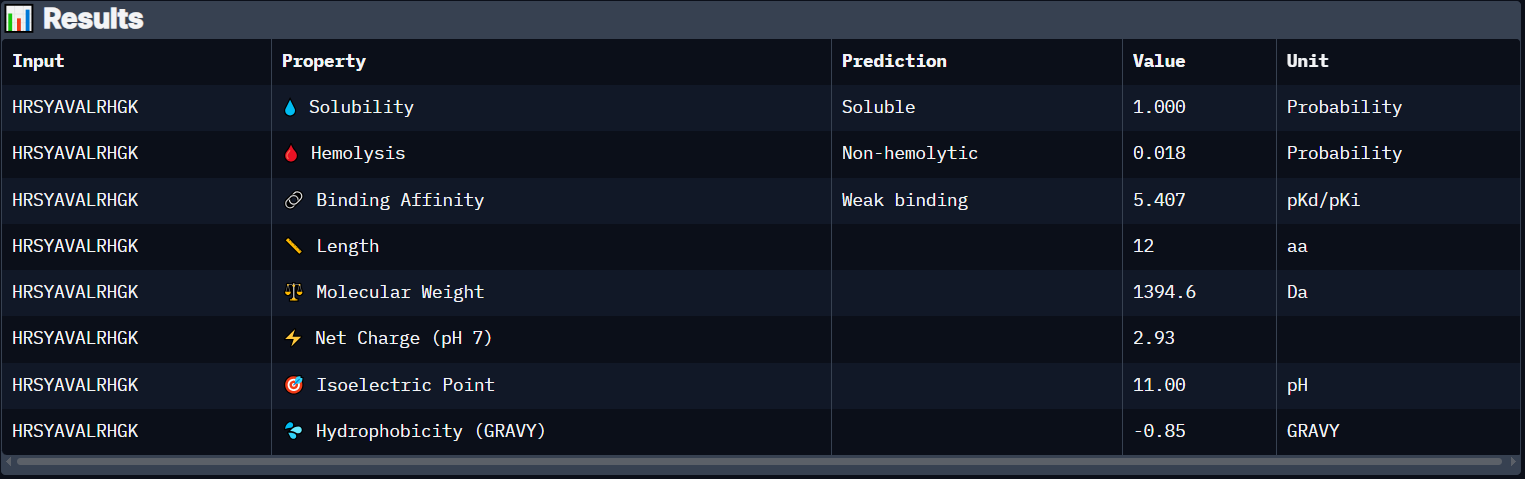
Peptide 1 has a very high solubility score and is also non hemolytic so it is generally safe, however it shows a weak binding affinity, The results also show that it has a isoelectric point of 11.00 explaining its positive net charge (2.93), hydrophilic (-0.85) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1394.6 Daltons.
Peptide 2 (HHSGPVAVRWKX)
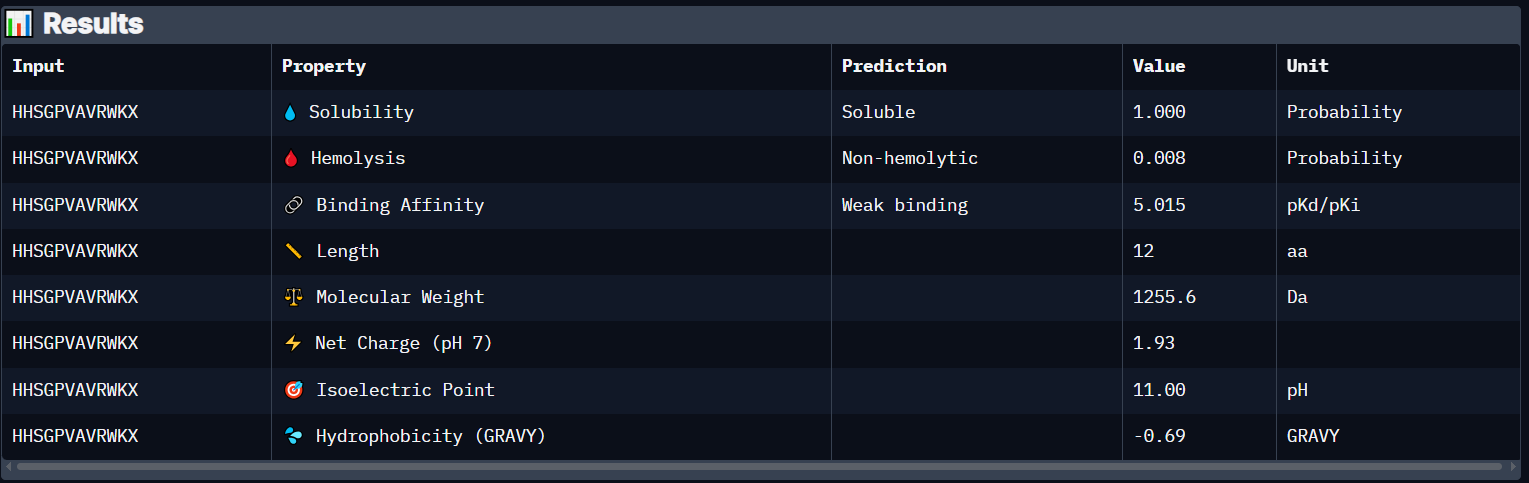
Peptide 2 has a very high solubility score and is also non hemolytic so it is generally safe, however it also shows a weak binding affinity, The results also show that it has a isoelectric point of 11.00 explaining its positive net charge (1.93), hydrophilic (-0.69) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1255.6 Daltons.
Peptide 3 (WRSPAAAVEHWX)
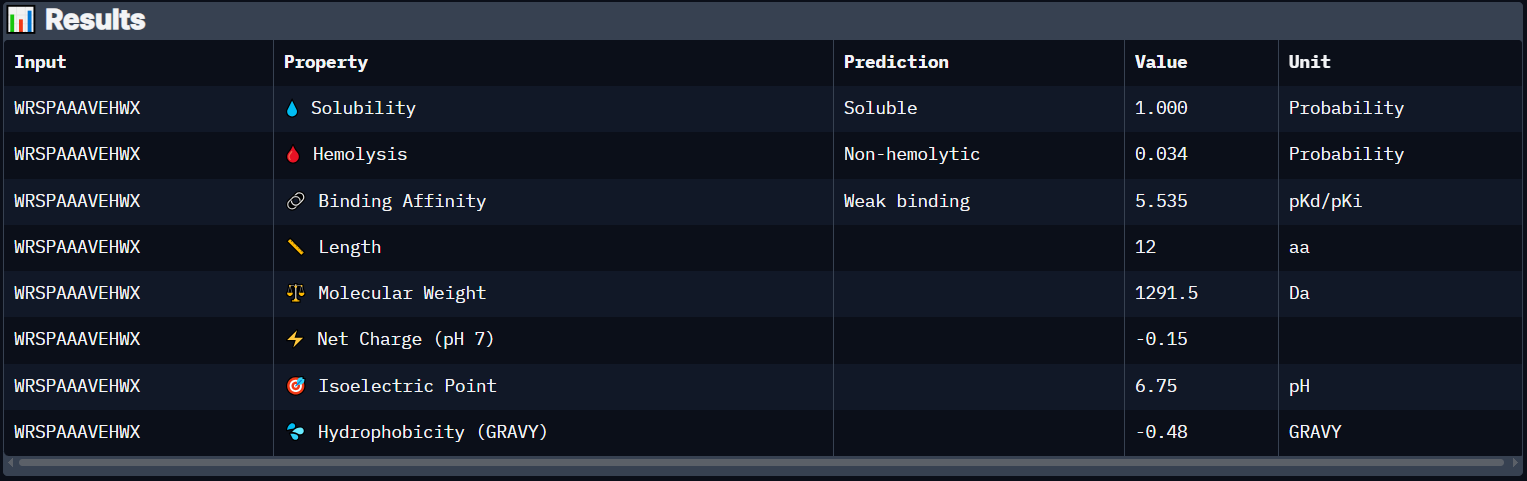
Peptide 3 has a very high solubility score and is also non hemolytic so it is generally safe, however it shows a weak binding affinity, The results also show that it has a isoelectric point of 6.75 explaining its negative (or almost neutral) net charge (-0.15), hydrophilic (-0.48) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1291.5 Daltons.
Peptide 4 (WRYGVVGVRLWE)
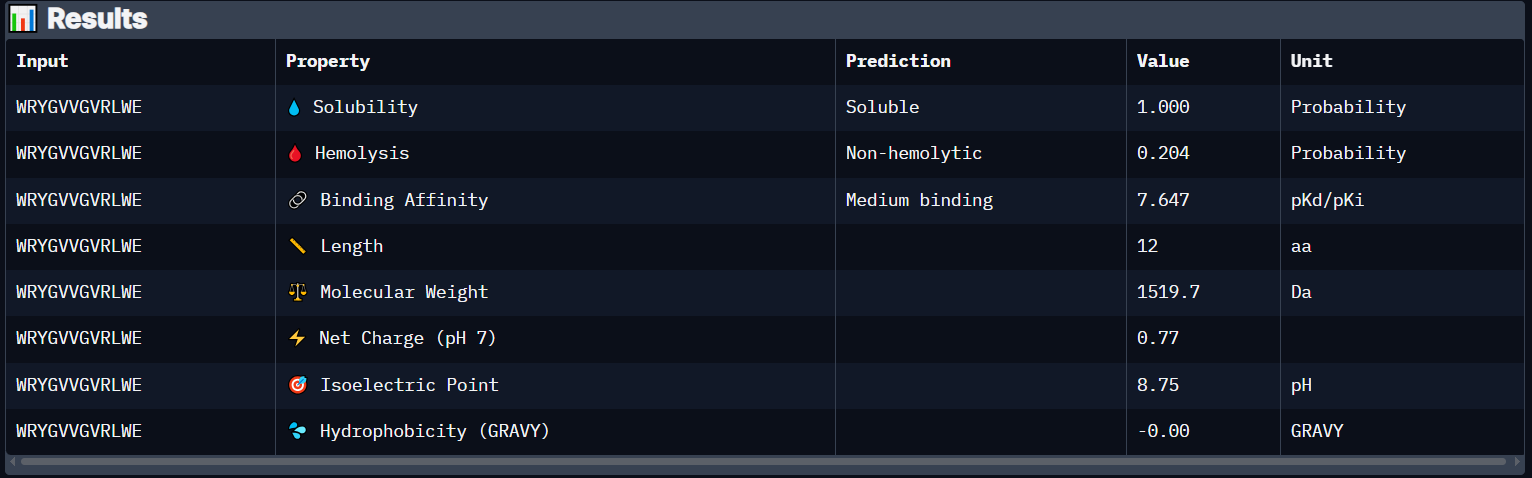
Peptide 4 has a very high solubility score and is also non hemolytic so it is generally safe, and it shows Medium binding affinity :D, The results also show that it has a isoelectric point of 8.75 explaining its positive net charge (0.77), amphiphilic (-0.00) so it binds well to both water or greasy non-polar pockets of the protein, and has a molecular weight of 1519.7 Daltons.
Overall it seems like all the generated Peptides are soluble and non hemolytic so they are safe to use, Peptide 4 seems to be the one with the highest chance of Binding because it has Medium Binding Affinity compared to all Weak Binding affinity of all other peptides, even though it didnt have the highest ipTM score, it got 0.47 while Peptide 1 got 0.54 which proves structural prediction and Shape complementarity is not the only factor affecting Protein-Peptide Binding.
So with that, i have decided to proceed with Peptide 4 WRYGVVGVRLWE since it has a higher Binding Affinity than all others
Part 4: Generate Optimized Peptides with moPPIt
For this part i have decided to pick the amino acid residues from 2-6, since our mutation is A4V so i picked it with two amino acids upstream and 2 downstream to try to bind to this specific area.
Here is the moPPIt generated peptides
| Index | Peptide | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|---|
| 6 | REYDQKQICKKL | 0.9428217448 | 0.8333333135 | 7.031765938 | 0.8390983939 |
| 7 | KKSKKQKELTCG | 0.9830000903 | 0.9166666865 | 6.922430038 | 0.7878084183 |
| 8 | IQQWETKGKRLK | 0.9603748098 | 0.7500000000 | 5.674941063 | 0.5862649679 |
I gave them indexes starting from 6 to account for the previously generated 4 peptides and the control one
To level the playing field for comparison, i decided to take those Peptides, and run them through AlphaFold and PeptiVerse and compare them with the pepMLM ones.
Peptide 6 (REYDQKQICKKL)
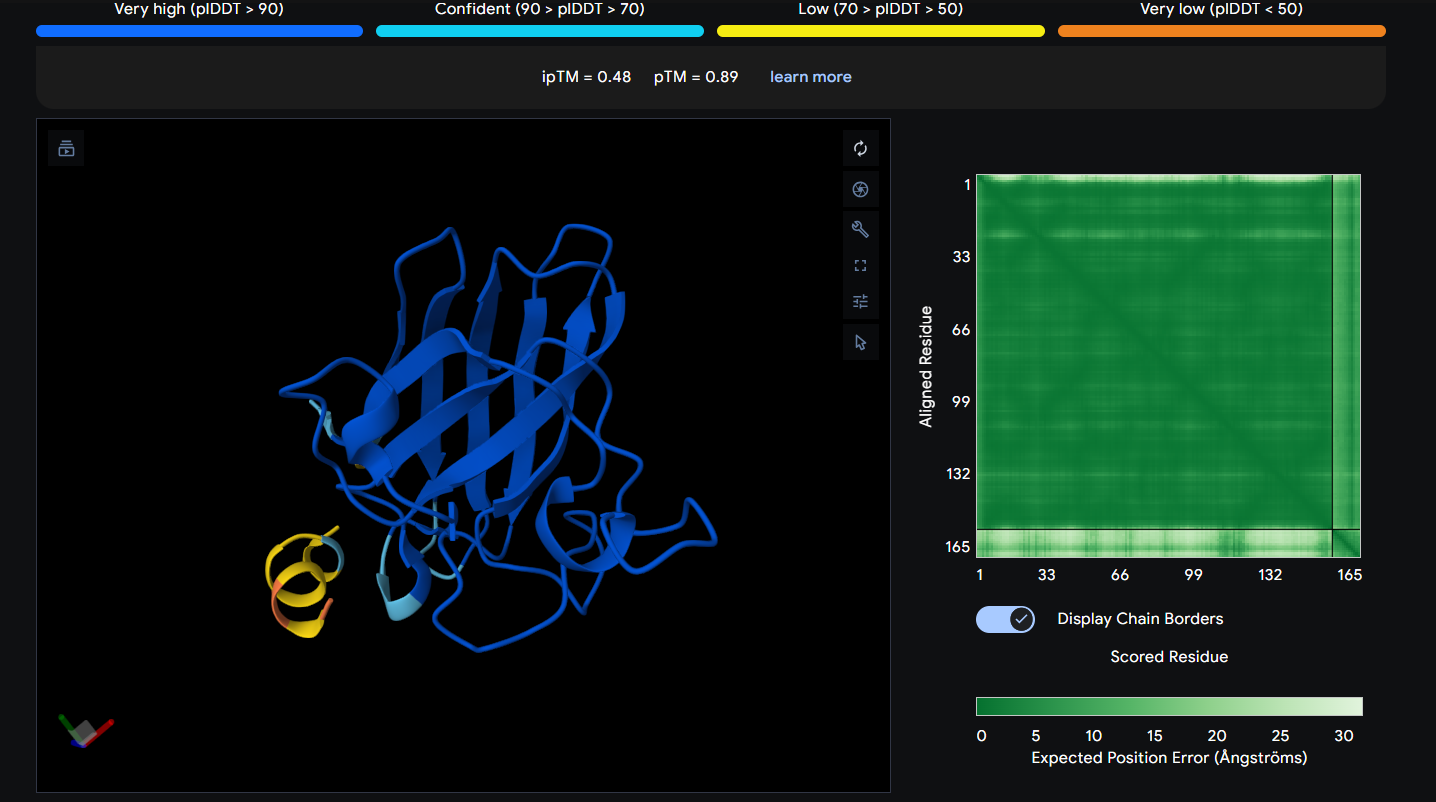
Peptide 6 seems to be floating away from the protein but slightly close to the N-Terminus region where our A4V mutation is, but it is not binded to it and the ipTM score is 0.48 so it falls in the failed prediction zone
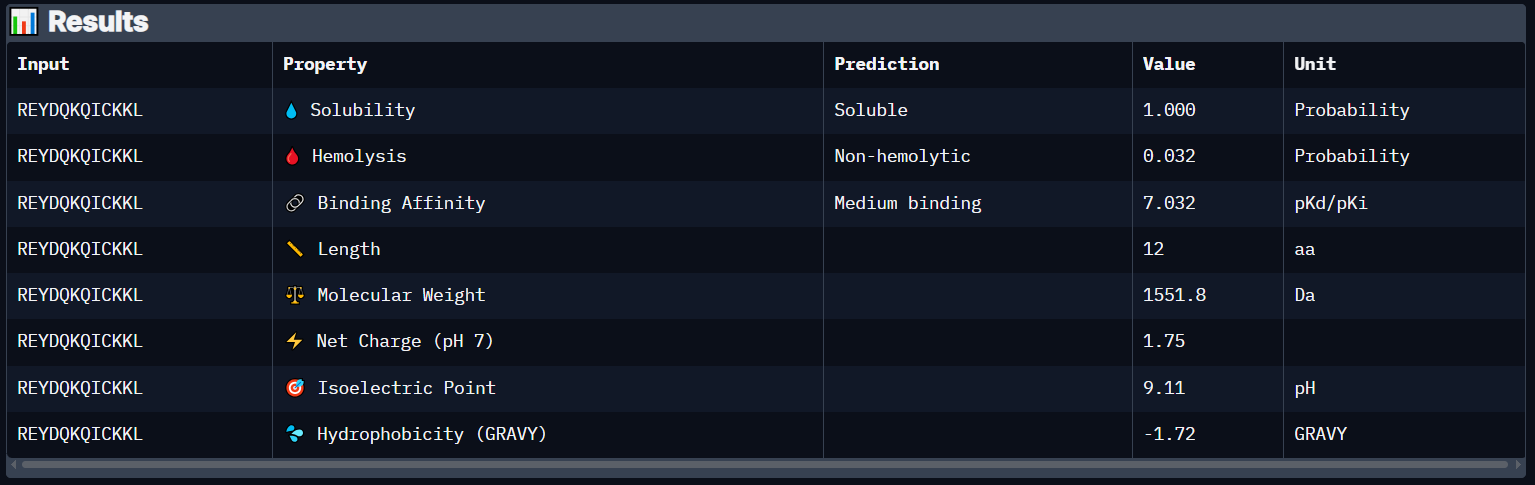
Peptide 6 has a very high solubility score and is also non hemolytic so it is generally safe, and it shows a medium binding affinity, The results also show that it has a isoelectric point of 9.11 explaining its positive net charge (1.75), hydrophilic (-1.72) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1551.8 Daltons.
Peptide 7 (KKSKKQKELTCG)
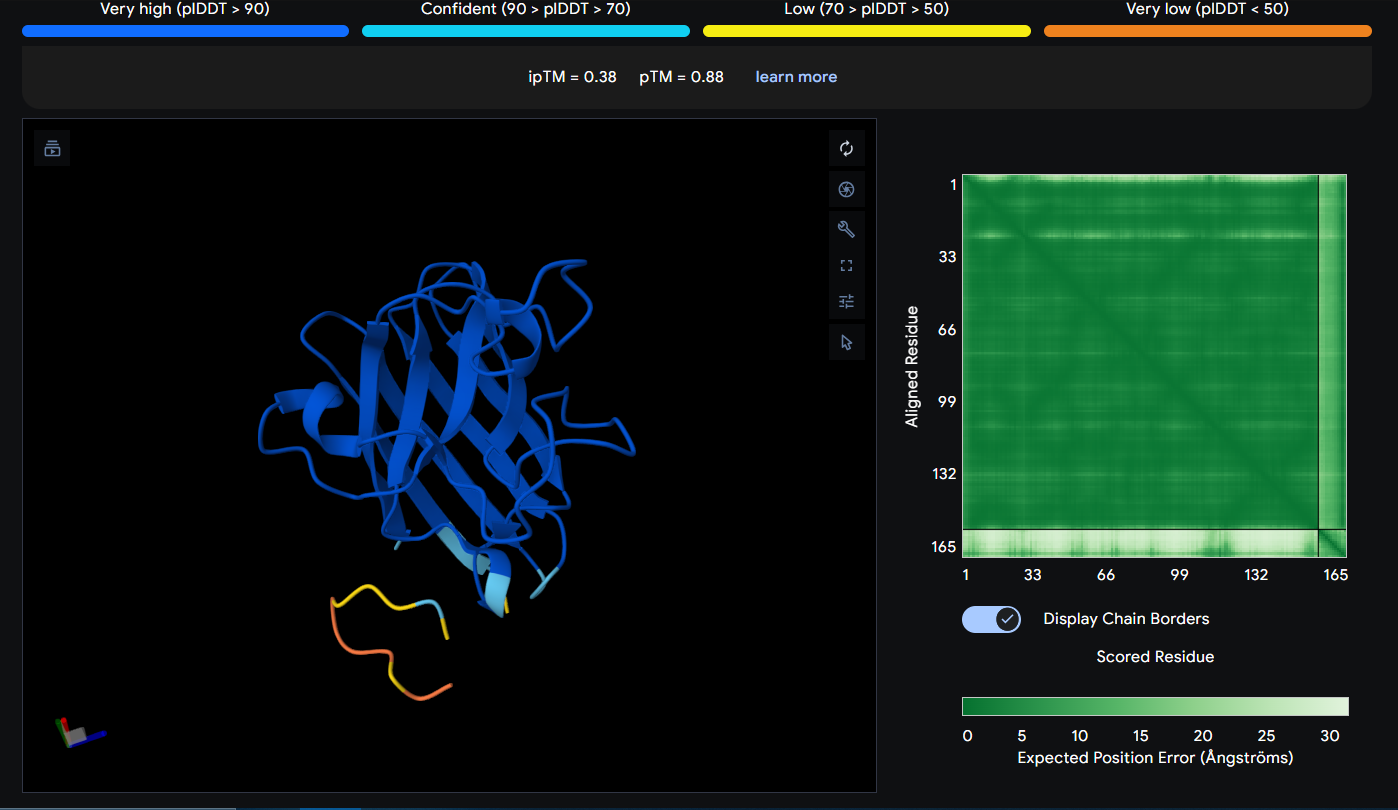
Peptide 7 seems to be floating away from the protein and also very slightly close to the N-Terminus and the mutation region as well, yet again it is not binded to it and the ipTM score is 0.38 so it falls in the failed prediction zone
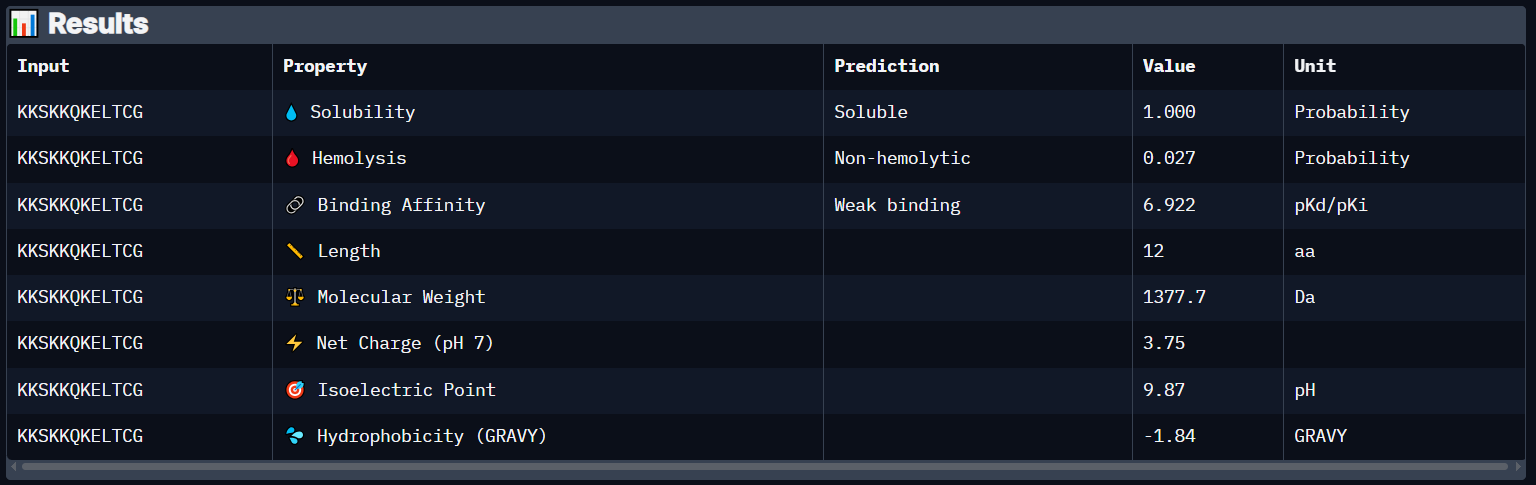
Peptide 6 has a very high solubility score and is also non hemolytic so it is generally safe, and it shows a weak binding affinity, The results also show that it has a isoelectric point of 9.87 explaining its positive net charge (3.75), hydrophilic (-1.84) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1377.7 Daltons.
Peptide 8 (IQQWETKGKRLK)
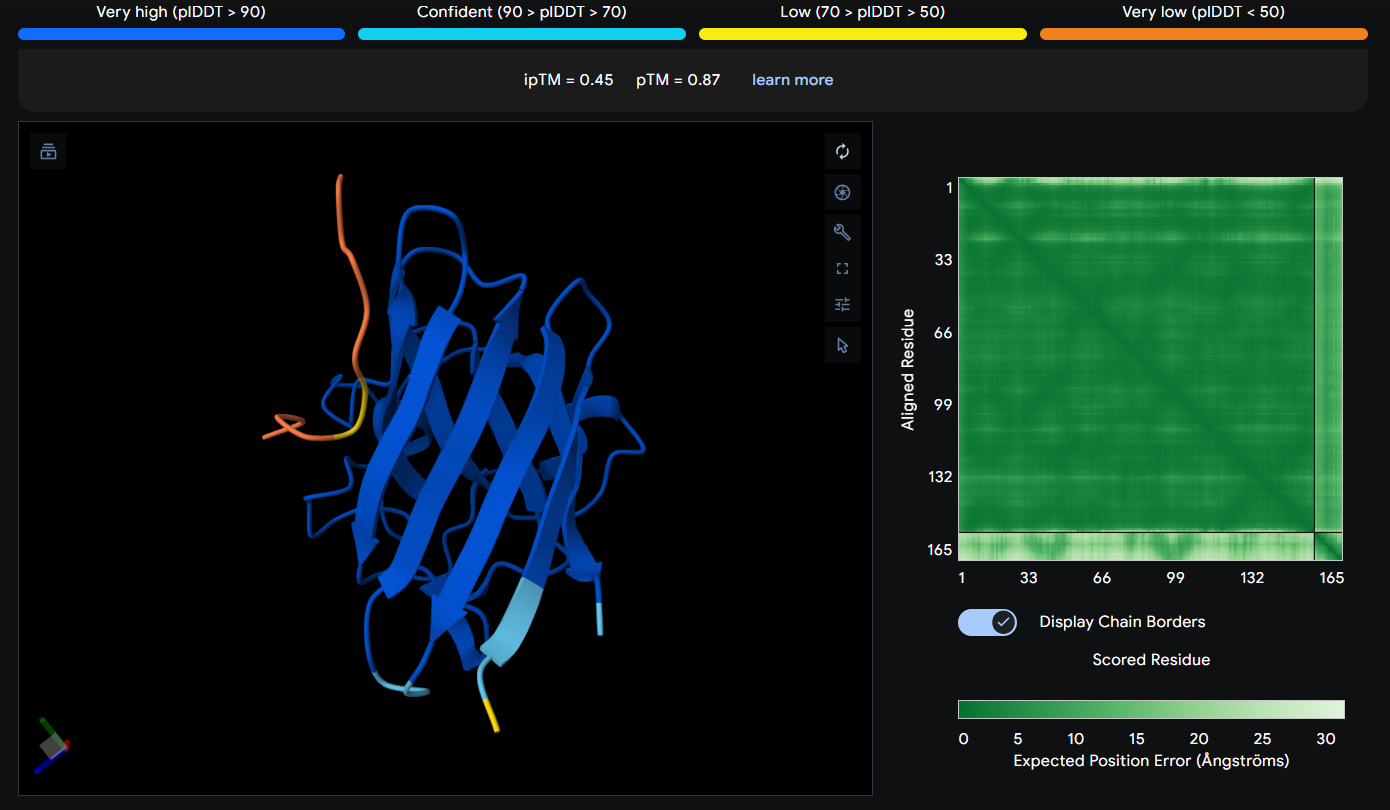
Peptide 8 seems to be floating away from the protein and also very far away from to the N-Terminus and the mutation region (which is weird), and again it is not binded to it and the ipTM score is 0.45 so it falls in the failed prediction zone
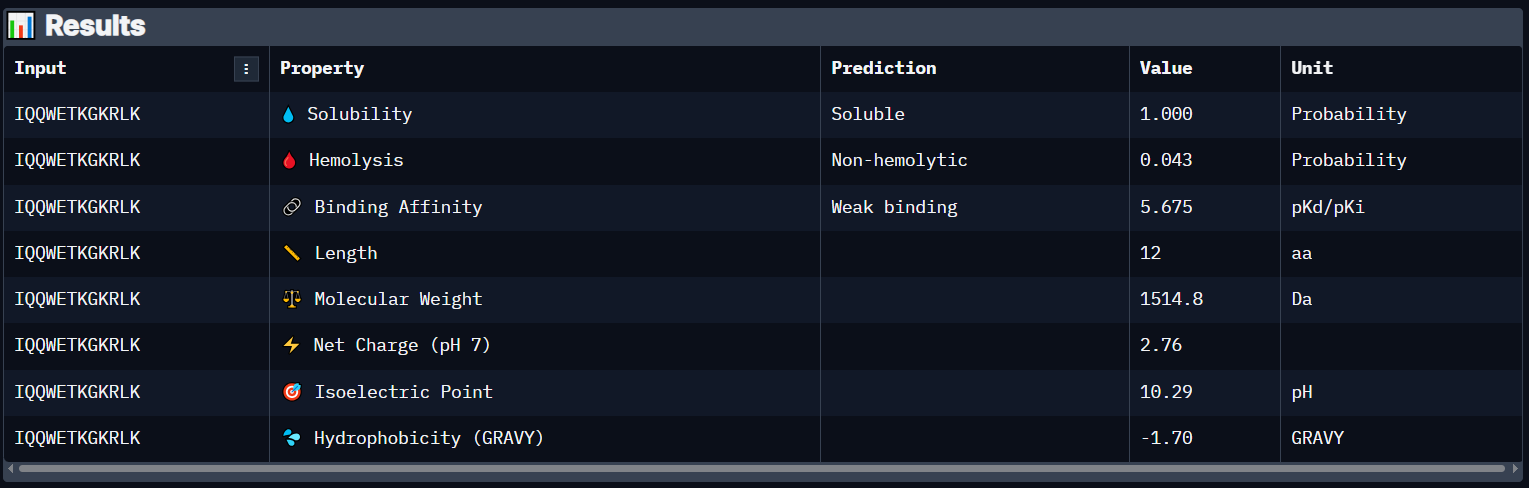
Peptide 6 has a very high solubility score and is also non hemolytic so it is generally safe, and it shows a weak binding affinity, The results also show that it has a isoelectric point of 10.29 explaining its positive net charge (2.76), hydrophilic (-1.70) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1514.8 Daltons.
While pepMLM generates peptides randomly and tries to somehow bind to the protein through the protein-protein interactions it has learned, moPPIt seems to follow a more Rational Design when designing those peptides and tries to target a specific region/residue to achieve the binding there. With that said, it seems like all my generated peptides have failed surpassing the AlphaFold threshold of an ipTM score of at least more than 0.6 and most of them seem to show a weak binding affinity on PeptiVerse except for Peptides 4 and 6 which actually showed medium binding affinity.
Evaluation of the generated peptides before clinical studies is very crucial and it requires bridging the massive gap between computational biology and actual human physiology, first these peptides need to be synthesized and then tested on human cell cultures we mainly need to confirm that it does its expected job which is preventing SOD1 from misfolding or aggregating and most importantly not to kill the cell in the process, Then we may test these on In Vivo Animal Models like an ALS Mouse Model, to monitor if these peptides can actually cross the Blood Brain Barrier (BBB) and reach the affected Neurons.
Additional Tests and Validations may include Immunogenetic Testings to make sure the Human Body doesn’t mark this synthetic peptide as a foreign invader, which can trigger a dangerous immune response or cause the body to generate neutralizing antibodies against the drug, also we may need to check if it needs chemical modifications to survive cellular proteases since peptides can be an easy target for them and this negatively affects the peptide’s half life.
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
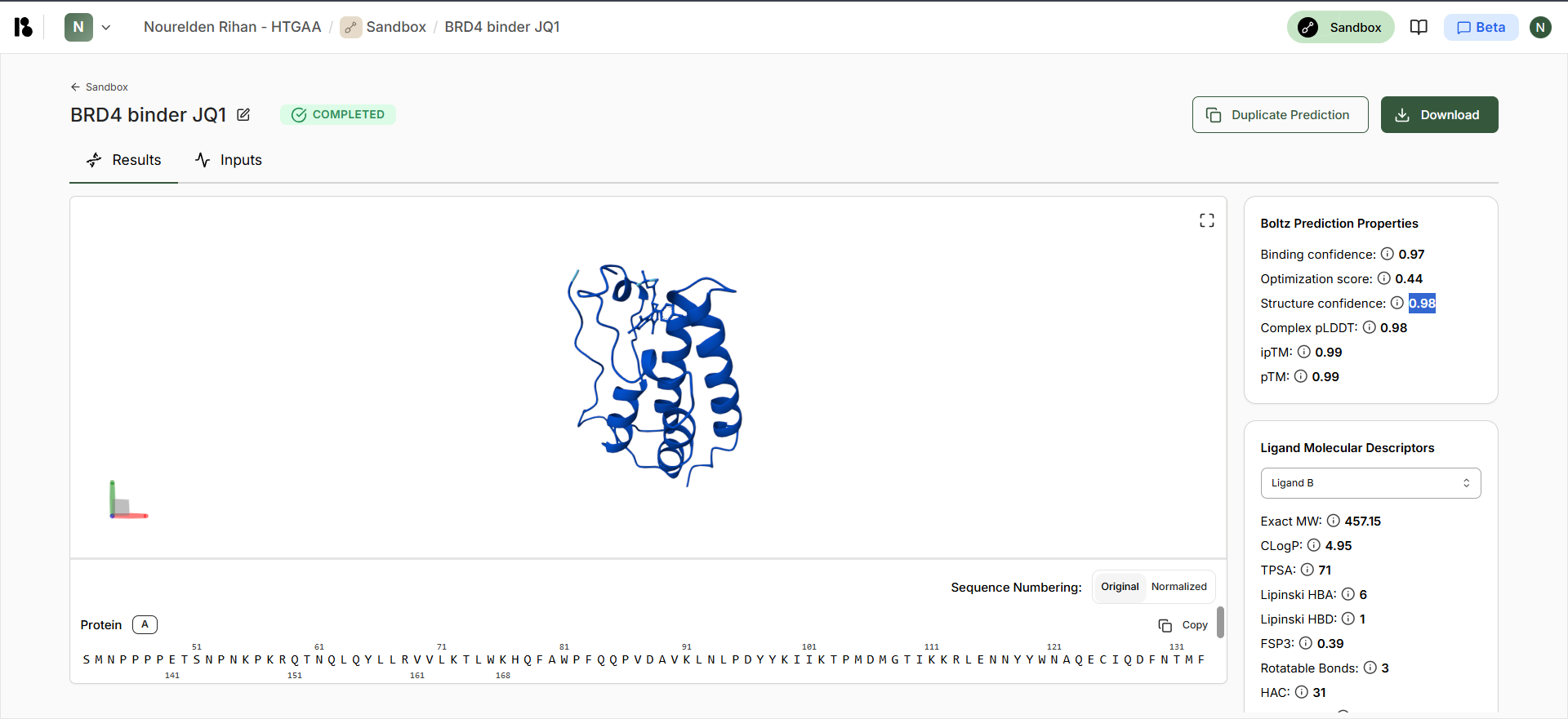
Boltz JQ1 Output
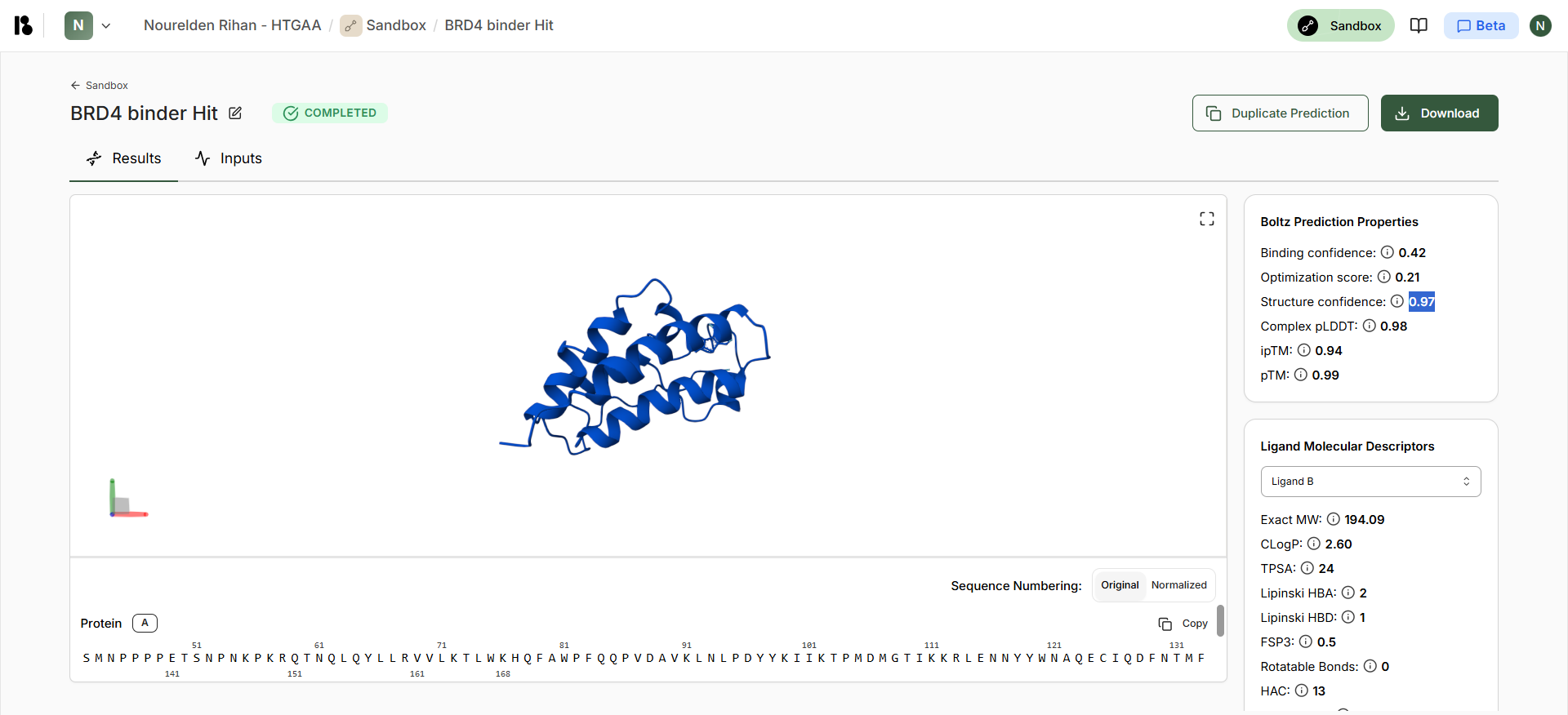
Boltz Hit Output
Boltz Lead Output
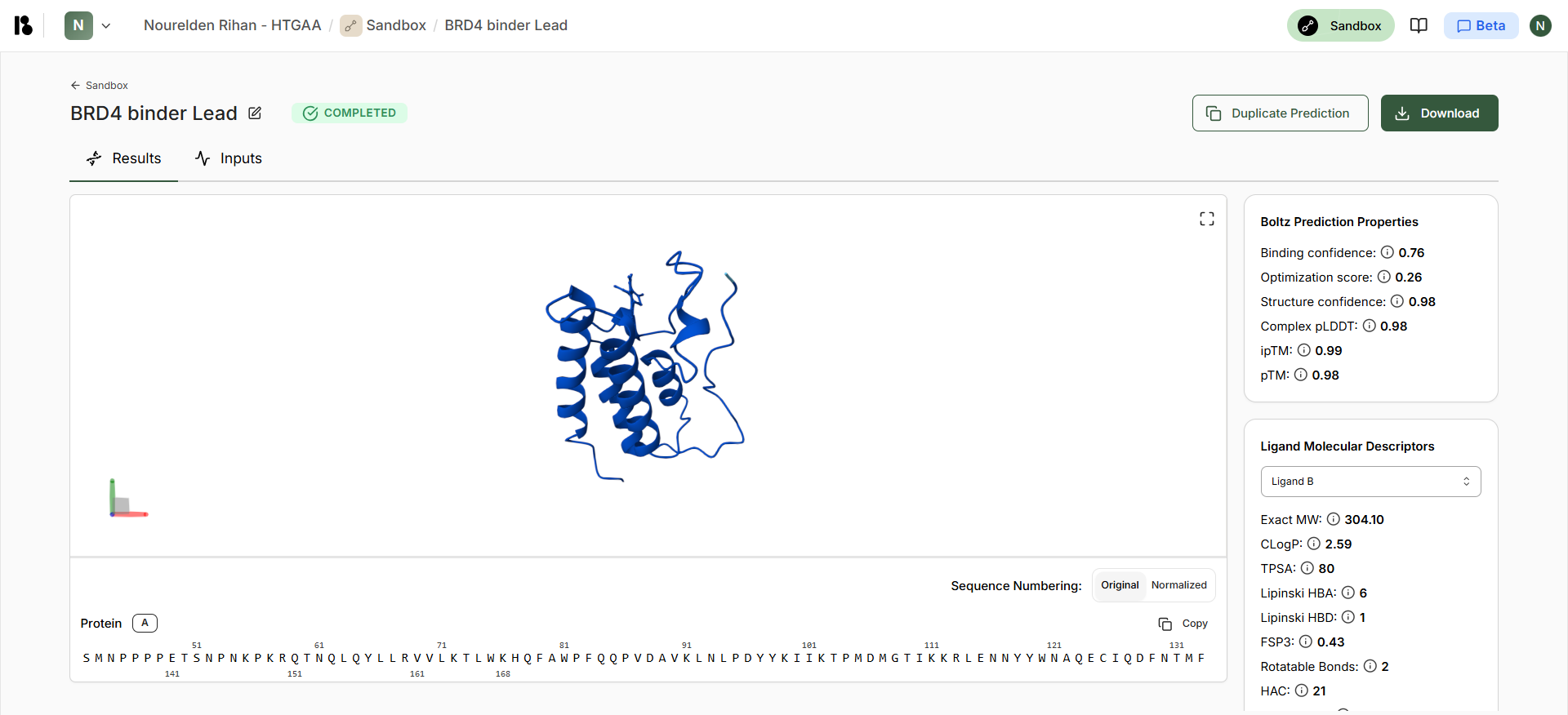
| Compound | Binding Confidence | Optimization Score | Structure Confidence |
|---|---|---|---|
| Hit | 0.42 | 0.21 | 0.97 |
| Lead | 0.76 | 0.26 | 0.98 |
| JQ1 | 0.97 | 0.44 | 0.98 |
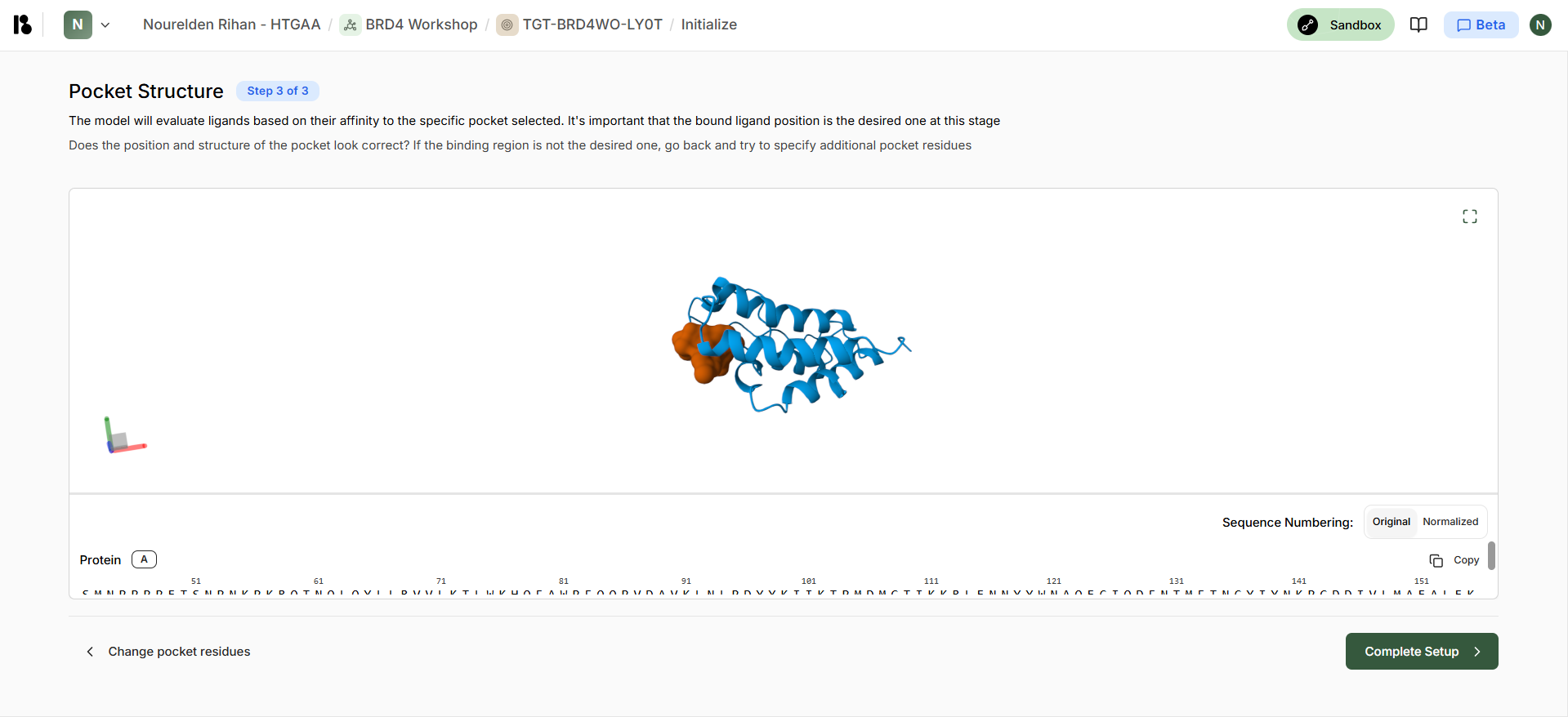
Boltz Predicted Pocket Structure
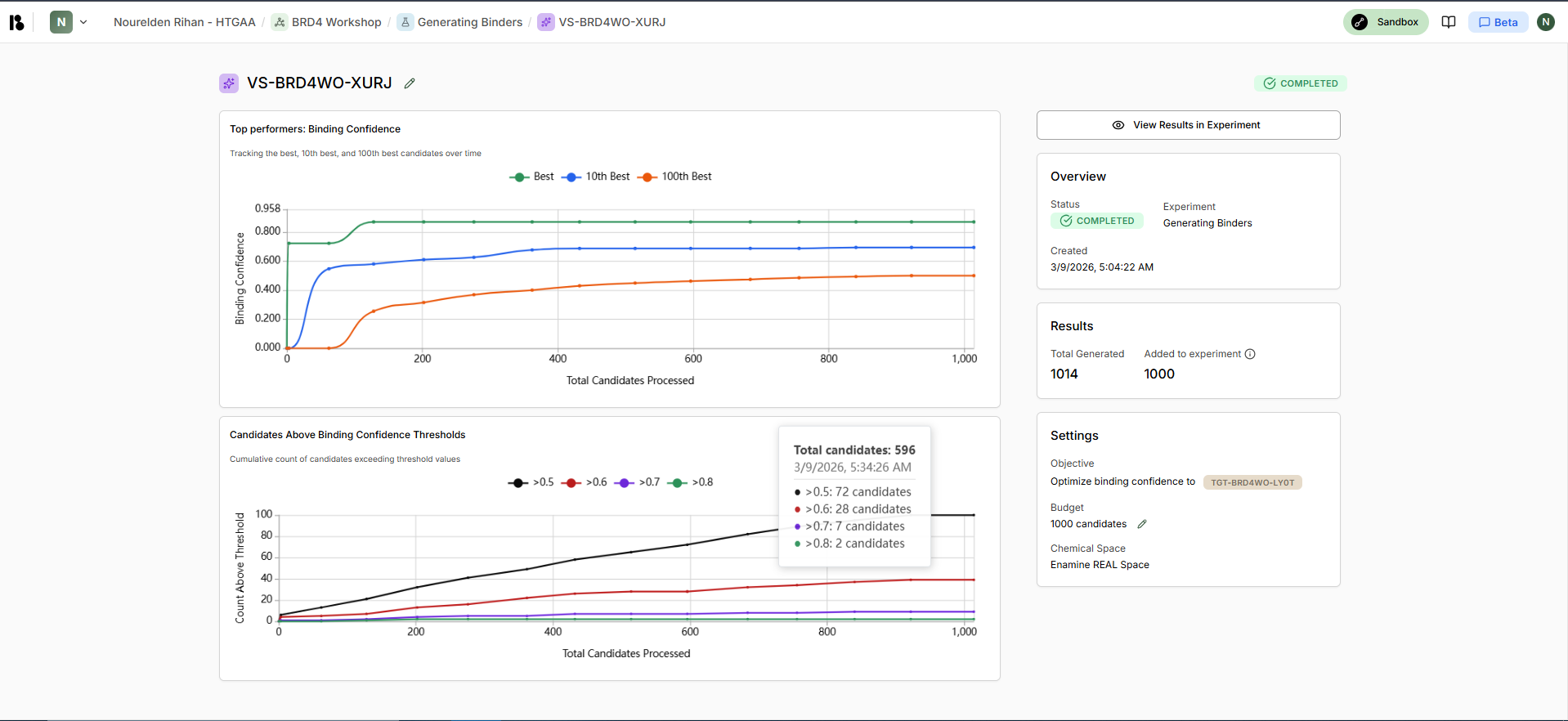
Boltz AI Generated Binders
Part C: Final Project: L-Protein Mutants
Option 1: Mutagenesis
Here is the Pictures of the Clustal Omega Multiple Sequence Alignment (MSA) Output

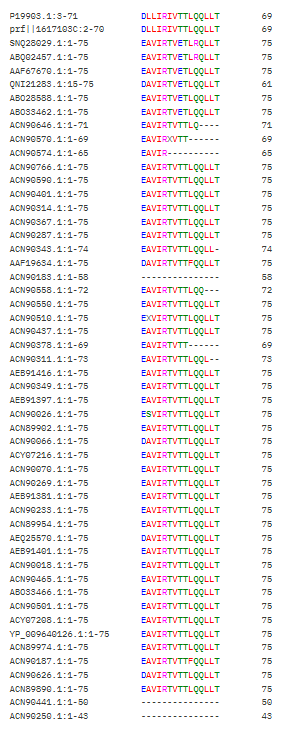
It seems like these Positions [21,25,28-29,33,35-37,40] have an “*” which means these have not changed at all and most probably are a totally conserved crucial region that should not be mutated, Positions [17,26,30] have a “:” which means they are highly conserved, but mutations that are very similar in shape, structure and chemical properties are tolerated, the rest seem to be more flexible.
Interesting Finding here, All the conserved regions seem to be in the Soluble Domain (1-40) that is responsible for DnaJ Interaction :D
This is the generated L Protein Mutation Heatmap
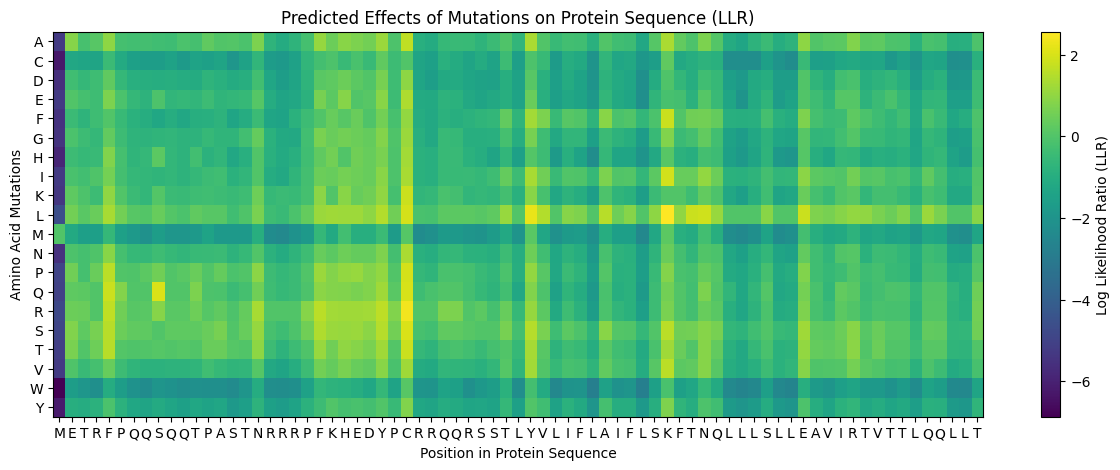
I tried to cross reference with the experimental data sheet but it was quite hard to do manually so i wrote a script and ran it on Here on Colab to just find the common ones across the two files and came up with 3 common ones, however even though these 3 had quite a high LLR Score, the experimental data showed a Lysis score of 0, and i believe this shows the limitation in the structure based modeling, it predicted structural stability but failed to account for the functional mechanism of lysis.
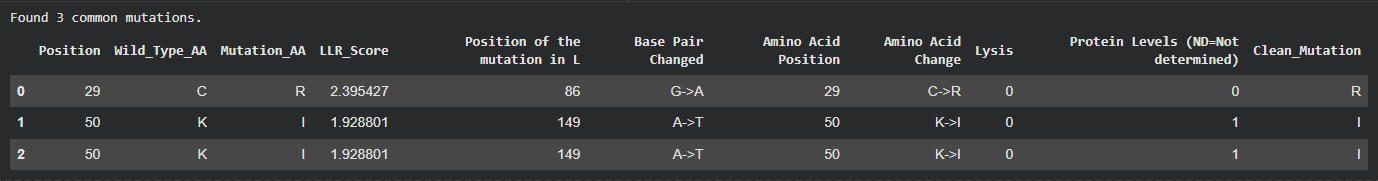
The common mutations identified by my algorithm
So Then i Filtered my generated mutations first to target the soluble domain (1-40) first while making sure to avoid the totally conserved regions [21,25,28-29,33,35-37,40], the highly conserved regions [17,26,30] and my 3 common matches to avoid choosing mutants that are either disrupting the protein or have been already experimented on with no lysis observed.
The two mutations i have decided to go with for the Soluble Domain are:
| Index | Position | Wild_Type_AA | Mutation_AA | LLR Score |
|---|---|---|---|---|
| 1 | 39 | Y | L | 2.24177968502044 |
| 2 | 9 | S | Q | 2.01432478427886 |
These have the highest LLR score in this region and avoid any conservative regions in the protein
Then i repeated the steps again, this time targeting the transmembrane domain (41-75) and followed the same criteria, and i picked these 2 mutations:
| Index | Position | Wild_Type_AA | Mutation_AA | LLR Score |
|---|---|---|---|---|
| 3 | 50 | K | L | 2.56146776676178 |
| 4 | 53 | N | L | 1.86493206024169 |
And Again because These have the highest LLR score in this region and avoid any conservative regions in the protein
For the Final 5th mutant i decided to pick this one:
| Index | Position | Wild_Type_AA | Mutation_AA | LLR Score |
|---|---|---|---|---|
| 5 | 52 | T | L | 1.81396758556365 |
Because this one still has a relatively high LLR Score and is in the area where the L Protein does not overlap with either the Coat Protein or the replicase ones.
so here is the full 5 Mutations table i chose:
| Index | Position | Wild_Type_AA | Mutation_AA | LLR Score |
|---|---|---|---|---|
| 1 | 39 | Y | L | 2.24177968502044 |
| 2 | 9 | S | Q | 2.01432478427886 |
| 3 | 50 | K | L | 2.56146776676178 |
| 4 | 53 | N | L | 1.86493206024169 |
| 5 | 52 | T | L | 1.81396758556365 |
AlphaFold Multimer Runs
Mutant 1 (Y39L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
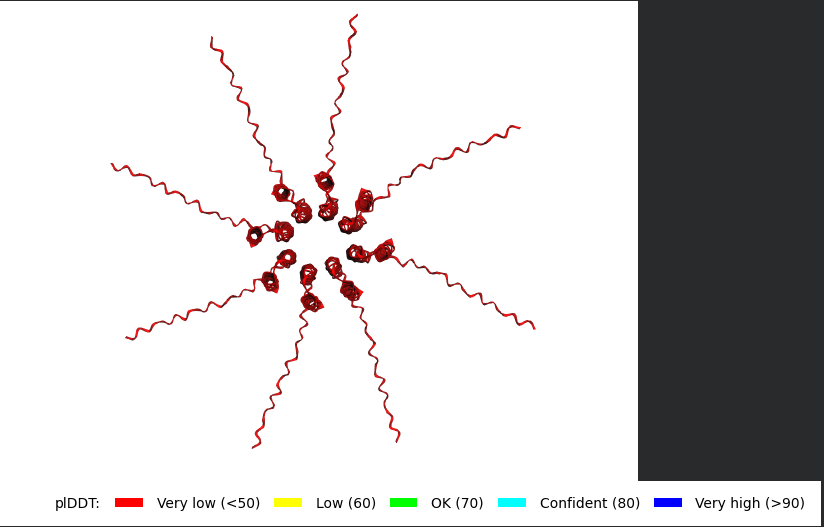
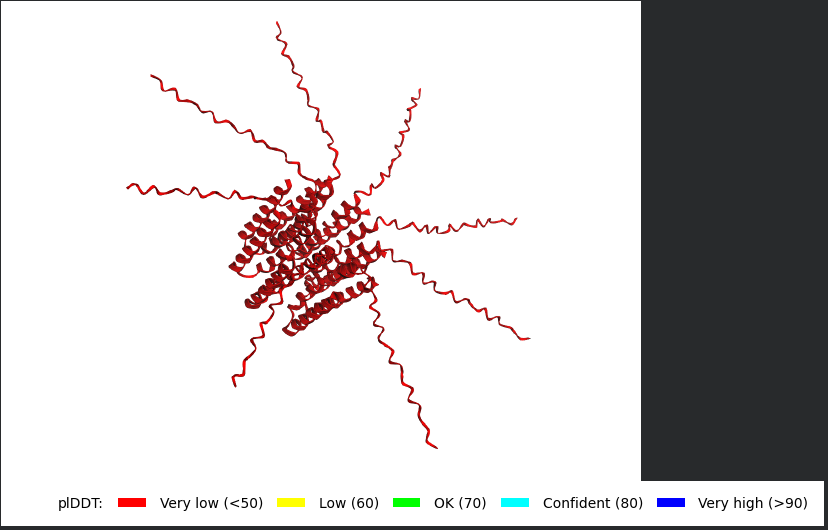
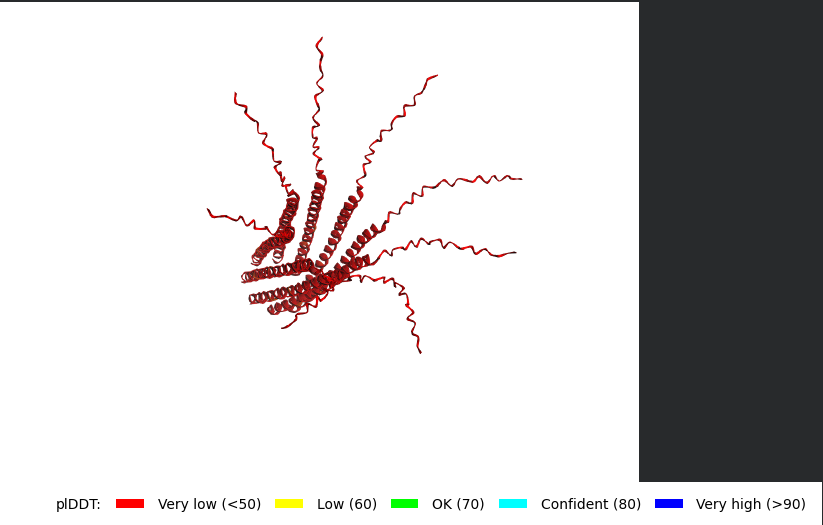
The L Proteins do form a cylinder like shape mimicking the transmembrane pore but the piDDT score are very low (<50) so this probably won’t express well or won’t express at all if done in wet lab :(.
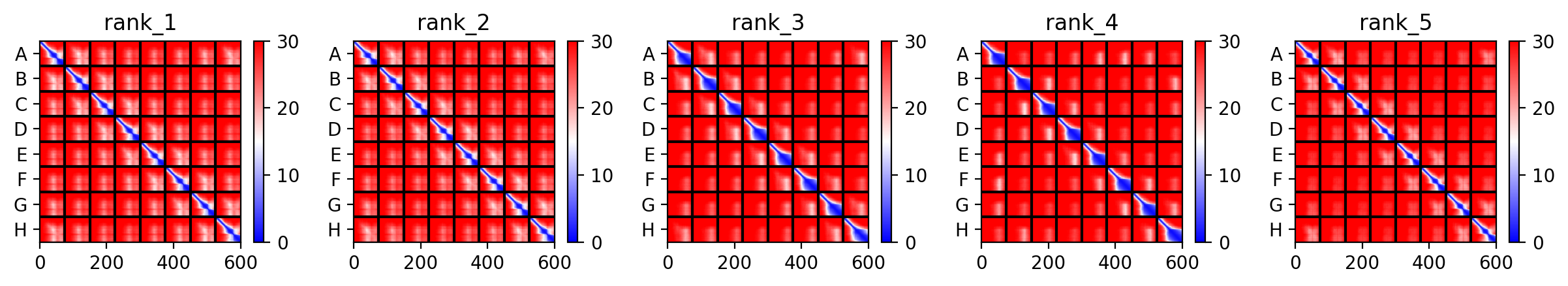
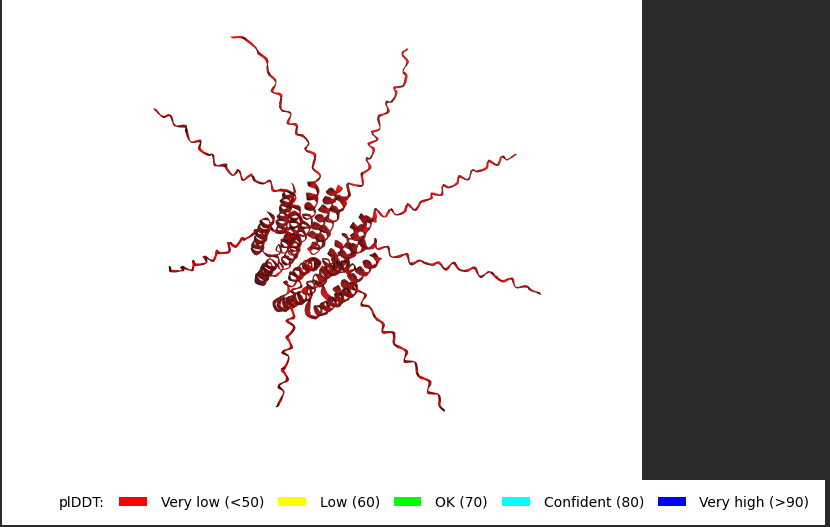
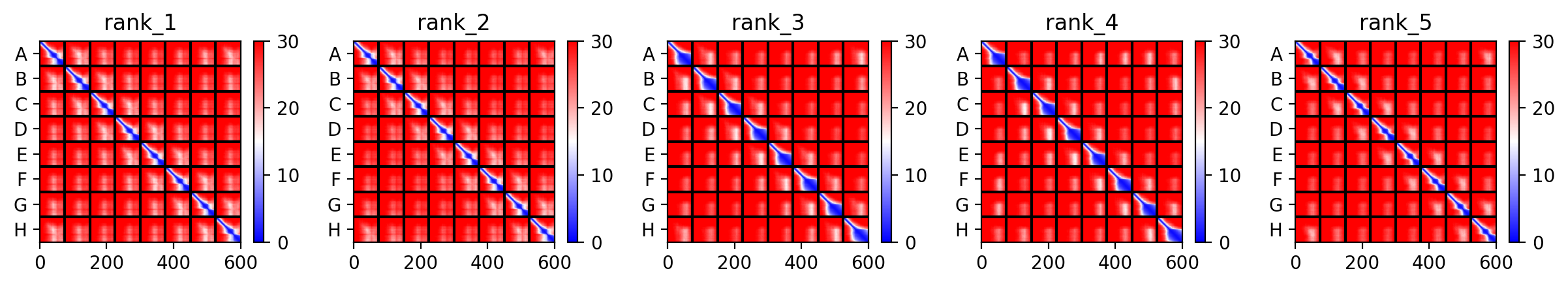
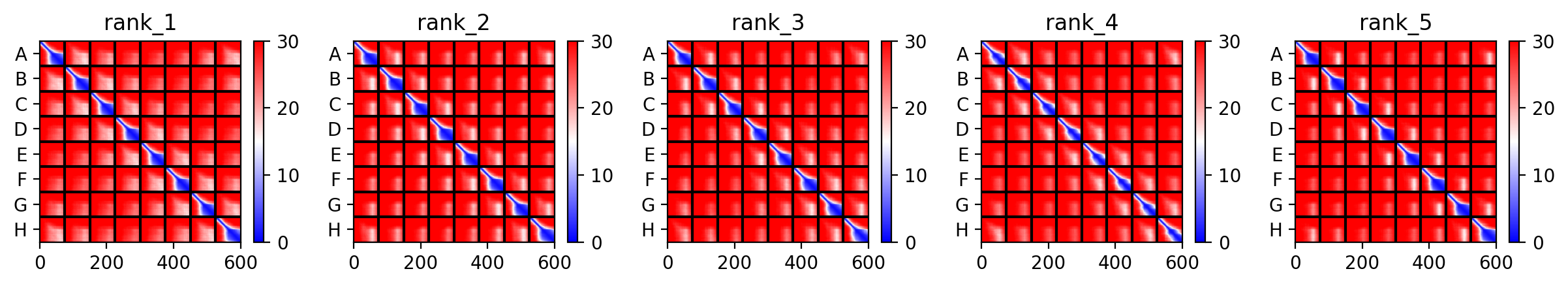
This is the Predicted Aligned Error (PAE), i asked AI (Gemini) how to interpret this and it said this:
So following this, it seems like each monomer folds correctly with high confidence yet their together-grouping is not reliable at all with very low confidence scores possibly hinting that the pore forming shape we saw might not actually happen (i hope i understood that correctly XD)
Mutant 2 (S9Q)
The Monomer Sequence:
METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Same exact findings for this mutant too :(.
Again, Same Here as well. (it is the same for all five mutants :(. )
Mutant 3 (K50L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
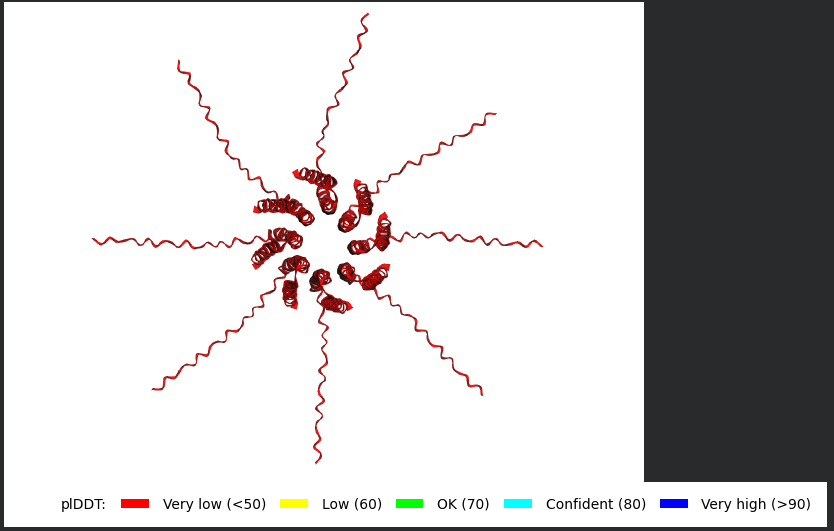
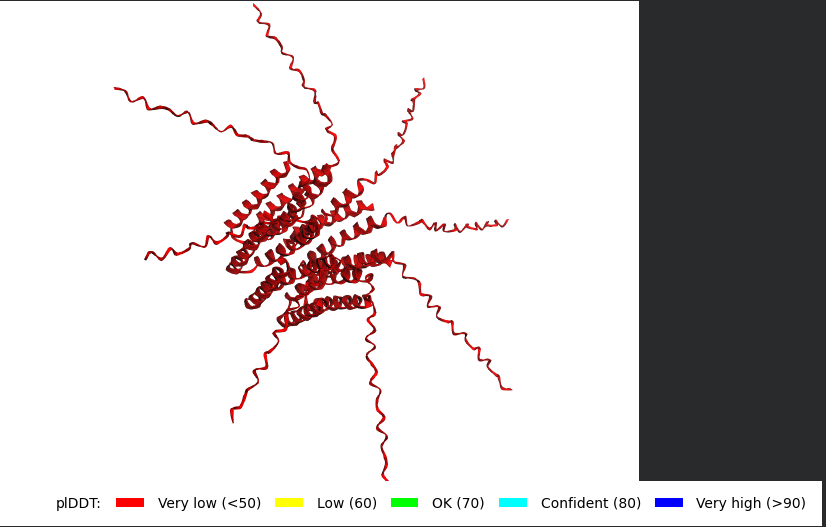
Mutant 4 (N53L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT
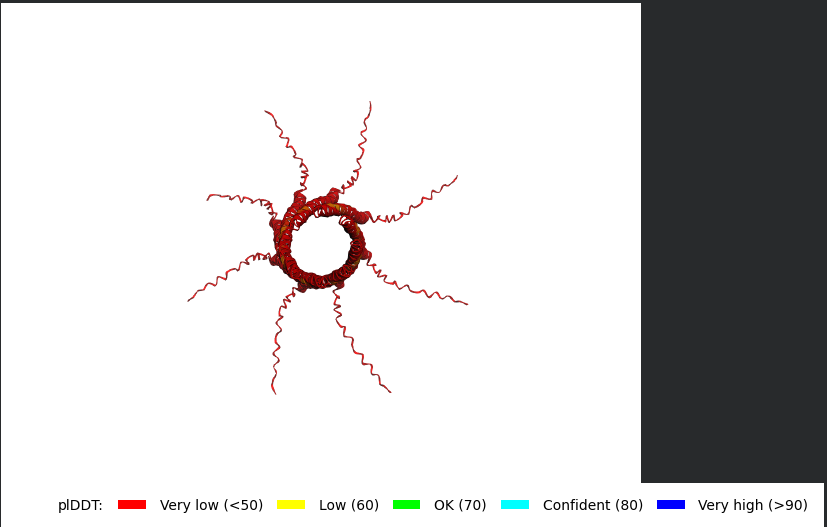
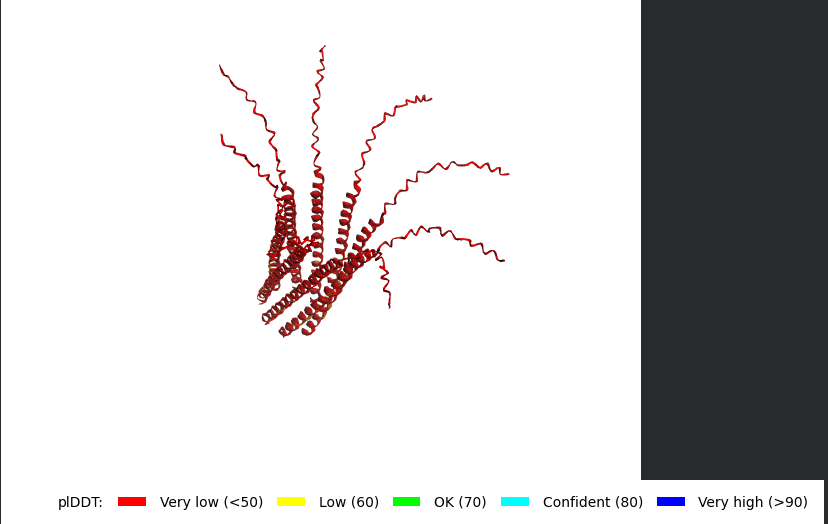
Mutant 5 (T52L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT
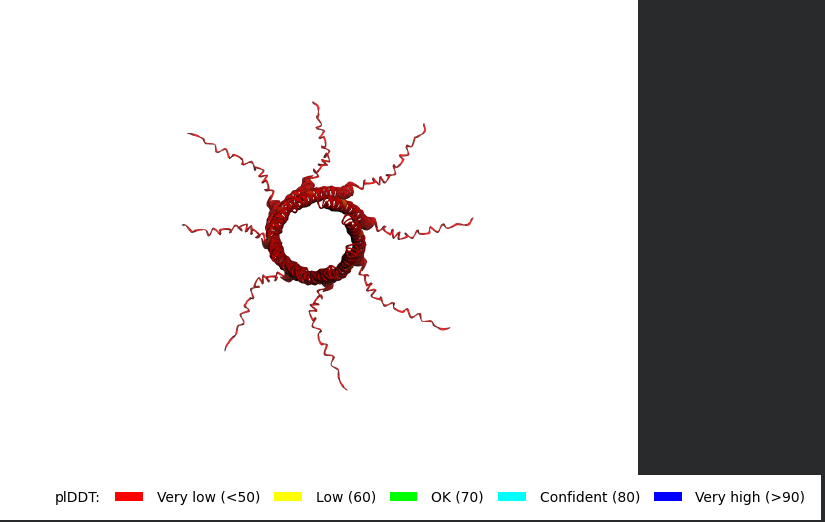
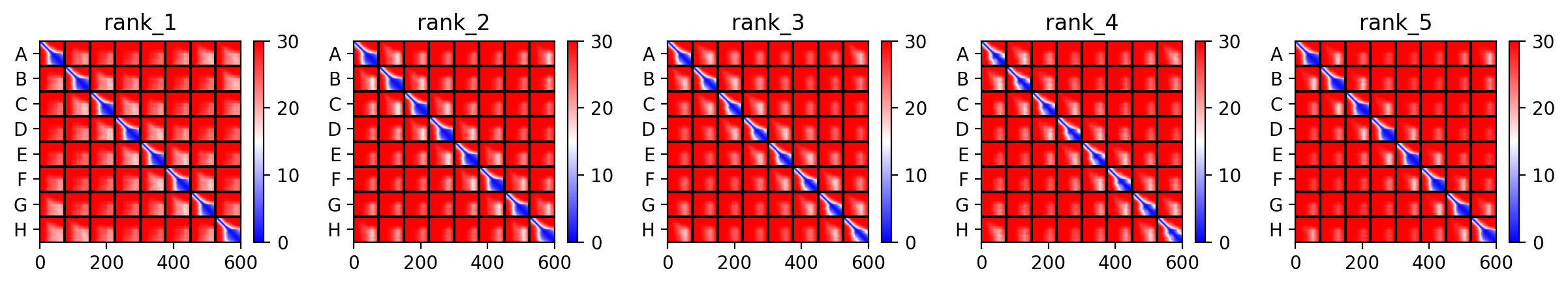
I Actually decided to pick a mutant from the Experimental Data Sheet and made sure it has been proven to have the Lysis Effect, my aim is to try to perform the AlphaFold Multimer step for it, to have a look at how different it might be, the mutant i picked was R30Q.
This is the monomer sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
This is the full Alphafold sequence i used:
METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
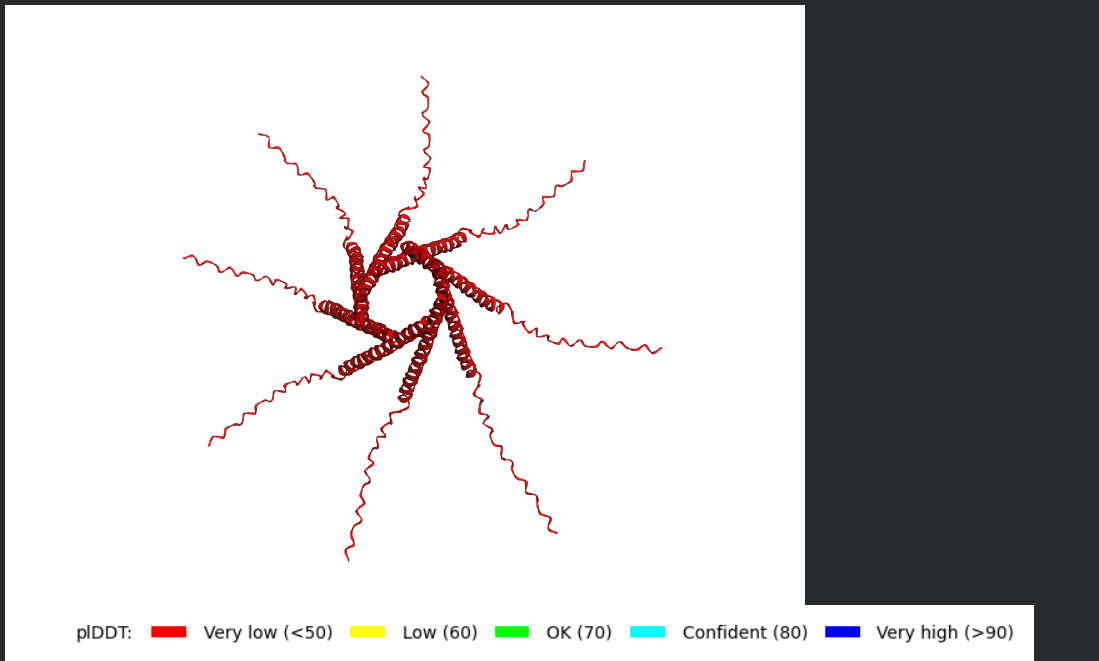

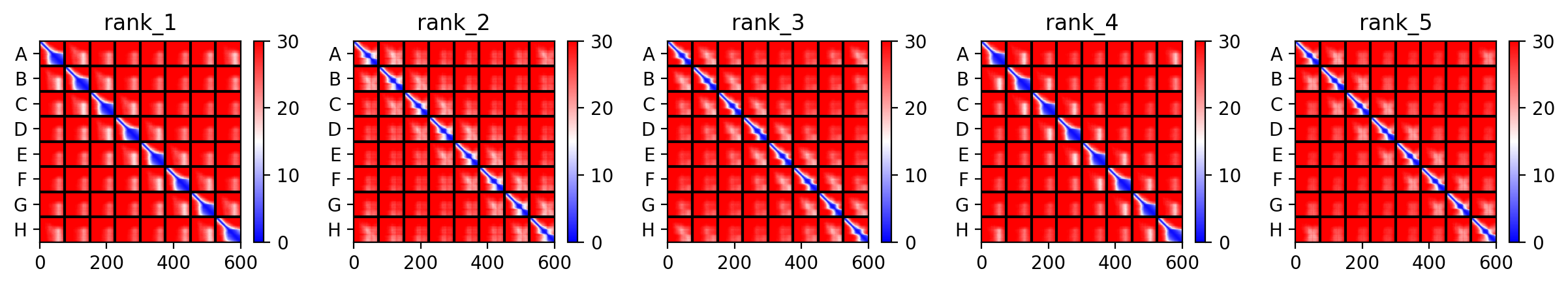
Well i got bad news and good news, the bad news is, the structre is also of very low confidence and the Predicted Aligned Error (PAE) Plot shows the same low confidence for the monomer’s interaction with each othe, the good news though is that this is the run of a mutant that has been experimentally validated and has the lysis effect and protein level determined, so maybe this gives me hope that my five mutants might actually stand a chance in a wet lab validation regardless of the very low confidence scores it got.