I am a Medical Student from Egypt, with a love for code and a heart for Synthetic Biology. I’m here to pivot from treating patients to engineering biology. I believe the most creative solutions happen at the intersection of disciplines.
Off the Clock: I am a nature enthusiast stuck in the city. I dream of going fishing and mountain hiking. If you’ve ever cast a line or summited a peak, please share your stories, I need the inspiration!
𓃠 Week 1 Homework 𓃠 1. First, describe a biological engineering application or tool you want to develop and why. I want to build a Biological 3D Printer :D It is a quite crazy idea, basically a biological 3D printer takes in a DNA file and prints it and expresses it right away and delivers the target product in vial.
𓃠 Week 3 Homework 𓃠 Assignment: Python Script for Opentrons Artwork This is an Ancient Egyptian Pharaoh Figure, this was made using this GUI Also here is the live link to check it out! :D
𓃠 Week 4 Homework 𓃠 Part A. Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Proteins make about 20%-30% of the composition of meat, so we can take 25% as the average, this means 500 x 0.25 = 125g protein, to determine molar mass, 1 Dalton is approximately equal to 1 gram per mole (g/mol), so an average amino acid weighs 100 g/mol which when divided by the molar mass of 100 daltons gives 1.25 moles of amino acids, then we can multiply this by Avogadro’s number (6.022x1023), we get 7.5275 x 1023 molecules of amino acids in the 500 grams of meat (this is extremely huge wow XD)
𓃠 Week 5 Homework 𓃠 Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM Here is the Human SOD1 sequence from Uniprot (P00441)
𓃠 Week 6 Homework 𓃠 Assignment: DNA Assembly 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion High-Fidelity PCR Master Mix is basically a pre-optimized solution containing every chemical component needed for DNA amplification except for the DNA template and the primers.
𓃠 Week 7 Homework 𓃠 Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? IANNs are more suited for biology since they are not constrained with the digital “0 and 1” appraoch, but can follow an Analog appraoch that is more realistic and suited for biology, because biology is not in an On/Off stage but it varies with different values and expression levels and they are more flexible in that they help in designing more Decision Boundaries without having to create new parts from the scratch and as we have seen with the Neuromorphic wizard, it can also tap and work in Advanced areas like “Dual Region” zones where a cell activates if inputs are strictly below a threshold Or strictly above a threshold, but remains totally inactive in the intermediate zone
𓃠 Week 9 Homework 𓃠 Homework Part A: General and Lecturer-Specific Questions General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis is basically taking the protein-making machinery out of a living cell and running the process in a test tube. The biggest advantage over traditional in vivo methods (which use living cells) is that you do not have to worry about keeping a cell alive. When you work with living cells, the cell membrane blocks you from easily changing the environment, and the cell’s natural life-cycles get in the way.
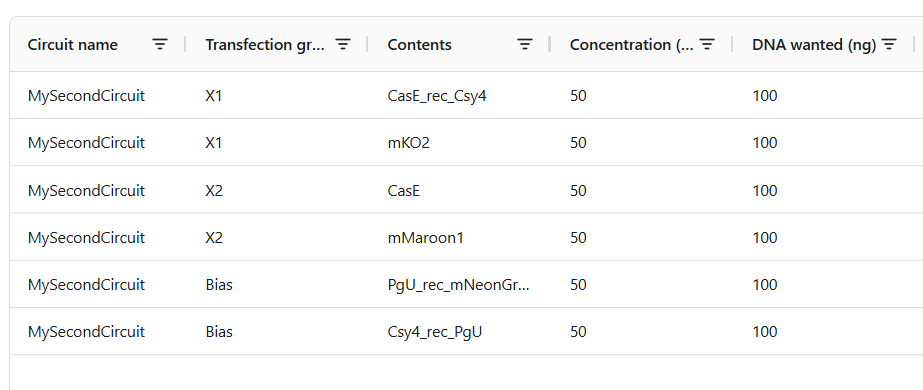
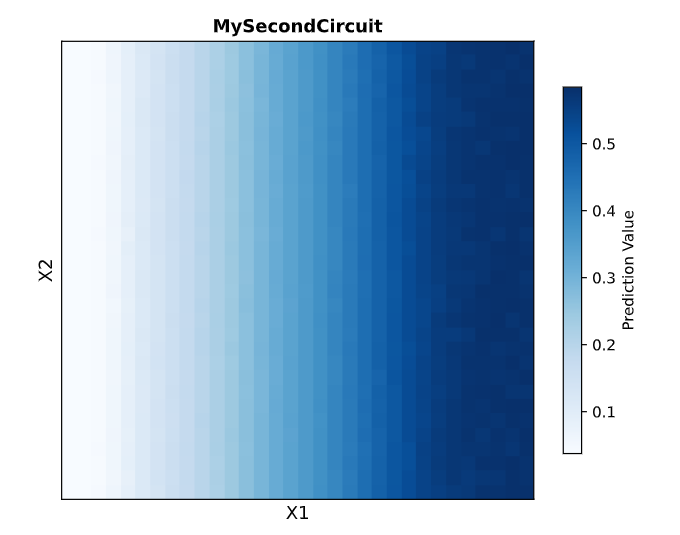
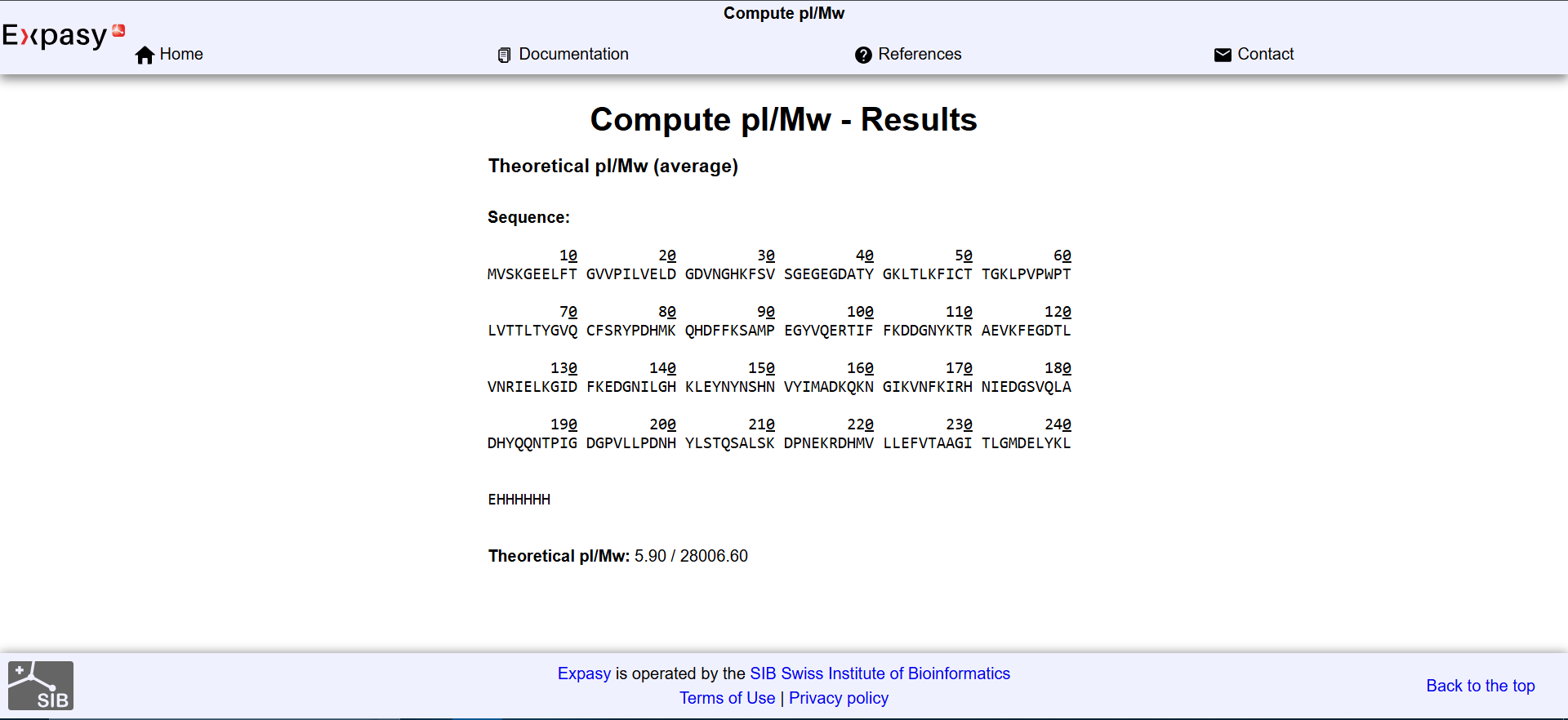
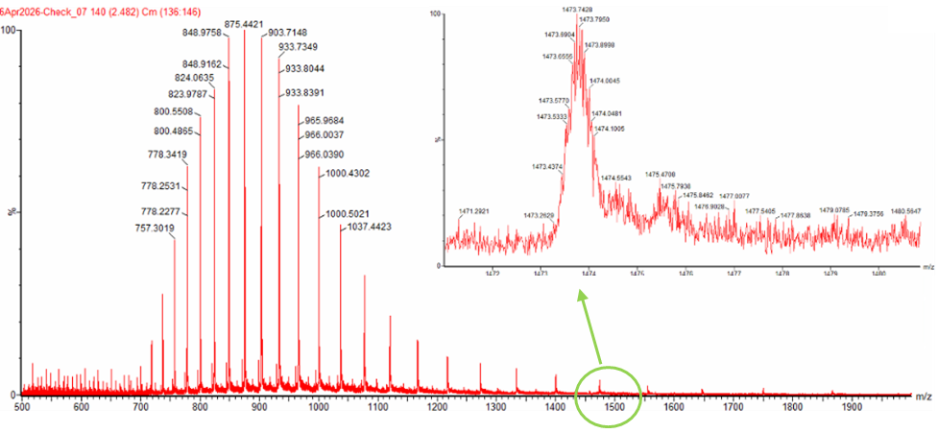
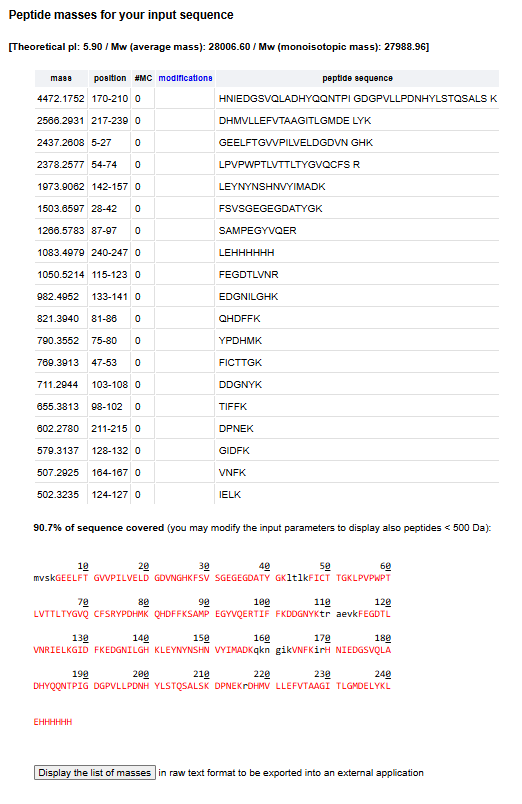
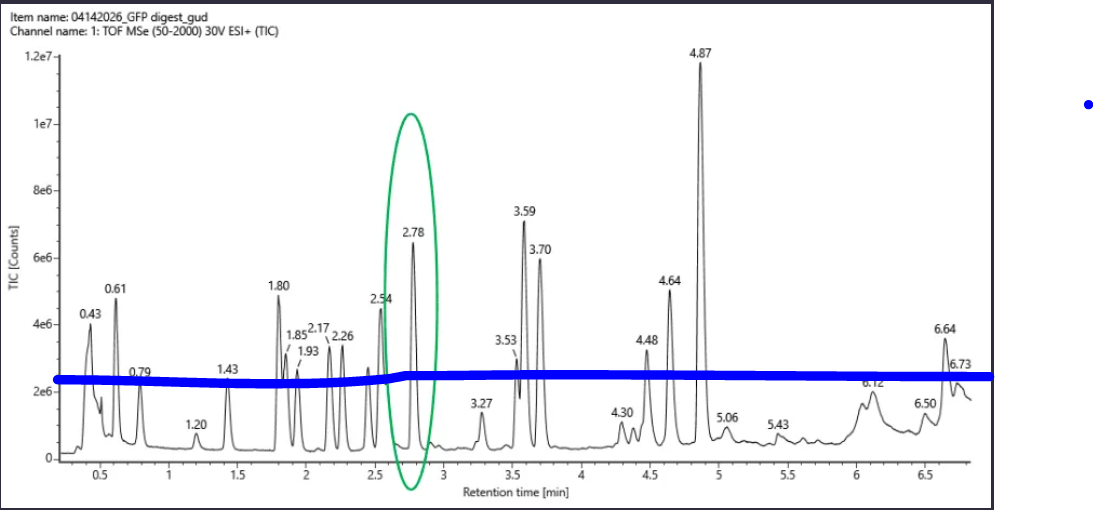
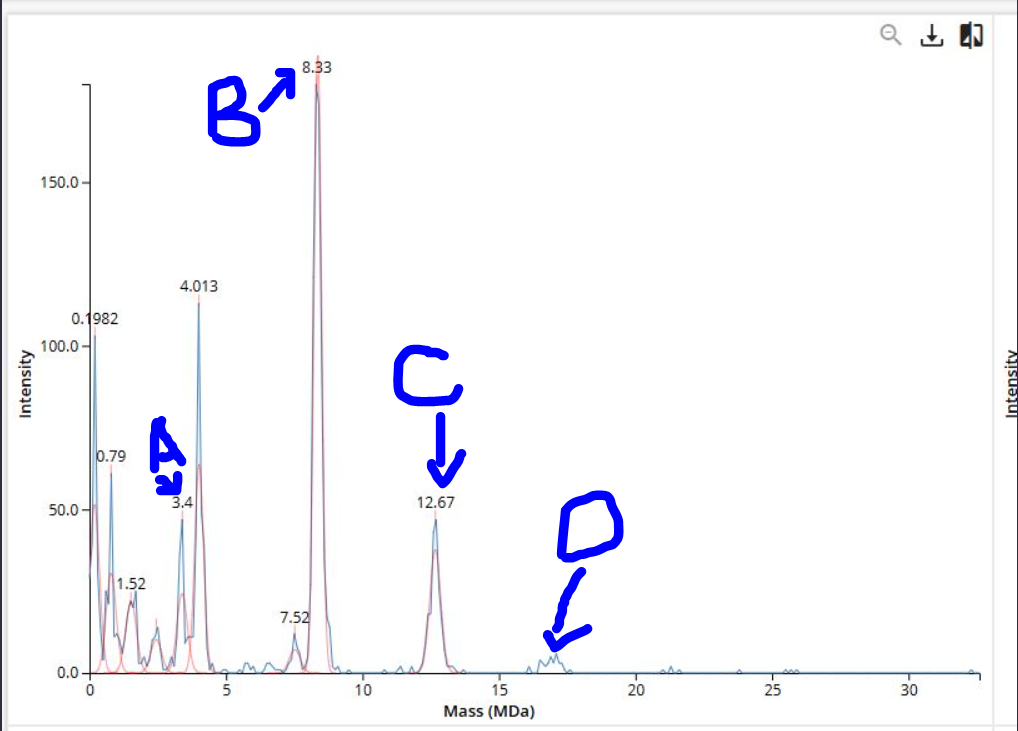
𓃠 Week 10 Homework 𓃠 Homework: Final Project α-Pinene titer: quantity of our target molecule produced per chassis per carbon source. Measured by GC-MS with dodecane overlay extraction from 96-well deep plates. AgPS protein identity and mass: purified AgPS-His6 protein (via Ni-NTA) can be characterized by intact protein LC-MS to confirm correct molecular mass and verify the His6 tag is present. Cell growth (OD₆₀₀): optical density at 600nm across all chassis × carbon source conditions. Measured on the PHERAstar FSX plate reader. Tells us if the strain is healthy while producing. Gene expression (mRNA level): RT-qPCR on the CFX Opus machine using primers targeting AgGPPS2 and AgPS transcripts. Confirms the genes are being transcribed in vivo. 𓋹 𓋹 𓋹 𓋹 𓋹 Homework: Waters Part I — Molecular Weight The Theoritical Molecular weight of the eGFP Sequence is 28006.60
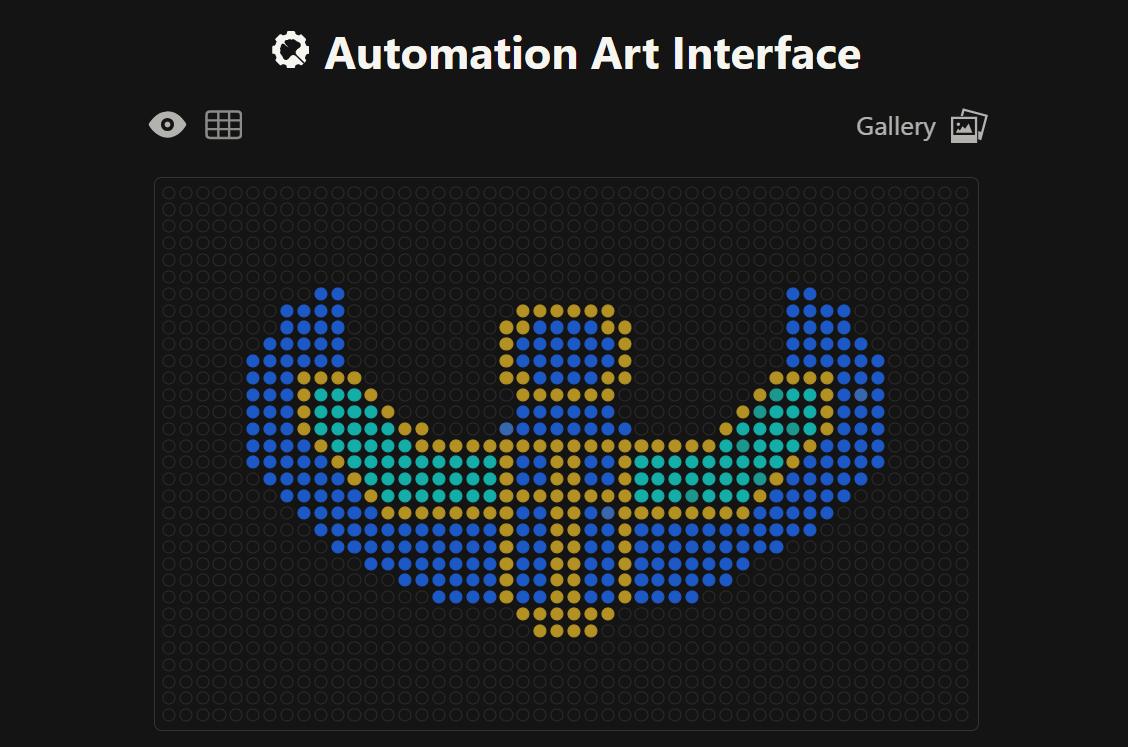
𓃠 Week 11 Homework 𓃠 Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork I just want to say, this was soooo fun! :D
and i truly love how it changed alot and all the virtual pixel fun wars i’ve had and truces and teamups, it was a really engaging and lovely experience
HTGAA Committed Listener (CL) Agreement I am a HTGAA Committed Listener, my responsibilities are:
Watching class lectures and recitations Participating in node reviews Developing and documenting my homework Actively communicating with other students and TAs on the forum Allowing HTGAA and BioClub to share my work (with attribution) Honestly reporting on my work, and appropriately attributing and citing the work of others (both human and non-human) Following locally applicable health and safety guidance Promoting a respectful environment free of harassment and discrimination Signed by committing this file to my documentation page/repository,
Subsections of Homework
Week 1 HW: Principles and Practices
𓃠 Week 1 Homework 𓃠
1. First, describe a biological engineering application or tool you want to develop and why.
I want to build a Biological 3D Printer :D
It is a quite crazy idea, basically a biological 3D printer takes in a DNA file and prints it and expresses it right away and delivers the target product in vial.
How it works: You download a DNA file (a plasmid sequence) for a specific function, like a molecule that smells like chocolate, or a protein that glows red, or a specific medication. You send the file to the printer, which synthesizes or assembles the genetic instruction, expresses it using a cell-free system or bacterial chassis, and delivers the purified product in a vial.
I want to build this because I want to democratize manufacturing. Right now, biology is locked in big labs. I want to bring ‘Bio-Production’ to the home, the remote clinic, or even a spaceship. If you can email a file, you should be able to print a cure (or a scent).
𓋹 𓋹 𓋹 𓋹 𓋹
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Main Goals include:
A. Preventing Malicious Use (Biosecurity): to make sure such printers will not be used to create harmful, toxic or dangerous products or molecules, maybe this can achieved through:
DNA Screening: We can screen the DNA sequence that is requested for printing, to check and prevent the process if it encodes for a dangerous molecule or a viral toxin
Reagent Lock-in: To make sure the reagents, proteins and raw materials in the printer are only viable inside the printer environment and cannot be extracted and used else where
B. End User Safety & Reliability (Biosafety): we need to make sure the printed molecule is exactly what it claims to be, and to make sure no mutation or contamination has occured
Proofreading Mechanism: we can apply a proofreading startegy during and after printing and synthesis to confirm similarity between the printed the DNA and the uploaded DNA file
Proper Elimination of undesired or faulty molecules: we need to be able to safely and properly discard or eliminate mutated or contaminated sequences once identified
𓋹 𓋹 𓋹 𓋹 𓋹
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
A. Action 1: International Regulation & Monitoring: This action can be pursued & applied through international health organizations like WHO and local alikes
Purpose: We can regulate what DNA sequences get printed through a digital signature that has to be approved by such organiztions, and the printer can only print these approved sequences
Design: We can have a main website similiar to the iGEM registry where users can submit new DNA sequences that they want to print and Organizations like the WHO can have a team of scientists + AI that periodically screen new DNA sequences that get submitted and either give it a signature for public use or flag it as Dangerous/Toxic and prevents its printing
Assumptions: We assume that bad people cannot manipulate the signature or bypass its checking before printing
Risks of Failure & Success:
4a. Failure: Risks include Hackers being able to bypass certain signatures and produce dangerous and harmful products through the printers or even hijacking the printers themselves to allow it to print without checking anything
4b. Success: If these regulations get applied so strictly, it could possibly lead to a lack of innovation and creativity and less DNA sequences the end users are able to print, Also local regulations would also mean some molecules might be allowed to be printed in one country and not allowed in another
B. Action 2: Companies Regulation: This Regulation is applied through the parent company that sells the printers
Purpose: We can regulate the usage printers reagents and chemicals and make sure it only uses certified or accepted reagents, more like printers cartridge system or coffee pods, mainly to make sure users can’t take the reagents and use it elsewhere
Design: The company can design the printer and cartridge so that they become dependant on each other, maybe the reagents need an activation factor that is only in the printer system or hardware so that without it, the reagents are dormant and cant be used
Assumptions: We assume the chemical “lock” is not breakable by other chemistry sets, we also assume users can afford the prices of the printer reagent cartridges
Risks of Failure & Success:
4a. Failure: Risks include bad people managaing to break the chemical formula and use the reagents in other probably malicious work
4b. Success: Monopoly, if one company produces those reagents and it can increase prices or stop selling to specifc countries or competitors for company gains
C. Action 3: Community Integration & Bio Bug Bounty Programs: This action involves both the companies and the community for a shared safe future
Purpose: Integrate the Community into the process through lectures and workshops about proper usage of the printers, how to deal with malfunctions, how to submit DNA designs and the application of Bug Bounty incentive programs to quickly and effictevly patch errors, faults or flaws in the printer’s software and hardware
Design: Incentives can be offered to people who can bypass certain security checks in the printer or print a certain toxic molecule that the print is not supposed to print, this helps turn possible hackers into quality assurance testers.
Assumptions: We assume the bounty hunters will report all findings and that the incentive given is enough to make sure they report to us.
Risks of Failure & Success:
4a. Failure: A bounty hunter discovers a major defect in the printer but recieves a bigger incentive from another competitor or bad groups and delivers the information to them instead
4b. Success: It is actually hard to think of one. maybe all these trials and errors and bug hunting will make the printer model known and allow for competitors to start companies and compete with us
𓋹 𓋹 𓋹 𓋹 𓋹
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Action 1
Action 2
Action 3
Enhance Biosecurity
• By preventing incidents
1
1
1
• By helping respond
3
n/a
2
Foster Lab Safety
• By preventing incident
1
2
2
• By helping respond
3
n/a
3
Protect the environment
• By preventing incidents
1
2
2
• By helping respond
3
n/a
2
Other considerations
• Minimizing costs and burdens to stakeholders
3
3
1
• Feasibility?
1
1
2
• Not impede research
2
2
2
• Promote constructive applications
2
1
1
𓋹 𓋹 𓋹 𓋹 𓋹
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties
Based on the scoring matrix, I recommend a Hardware-First, Software-Verified Hybrid Strategy, prioritizing Action 1 (Digital Screening) as the immediate standard, supported by Action 2 (Reagent Lock).
Hardware is the Hardest Barrier, Software can be hacked, and Bounties are reactive and takes a while to build community knowledge. Physical reagent cartridges is a very robust fail-safe for preventing accidental contamination or malicious misuse by non-experts, however reagents can be found elsewhere and maybe chemically designed to imitate the proposed solutions, so having signatured DNA sequences remain the most reliable approach.
These actions however carry tradeoffs that include lack of creative space for other users to design DNA sequences they like and have to go through a rigrous application process to get approved and signatured, and the Reagent Lock risks Company Monopoly if it is allowed to be the sole producer of these types of reagents.
𓋹 𓋹 𓋹 𓋹 𓋹
6. Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
One of the ethical concerns that arose to me is how “Open Source” in this setting can actually do more harm than good, if a lab builds a toxic molecule or a virus using the printer in a closed setting, we can basically lock up and quarantine the lab, but with open source, if the process goes online on the internet, there is no going back, and now anyone can have it. this ties back to the importance of the proposed Signature only printing action, as it can help mitigate the effects of distributed dangerous DNA sequences and trying to print it, maybe also we will need to make these printers Online only, so proper monitoring of what is being printed and by whom, of course users must be informed of this and be allowed to either accept such terms to buy the printer and use our products or refuse, and in the case of refusal, and taking biosecurity into consideration, the products should not be sold to the user nor permitted for use in this case
𓋹 𓋹 𓋹 𓋹 𓋹
𓃠 Week 2 Lecture Prep 𓃠
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate for polymerase is 1:106.
The Human Genome is ~ 3.2 Gbp, doing the math means that it makes 3200 errors each time which is alot.
Biology deals with these errors through proofreading and corrections, one example is the MutS Repair System where it fixes the mismatches and repairs them using DNA polymerase and Ligase
𓋹 𓋹 𓋹 𓋹 𓋹
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An Average human protein can be up to 1036 bp, each 3 (called a codon) codes for an amino acid, but amino acids can be coded by many combinations in those codons, making the different ways to code for the average human protein way too much and increases exponentially.
In practice, the many of these theoretical coding sequences do not work well since the nucleotide sequence determines the physical behavior of the mRNA molecule. One of the reasons is formation of mRNA secondary structures, such as hairpins and loops, which are governed by the “Minimum Free Energy” of the sequence. Other reasons include that specific nucleotide patterns can create recognition sites for cellular enzymes like RNase, leading to in vivo cleavage and the destruction of the mRNA before it can be translated
𓋹 𓋹 𓋹 𓋹 𓋹
3. What’s the most commonly used method for oligo synthesis currently?
The phosphoramidite method performed via Solid-Phase Synthesis of Oligos is the current most common method. It starts with phosphoramidite Coupling then Capping of unreacted sites followed by Oxidation and then Deblocking. These steps are then repeated for as much times needed for the synthesis
𓋹 𓋹 𓋹 𓋹 𓋹
4. Why is it difficult to make oligos longer than 200nt via direct synthesis?
(Had to look up more info about this topic using AI)
It mainly returns to difficulties regarding Yield, Truncation Products and Depurination. The longer the synthesized chain gets, the lower the yield gets, and it is not uniform but exponential. Additionally Long Chains accumalate Truncation Products which may happen because of errors or depurination from acids added during the synthesis stage.
On the bright side, Twist Bioscience has came up with an Enhanced Process & Chemistry allowing PCR yield to go up to >10 fold increase, 1:2000 error rate and acheiving a 500bp Oligos
𓋹 𓋹 𓋹 𓋹 𓋹
5. Why can’t you make a 2000bp gene via direct oligo synthesis?
This touches back to the previous question, with exponential Yield Decay, a 2000bp gene would have a very tiny yield, making most of the produced bases possibly junk, also the depurination issue arises, because the base pairs at the very beginning of the chain will need to resist and endure the acids added in the Deblocking phase up to 2000 times which is difficult.
The current process of creating the 2000bp gene would creating multiple oligos (200bp - 500bp) and stitching them together using DNA ligase.
𓋹 𓋹 𓋹 𓋹 𓋹
6. [(Advanced students)] Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or devise one of your own:
The Idea: I propose creating a “High-Performance” Red Blood Cell that acts like a backup oxygen tank for hikers, firefighters or Rescue teams in extreme conditions. We can engineer the RBCs’ Heamoglobin to be much more sensetive to markers that arise in low oxygen or hypoxic settings.
To work it should have a biosensor that can detect Lactic Acid, which is the acid that gets produced in low oxygen settings and causes the burning sensation in your muscles when you run and once the cell senses high acid levels, the engineered haemoglobin gets activated and lets go of even more oxygen that it normally would creating a higher oxygen surge instantly into the muscles or brain to properly and rapidly accomodate to the new environment.
Since these cells have no nucleus or DNA (Enuculated), they are just temporary “smart delivery bags”, They circulate for a few weeks to keep the hiker safe, then naturally die off without changing the hiker’s permanent genetics.
This also solves the “Natural Acclimation Time” problem mentioned in the abstract. Instead of waiting weeks for the body to get used to high altitude, or low oxygen environments, a person can receive a transfusion of these “Oxygen Turbo Cells” and be ready immediately to tackle these extreme environments and this applies to what the abstract mentioned as “enhance physiological resilience”.
AI citation: i used Gemini to help with idea validation and properly tying it back to match the requirements of the Grant Program
𓋹 𓋹 𓋹 𓋹 𓋹
Week 2 HW: DNA Read, Write, & Edit
𓃠 Week 2 Homework 𓃠
Part 1: Benchling & In-silico Gel Art
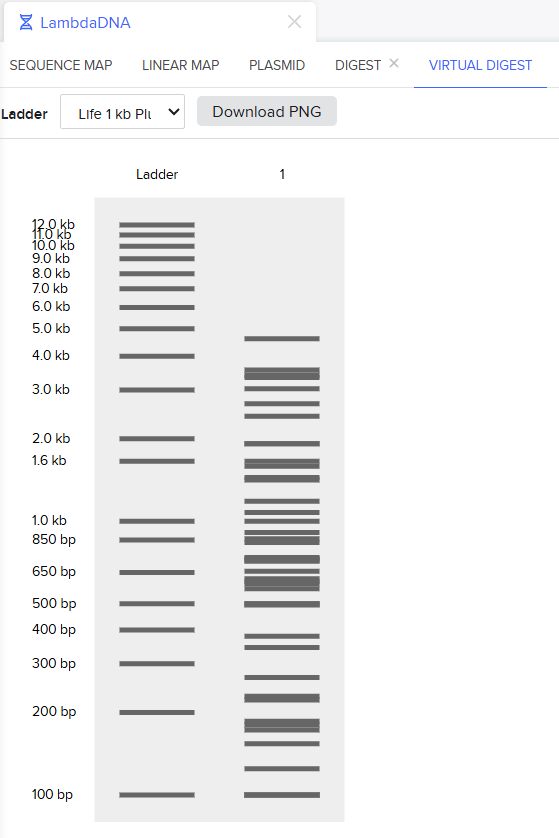
Simulate Restriction Enzyme Digestion with the following Enzymes:
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
I tried to do a smiley face and it turned out so bad but i love it XD!
Many thanks to Ronan for his Website, it really helped alot make this much faster!
𓋹 𓋹 𓋹 𓋹 𓋹
Part 3: DNA Design Challenge
3.1. Choose your protein.
Hemocyanin.
It is a giant, copper-based protein that functions as the respiratory pigment for many mollusks and arthropods, but the coolest thing is that when their blood is oxygenated it turns into Blue :D.
Unlike our iron-based hemoglobin which is packed inside cells, hemocyanin floats freely in the hemolymph of animals like the Atlantic Horseshoe Crab and the Keyhole Limpet. It is a medical powerhouse for humans; its massive, alien structure provokes a strong immune response, making it an effective immunotherapy treatment for bladder cancer and a crucial carrier protein for vaccines (helping the body recognize small drug molecules) and more (1).
Here is the Hemocyanin Protein Sequence
>sp|P04253|HCY2_LIMPO Hemocyanin II OS=Limulus polyphemus OX=6850 PE=1 SV=2 TLHDKQIRVCHLFEQLSSATVIGDGDKHKHSDRLKNVGKLQPGAIFSCFHPDHLEEARHLYEVFWEAGDFNDFIEIAKEARTFVNEGLFAFAAEVAVLHRDDCKGLYVPPVQEIFPDKFIPSAAINEAFKKAHVRPEFDESPILVDVQDTGNILDPEYRLAYYREDVGINAHHWHWHLVYPSTWNPKYFGKKKDRKGELFYYMHQQMCARYDCERLSNGMHRMLPFNNFDEPLAGYAPHLTHVASGKYYSPRPDGLKLRDLGDIEISEMVRMRERILDSIHLGYVISEDGSHKTLDELHGTDILGALVESSYESVNHEYYGNLHNWGHVTMARIHDPDGRFHEEPGVMSDTSTSLRDPIFYNWHRFIDNIFHEYKNTLKPYDHDVLNFPDIQVQDVTLHARVDNVVHFTMREQELELKHGINPGNARSIKARYYHLDHEPFSYAVNVQNNSASDKHATVRIFLAPKYDELGNEIKADELRRTAIELDKFKTDLHPGKNTVVRHSLDSSVTLSHQPTFEDLLHGVGLNEHKSEYCSCGWPSHLLVPKGNIKGMEYHLFVMLTDWDKDKVDGSESVACVDAVSYCGARDHKYPDKKPMGFPFDRPIHTEHISDFLTNNMFIKDIKIKFHE
𓋹 𓋹 𓋹 𓋹 𓋹
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
I decided to use Benchling’s Codon Optimization for E. Coli K-12. Codon Optimization is important because it uses the amino acids more native to the chosen organism which boosts speed and efficiency of translation and also if it is not done the organism might not have enough complementary tRNA anti codon molecules to synthesize this specific amino acid.
When this protein is introduced to the E. coli K-12, it can start transcribing this DNA into an mRNA then this mRNA can be translated into a protein so that the bacteria can use it, and since we have codon optimized it for the e coli, we should expect a smooth translation process without any stalling.
𓋹 𓋹 𓋹 𓋹 𓋹
3.5. [Optional] How does it work in nature/biological systems?
1) Describe how a single gene codes for multiple proteins at the transcriptional level.
Alternative Splicing. A single Gene in Eukaryotes can code for multiple proteins through a process called Alternative Splicing, It takes on many forms, like Exon Skipping, where a certain exon might be skipped possibly altering the protein function, or Alternative 5’ or 3’ Splicing, where some exons can be longer or shorter hence affecting the number of produced amino acids altering the function too, another way is intron retention where some introns are not spliced out which can introduce a stop codon and can cause the protein to decay using the Nonsense mediated decay pathway.
𓋹 𓋹 𓋹 𓋹 𓋹
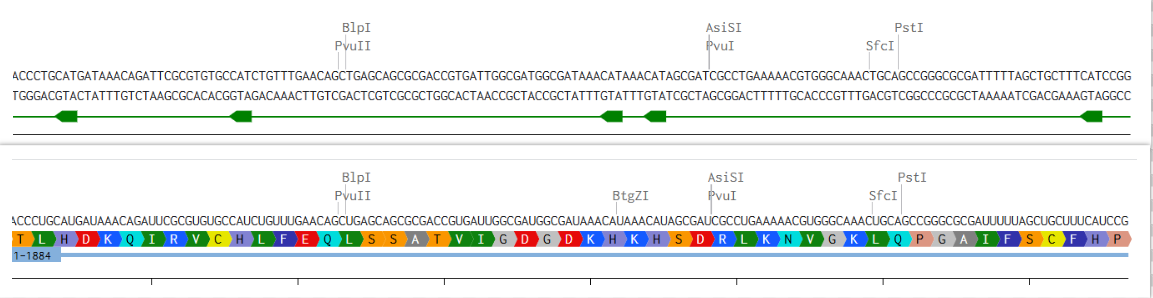
2) Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
Here is a Picture of the very first few bases of Hemocyanin II DNA, RNA & Amino Acid
𓋹 𓋹 𓋹 𓋹 𓋹
Part 4: Prepare a Twist DNA Synthesis Order
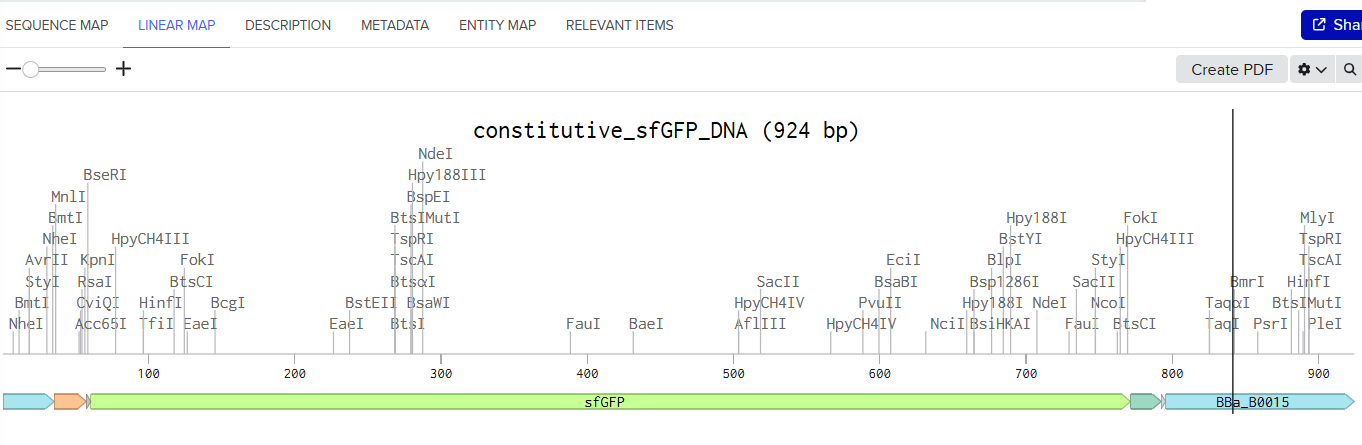
Here is a screenshot of my Linear map of Constitutive sfGFP DNA and here is the Benchling Link
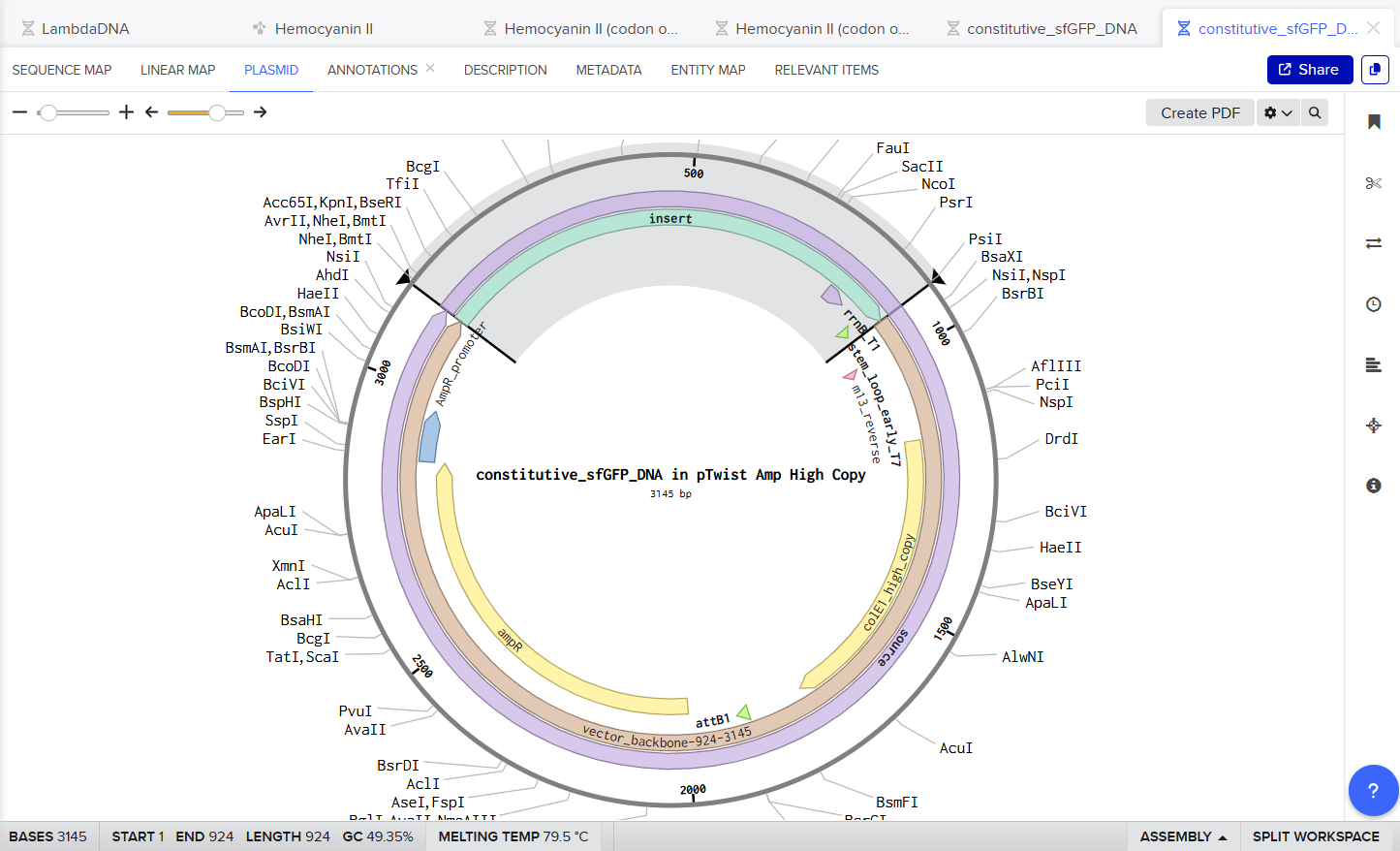
Here is a screenshot of my Twist Ready Plasmid :D
𓋹 𓋹 𓋹 𓋹 𓋹
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I’d like to sequence and read the genome of the Axolotl (Ambystoma mexicanum) to learn more about the process of its limb regeneration and how it happens, and figure out if other species have these genes too whether Human, Marine or Plants.
𓋹 𓋹 𓋹 𓋹 𓋹
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use PacBio SMRT sequencing. This is a third-generation technology, which is perfect because the Axolotl genome is very huge (32 billion bases!) and full of repetitive parts that confuse older, short-read machines. Third-generation sequencing reads single molecules of DNA in real-time, giving us the long reads necessary to bridge those gaps and locate the regeneration genes i care about.
For the process, I’d follow the SMRTbells process where I’d start with really long strands of high-quality DNA and attach hairpin adapters to both ends, turning them into circular loops. These loops go into tiny wells where a polymerase enzyme runs around the circle, adding bases. As each base (A, C, T, G) is added, it gives off a specific colored flash of light. The machine records these flashes to decode the sequence. The final output is HiFi reads: extremely long, highly accurate digital sequences that we can assemble into a full map.
𓋹 𓋹 𓋹 𓋹 𓋹
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I want to synthesize the Dsup (Damage Suppressor) gene found in Tardigrades, Tardigrades are famous for surviving the vacuum of space and massive radiation because this specific protein wraps around their DNA like a physical shield to prevent damage. By synthesizing this gene and inserting it into human cells or gut bacteria or plants, I want to see if we can “borrow” this superpower to protect astronauts from lethal cosmic radiation on the long space journies, effectively genetically engineering a radiation protection.
𓋹 𓋹 𓋹 𓋹 𓋹
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
For the DNA synthesis technology, maybe i will rely either on the current common Phosphoramidite DNA synthesis or use an Enzymatic Synthesis using Terminal deoxynucleotidyl transferase (TdT), or maybe an even more easier and more quick method is to just use Twist Bioscience and order the gene right away :D
Basically the steps for phosphoramidite synthesis process would start first with deblocking of the nucleotide then it couples using phosphoramidite, after that we do Capping for the unreacted sites to prevent any faulty chains to continue growing, and then oxidation to seal and empower the bond of our newly added nucleotide, then the process is repeated until we get a chain of N bases, then these Oligos are stitched and assembled together using methods like Gibson Assembly, Golden Gate Assembly or the recently announced Sidewinder(2) way (which is pretty cool :D)
Phosphoramidite Synthesis currently faces the major issue of Exponentially Decaying Yield when the synthesized chain gets longer however Twist Bioscience seems to be doing a really great job regarding this especially for their achievement of direct synthesis of the first 700mer Oligo using “Enhanced Chemistry”. For DNA Assembly currenty Sidewinder(2) seems like a very promising tool that can achieve high accuracy and avoids many of the errors that can happen from long, repititve or high GC content Oligos that methods like Gibson Assembly or Golden Gate Assembly used to suffer from.
𓋹 𓋹 𓋹 𓋹 𓋹
5.3 DNA Edit
(i) What DNA would you want to edit and why?
Quite a crazy idea but i would like to edit the DNA of the E. Coli K-12 and install regeneration genes from the Axolotl to allow the e coli to be able to regrow and detach vesicles containing molecules that it can produce or has been metabolically engineered to produce. this can allow it to match some Yeast ability for storing produced molecules especially hydrophobic molecules. These vesicles can be engineered to float to the top of flask or fermentation tanks which would allow easy extraction and purification.
𓋹 𓋹 𓋹 𓋹 𓋹
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-Cas9 to edit the genome. The process starts with designing a Guide RNA (gRNA) that acts as a GPS coordinate for the specific location in the E. coli genome where I want to make the edit, then introduce a plasmid into the cells containing the Cas9 enzyme, the gRNA, and a Donor DNA template (which carries the Axolotl genes). The Cas9 enzyme cuts the bacterial DNA at the target site, and the cell repairs this cut using the Donor template, successfully “pasting” the new regeneration genes, this process is called Homology-Directed Repair.
However, this method has limitations, mainly of efficiency and payload size. Inserting large, complex gene pathways is much harder than making small mutations, the larger the DNA insert, the less likely the bacteria are to accept it. There is also the risk of off-target effects, where the enzyme cuts the wrong part of the genome, potentially killing the cell, Also sometimes the Homology-Directed Repair may not be efficient and may introduce mutations that can render the insert non functional
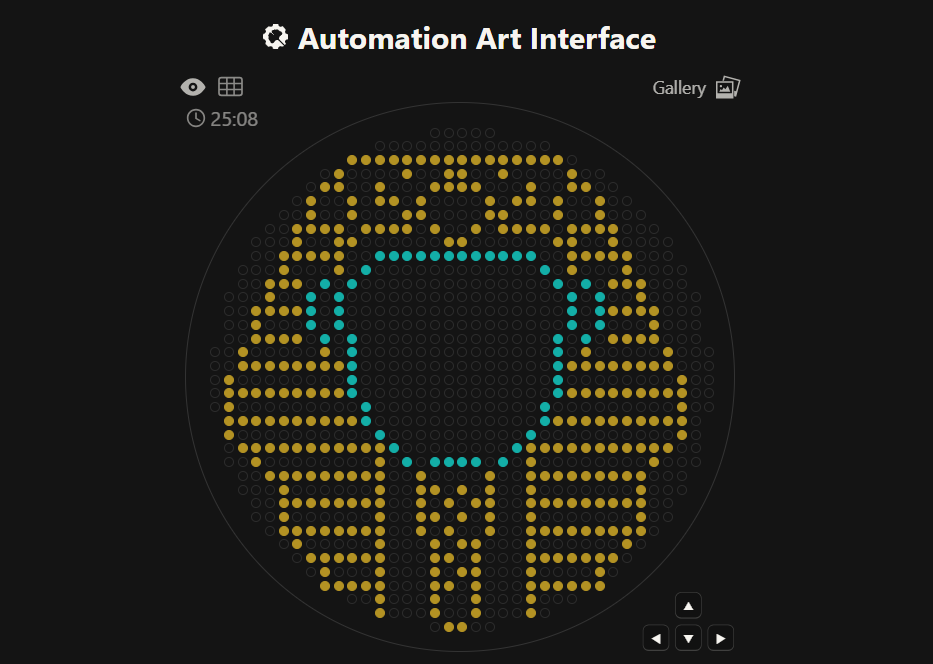
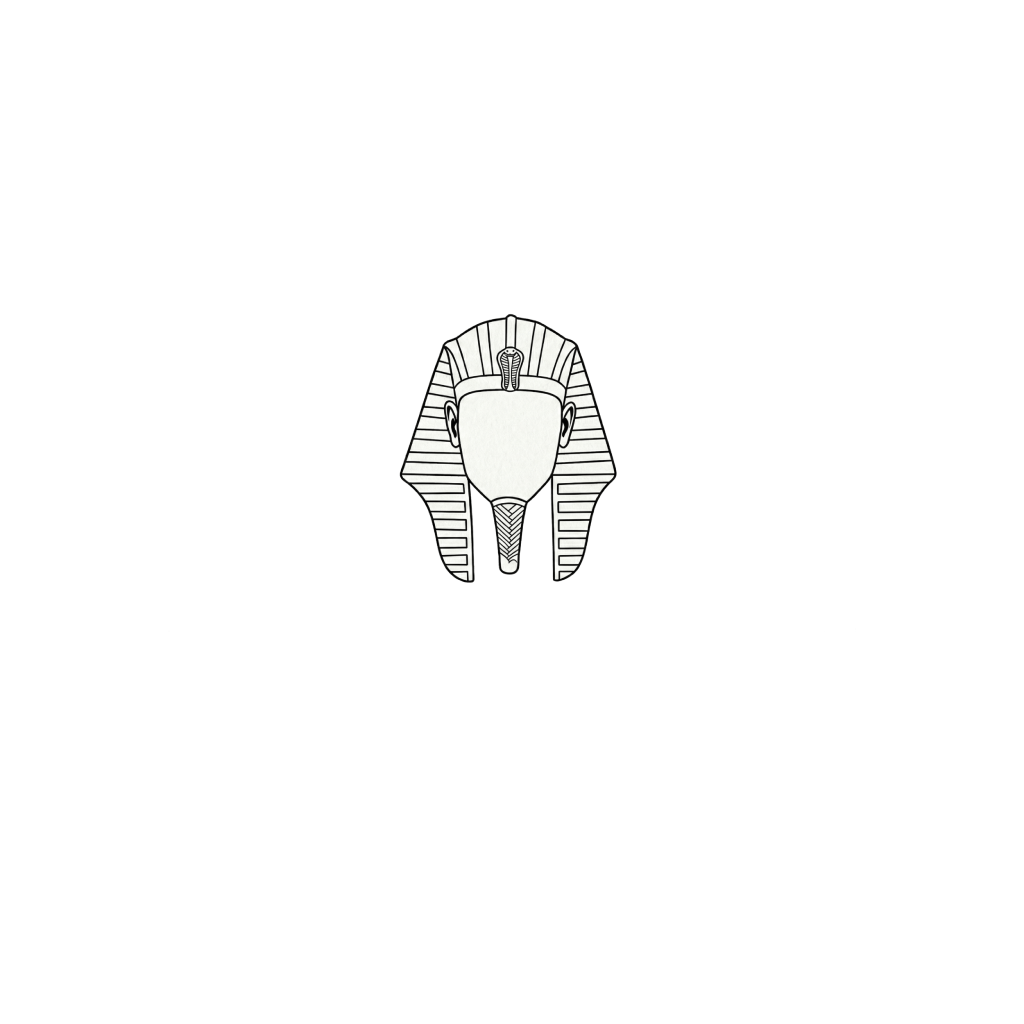
This is an Ancient Egyptian Pharaoh Figure, this was made using this GUI
Also here is the live link to check it out! :D
𓋹 𓋹 𓋹 𓋹 𓋹
Note
This section is going to be just documentation about how i managed to draw the Ancient Egyptian Pharaoh Figure, to view the rest of the homework, You can skip to Post-Lab Questions
So i started out with this image which i generated using Gemini a long while ago.
I wanted to draw it using the Opentrons OT-2 but i had no idea how + i am not a very good pixel artist.
So i decided to use AI to help me with this, first i went to Ronan’s GUI (Thanks Again Ronan! :D) and i roughly counted there was (on default settings) 36x36 pixels (i was off by a little number btw XD)
So i went back to Gemini and prompted it to draw my the image on a 36x36 pixel grid and this was the output
It was pretty good to me, but i need coordinates for the Opentrons API, i tried to roughly copy it on Ronan’s GUI but i failed misreably and wasn’t accurate
So when i was searching online for tools to like give some sort of coordinates to this (and i didn’t find XD), i stumbled on this website called pixilart, I went in and generated a 36x36 pixel grid and guess what? I drew it from scratch using Gemini’s image as the reference and i added some tweaks myself to it too :D
It took alot of effort trying to focus on those pixels to get a picture perfect copy of it, but in the end the mission was a success :D
Now that i could somehow get x/y coordinates, i wanted to redo this on Ronan’s GUI so that the coordinates i give to Opentrons are accurate, so now i had to take my new reference and map it there.
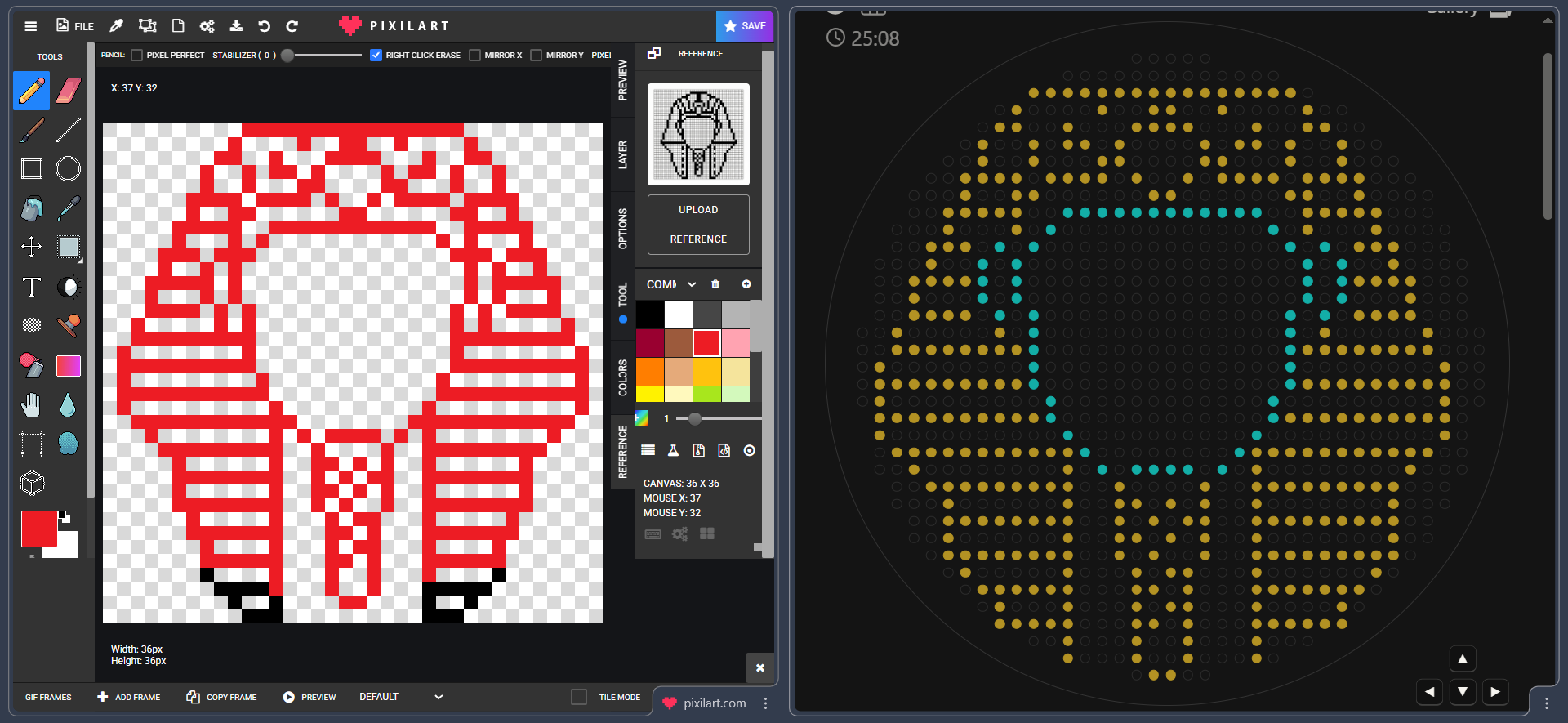
I tried to upload the image but it didn’t map it well at all, maybe because of the pixel grids, so i had to do it manually XD
Here is a screenshot of me mapping every single pixel on Ronan’s GUI and marking it in red on pixilart so i don’t mix something up :D
It took sometime but the results were totally worth it :D
This way i took the generated coordinates and went the Opentrons Google Colab Notebook and used Gemini to write the actual function because i was too tired after all the drawing XD, and this was the result! :D
Here is some other designs that i did too while in the Autonomous Cloud Lab Stream where we were printing Fluorescent Artwork designs
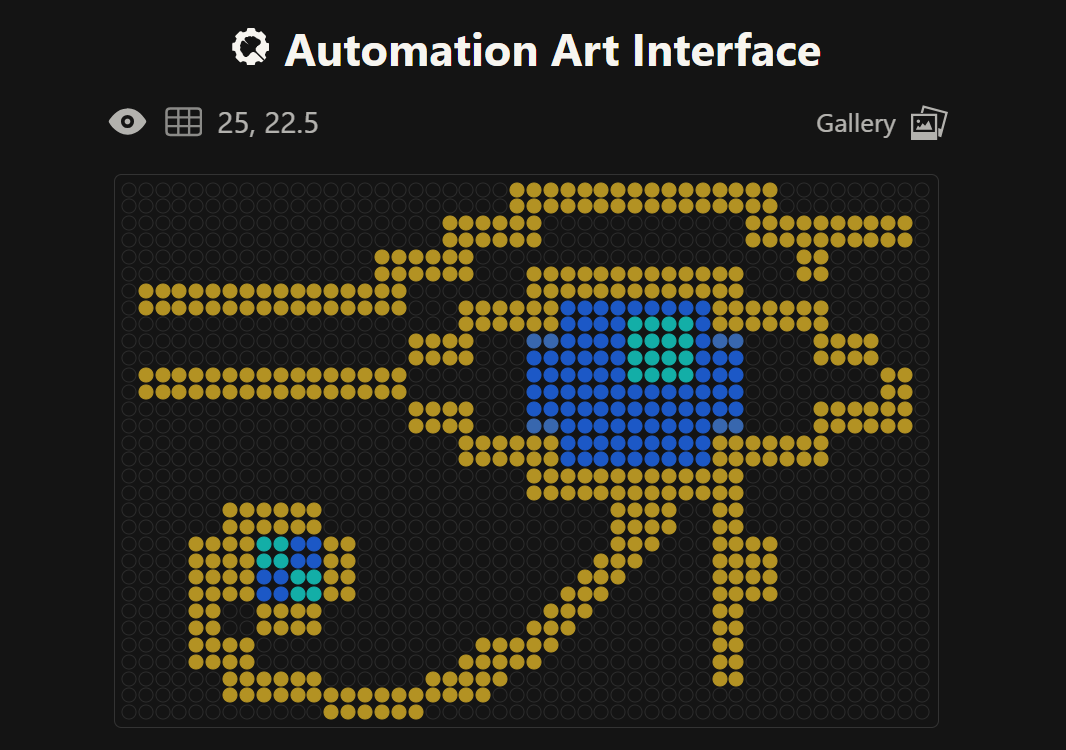
This is the Eye of Ra, a very common and famous Ancient Egyptian Symbol
This is a Winged Scarab, which is also a very well known Ancient Egyptian Symbol
𓋹 𓋹 𓋹 𓋹 𓋹
Post-Lab Questions — DUE BY START OF FEB 24 LECTURE
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Paper: Development of a Modular Lab Automation System with Applications to Animal and Bacteria Cell Culture(1)
This paper is a perfect example of why automation is critical for modern synthetic biology. They identify the key challenges of manual lab work which includes that complex protocols are tedious, prone to human error, and suffers from a lack of reproducibility. and To solve this, so they developed a modular lab automation system. They validated their system through automating cell culture, for both simple bacteria (prokaryotic) and complex animal cells (eukaryotic).
The most important part of their project isn’t just the robot, it’s their focus on transparency, quality control, and community reuse. They created a system that automatically generates Jupyter Notebooks as a “full protocol execution report,” which makes the experiments perfectly documented and reusable. They also described how this modular system is the first step toward “self-driving labs,” where AI and machine learning models can design and run their own experiments, possibly creating a fully automated DBTL cycle.
𓋹 𓋹 𓋹 𓋹 𓋹
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more.
Automation Tools can help me quickly and efficiently do many tasks and test different scenarios with minimal errors, for example in Idea 1 i can use Automation Tools to test out different circuits with different promotors to test them and check out which on is the most optimal and which ones burden the cell too much, same with Idea 2 where i can test different Inducer Ratios and measure production titers in a quick way
𓋹 𓋹 𓋹 𓋹 𓋹
Final Project Ideas
The presentation slides can be found at the Slides Deck
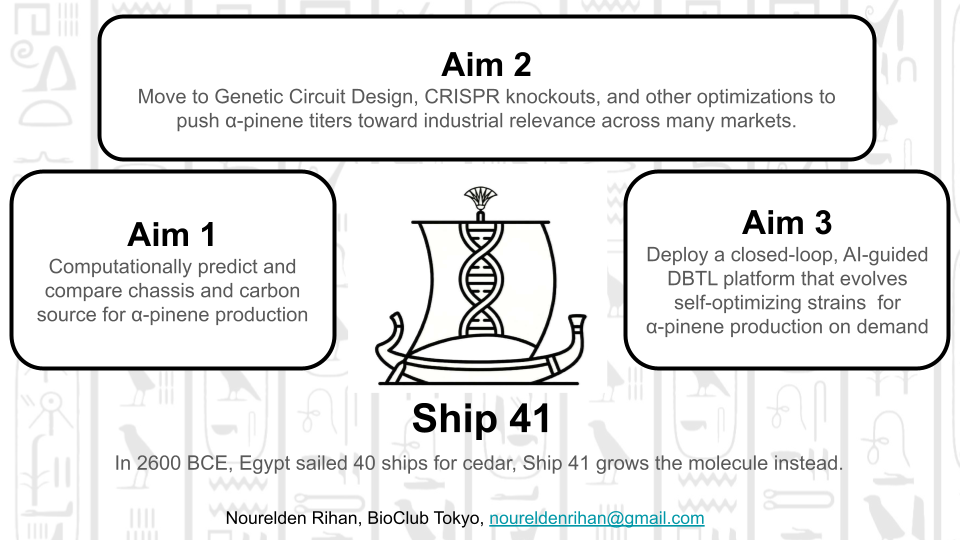
Idea 1: The Malaria Machine: Computational Optimization of Artemisinic Acid Biosynthesis
The Problem: Artemisinin, the frontline treatment for malaria which still kills ~600,000 people annually, relies on plant extraction from Artemisia annua — an inherently slow, geographically limited, and climate-vulnerable supply chain that cannot meet global demand consistently.
The Mechanism: This project computationally engineers a microbial factory (E. coli or yeast) to overproduce artemisinic acid, the direct biosynthetic precursor to artemisinin, by modeling the mevalonate pathway using flux balance analysis, systematically evaluating knockout and overexpression strategies, and designing optimized genetic circuits for the winning candidate.
The Impact: By delivering a fully documented, wet-lab-ready computational blueprint for high-yield artemisinic acid biosynthesis, this project contributes toward a scalable, plant-independent, and globally accessible supply of the world’s most critical antimalarial compound.
𓋹 𓋹 𓋹 𓋹 𓋹
Idea 2: The Bio-Propulsion Model: Engineering Off-World Propellant
The Problem: High-density aerospace propellants (like JP-10) are entirely petroleum-derived, making them unsustainable on Earth and physically impossible to drill for during deep space missions.
The Mechanism: This project computationally optimizes a microbial factory, utilizing Metabolic pathway, to metabolically convert raw carbon into alpha-pinene.
The Impact: Because alpha-pinene can be chemically dimerized into a direct, high-energy biological equivalent of JP-10 rocket fuel, this creates a scalable, closed-loop propulsion supply for advanced aerospace applications.
𓋹 𓋹 𓋹 𓋹 𓋹
Idea 3: The Coral Distress Beacon: Early-Warning Marine Biosensors
The Problem: Corals release specific chemical stress markers, such as Reactive Oxygen Species (ROS), immediately before undergoing heat-induced bleaching.
The Mechanism: This project proposes engineering a marine microbe biosensor with a targeted genetic logic circuit designed to detect these exact ROS molecules and output a highly visible fluorescent signal.
The Impact: By deploying this living “distress beacon,” marine biologists gain a realtime, colorful early warning system, allowing them to intervene and protect the reef before the ecological damage becomes irreversible.
𓋹 𓋹 𓋹 𓋹 𓋹
Extra Ideas
Idea 4: BioDesalination: Engineering a Living Portable Freshwater System
The Problem: Existing desalination technologies are energy-hungry, expensive and dependent on infrastructure which sometimes makes it inaccessible to coastal communities, small vessels and off-grid environments that need them the most
The Mechanism: This project aims to engineer a microorganism with optimized ion transport and osmoregulation pathways to actively convert saltwater into freshwater through biological membrane processes. It begins computationally, modeling genetic modifications and pathway optimization, with a clear roadmap toward experimental validation.
The Impact: A living, self-sustaining, portable biodesalination system that requires no external energy infrastructure, deployable anywhere there is saltwater and sunlight
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Proteins make about 20%-30% of the composition of meat, so we can take 25% as the average, this means 500 x 0.25 = 125g protein, to determine molar mass, 1 Dalton is approximately equal to 1 gram per mole (g/mol), so an average amino acid weighs 100 g/mol which when divided by the molar mass of 100 daltons gives 1.25 moles of amino acids, then we can multiply this by Avogadro's number (6.022x1023), we get 7.5275 x 1023 molecules of amino acids in the 500 grams of meat (this is extremely huge wow XD)
𓋹 𓋹 𓋹 𓋹 𓋹
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because when we eat them, we do not inherit their DNA or Genomic composition, we just digest them and use the amino acids and other nutrients from their catabolism to build our own self using our own Genome instructions
𓋹 𓋹 𓋹 𓋹 𓋹
3. Why are there only 20 natural amino acids?
The currently settled on 20 Amino acids provide a sweet spot between the most chemical diversity with the fewest components to a build functioning complex system, however we dont seem to be only stuck with 20, as there are 2 other natural amino acids Selenocysteine is one of them, it is used by humans and bacteria for precise antioxidant enzymes, the other is Pyrrolysine which is found strictly in ancient methane-producing archaea.
𓋹 𓋹 𓋹 𓋹 𓋹
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, you definetly can, actually non natural amino acids have been created before, usually by chemically modifying the “R-group” side chain to create proteins with other powers like heat resistance, fluorescence, or adhesive properties. Maybe we can design new amino acids that can glow different colors depending on how powerful radiation is in a certain place.
𓋹 𓋹 𓋹 𓋹 𓋹
5. Where did amino acids come from before enzymes that make them, and before life started?
(Had to use AI for this one) Amino acids were already being mass produced by high energy physics and geology, forming spontaneously whenever simple carbon compounds interact with energy, some common sources were Atmospheric Sparks (Lightning), Cosmic Delivery (Meteorites), and Deep-Sea Vents (Geothermal Heat).
𓋹 𓋹 𓋹 𓋹 𓋹
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
it will form a Left-Handed helix, since D-amino Acids are mirrors of the L-amino acids that make Right handed helices
𓋹 𓋹 𓋹 𓋹 𓋹
8. Why are most molecular helices right-handed?
Because more than 99% of the amino acids in the body are in the L-form (Left-handed), as they are the exclusive building blocks for proteins and enzymes, so most of the produced helices will be right handed.
𓋹 𓋹 𓋹 𓋹 𓋹
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Unlike alpha helices, which are self contained cylinders, a single beta strand is structurally incomplete. Its edges have exposed hydrogen bond donors and acceptors that act like open Velcro hooks. If these edges are not protected by the protein’s own fold, they will inevitably recruit strands from neighboring proteins to satisfy these bonds, triggering a stacking event.
The primary driving force for beta sheet aggregation is the thermodynamic stability gained from intermolecular hydrogen bonding combined with the hydrophobic effect.
𓋹 𓋹 𓋹 𓋹 𓋹
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Proteins normally fold into delicate 3D shapes to do their jobs, but keeping those shapes takes constant effort. If a protein gets damaged, it can collapse into a “β-sheet”, a misfolded shape that acts like a microscopic zipper. These sheets lock together so tightly and squeeze out so much water that the body’s natural recycling system literally cannot break them apart. That extreme, indestructible stability is exactly why they clump up in the brain and cause diseases.
However, that same indestructible nature makes them an incredible raw material. Because these microscopic β-sheets are stronger than steel and completely waterproof, scientists are now programming cells to grow harmless, synthetic versions of them in the lab. We can harvest these lab grown amyloids to build strong biofibers.
𓋹 𓋹 𓋹 𓋹 𓋹
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
crtI (phytoene desaturase), it is a bacterial enzyme that handles the multi-step conversion of colorless phytoene into the bright red pigment lycopene. i mainly chose it because i was working on a metabolic engineering project recently on lycopene production so i thought it would be cool to pick crtI and explore it more
The length of the protein is 492 aminoacids, with Leucine (L) being the most common amino acid which appears 57 times.
Uniprot ID P21685 and it has 242 homologs.
𓋹 𓋹 𓋹 𓋹 𓋹
Identify the structure page of your protein in RCSB
The structure for Phytoene Dehydrogenase was deposited on Jan 26th 2012, it has a good resolution of 2.4 Å
Apart from the Main Protein Polymer, it has other components including Ligands, Water and Ions.
𓋹 𓋹 𓋹 𓋹 𓋹
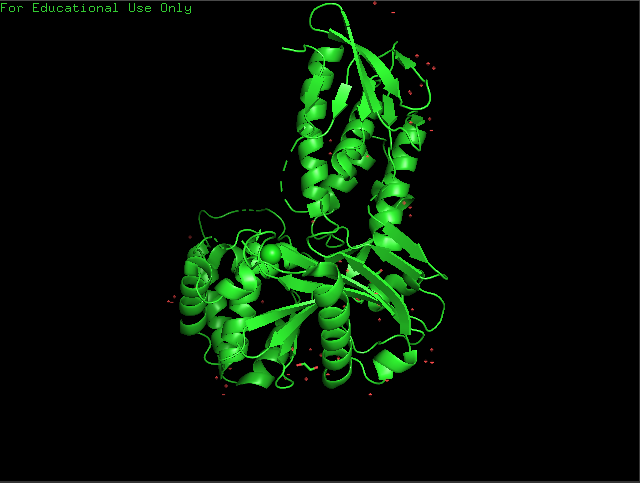
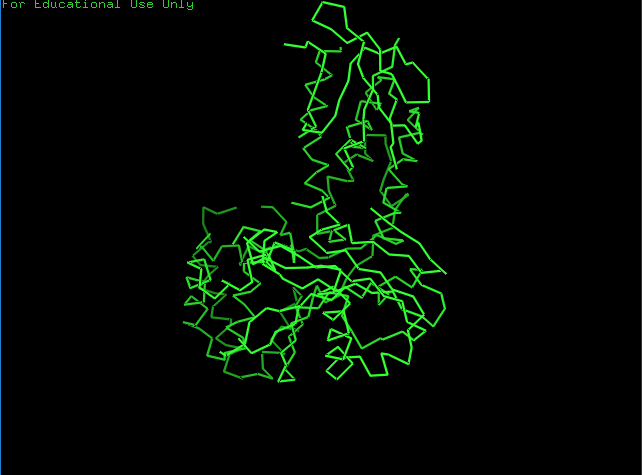
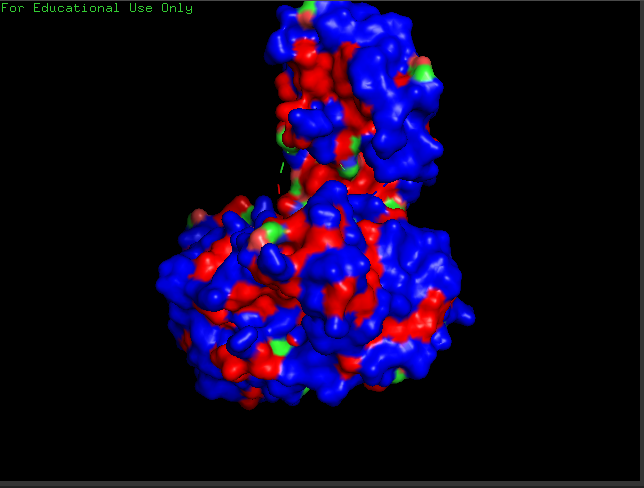
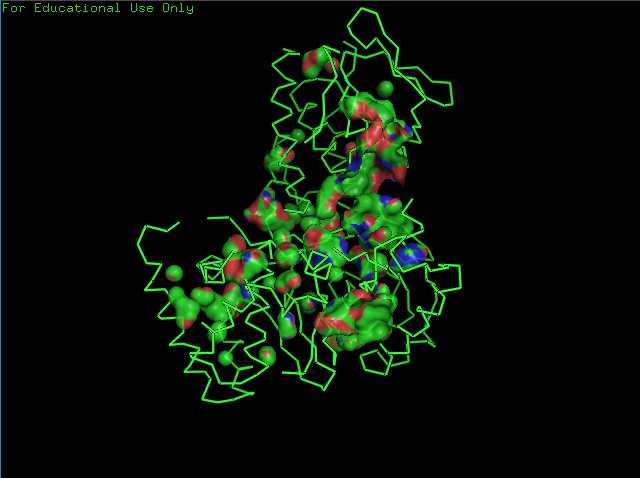
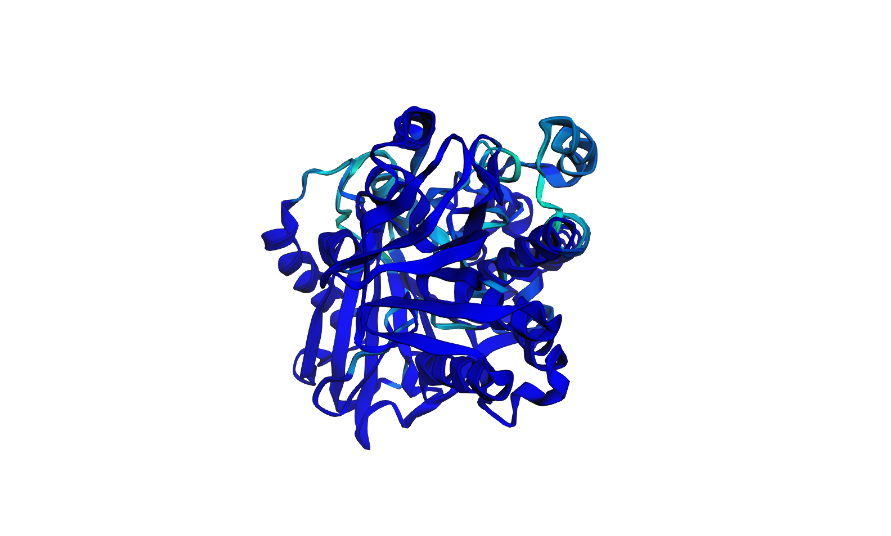
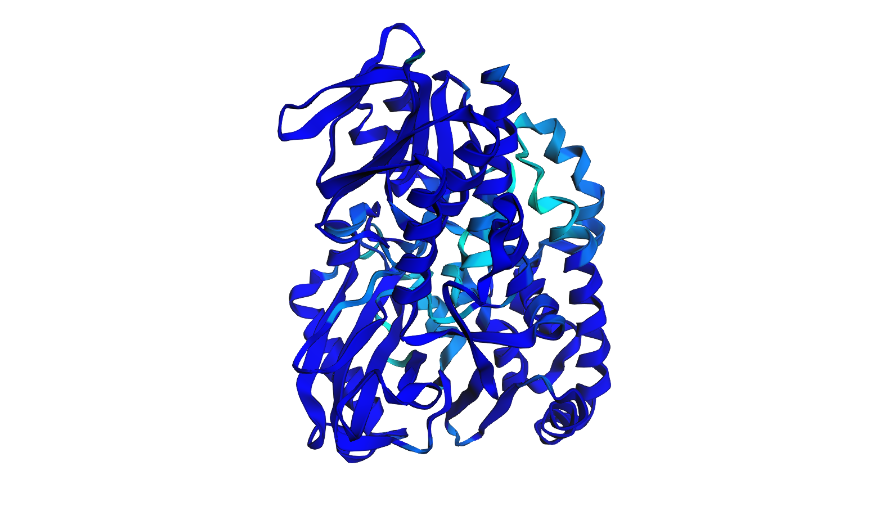
Open the structure of your protein in any 3D molecule visualization software.
Cartoon View
Ribbon View
Ball & Sticks View
crtI Residues, Blue are hydrophilic and red are hydrophobic, it seems to have more hydrophilic residues than hydrophobic ones
crtI Cavities
𓋹 𓋹 𓋹 𓋹 𓋹
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
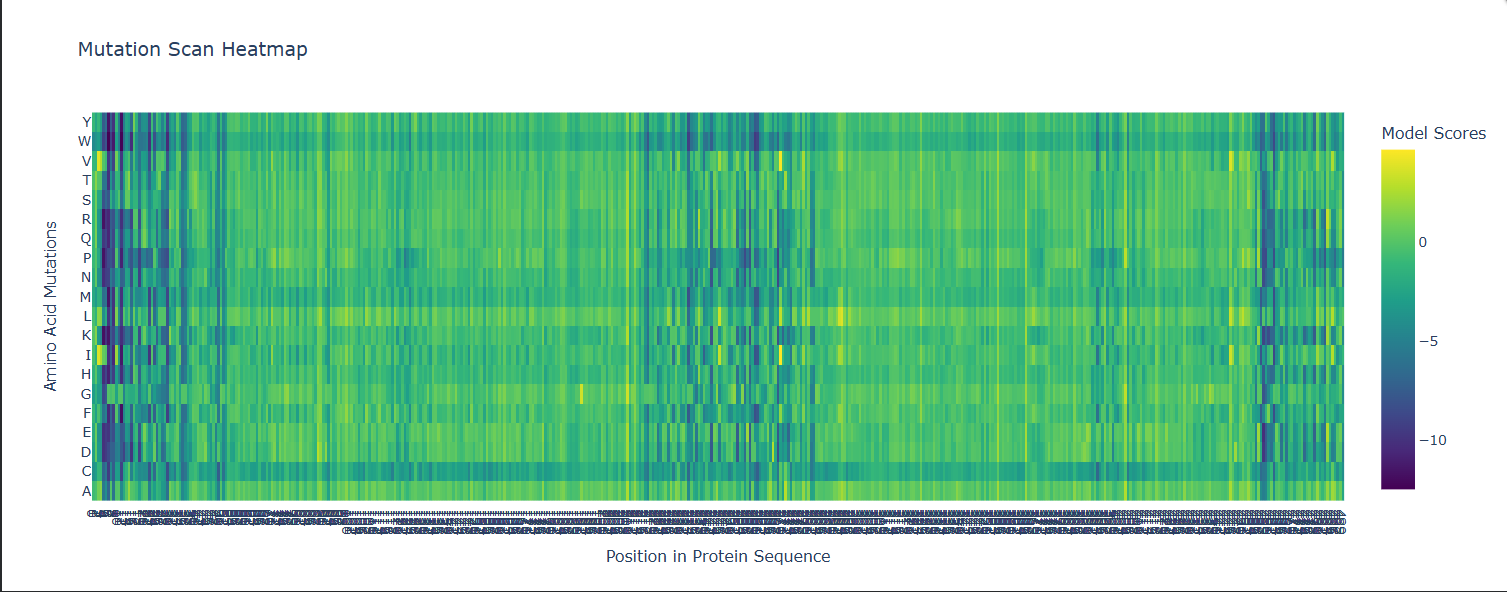
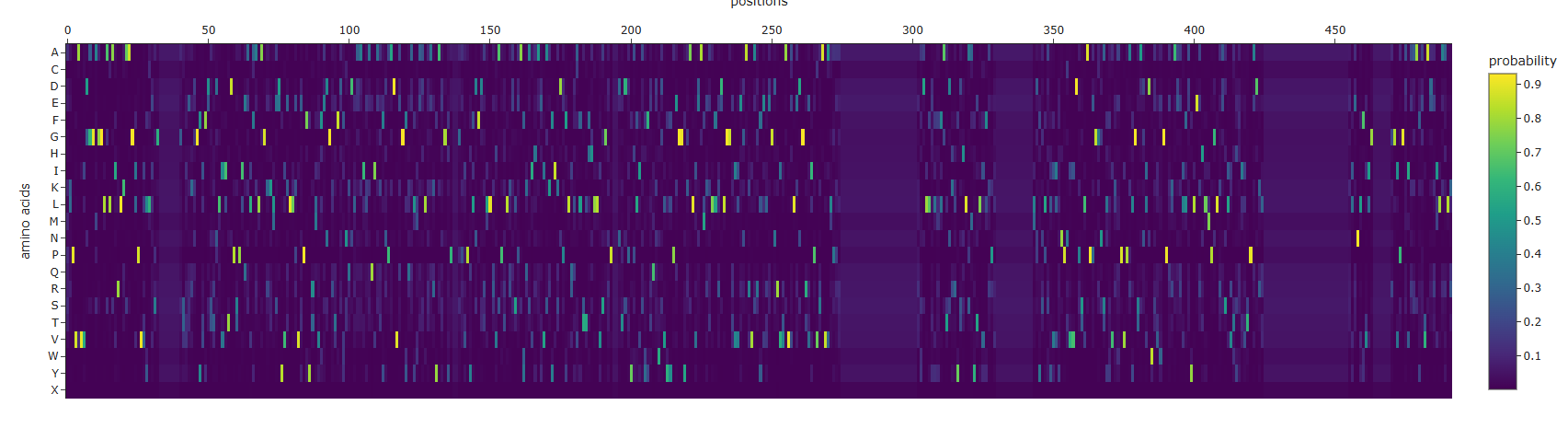
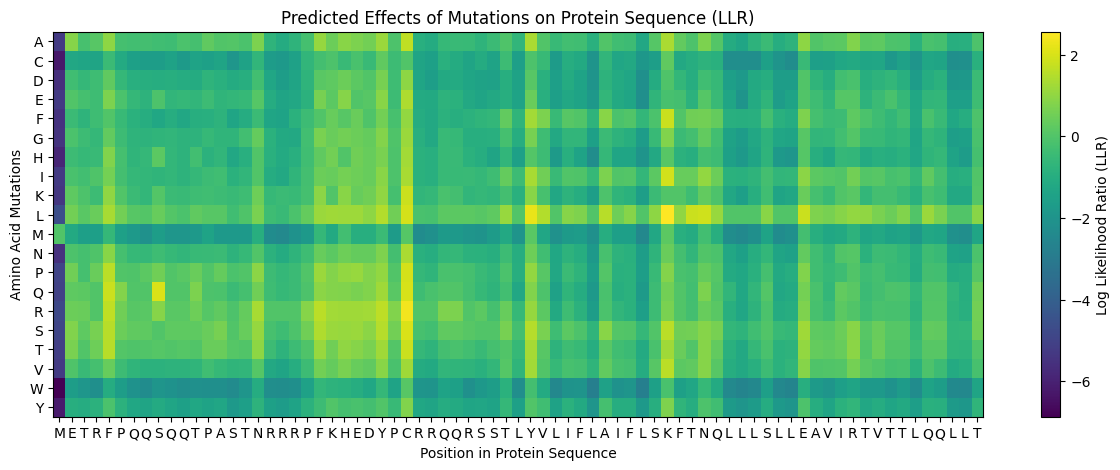
Some Patterns that I identified include that some amino acids in the crtI protein, especially those between positions 4-8 seem to have the lowest scores for mutations possibly highlighting how important or cruical these are for the protein’s stability and function.
On the other hand, some amino acids show high and positive scores when mutated to other amino acids, potentially showing better stability or favorable changes, such as positions 354 & 365 seem to be having a high score for each differtiated amino acid.
Position 269 seems to have the highest score when mutated to Valine (V) or Isoleucine (I).
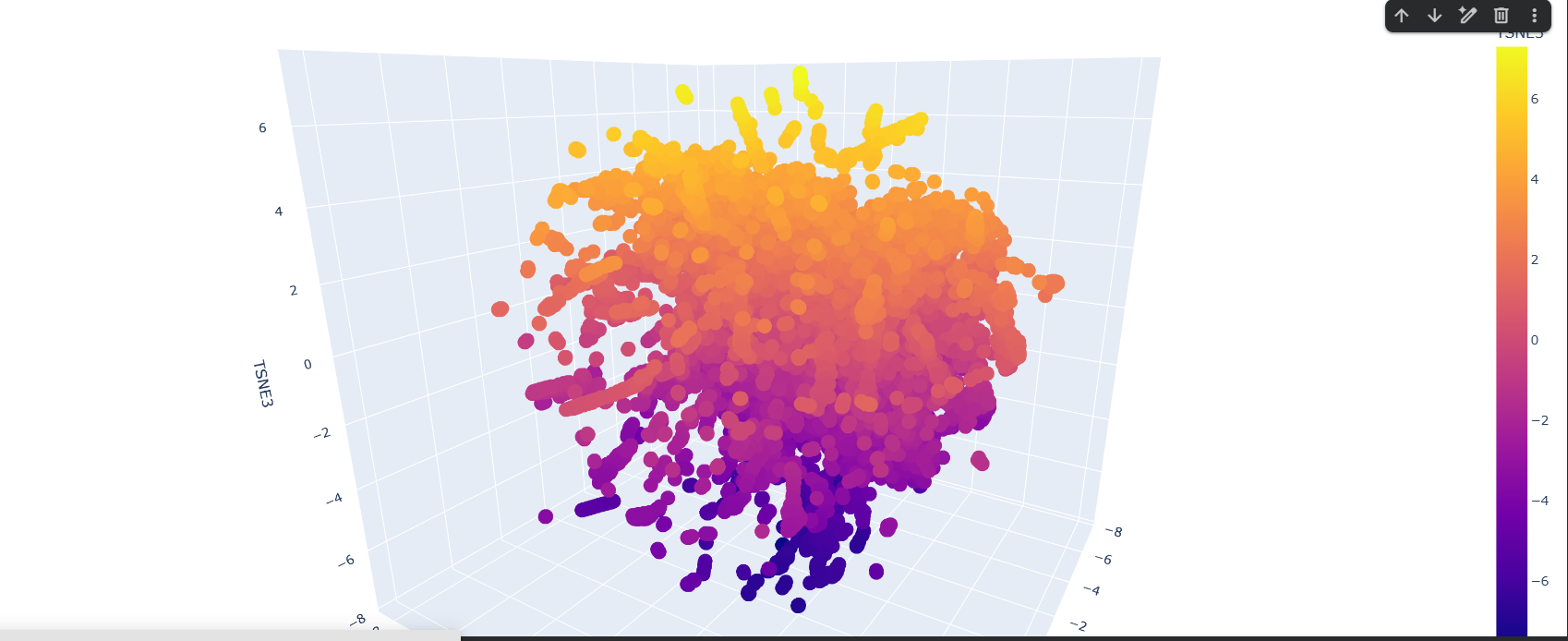
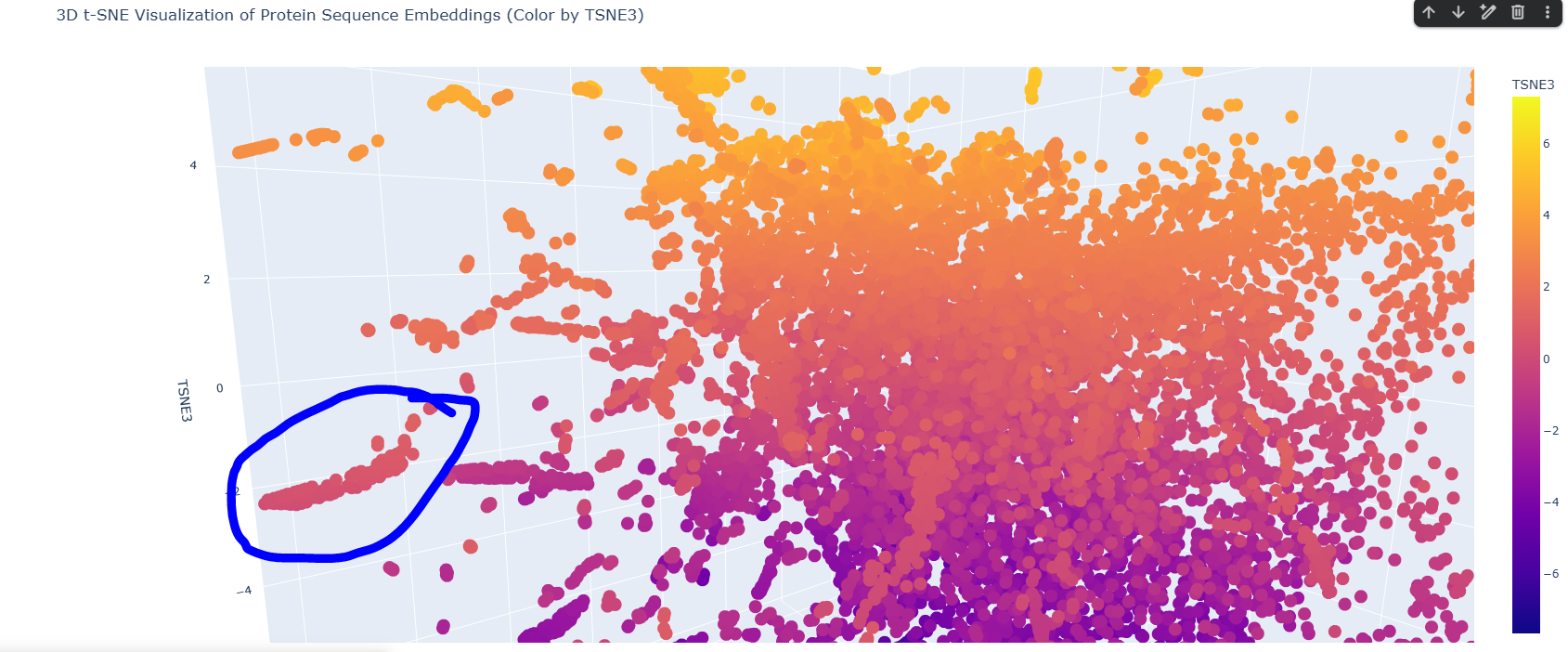
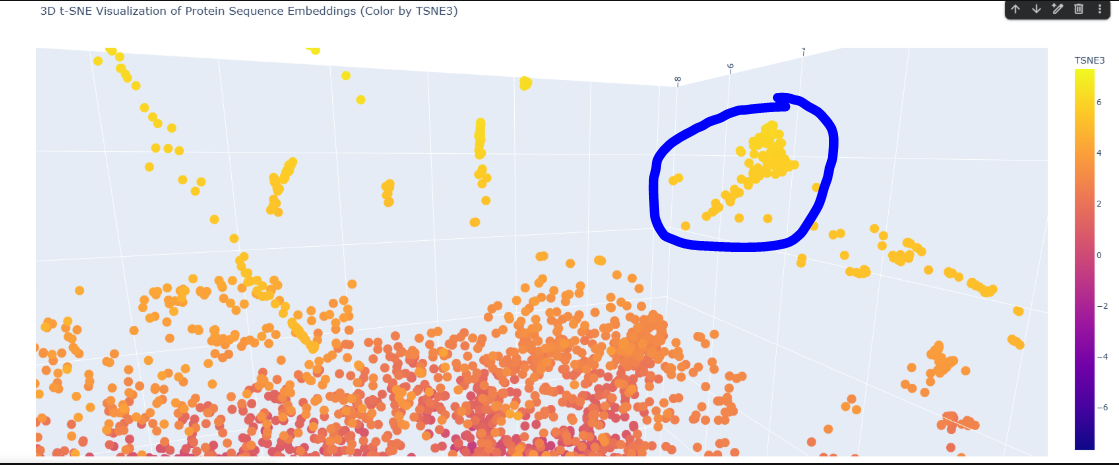
Here is the TSNE map, i hightlighted my crtI protein in black.
I found this neighborhood cluster that hosts many dehydrogenase proteins like Retinal Dehydrogenase & Putative dehydrogenase, and other Reductase proteins too
Here in this cluster i found many microbial cytochrome proteins and some photosystem proteins, and other mitochondiria and ATP related ones
𓋹 𓋹 𓋹 𓋹 𓋹
C2. Protein Folding
This is how the normal crtI folded
Here i tried swapping amino acids 10-13 to be all Valine (V) and here is the result
Here i actually picked two of the lowest scores from the mutation scan heatmap and decided to switch positions 4 and 5 which were Threonine (T) and switched them with Glutamic acid (E) which had a very low negative scores of -9.97 and -9.39 for positions 4 and 5 respectively
Here i tried to go a bit crazy and removed 30 amino acids, in sets of 10 randomly across the protein to see what effect this could have on it
𓋹 𓋹 𓋹 𓋹 𓋹
C3. Protein Generation
Inverse Folded Protein Sequence:
ALPVAVVDGGAGGLALAIRLKAAGLPVVLLESGXXXXXXXGSVEKDGFIFDTTDLIITDPSPIEALFALAGKKLEDYVKLLKVEPFYRMVFENGRTFDFNQDLAAILAQIAKFNPADVAGFQALMAALRARYAEGYPXXGPVPYLDFDRLLRVAPTLRESPAYKAIHAEIAKYIKDPFLQLALTTFHLLVSGRPXXDTDPYHLISYFTQDWDVYYPEGGYKALVEAMKTLLRDLGGTIVEGARVARFELEGNRVVAVVLEDGRVIPVSAVALPPAXXXXXXXXXXXXXXXXXXXXXXXXXXXRYDLLELFFATDKRYDHLAMYTLVFKPVXXXXXXXXXXXXXLDYSLALVIYNPNVVDPSLAPEGGNSLYVKAPVPALGSANIDWSVWGPEKAEELLAYLEAHLMPGLRASLVTHAIVTPADKLXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXELENLFVIGXXXXXXGGIPGAIAAAFEVADRILAALK
I generated an AA Probaility Heat map and the ESM Fold for the ProteinMPNN produced sequence, it actually doesn’t really hold the same shape as the original one, when i checked the output of the ProteinMPNN on colab it was this:
>T=0.1, sample=0, score=0.8096, seq_recovery=0.4642 ALPVAVVDGGAGGLALAIRLKAAGLPVVLLESGXXXXXXXGSVEKDGFIFDTTDLIITDPSPIEALFALAGKKLEDYVKLLKVEPFYRMVFENGRTFDFNQDLAAILAQIAKFNPADVAGFQALMAALRARYAEGYPXXGPVPYLDFDRLLRVAPTLRESPAYKAIHAEIAKYIKDPFLQLALTTFHLLVSGRPXXDTDPYHLISYFTQDWDVYYPEGGYKALVEAMKTLLRDLGGTIVEGARVARFELEGNRVVAVVLEDGRVIPVSAVALPPAXXXXXXXXXXXXXXXXXXXXXXXXXXXRYDLLELFFATDKRYDHLAMYTLVFKPVXXXXXXXXXXXXXLDYSLALVIYNPNVVDPSLAPEGGNSLYVKAPVPALGSANIDWSVWGPEKAEELLAYLEAHLMPGLRASLVTHAIVTPADKLXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXELENLFVIGXXXXXXGGIPGAIAAAFEVADRILAALK
The sequence recovery was 0.4642, i tried to regenerate it again and got 0.4840, still both are under 50% so that explains why the produced Fold is quite different
𓋹 𓋹 𓋹 𓋹 𓋹
Part D. Group Brainstorm on Bacteriophage Engineering
Primary Goal: Increase the structural stability of the MS2 bacteriophage lysis protein (L) while preserving its ability to lyse bacterial cells.
Secondary Goal: Decrease the protein’s reliance on the host chaperone DnaJ, which could allow the lysis protein to function more efficiently and independently in engineered systems
Design Focus: The strategy involves stabilizing the transmembrane and oligomerization regions, protecting essential functional motifs (such as the L48–S49 motif), and modifying the N-terminal region to bypass DnaJ dependence
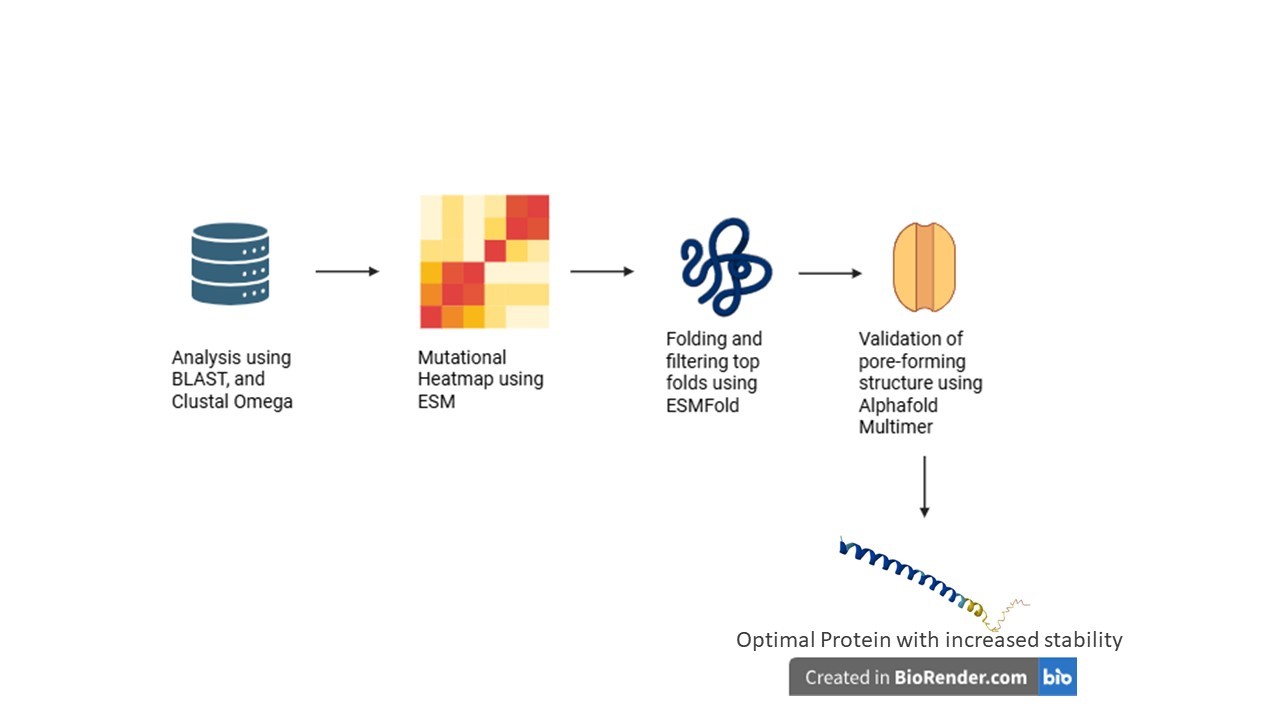
Computational Pipeline
The project utilizes a multi-step computational protein engineering pipeline to rationally design mutations:
Homolog Discovery (BLAST): Identifying related lysis proteins to find evolutionarily conserved residues and natural sequence variations
Multiple Sequence Alignment (Clustal Omega): Mapping essential structural regions and differentiating between highly conserved zones (to be protected) and mutable sites
In Silico Mutagenesis (ESM): Using protein language models to generate mutation heatmaps and rationally select amino acid substitutions that improve protein fitness and stability
Structure Prediction (ESMFold): Modeling the 3D structures of promising mutants to ensure the essential transmembrane helix is not distorted
Complex Prediction (AlphaFold Multimer): Evaluating whether mutated proteins can successfully form the required oligomeric pore complex (>10 subunits) and assessing if N-terminal mutations successfully reduce interactions with DnaJ
Expected Outcomes and Applications
The pipeline is expected to yield MS2 L variants with enhanced structural stability, proper transmembrane insertion, lower aggregation risks, and reduced DnaJ dependency
These optimized proteins have potential downstream applications in synthetic phage engineering, antimicrobial protein development, and bacterial ghost cell production
Challenges and Future Validation
Key computational challenges include the limited training data for small transmembrane toxins (as models primarily focus on globular proteins), the poor database annotation of single-gene lysis proteins (amurins), and the risk of over-stabilizing the protein, which could impede proper membrane insertion or functional oligomerization
Future steps will involve experimentally expressing the computationally identified mutants in E. coli to validate protein stability, lysis timing, and DnaJ independence
Schematic
You can check out the fully detailed Project Proposal here
𓋹 𓋹 𓋹 𓋹 𓋹
Week 5 HW: Protein Design Part ii
𓃠 Week 5 Homework 𓃠
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
Here is the Human SOD1 sequence from Uniprot (P00441)
Some of the peptides had X in it and Alphafold seems to reject it, i asked Gemini and it mentioned it should be swapped, Safe Options include Alanine (A) or Glycine (G) or a Rational swap where i can choose a hydrophobic Leucine (L) or Valine (V) or a hydrophilic Lysine (K) or Arginine (R) swap that depends on the pocket i am binding to, for now though i have decided to go with an Alanine (A) swap for simplicity.
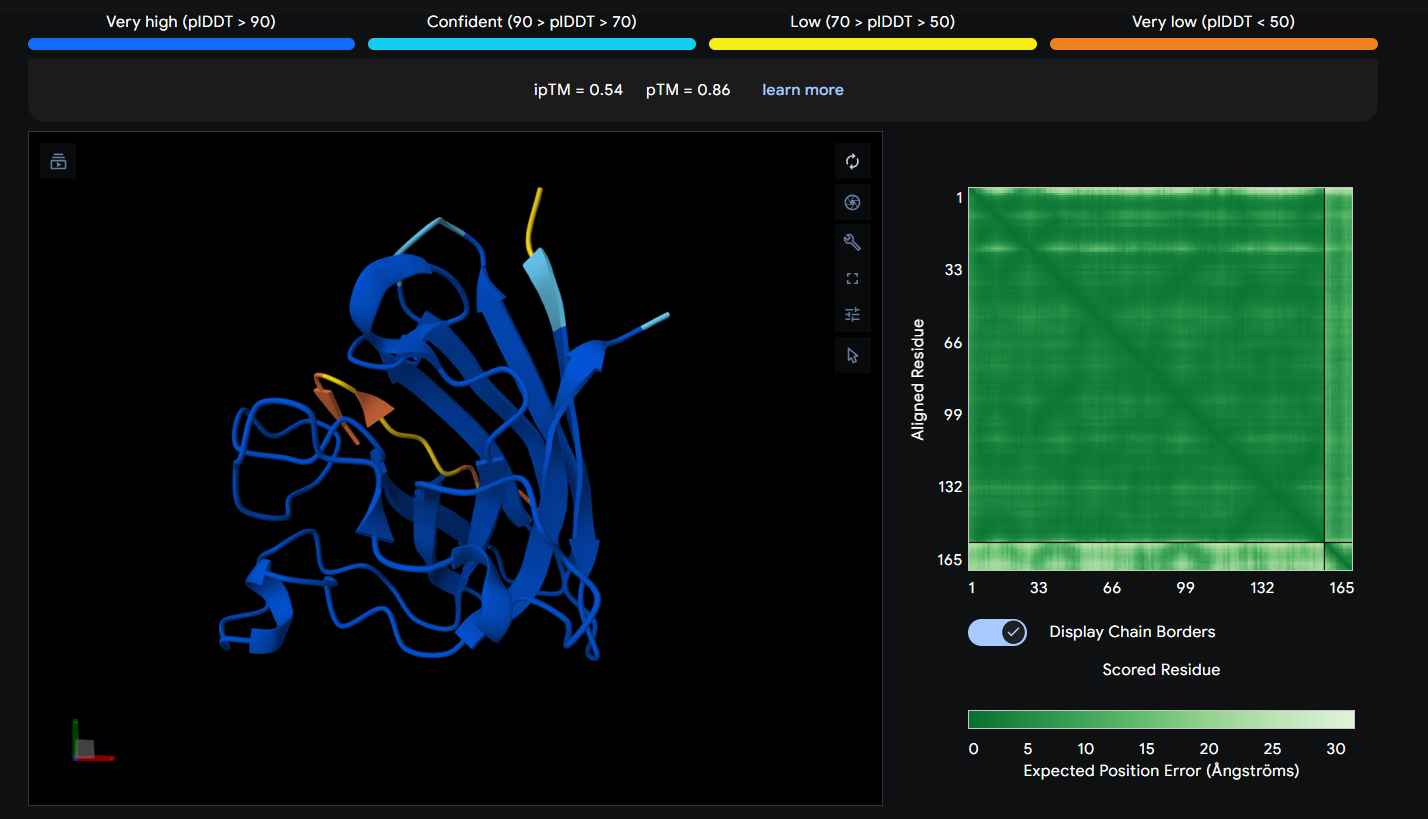
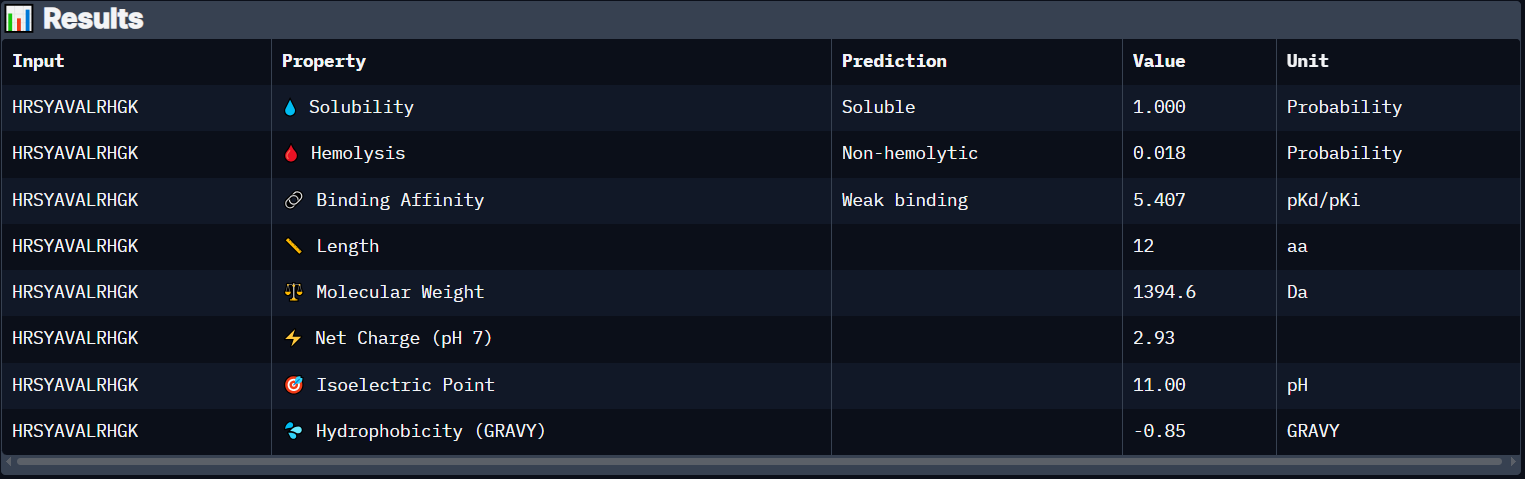
Peptide 1 (HRSYAVALRHGK)
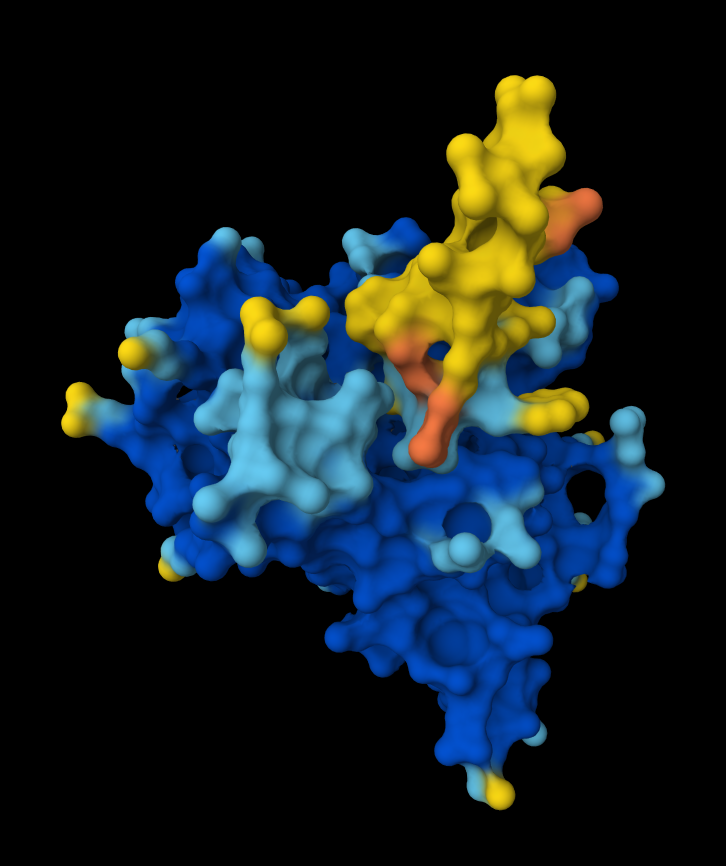
The Peptide seems to be floating really close near a side of the Dimer Interface but it is not totally sticking to it
Here in this Molecular Surface View, it shows that the Peptide is floating and not touching the protein surface, yet it looks to be fitting into the protein surface groove well.
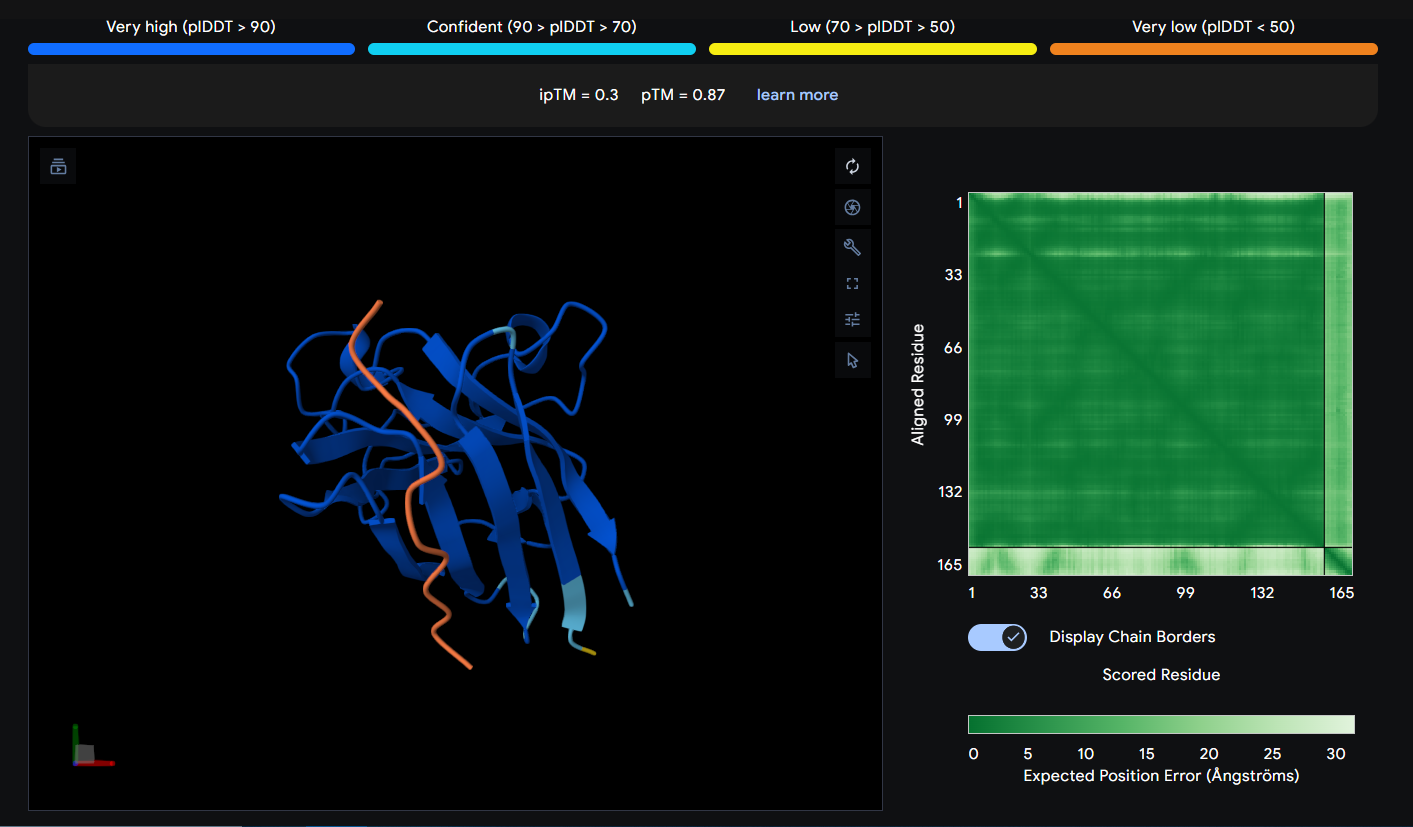
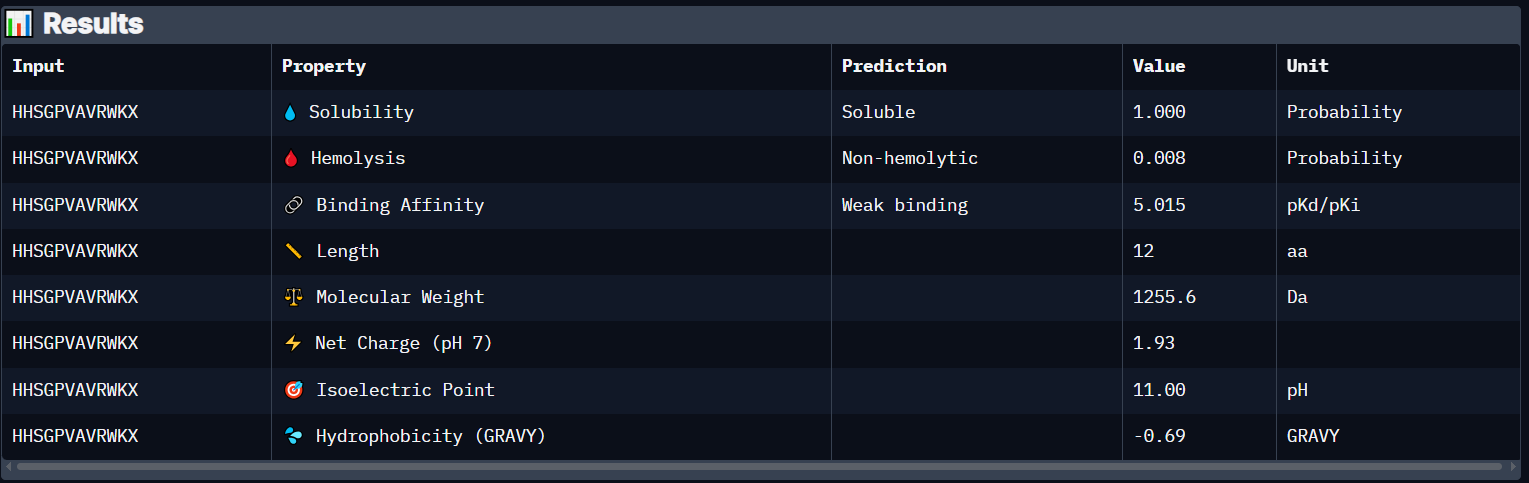
Peptide 2 (HHSGPVAVRWKX)
This Peptide seems to be floating near the Dimer Interface but not actually stuck to it.
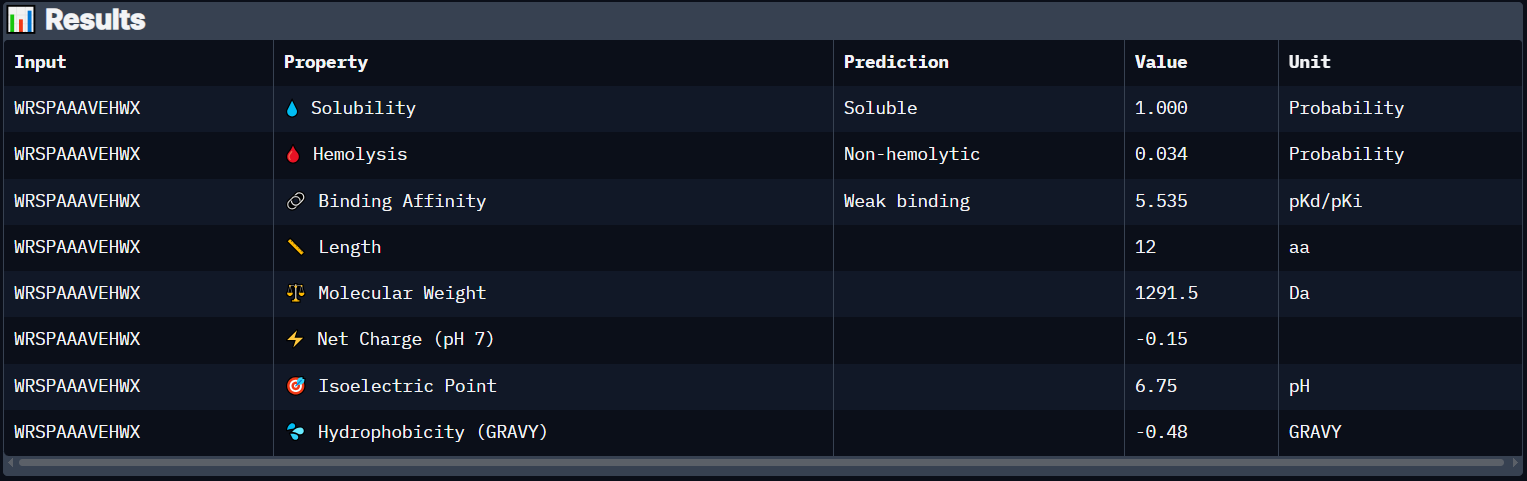
Peptide 3 (WRSPAAAVEHWX)
The Peptide seems to be floating away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface.
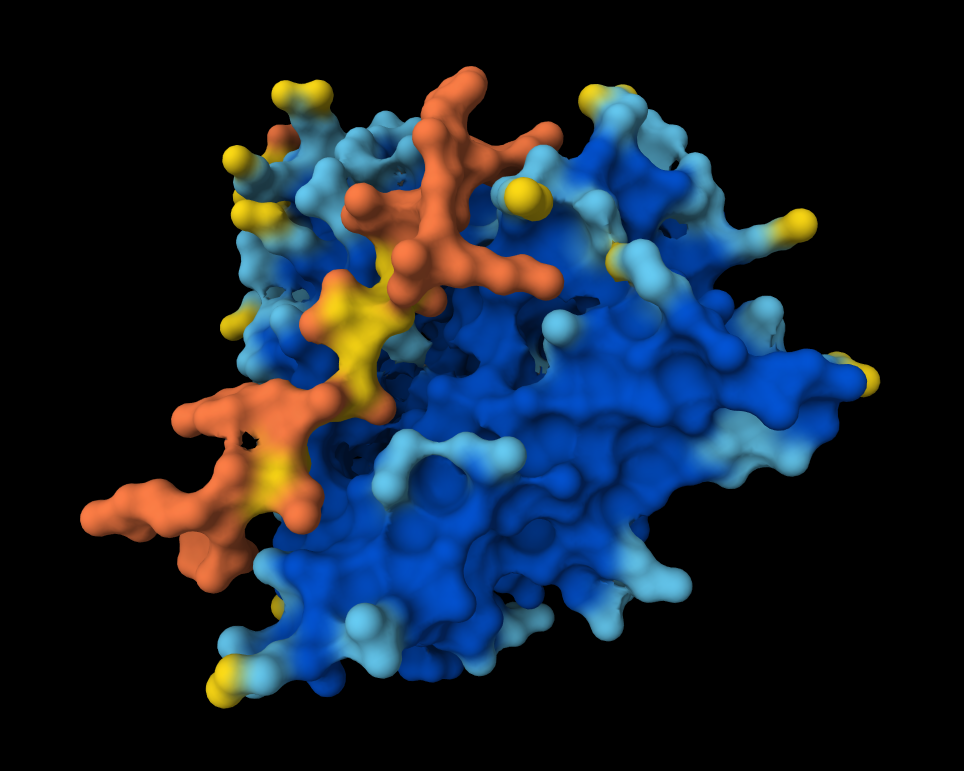
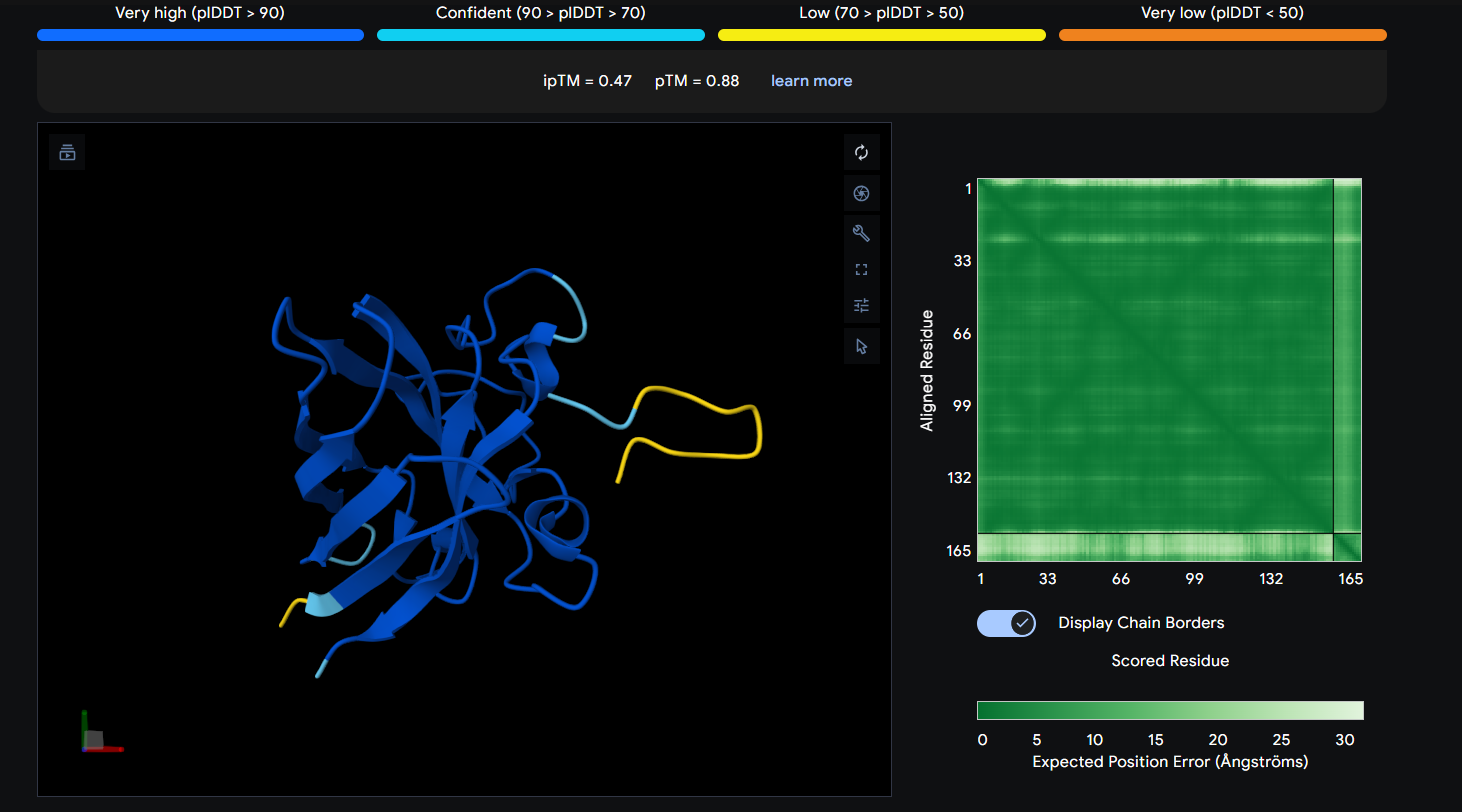
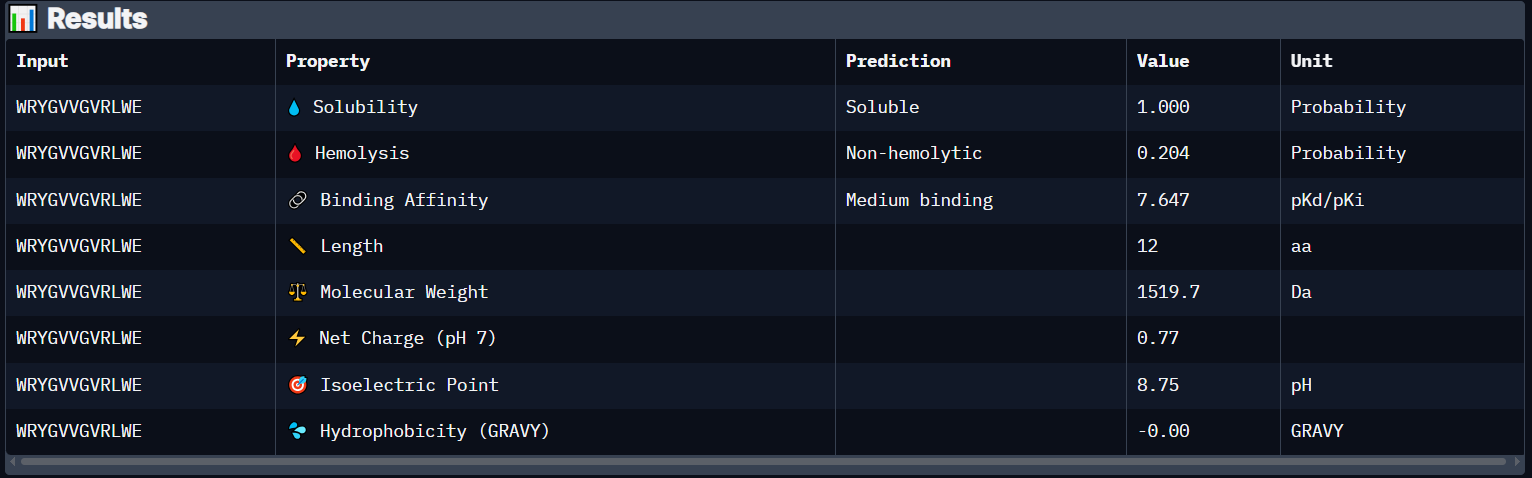
Peptide 4 (WRYGVVGVRLWE)
The Peptide seems to be buried into a groove into the protein, yet does not engaging with the N-terminus, the beta barrel or the Dimer Interface.
This image shows clearly how the peptide seems to be holding into a groove into the protein.
The Peptide seems to be floating away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface.
Index
Peptide
ipTM Score
1
HRSYAVALRHGK
0.54
2
HHSGPVAVRWKX
0.3
3
WRSPAAAVEHWX
0.34
4
WRYGVVGVRLWE
0.47
5
FLYRWLPSRRGG
0.41
The ipTM score tells you how confident AlphaFold is that these two chains actually interact. It ranges from 0 to 1. Generally, and as noted in AlphaFold, Scores beyond 0.8 are confident high-quality predictions) while below 0.6 are a failed prediction, in between is considered a grey zone where predictions could be correct or incorrect.
Looking at my ipTM scores, it seems like all the generated peptides fall into the failed prediction zone, with Peptide 1 almost surpassing it with a score of 0.54, Peptides 1 (0.54) & 4 (0.47) seem to have a higher score than the control Peptide 5, while Peptides 2 (0.3) & 3 (0.34) have very low scores.
𓋹 𓋹 𓋹 𓋹 𓋹
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Peptide 1 (HRSYAVALRHGK)
Peptide 1 has a very high solubility score and is also non hemolytic so it is generally safe, however it shows a weak binding affinity, The results also show that it has a isoelectric point of 11.00 explaining its positive net charge (2.93), hydrophilic (-0.85) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1394.6 Daltons.
Peptide 2 (HHSGPVAVRWKX)
Peptide 2 has a very high solubility score and is also non hemolytic so it is generally safe, however it also shows a weak binding affinity, The results also show that it has a isoelectric point of 11.00 explaining its positive net charge (1.93), hydrophilic (-0.69) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1255.6 Daltons.
Peptide 3 (WRSPAAAVEHWX)
Peptide 3 has a very high solubility score and is also non hemolytic so it is generally safe, however it shows a weak binding affinity, The results also show that it has a isoelectric point of 6.75 explaining its negative (or almost neutral) net charge (-0.15), hydrophilic (-0.48) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1291.5 Daltons.
Peptide 4 (WRYGVVGVRLWE)
Peptide 4 has a very high solubility score and is also non hemolytic so it is generally safe, and it shows Medium binding affinity :D, The results also show that it has a isoelectric point of 8.75 explaining its positive net charge (0.77), amphiphilic (-0.00) so it binds well to both water or greasy non-polar pockets of the protein, and has a molecular weight of 1519.7 Daltons.
Overall it seems like all the generated Peptides are soluble and non hemolytic so they are safe to use, Peptide 4 seems to be the one with the highest chance of Binding because it has Medium Binding Affinity compared to all Weak Binding affinity of all other peptides, even though it didnt have the highest ipTM score, it got 0.47 while Peptide 1 got 0.54 which proves structural prediction and Shape complementarity is not the only factor affecting Protein-Peptide Binding.
So with that, i have decided to proceed with Peptide 4 WRYGVVGVRLWE since it has a higher Binding Affinity than all others
𓋹 𓋹 𓋹 𓋹 𓋹
Part 4: Generate Optimized Peptides with moPPIt
For this part i have decided to pick the amino acid residues from 2-6, since our mutation is A4V so i picked it with two amino acids upstream and 2 downstream to try to bind to this specific area.
Here is the moPPIt generated peptides
Index
Peptide
Hemolysis
Solubility
Affinity
Motif
6
REYDQKQICKKL
0.9428217448
0.8333333135
7.031765938
0.8390983939
7
KKSKKQKELTCG
0.9830000903
0.9166666865
6.922430038
0.7878084183
8
IQQWETKGKRLK
0.9603748098
0.7500000000
5.674941063
0.5862649679
I gave them indexes starting from 6 to account for the previously generated 4 peptides and the control one
To level the playing field for comparison, i decided to take those Peptides, and run them through AlphaFold and PeptiVerse and compare them with the pepMLM ones.
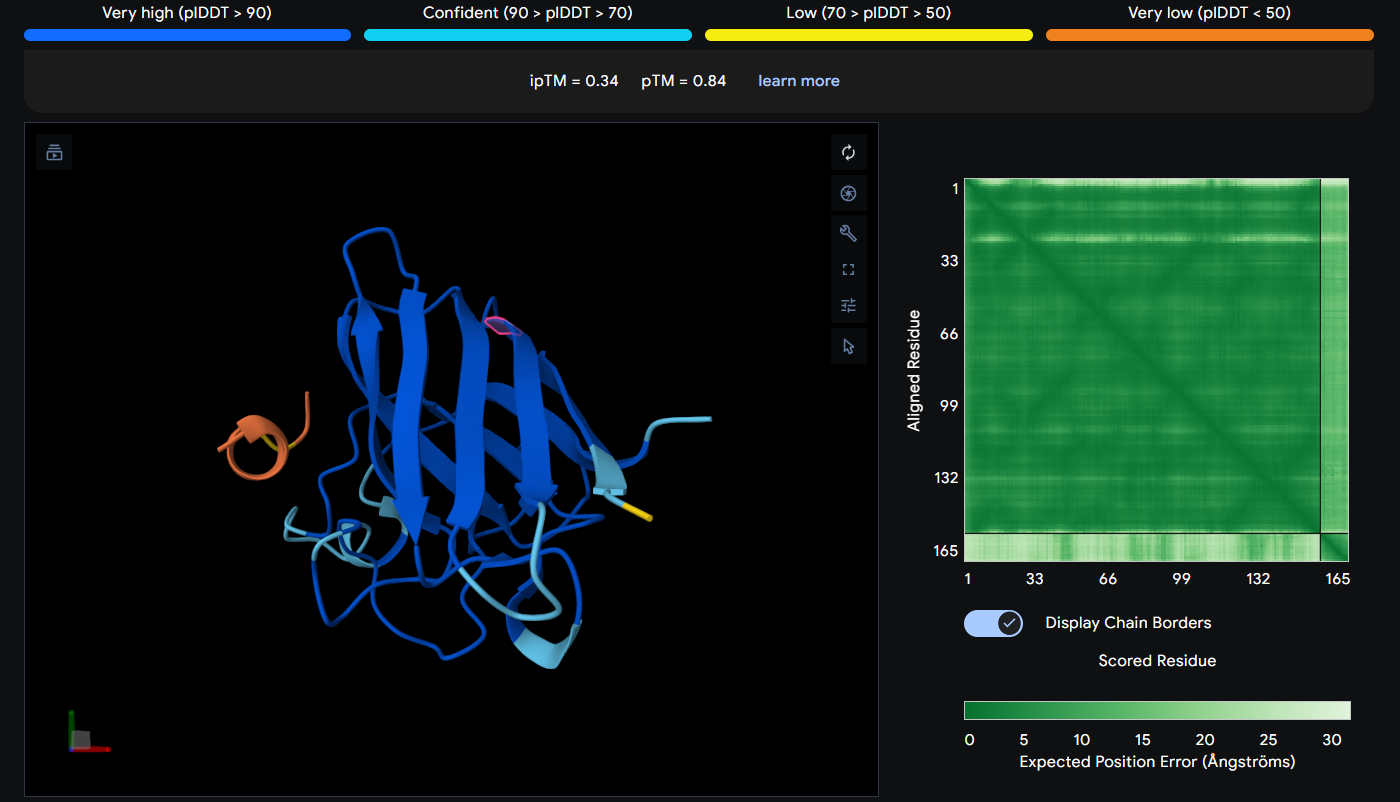
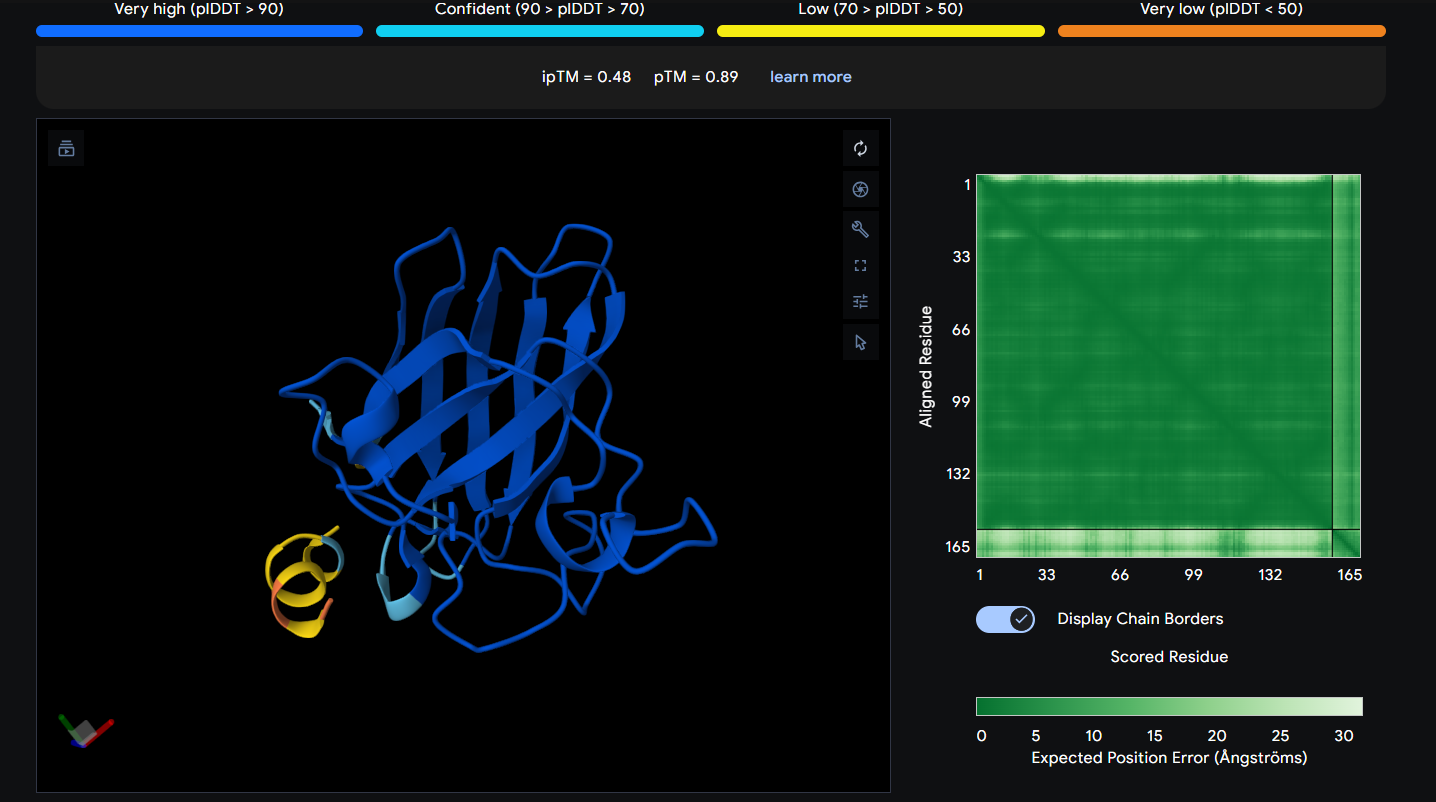
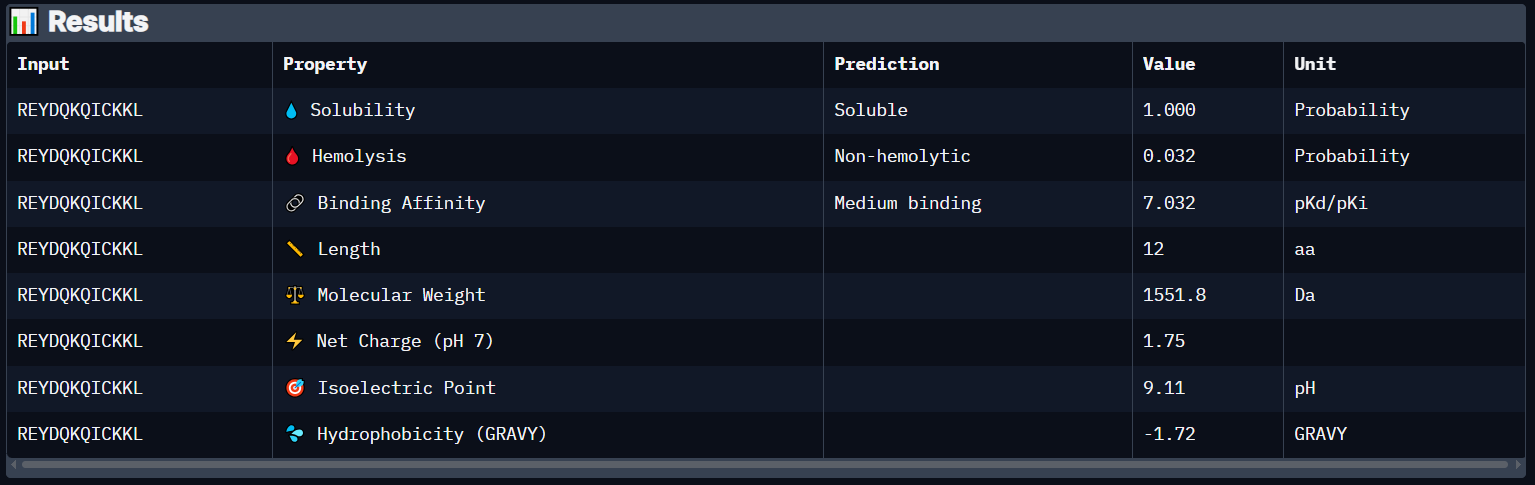
Peptide 6 (REYDQKQICKKL)
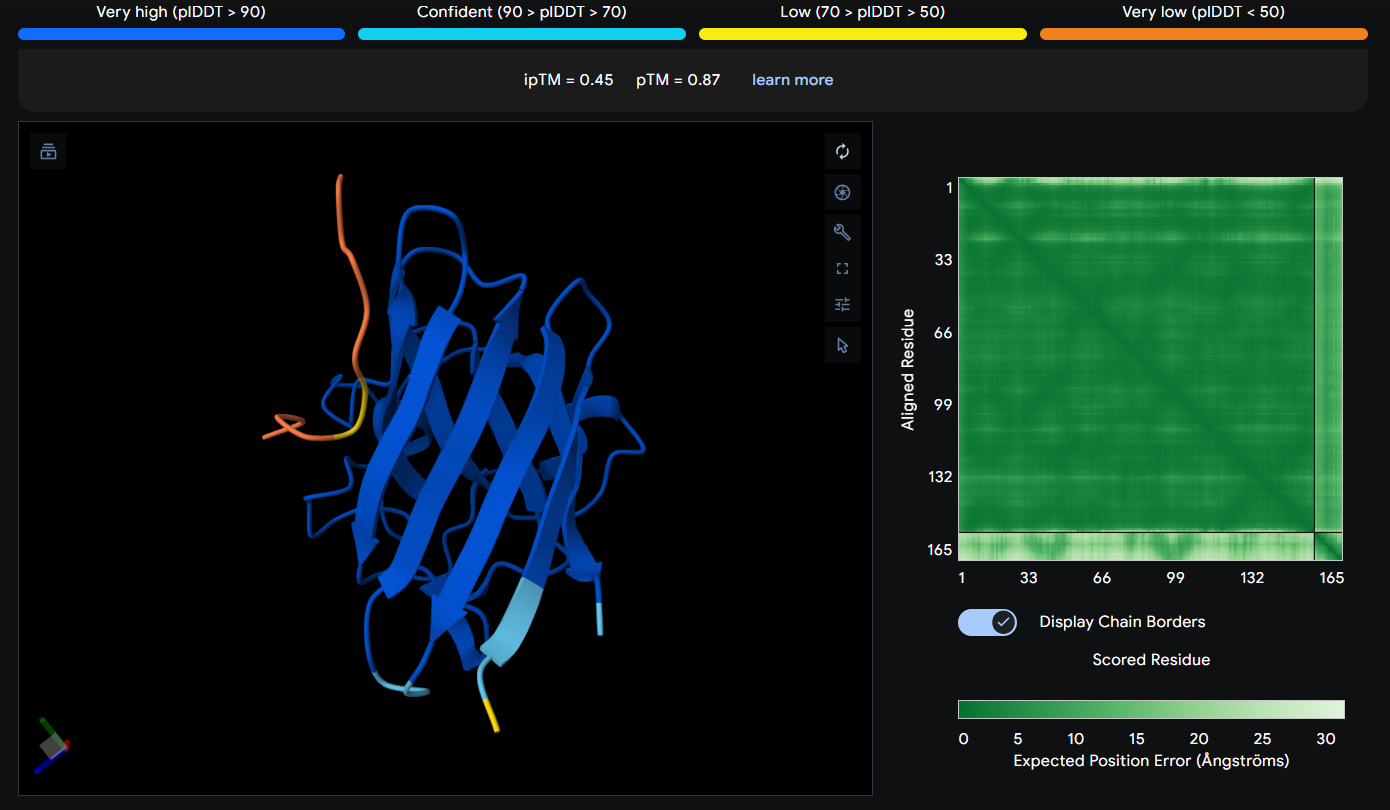
Peptide 6 seems to be floating away from the protein but slightly close to the N-Terminus region where our A4V mutation is, but it is not binded to it and the ipTM score is 0.48 so it falls in the failed prediction zone
Peptide 6 has a very high solubility score and is also non hemolytic so it is generally safe, and it shows a medium binding affinity, The results also show that it has a isoelectric point of 9.11 explaining its positive net charge (1.75), hydrophilic (-1.72) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1551.8 Daltons.
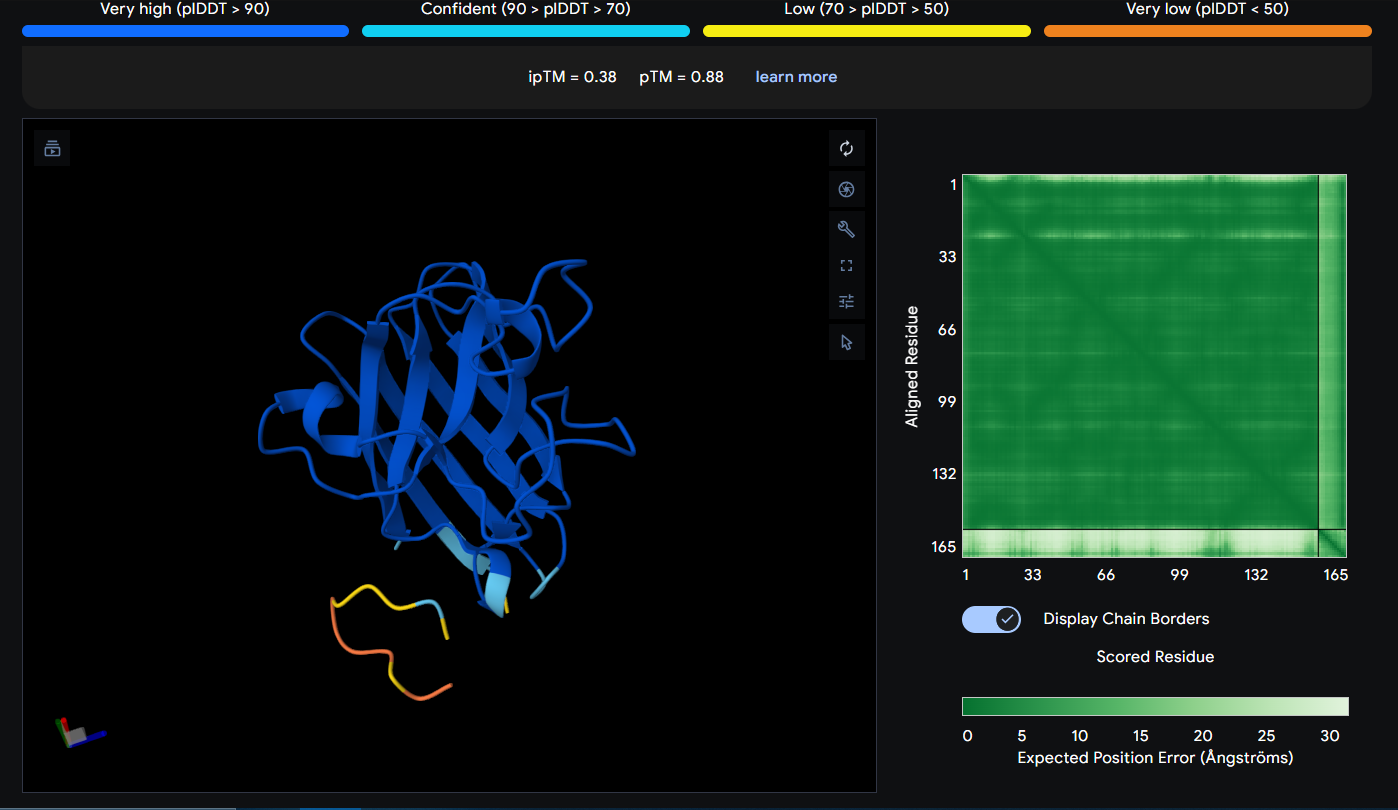
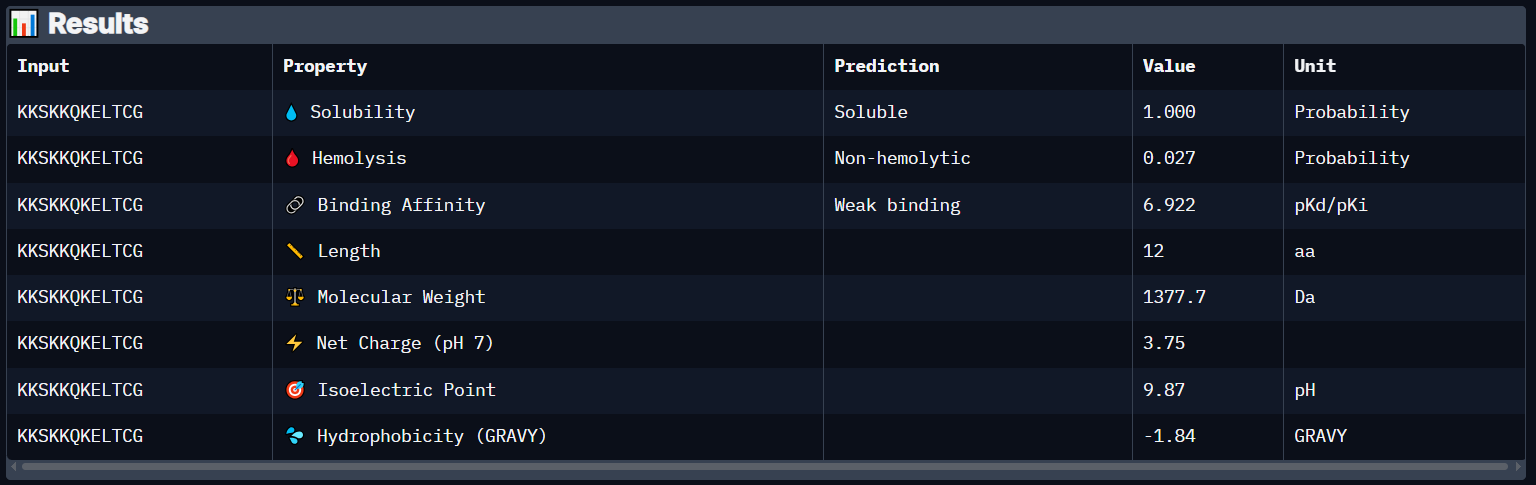
Peptide 7 (KKSKKQKELTCG)
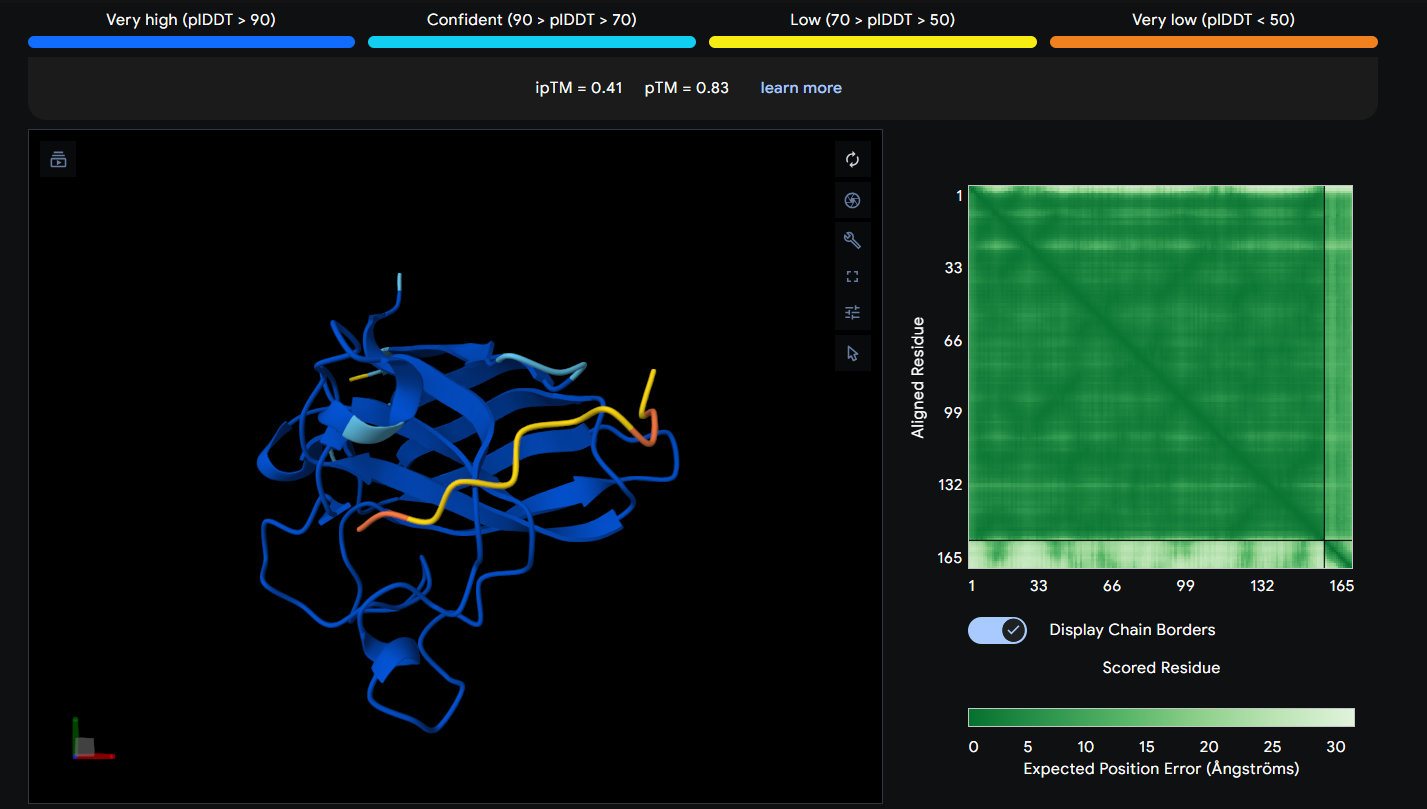
Peptide 7 seems to be floating away from the protein and also very slightly close to the N-Terminus and the mutation region as well, yet again it is not binded to it and the ipTM score is 0.38 so it falls in the failed prediction zone
Peptide 6 has a very high solubility score and is also non hemolytic so it is generally safe, and it shows a weak binding affinity, The results also show that it has a isoelectric point of 9.87 explaining its positive net charge (3.75), hydrophilic (-1.84) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1377.7 Daltons.
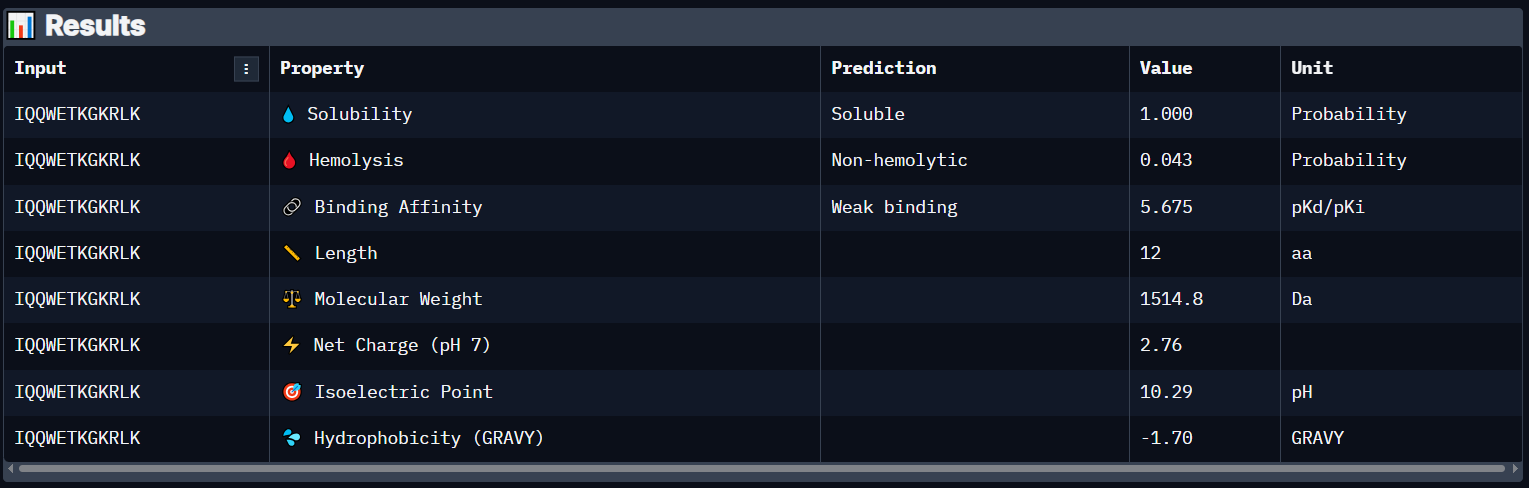
Peptide 8 (IQQWETKGKRLK)
Peptide 8 seems to be floating away from the protein and also very far away from to the N-Terminus and the mutation region (which is weird), and again it is not binded to it and the ipTM score is 0.45 so it falls in the failed prediction zone
Peptide 6 has a very high solubility score and is also non hemolytic so it is generally safe, and it shows a weak binding affinity, The results also show that it has a isoelectric point of 10.29 explaining its positive net charge (2.76), hydrophilic (-1.70) so it binds well to hydrophilic or polar pockets of the protein, and has a molecular weight of 1514.8 Daltons.
While pepMLM generates peptides randomly and tries to somehow bind to the protein through the protein-protein interactions it has learned, moPPIt seems to follow a more Rational Design when designing those peptides and tries to target a specific region/residue to achieve the binding there. With that said, it seems like all my generated peptides have failed surpassing the AlphaFold threshold of an ipTM score of at least more than 0.6 and most of them seem to show a weak binding affinity on PeptiVerse except for Peptides 4 and 6 which actually showed medium binding affinity.
Evaluation of the generated peptides before clinical studies is very crucial and it requires bridging the massive gap between computational biology and actual human physiology, first these peptides need to be synthesized and then tested on human cell cultures we mainly need to confirm that it does its expected job which is preventing SOD1 from misfolding or aggregating and most importantly not to kill the cell in the process, Then we may test these on In Vivo Animal Models like an ALS Mouse Model, to monitor if these peptides can actually cross the Blood Brain Barrier (BBB) and reach the affected Neurons.
Additional Tests and Validations may include Immunogenetic Testings to make sure the Human Body doesn’t mark this synthetic peptide as a foreign invader, which can trigger a dangerous immune response or cause the body to generate neutralizing antibodies against the drug, also we may need to check if it needs chemical modifications to survive cellular proteases since peptides can be an easy target for them and this negatively affects the peptide’s half life.
𓋹 𓋹 𓋹 𓋹 𓋹
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
It seems like these Positions [21,25,28-29,33,35-37,40] have an “*” which means these have not changed at all and most probably are a totally conserved crucial region that should not be mutated, Positions [17,26,30] have a “:” which means they are highly conserved, but mutations that are very similar in shape, structure and chemical properties are tolerated, the rest seem to be more flexible.
Interesting Finding here, All the conserved regions seem to be in the Soluble Domain (1-40) that is responsible for DnaJ Interaction :D
This is the generated L Protein Mutation Heatmap
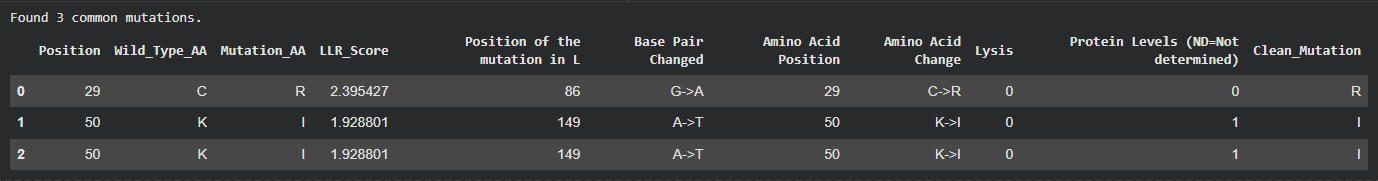
I tried to cross reference with the experimental data sheet but it was quite hard to do manually so i wrote a script and ran it on Here on Colab to just find the common ones across the two files and came up with 3 common ones, however even though these 3 had quite a high LLR Score, the experimental data showed a Lysis score of 0, and i believe this shows the limitation in the structure based modeling, it predicted structural stability but failed to account for the functional mechanism of lysis.
The common mutations identified by my algorithm
So Then i Filtered my generated mutations first to target the soluble domain (1-40) first while making sure to avoid the totally conserved regions [21,25,28-29,33,35-37,40], the highly conserved regions [17,26,30] and my 3 common matches to avoid choosing mutants that are either disrupting the protein or have been already experimented on with no lysis observed.
The two mutations i have decided to go with for the Soluble Domain are:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
1
39
Y
L
2.24177968502044
2
9
S
Q
2.01432478427886
These have the highest LLR score in this region and avoid any conservative regions in the protein
Then i repeated the steps again, this time targeting the transmembrane domain (41-75) and followed the same criteria, and i picked these 2 mutations:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
3
50
K
L
2.56146776676178
4
53
N
L
1.86493206024169
And Again because These have the highest LLR score in this region and avoid any conservative regions in the protein
For the Final 5th mutant i decided to pick this one:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
5
52
T
L
1.81396758556365
Because this one still has a relatively high LLR Score and is in the area where the L Protein does not overlap with either the Coat Protein or the replicase ones.
so here is the full 5 Mutations table i chose:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
1
39
Y
L
2.24177968502044
2
9
S
Q
2.01432478427886
3
50
K
L
2.56146776676178
4
53
N
L
1.86493206024169
5
52
T
L
1.81396758556365
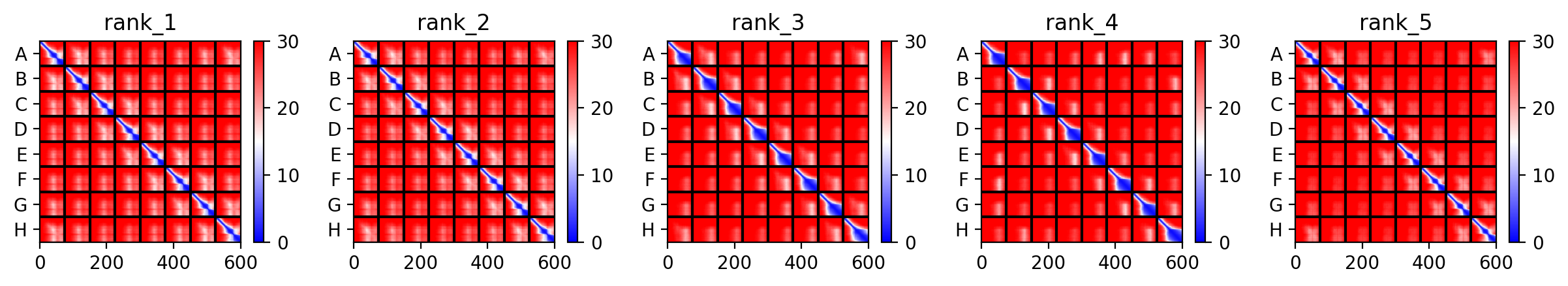
AlphaFold Multimer Runs
Mutant 1 (Y39L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
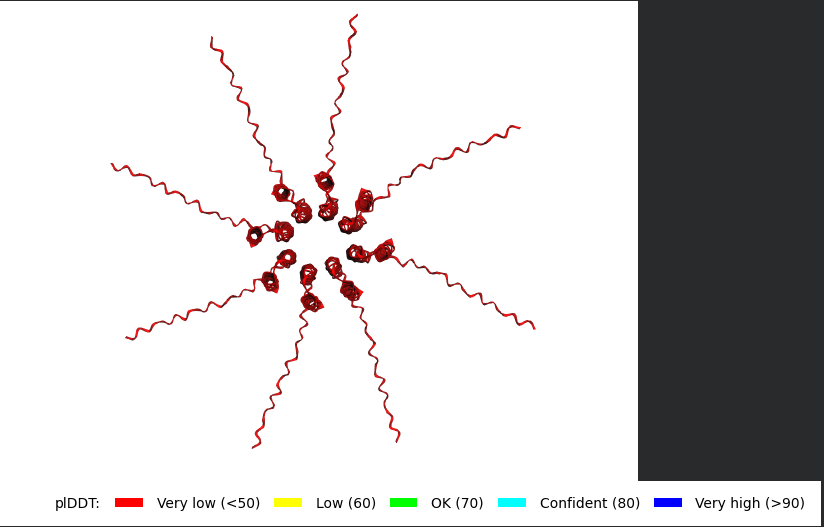
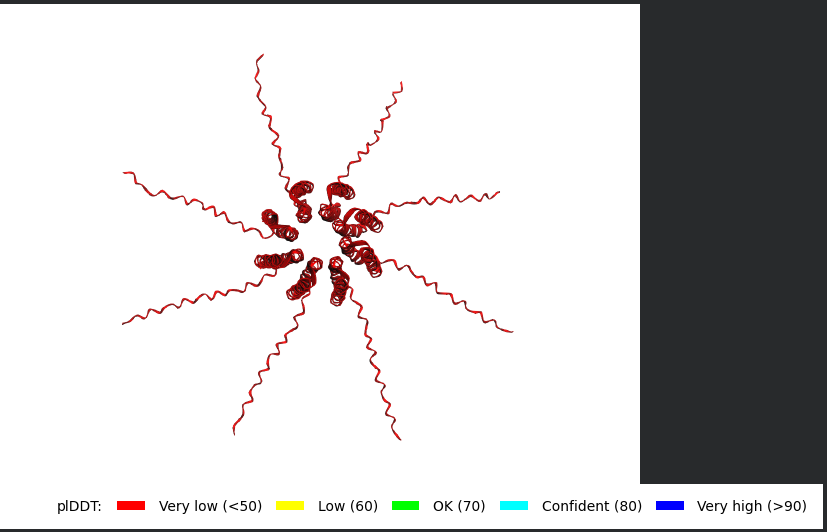
The L Proteins do form a cylinder like shape mimicking the transmembrane pore but the piDDT score are very low (<50) so this probably won’t express well or won’t express at all if done in wet lab :(.
This is the Predicted Aligned Error (PAE), i asked AI (Gemini) how to interpret this and it said this:
So following this, it seems like each monomer folds correctly with high confidence yet their together-grouping is not reliable at all with very low confidence scores possibly hinting that the pore forming shape we saw might not actually happen (i hope i understood that correctly XD)
Mutant 2 (S9Q)
The Monomer Sequence:
METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Same exact findings for this mutant too :(.
Again, Same Here as well. (it is the same for all five mutants :(. )
Mutant 3 (K50L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
Mutant 4 (N53L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT
Mutant 5 (T52L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT
𓋹 𓋹 𓋹 𓋹 𓋹
I Actually decided to pick a mutant from the Experimental Data Sheet and made sure it has been proven to have the Lysis Effect, my aim is to try to perform the AlphaFold Multimer step for it, to have a look at how different it might be, the mutant i picked was R30Q.
This is the monomer sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
This is the full Alphafold sequence i used:
METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
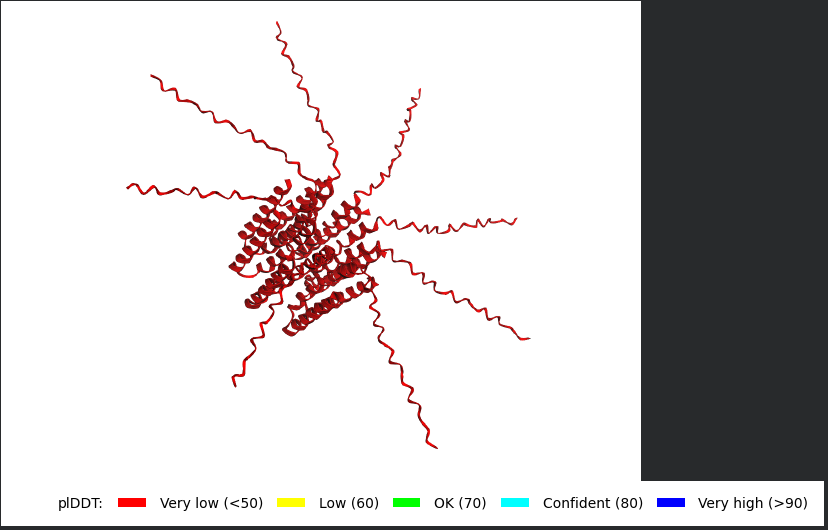
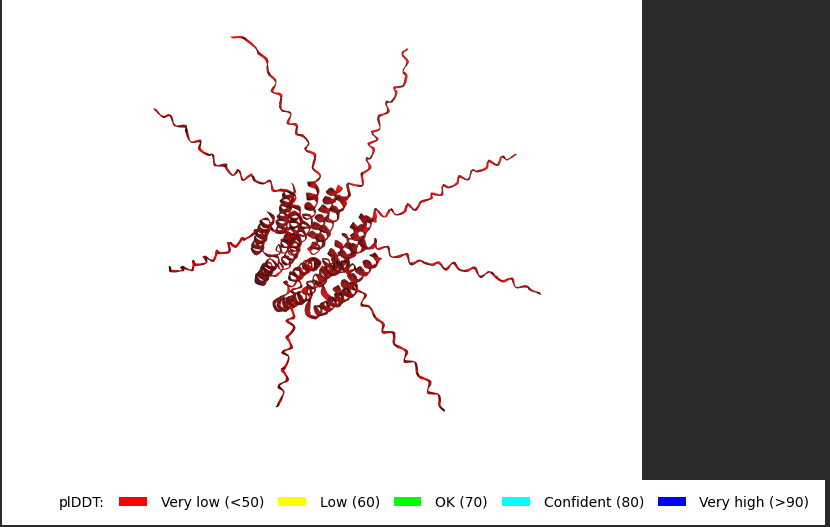
Well i got bad news and good news, the bad news is, the structre is also of very low confidence and the Predicted Aligned Error (PAE) Plot shows the same low confidence for the monomer’s interaction with each othe, the good news though is that this is the run of a mutant that has been experimentally validated and has the lysis effect and protein level determined, so maybe this gives me hope that my five mutants might actually stand a chance in a wet lab validation regardless of the very low confidence scores it got.
𓋹 𓋹 𓋹 𓋹 𓋹
Week 6 HW: Genetic Circuits Part i
𓃠 Week 6 Homework 𓃠
Assignment: DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix is basically a pre-optimized solution containing every chemical component needed for DNA amplification except for the DNA template and the primers.
It mainly consistes of:
Phusion High-Fidelity DNA Polymerase: This is the main enzyme that synthesizes the new DNA strand
Deoxynucleotide Triphosphates (dNTPs): These are the nucleotide building blocks that the DNA Polymerase use to synthesize the new DNA strand
Magnesium Chloride (MgCl2): This is an essential cofactor for the DNA polymerase, helps it with stabilizing the negatively charged alpha-phosphate of the dNTPs, reducing electrostatic repulsion and allowing primers to anneal more effectively to the template
Reaction Buffers: These maintain a stable chemical environment for the DNA Polymerase, it includes:
High Fidelty Buffer: The default buffer optimized to provide the highest possible sequence accuracy.
GC Buffer: This buffer is has special additives additives to help denature high GC content templates
pH Buffer: This buffer includes buffering agents that keep the reaction at a specific pH (8.8–9.3) to prevent DNA damage and maintain enzyme activity.
𓋹 𓋹 𓋹 𓋹 𓋹
2. What are some factors that determine primer annealing temperature during PCR?
The main two factors are the GC content of the primer, the more Gs and Cs, the higher the temperature, also the Primer Length, the longer the primer the more hydrogen bonds holding the strand and the higher the chance there will be GC regions too, one more factor is salt concentrations, High salt concentrations neutralize the negatively charged DNA backbone, stabilizing the bonds in the process and raising the melting temperature.
𓋹 𓋹 𓋹 𓋹 𓋹
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
They both do generate linear DNA, but the processs is different:
PCR generates the linear fragment through building DNA from scratch, you can pick the starting area you want through primer design, the end product usually has a blunt end and you end up with a huge amount of your target area since PCR also has exponential amplification
Restriction Enzyme Digests however generates the fragment through cutting existing DNA, you usually cant pick the starting area since each restriction enzyme has a specific area where it can make a cut, and depending on the used enzyme, you might end up with sticky or blunt ends, and you usually end up with just the amoung of your target fragment that depends on the starting amount.
𓋹 𓋹 𓋹 𓋹 𓋹
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
For Gibson Assembly the main thing you need to focus on is having overlapping sequences (20-40 bp) on both ends of your target DNA with whatever backbone it is going to be inserted in, so that the Gibson mix can chew back these overlaps and anneal them and start stitching them together.
𓋹 𓋹 𓋹 𓋹 𓋹
5. How does the plasmid DNA enter the E. coli cells during transformation?
There is two ways:
Chemical Transformation (Heat Shock): This is the most common method and is done using Calcium Chloride where the rapid heat change causes thermal imbalance across the membrane, causing pores to open in the bacterial cell wall so that the plasmid can enter.
Electroporation: This method uses a quick pulse of electricity and is more efficient for large plasmid, the idea is similar, High voltage electric pulses are sent and it disrupts the cell membrane causing pores so the plasmid can enter.
𓋹 𓋹 𓋹 𓋹 𓋹
6. Describe another assembly method in detail (such as Golden Gate Assembly)
I will talk in depth about Gibson Assembly
So Gibson Assembly is a molecular cloning method that allows seamless joining of multiple DNA fragments in a one pot reaction without requiring restriction enzymes or leaving behind scar sites, and it all mainly depends on having overlapping ends (20-40 bp) with the fragment next to it, and these overlapping ends can be designed using PCR primers.
Here is how the One Pot reaction happens:
The Gibson Master Mix is added and the temperature is set to around 50 degrees
T5 Exonuclease chews back the DNA from the 5’ ends exposing the overlaps we designed, this now makes them stick together and anneal
Phusion DNA Polymerase then fills those gaps using the overlapping strand as a template
Taq DNA Ligase it seals the gaps and nicks between the sugar-phosphate backbone to create a single continous strand of DNA
Gibson Assembly is very good in that it is Fast, Seamless and can assemble up to 15-20 fragments at once and doesnt care about internal restriction sites, however the overlapping primers is expensive, and the reaction can fail if the overlaps create stable hairpins or secondary structures
Benchling Workflow
I will do a simple experiment where i stitch a GFP Protein to a plasmid.
First we need to retreive the GFP DNA sequence from ENA.
This is the Link ot the Fasta file and Here is the DNA sequence:
TACACACGAATAAAAGATAACAAAGATGAGTAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAATGGGCACAAATTTTCTGTCAGTGGAGAGGGTGAAGGTGATGCAACATACGGAAAACTTACCCTTAAATTTATTTGCACTACTGGAAAACTACCTGTTCCATGGCCAACACTTGTCACTACTTTCTCTTATGGTGTTCAATGCTTTTCAAGATACCCAGATCATATGAAACAGCATGACTTTTTCAAGAGTGCCATGCCCGAAGGTTATGTACAGGAAAGAACTATATTTTTCAAAGATGACGGGAACTACAAGACACGTGCTGAAGTCAAGTTTGAAGGTGATACCCTTGTTAATAGAATCGAGTTAAAAGGTATTGATTTTAAAGAAGATGGAAACATTCTTGGACACAAATTGGAATACAACTATAACTCACACAATGTATACATCATGGCAGACAAACAAAAGAATGGAATCAAAGTTAACTTCAAAATTAGACACAACATTGAAGATGGAAGCGTTCAACTAGCAGACCATTATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCCACACAATCTGCCCTTTCGAAAGATCCCAACGAAAAGAGAGACCACATGGTCCTTCTTGAGTTTGTAACAGCTGCTGGGATTACACATGGCATGGATGAACTATACAAATAAATGTCCAGACTTCCAATTGACACTAAAGTGTCCGAACAATTACTAAAATCTCAGGGTTCCTGGTTAAATTCAGGCTGAGATATTATTTATATATTTATAGATTCATTAAAATTGTATGAATAATTTATTGATGTTATTGATAGAGGTTATTTTCTTATTAAACAGGCTACTTGGAGTGTATTCTTAATTCTATATTAATTACAATTTGATTTGACTTGCTCAAA
Now Let’s Create the DNA Entry for GFP on Benchling
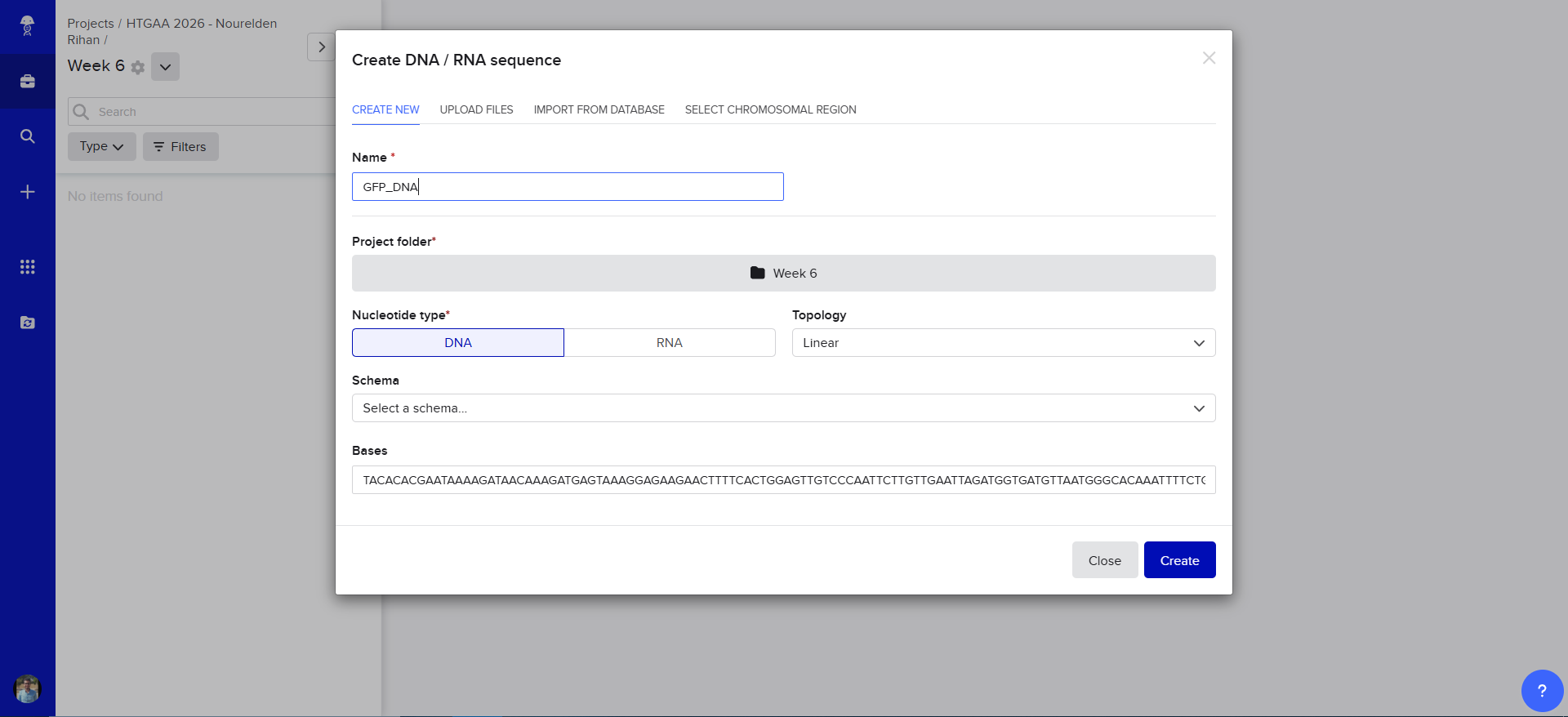
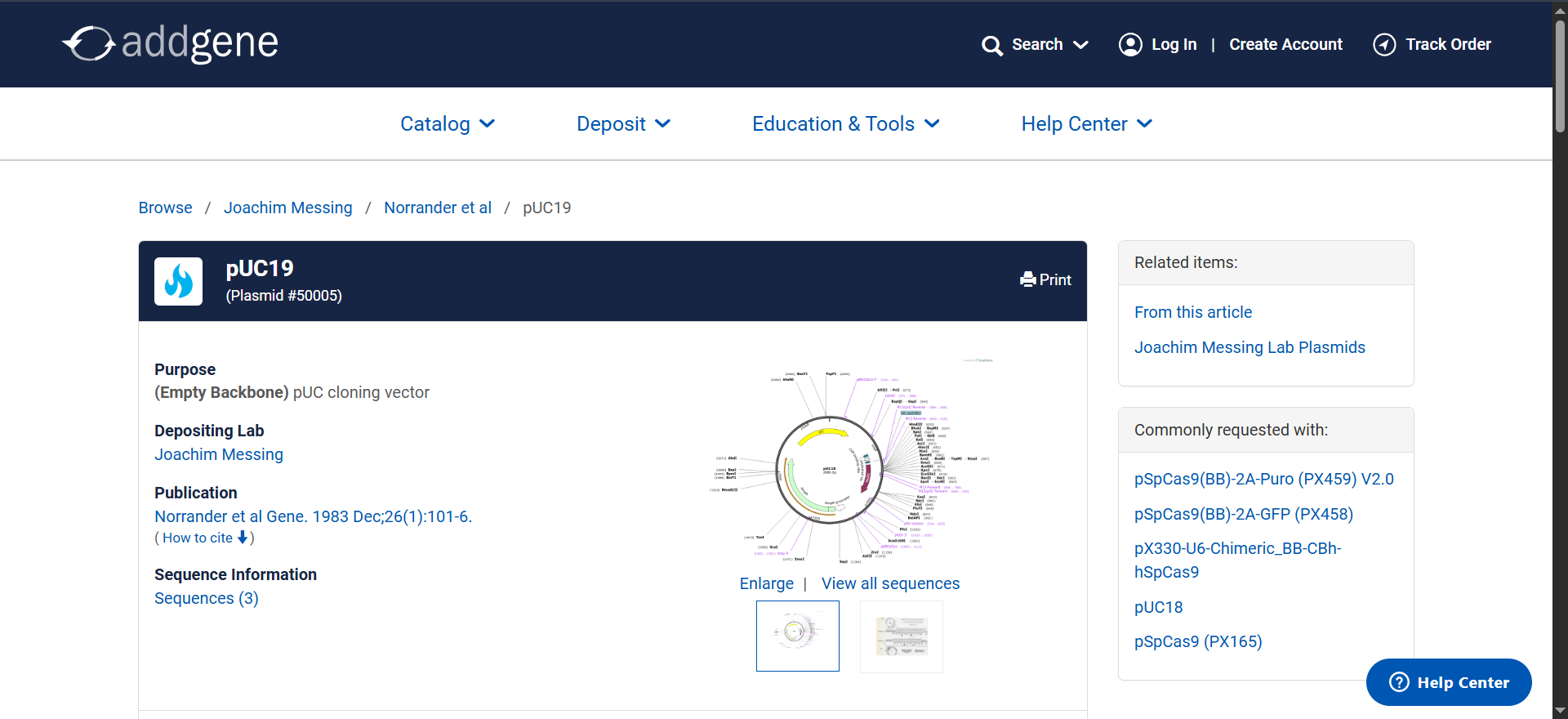
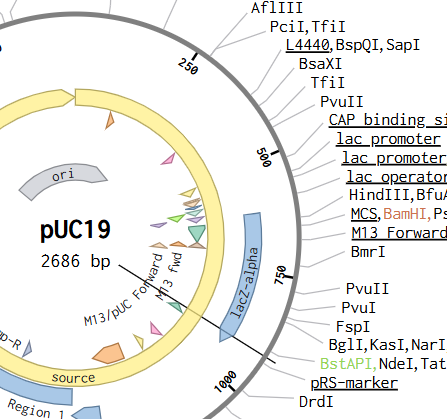
Now we need to find a proper plasmid to use, Gemini suggested i use pUC19 so i looked it up on Addgene
It is a High Copy cloning vector, 2686 bp backbone, with Ampicillin resistance for selection
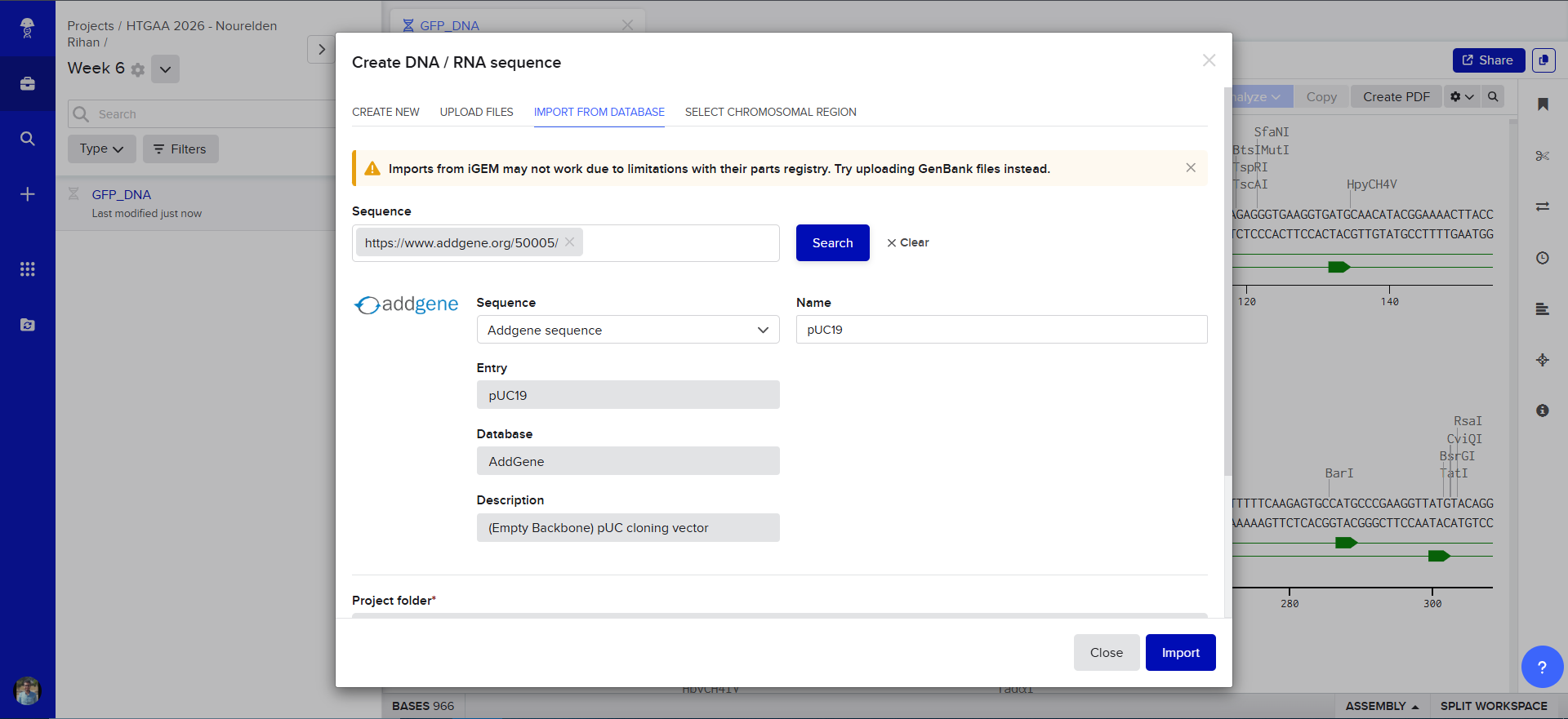
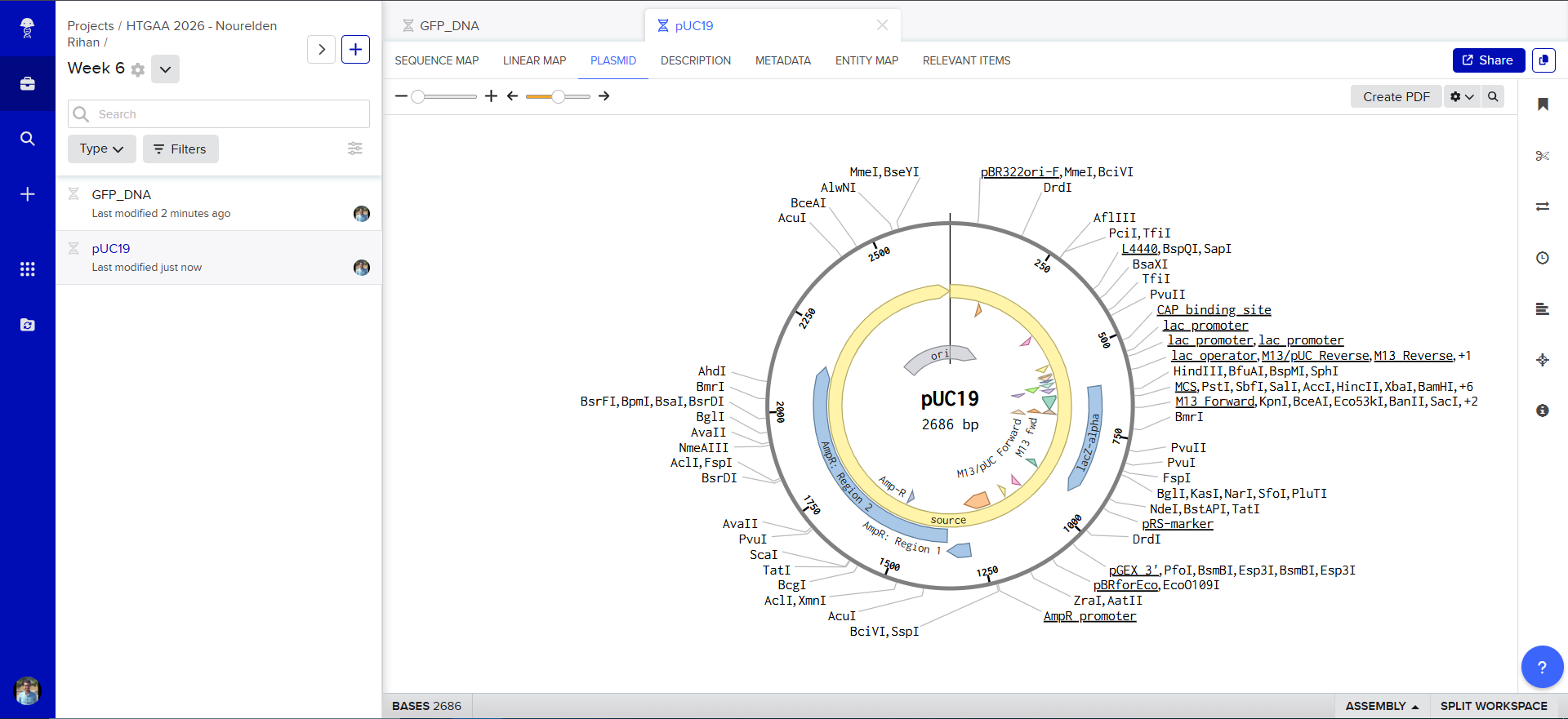
using the Addgene Link https://www.addgene.org/50005/ for pUC19 i imported it into Benchling
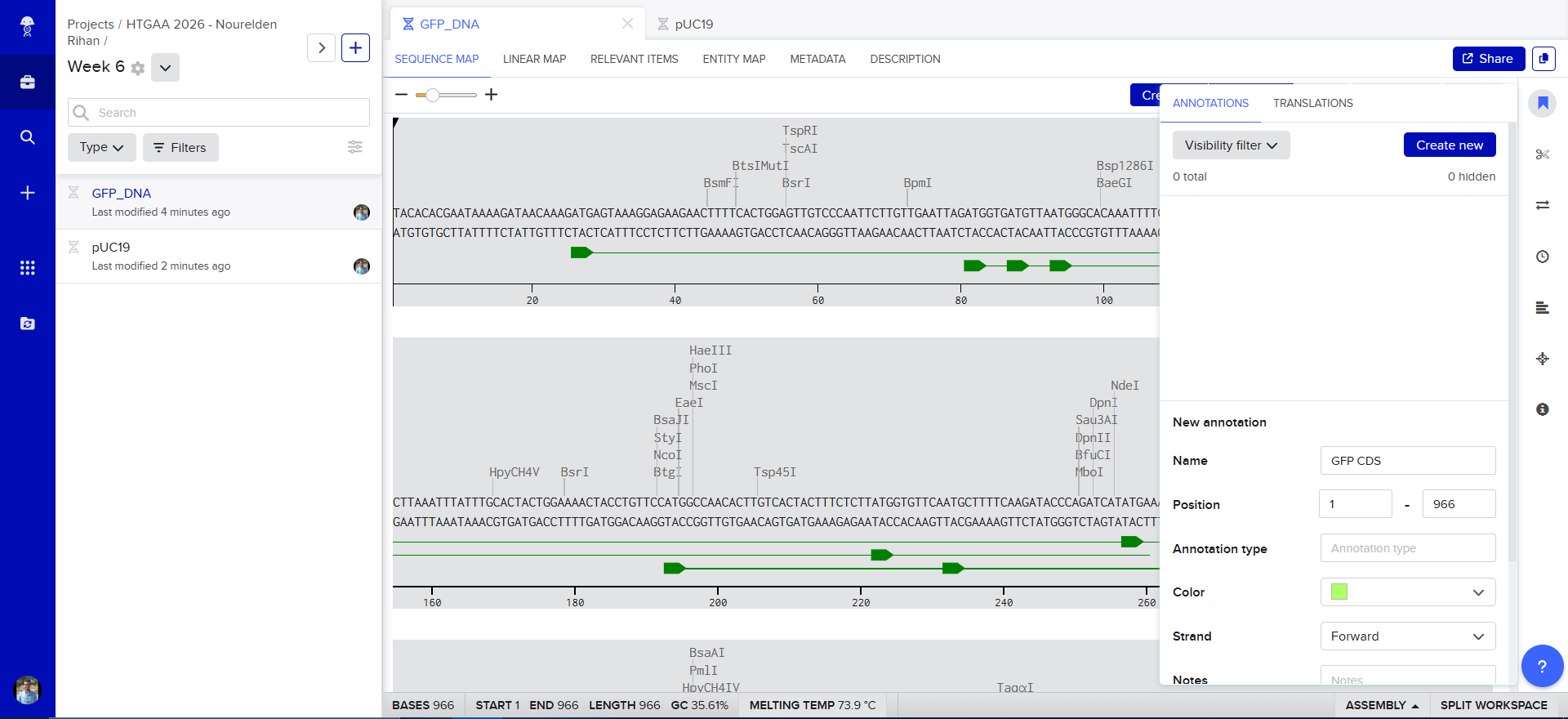
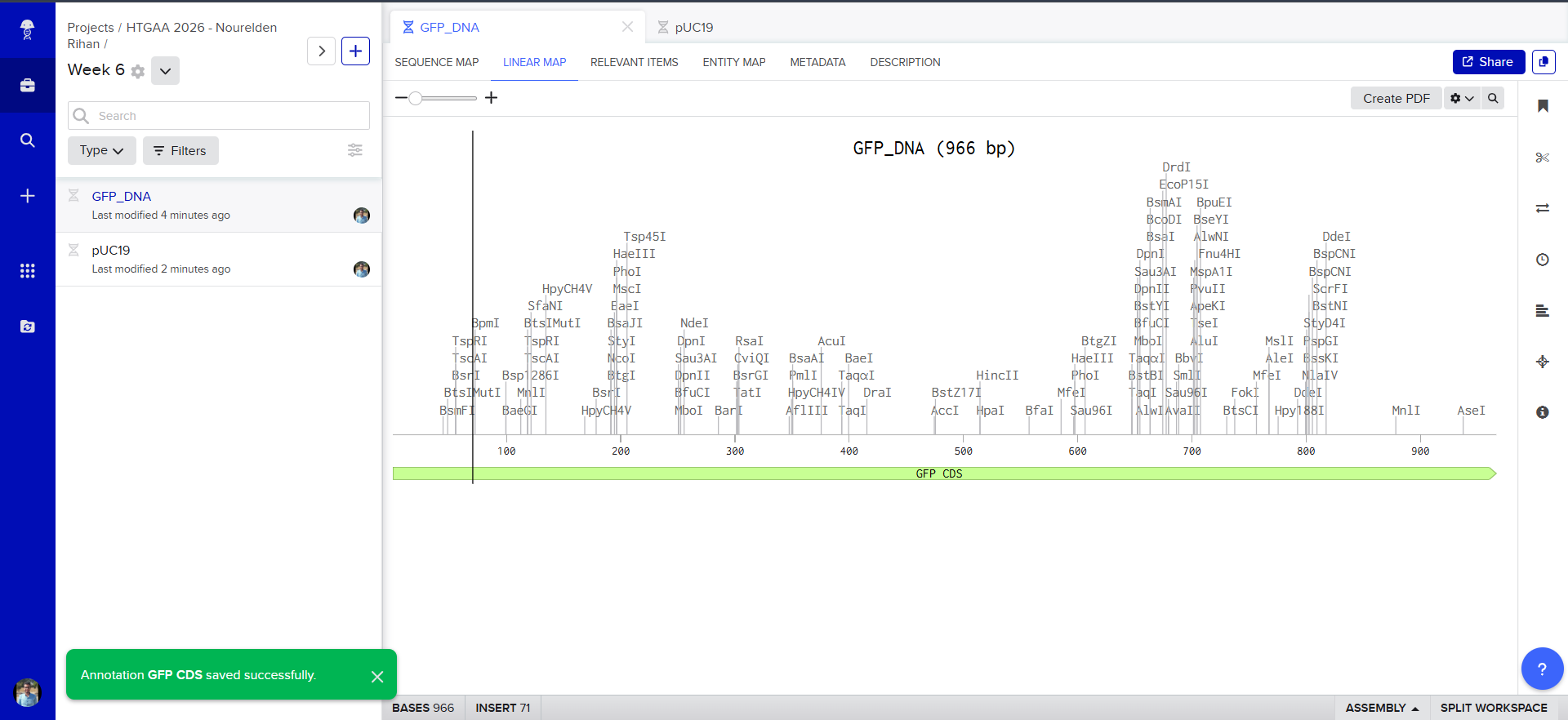
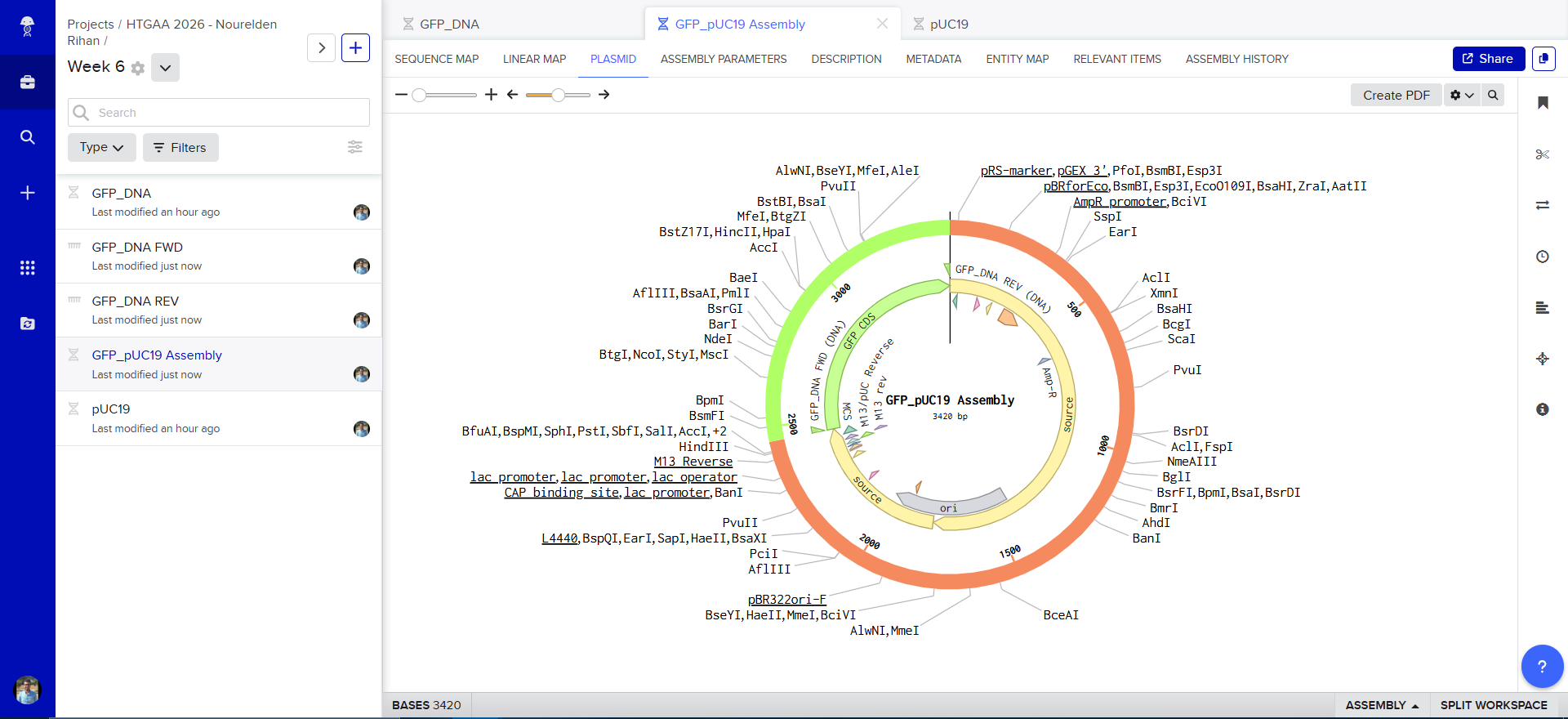
The Plasmid Looked cool with its annotations and was easily readible
so i decided to go in and annotate my GFP too, and since it was just the GFP coding sequence, the process was striaghforward, i just selected it all and named the annotation GFP CDS.
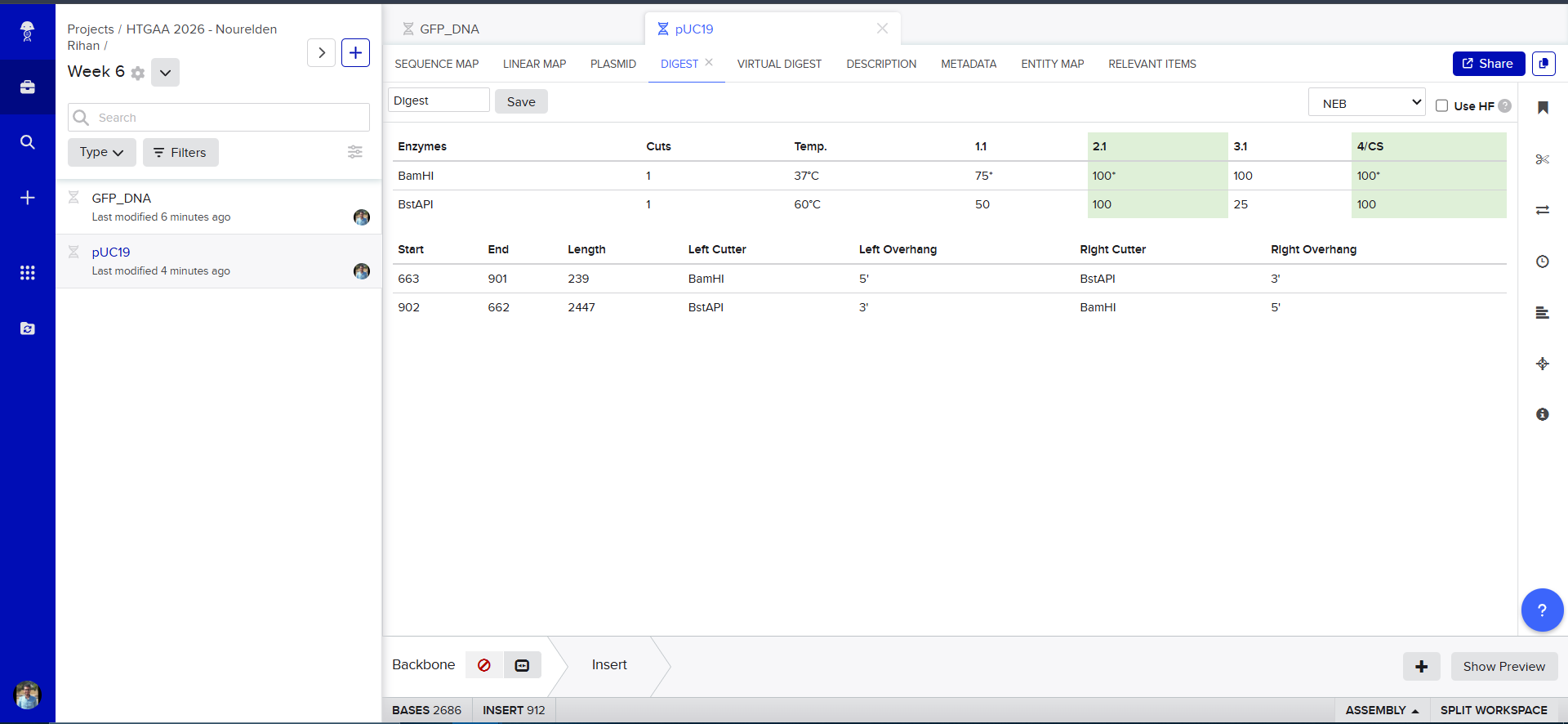
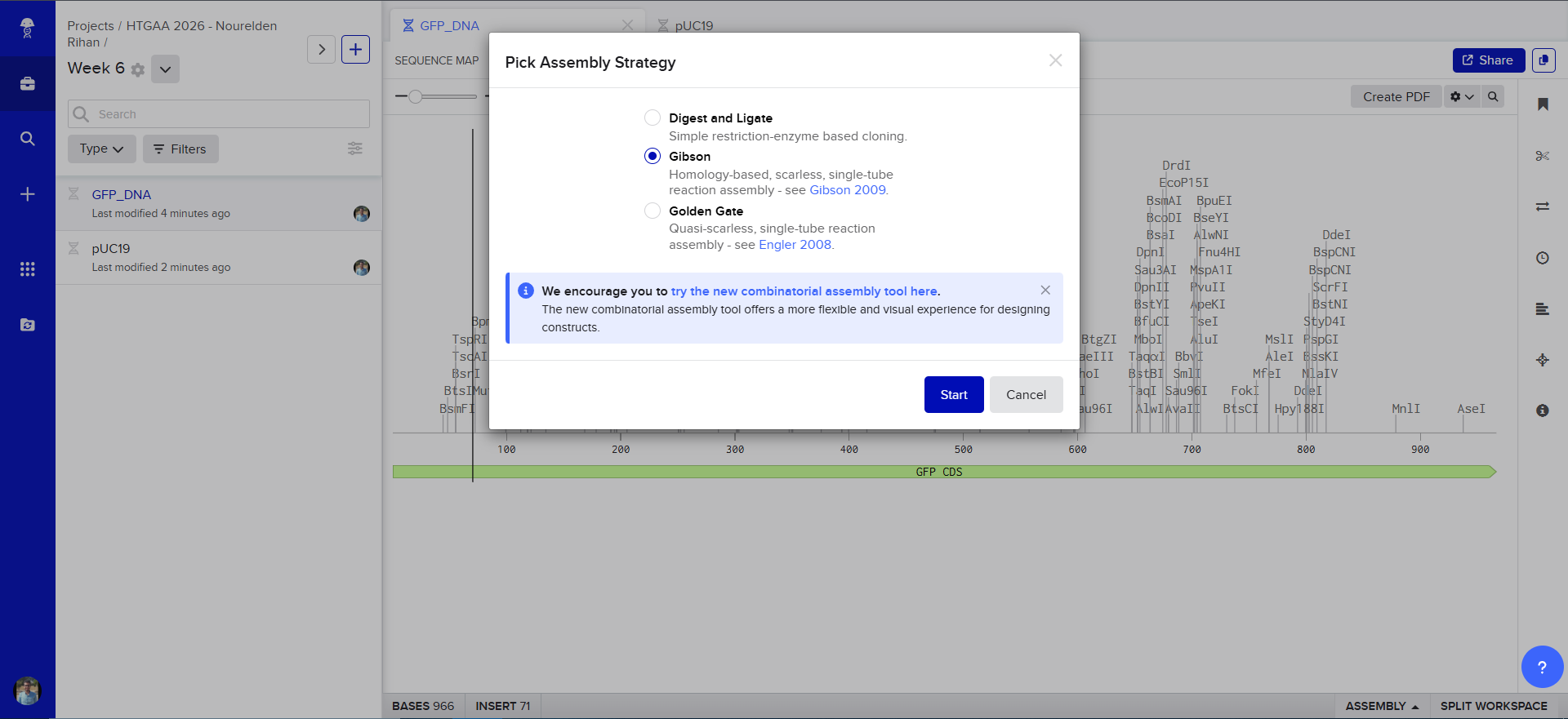
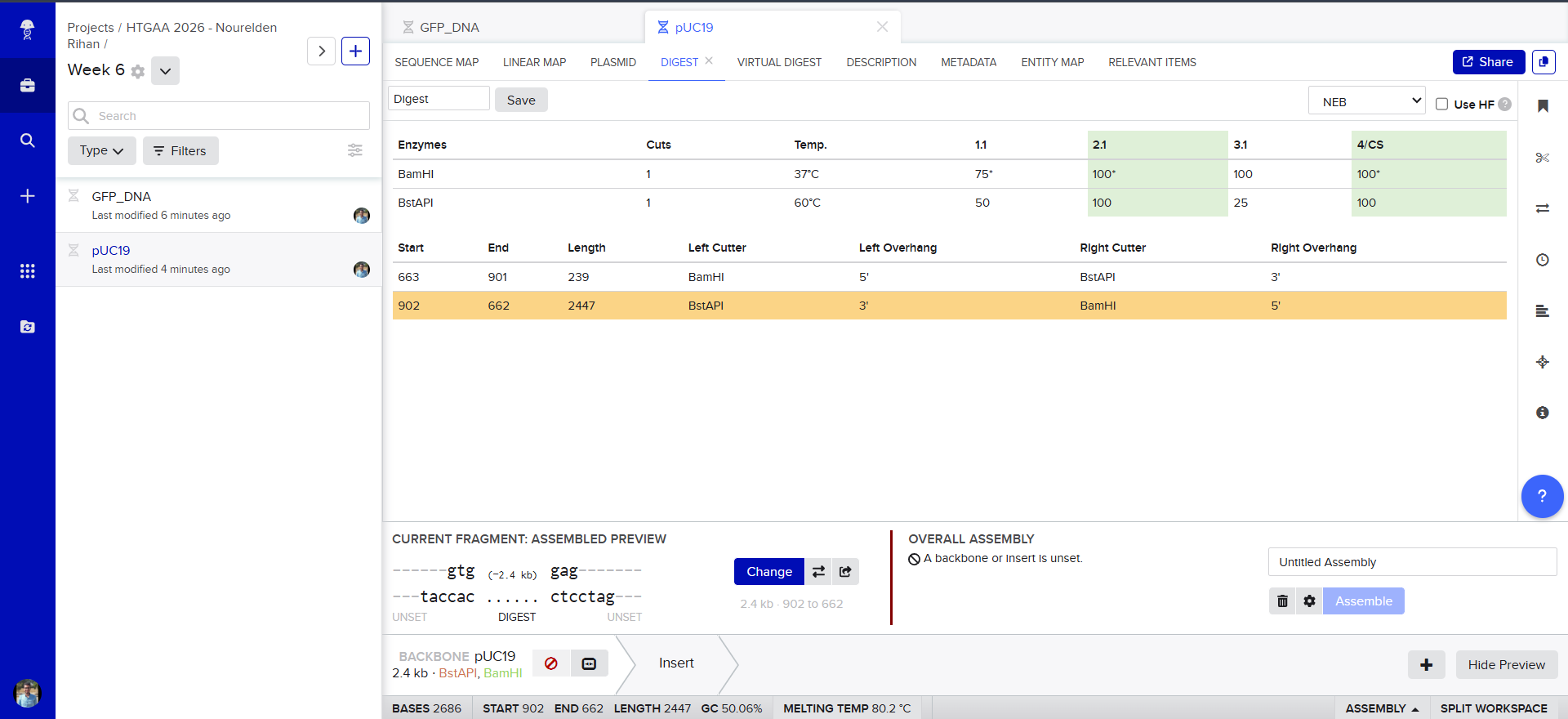
Now we need to cut and Linearize our vector to do Gibson Assembly, so i chose these two restricion enzymes, they are BamHI and BstAPI, why? because these only cut once and inside the Multiple Cloning Site (MCS)
And this is the result of the digest, we have two parts, the discarded small part (239 bp) and our linearized backbone (2447 bp)
And Now we do Gibson Assembly, first through the assembly wizard
then we select our linearized backbone as the “Backbone”
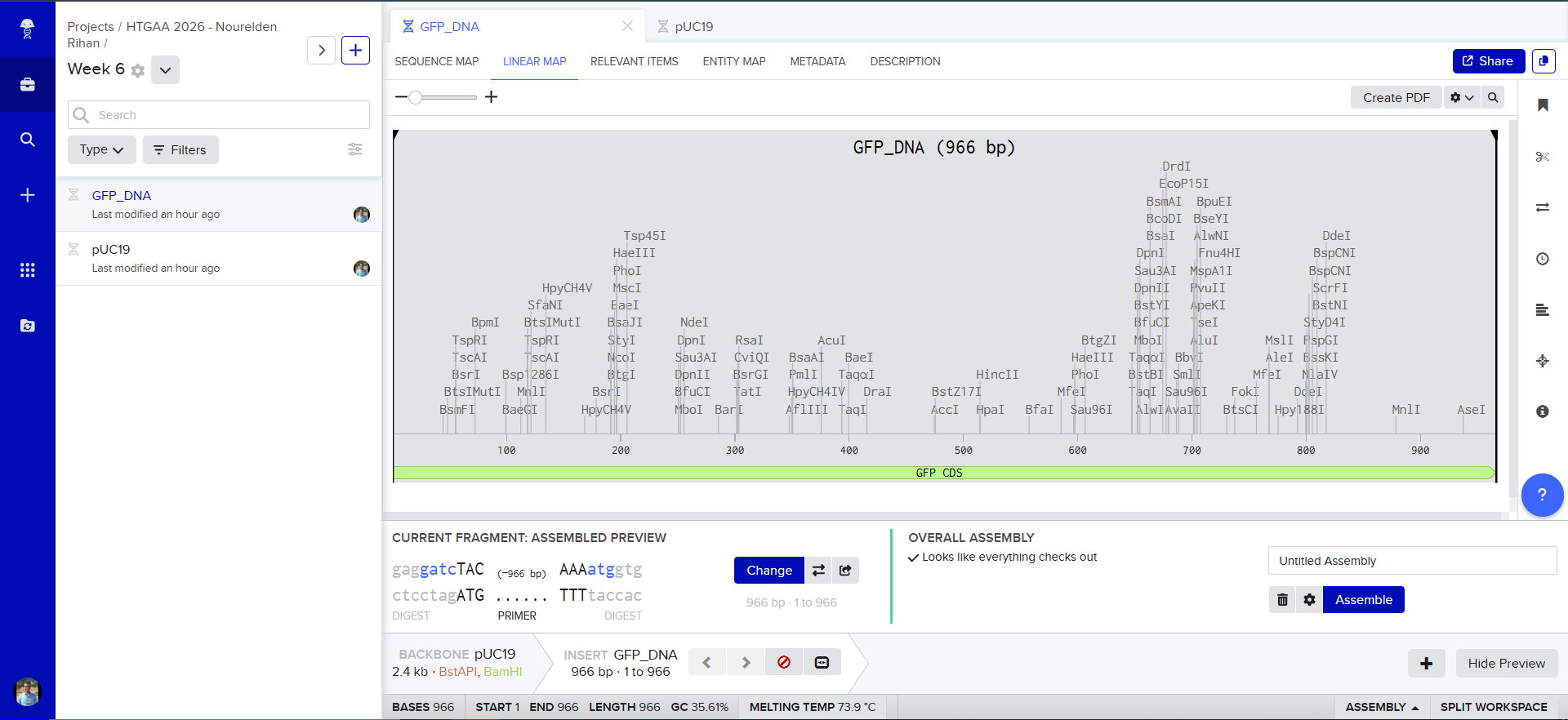
then we select all of our GFP as the “insert”
and tadaaaa! here is our final construct
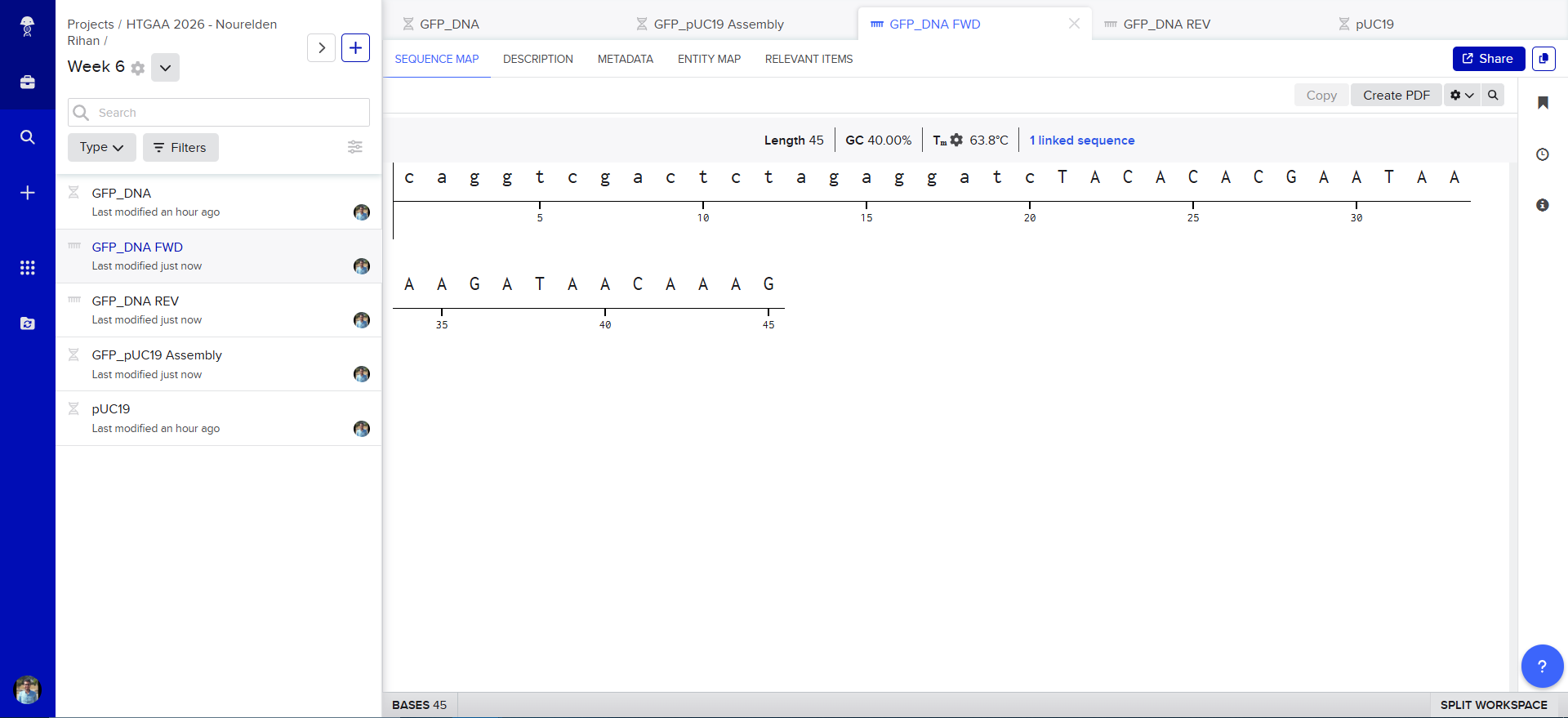
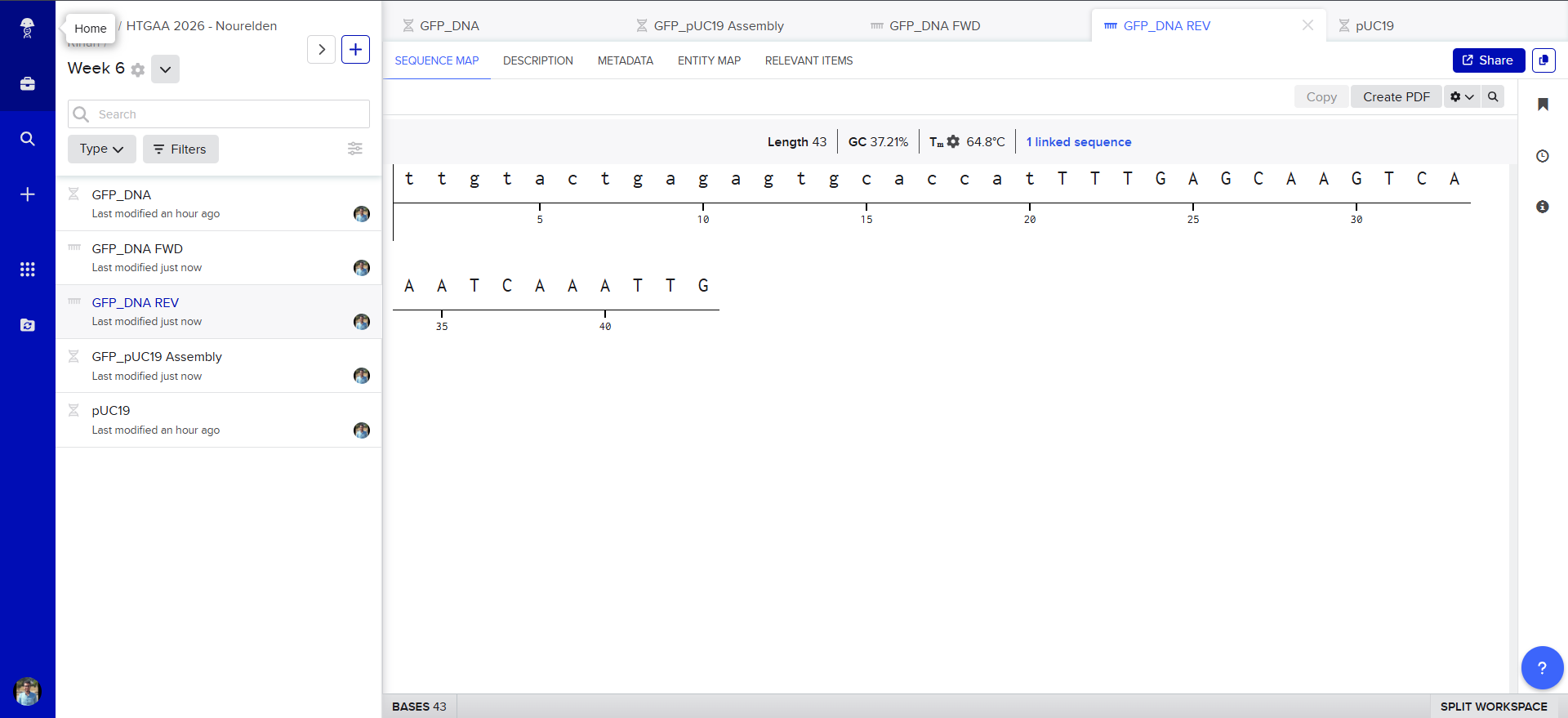
Also Benchling has generated the Forward and Reverse PCR Primers needed for the Reaction, here is the forward primer
caggtcgactctagaggatcTACACACGAATAAAAGATAACAAAG
and here is the reverse primer
ttgtactgagagtgcaccatTTTGAGCAAGTCAAATCAAATTG
𓋹 𓋹 𓋹 𓋹 𓋹
Assignment: Asimov Kernel
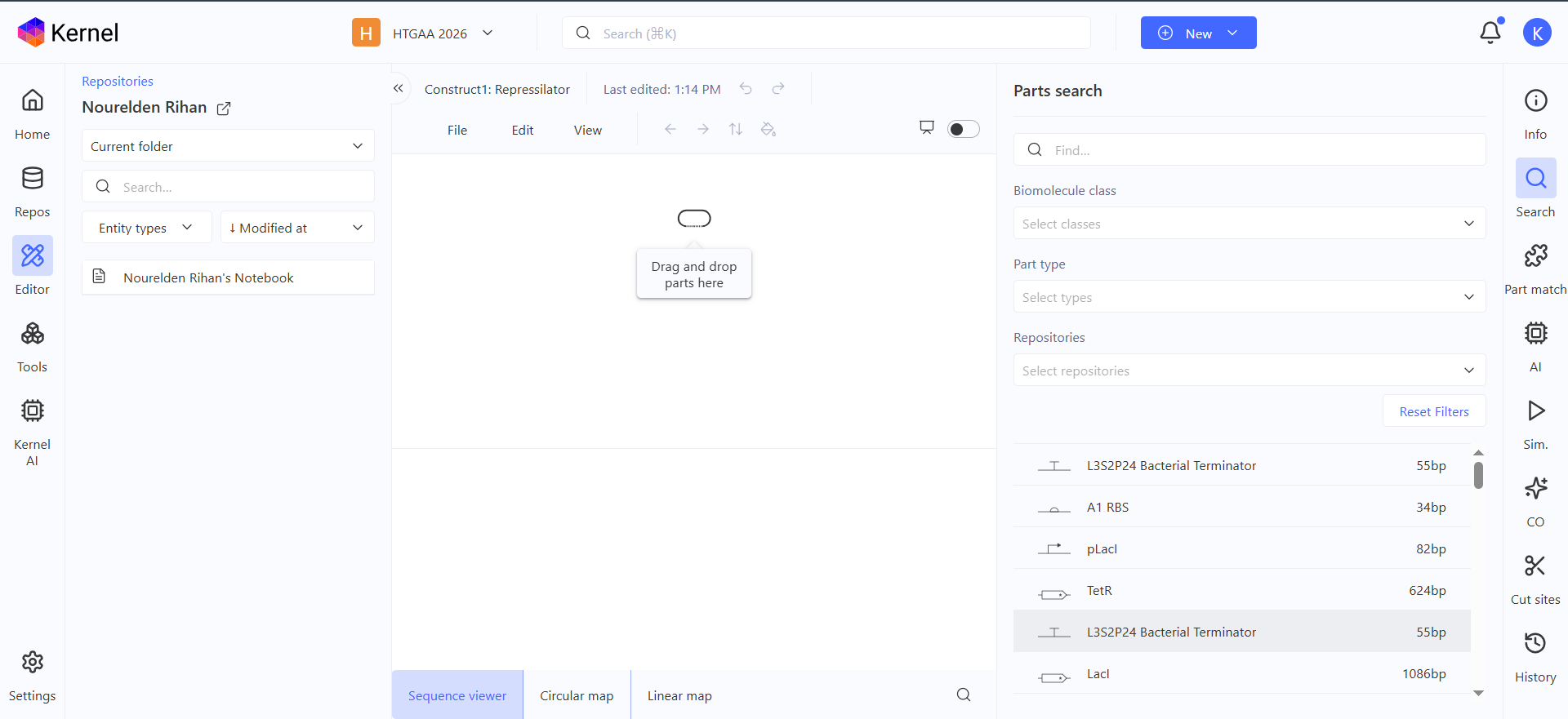
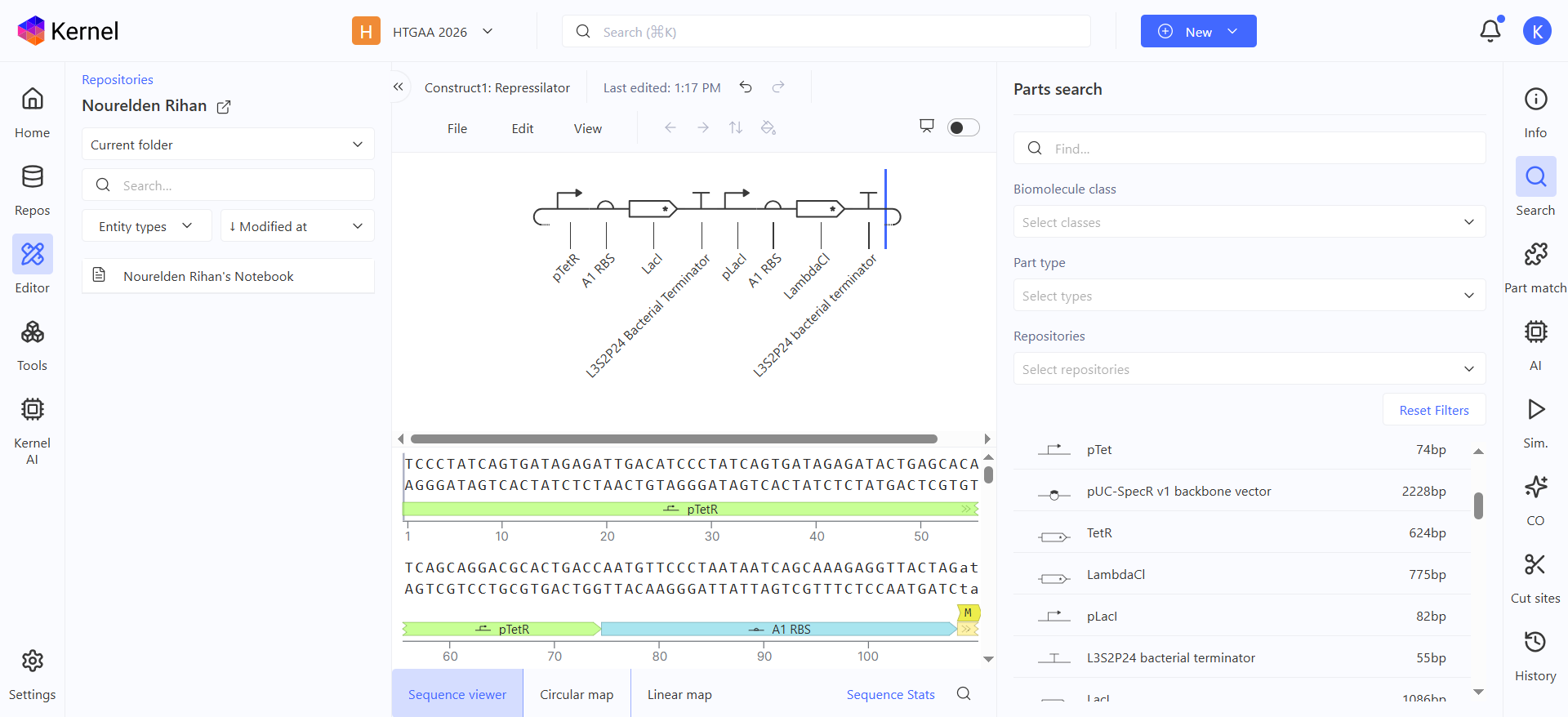
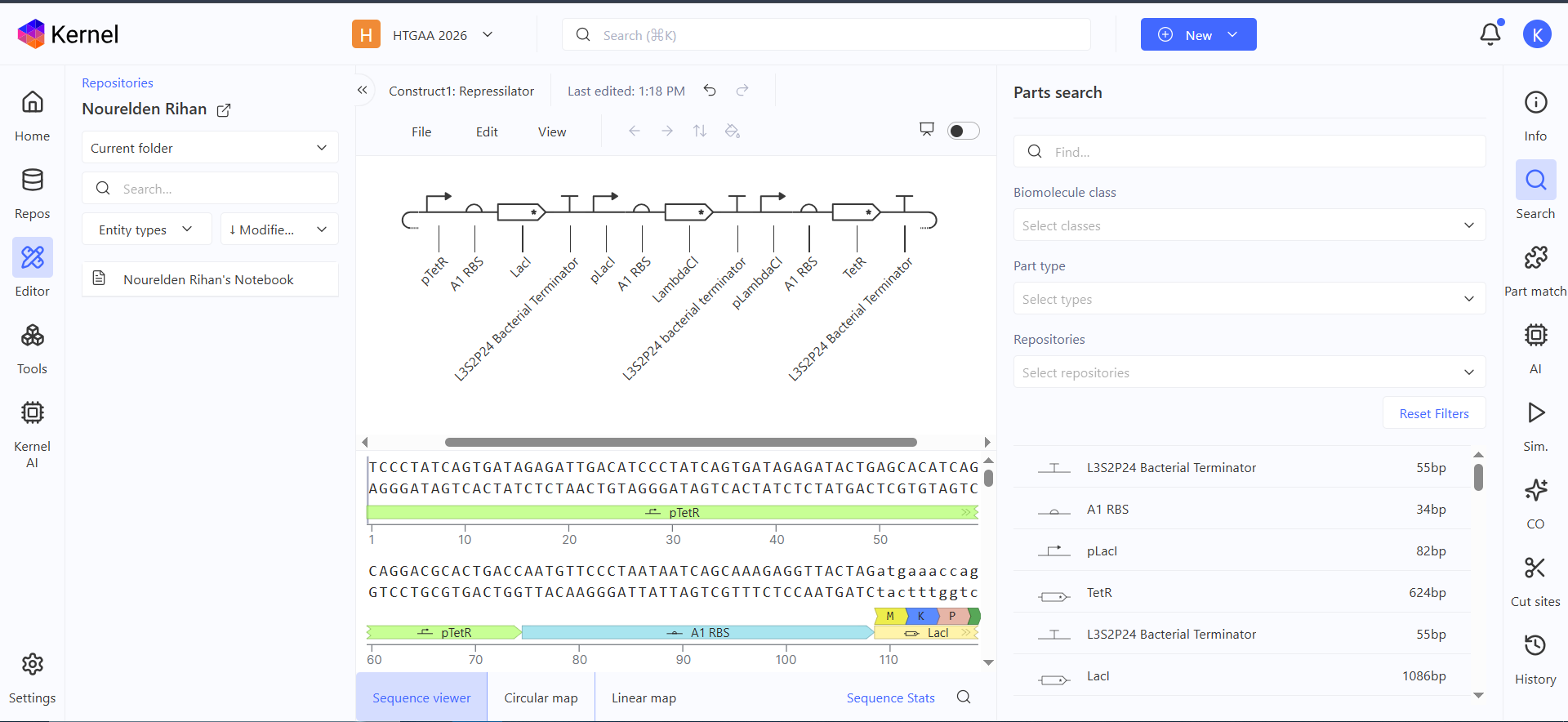
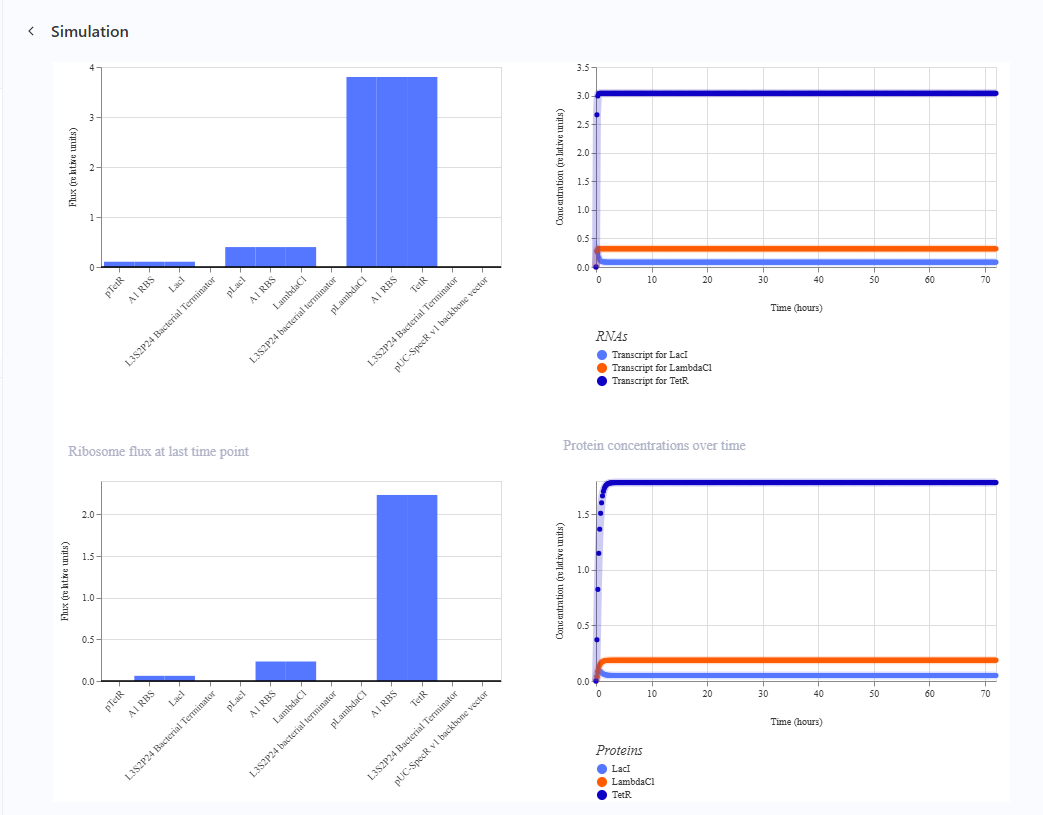
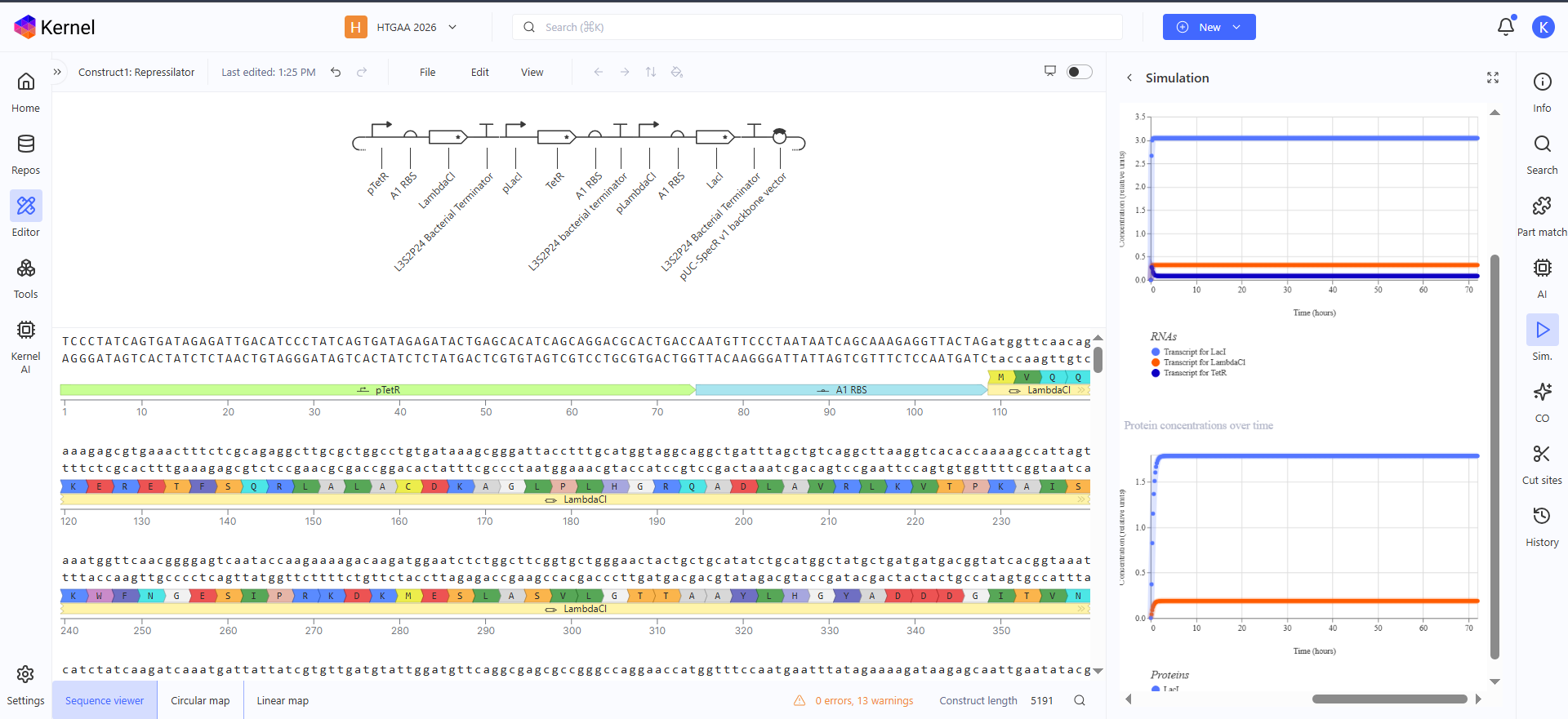
Here is the represilator workflow, i tried to follow the same model that Traci did in the Lecture
This was the simulation i got which is totally different than the Bacterial Demos one :(
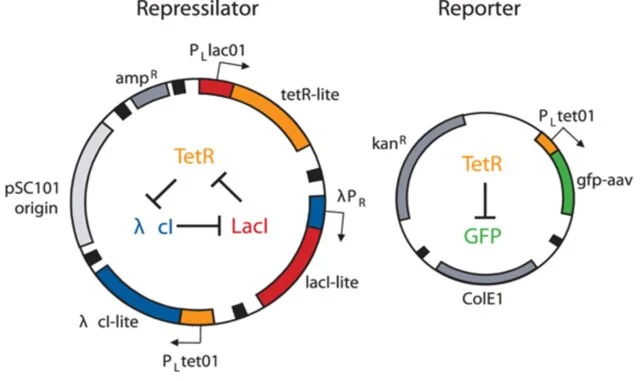
I thought i did it wrong so i looked up this represilator visual
i tried switching up my components to match it but as shown here, the graph is still the same :(
𓋹 𓋹 𓋹 𓋹 𓋹
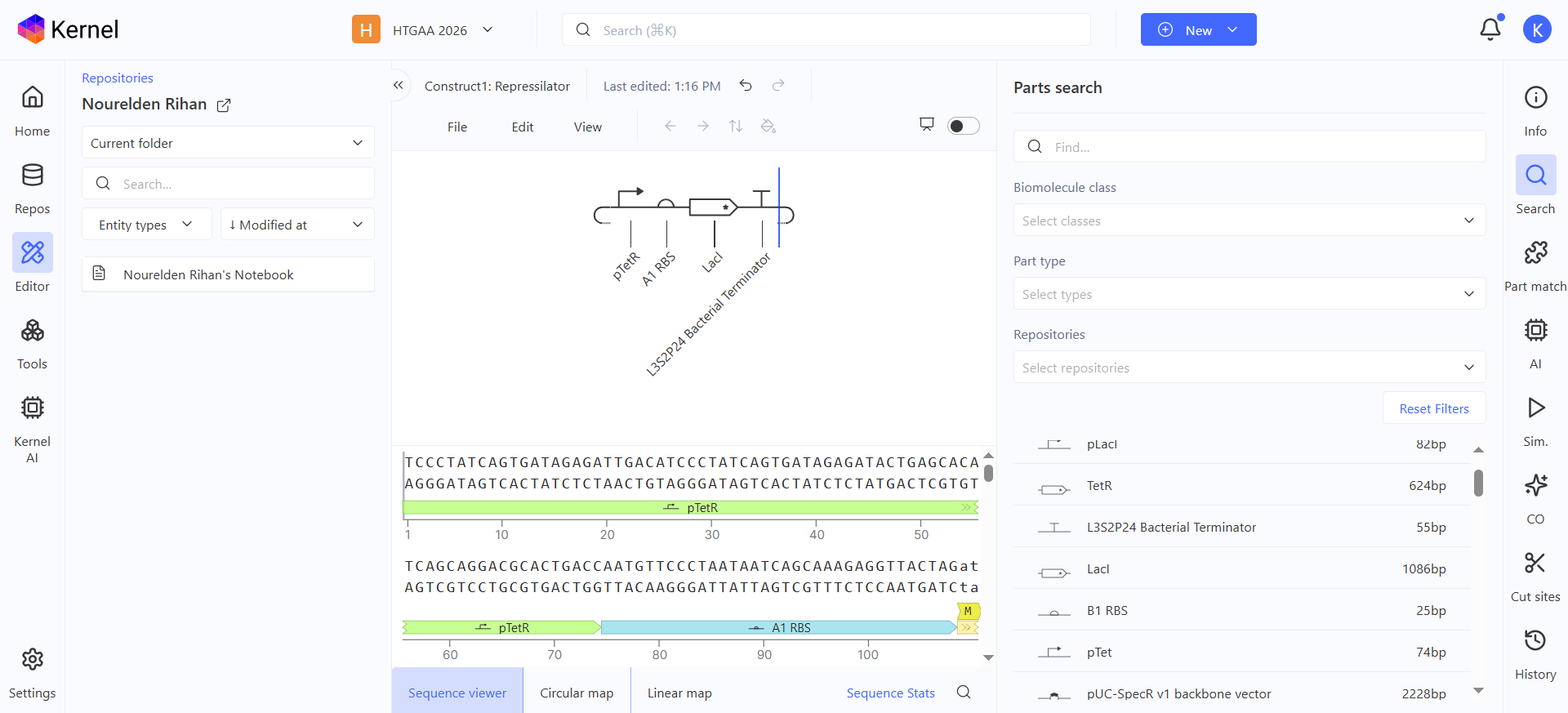
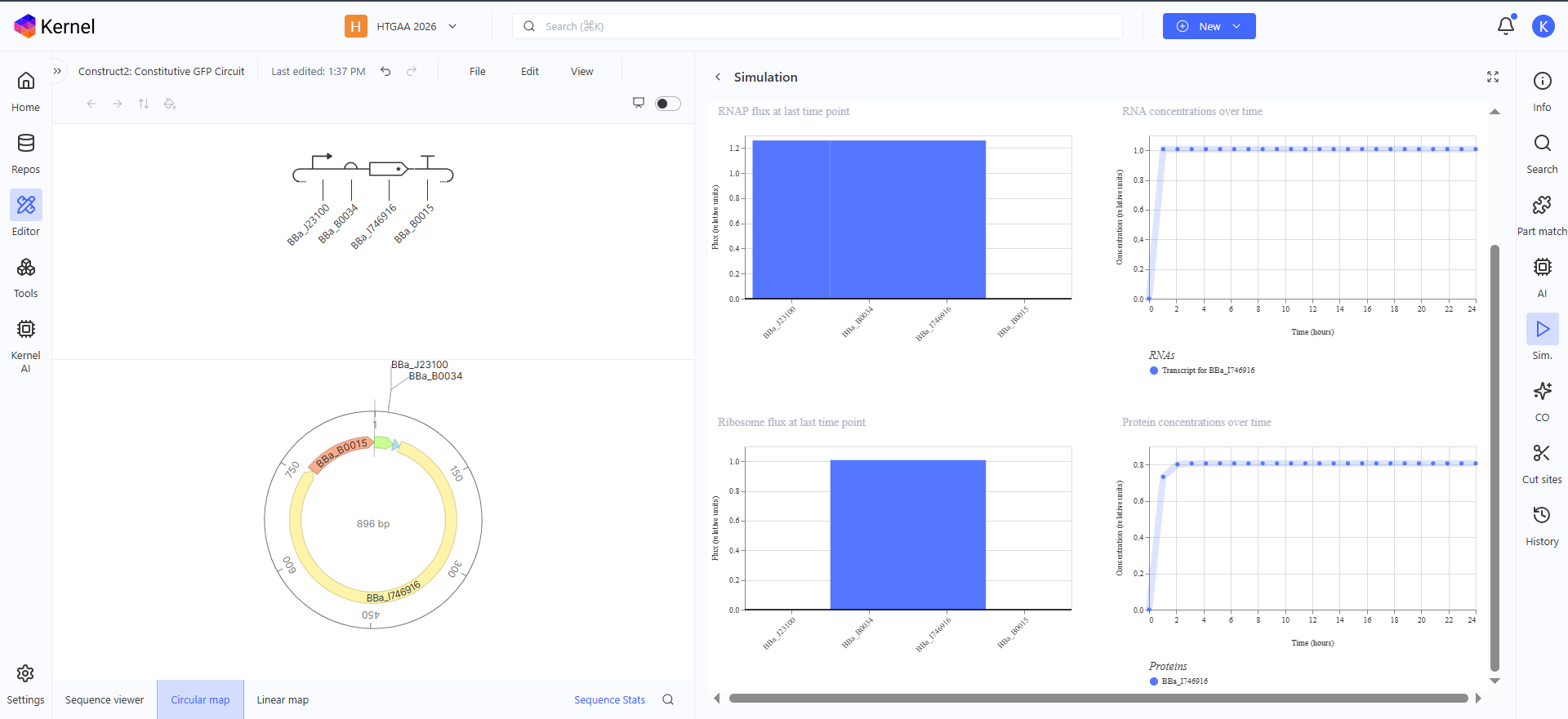
Construct 1: Constitutive GFP Circuit
This is a very simple GFP producing genetic circuit with a constitutive promoter for continous production
𓋹 𓋹 𓋹 𓋹 𓋹
Construct 2: Inducible RFP Circuit
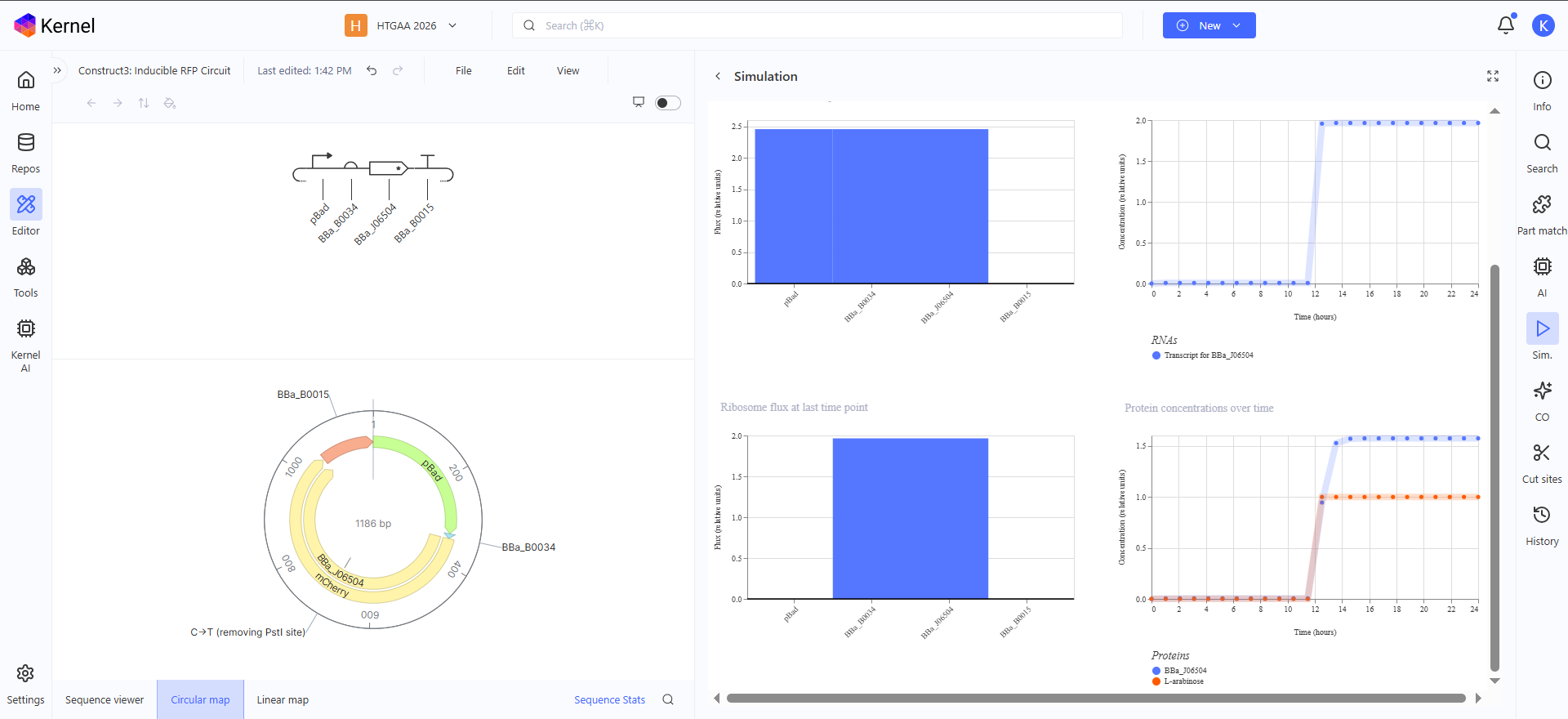
This is an Arabinose controlled RFP producing genetic circuit, in the simulation graph here, i added L-Arabinose at hour 12 so show the increasing production of RFP starting from this point
𓋹 𓋹 𓋹 𓋹 𓋹
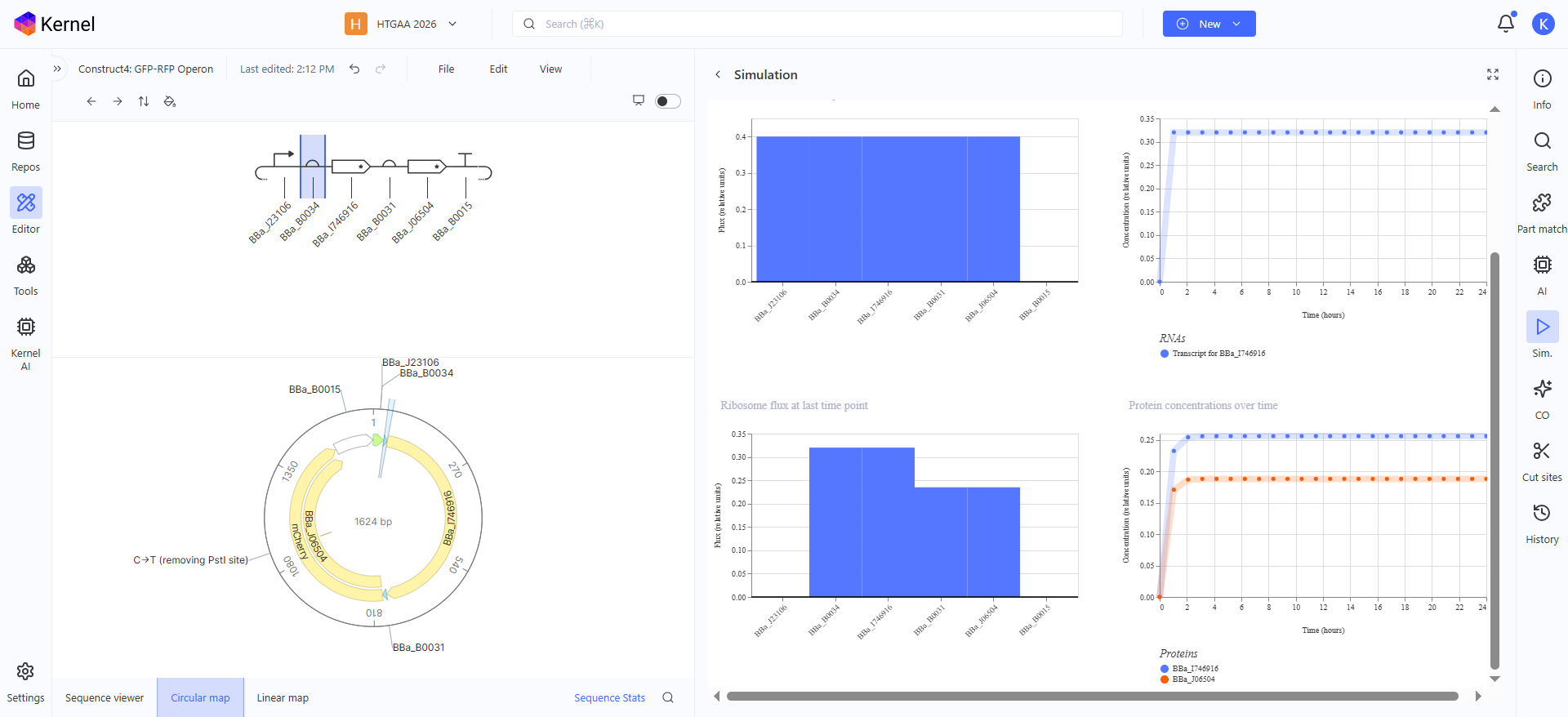
Construct 3: GFP-RFP Operon
This genetic circuit is an Operon of both GFP and RFP, using a single promotor and a single terminator and in between are both the GFP CDS and its RBS and the RFP CDS with its RBS, to make the simulation graph clearer in the production concentrations, i gave the RFP a weaker RBS than the GFP one so now in the graph you can see the protein concentration of RFP is a bit less than GFP
𓋹 𓋹 𓋹 𓋹 𓋹
Week 7 HW: Genetic Circuits Part ii
𓃠 Week 7 Homework 𓃠
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs are more suited for biology since they are not constrained with the digital “0 and 1” appraoch, but can follow an Analog appraoch that is more realistic and suited for biology, because biology is not in an On/Off stage but it varies with different values and expression levels and they are more flexible in that they help in designing more Decision Boundaries without having to create new parts from the scratch and as we have seen with the Neuromorphic wizard, it can also tap and work in Advanced areas like “Dual Region” zones where a cell activates if inputs are strictly below a threshold Or strictly above a threshold, but remains totally inactive in the intermediate zone
𓋹 𓋹 𓋹 𓋹 𓋹
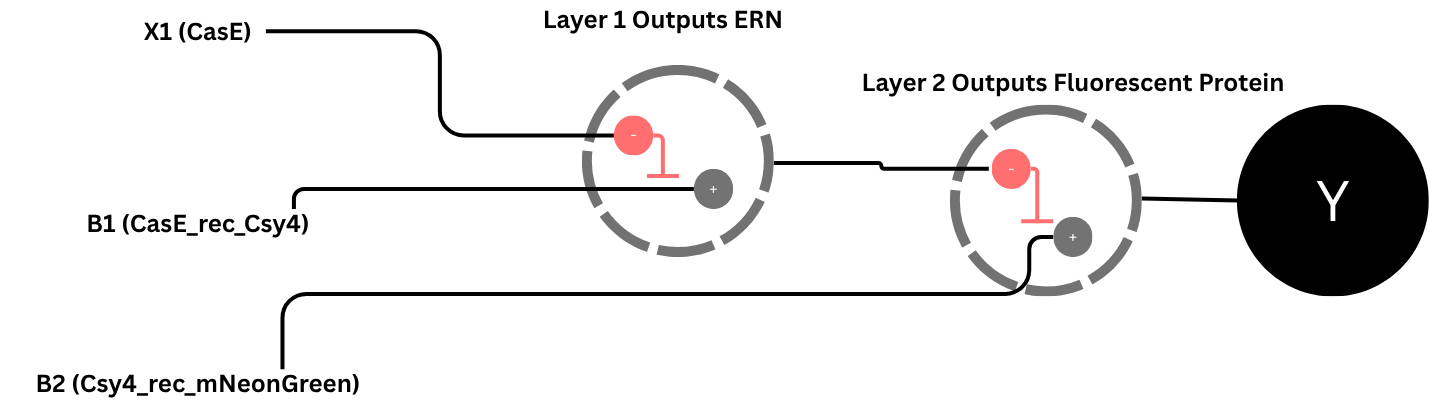
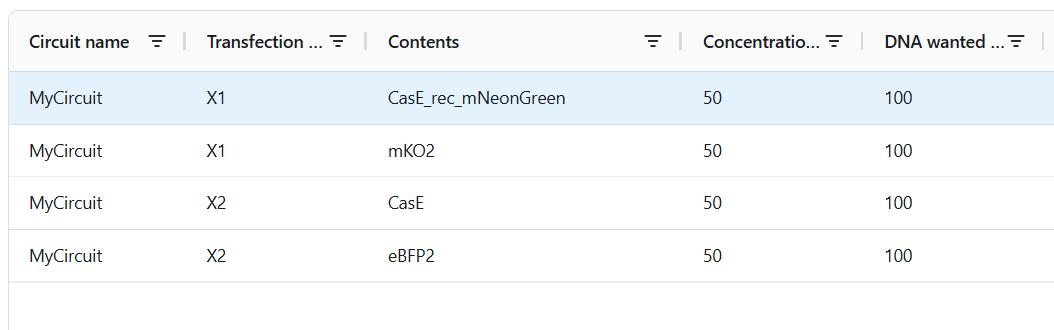
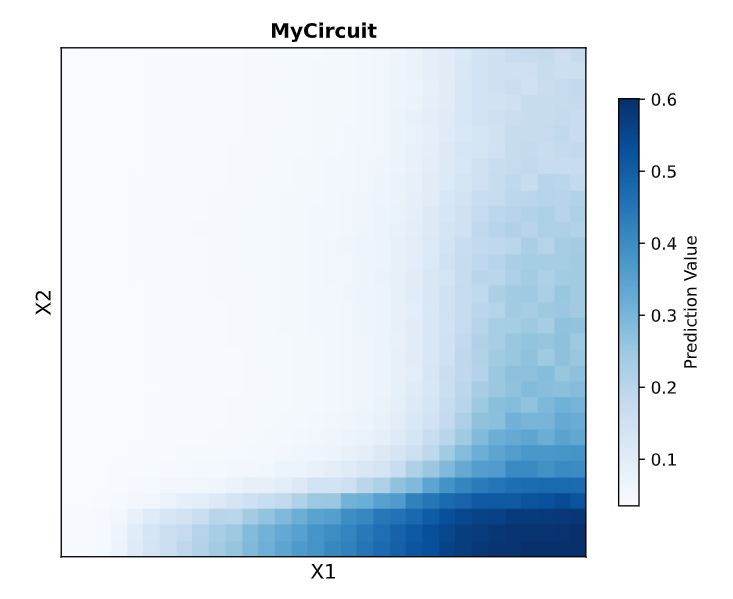
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Useful Applications for IANNs include that it can detect multiple inputs with varying ranging differencies, allowing it to sense and act on subtle expression differences and The network’s decision boundaries can be uniquely tuned to trigger only when the inputs fall into a highly specific area or threshold matching the random, analog nature of biology and on the other hand the network would make no action or keep the outputs at 0 when outside the decision boundaries
Limitations include that using endoribonucleases (ERNs) that bound to the RNA to prevent translation can be biologically expensive since it doesn’t destroy the RNA, it just holds on to it for a long while until it degrades, another one is that the bigger the circuits and the logic gets, probably the more unique ERNs you will need, and this can be tough but it is very important to prevent different branches of the network from accidentally destroying the wrong RNA targets
𓋹 𓋹 𓋹 𓋹 𓋹
3. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
𓋹 𓋹 𓋹 𓋹 𓋹
Lab (Week 7) — Neuromorphic Circuits
For the Lab, After spending countless hours trying to understand how these work and the different components and how to interpret them on the graph, I created this summary to explain what i understood exactly.
Our two main building blocks are colors (positive weights) and Endoribonucleases or ERNs (negative weights). An ERN’s main job is to degrade whatever RNA has a motif/tag that is specific to it. So in the Part Names excel file, you will see parts named like CasE_rec_mNeonGreen. This means that this is an RNA instruction to produce the mNeonGreen protein, but it has a target tag for CasE. If CasE is present, it sticks to that tag and cleaves the RNA, preventing the translation of mNeonGreen. The same concept follows for the other parts as well!
Then comes the prediction/graph part. You can split the graph into 4 quadrants to make it easier to understand. The Upper Left is where our X2 circuit is highly expressed and X1 circuit is not. The Upper Right is where both X2 and X1 are highly expressed. The Lower Right is where X1 is expressed and X2 is not, and the Lower Left is where neither are expressed.
Here is a simple example to clarify everything:
In X1, we put CasE_rec_mNeonGreen: This is supposed to produce the mNeonGreen protein whenever CasE is not available.
In X2, we put CasE: This means in areas where X2 is available, the CasE will destroy the green RNA, so we won’t see the mNeonGreen protein.
If you plot this, because X1 brings the color and X2 brings the ERNs, the cells will only glow in the Lower Right quadrant (where X1 is high and X2 is low)!
After That I tried to create this Circuit, my aim was to produce a graph that is totally blue on the right and fades to white on the left
This is my Setup, let me explain it
It relies on two main biases:
PgU_rec_mNeonGreen which produces the Green color all over the graph and has the PgU tag
Csy4_rec_PgU which cleaves the Green color RNA that has the PgU tag
so far we are supposed to have nothing produced since our second bias Csy4_rec_PgU sequesters our first bias PgU_rec_mNeonGreen, then comes in our X1 and X2.
Remember X1 controls the right half of the graph, which is the part we want filled with color, so X1 should probably break our second bias Csy4_rec_PgU to let our first bias PgU_rec_mNeonGreen produce the color
X1: CasE_rec_Csy4 this sequesters our second bias Csy4_rec_PgU so now the color is available on the right side
This effectively produces our desired graph, nothing else is needed but since we need to have an X2, we can make it so that it stops X1 letting the second bias be alive again Csy4_rec_PgU to stop the coloring, but we need to do it in an equal way so that X2 doesn’t overpower X1 and removes the color from the upper right quadrant
X2: CasE this effecitevly sequesters X1 on the left side and is as equal to it on the right side so it doesn’t whiten that part
I hope i still remember how to explain this when the next BioClub Tokyo homework review comes lol XD
𓋹 𓋹 𓋹 𓋹 𓋹
Assignment Part 2: Fungal Materials
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials, such as mycelium, are currently used to create alternative leathers for fashion, luxury packaging, and household thermal insulation, Because they take the shape of the molds they grow in, they are also used to fabricate solid objects like planters, furniture, and building bricks for structures and remote habitats
Advantages include that they are incredibly lightweight and deeply insulating as Ren mentioned a thin sheet of mycelium can block the heat of a blowtorch and they are also sustainable and can be grown entirely on agricultural waste like wood chips, grain, or hay
Disadvantages include Vulnerability and Contamination during growth and deformation which is to make the final material safe to handle (inert), it must be baked, which can cause the material to dehydrate and shrink which might break the desired shape
𓋹 𓋹 𓋹 𓋹 𓋹
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
We Engineer Fungi for many reasons including Biomanufacturing therapeutics and enzymes, Grow sustainable materials and foods or building bricks and tools
Advantages of fungi over bacteria includes fungi having secretion capacities 10 to 1,000 times higher than bacterial hosts, also they are able to survive harsh and extreme environmental conditions that would destroy bacterial cultures, including highly acidic pH levels, severe dryness, and toxic pollutant surges, also while bacteria typically require a continuous water phase to grow, filamentous fungi build intricate 3D networks of branching hyphae (mycelium) which in huge industrial bioreactors are more efficient to capture and degrade hydrophobic molecules that wet bacterial biofilms struggle to absorb
𓋹 𓋹 𓋹 𓋹 𓋹
Assignment Part 3: First DNA Twist Order
𓋹 𓋹 𓋹 𓋹 𓋹
Week 9 HW: Cell Free Systems
𓃠 Week 9 Homework 𓃠
Homework Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis is basically taking the protein-making machinery out of a living cell and running the process in a test tube. The biggest advantage over traditional in vivo methods (which use living cells) is that you do not have to worry about keeping a cell alive. When you work with living cells, the cell membrane blocks you from easily changing the environment, and the cell’s natural life-cycles get in the way.
By removing the cell entirely, you get an open system. This gives you much more flexibility and control over the experimental variables because you can easily add, remove, or change ingredients in the test tube without worrying about burdening the cell.
Because of this level of control, cell-free expression is much more beneficial than using live cells in a few specific cases like producing toxic proteins, If a protein is naturally toxic, trying to grow it in a living cell will just kill the cell before the process is finished, unlike cell-free systems that don’t have a life to lose, so it can successfully produce it, another example is using unnatural ingredients, If you want to build a protein using artificial or unnatural amino acids, living cells will usually reject them. In a cell-free test tube, you can easily force the system to accept and use these custom parts.
𓋹 𓋹 𓋹 𓋹 𓋹
Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free expression system is made up of five main components that work together to produce a protein outside of a living cell:
Cell Extract: This is the biological “machinery” (like ribosomes and enzymes) harvested from crushed cells. Its role is to actually read the instructions and assemble the protein.
Genetic Template (DNA or RNA): This acts as the blueprint. It provides the specific instructions for which protein the machinery needs to build.
Amino Acids: These are the raw materials or building blocks that the machinery links together to form the final protein.
Energy Source: The process requires power to run.
Reaction Buffer: This is a mixture of salts and chemicals (like magnesium) that maintains the correct pH and environment.
𓋹 𓋹 𓋹 𓋹 𓋹
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical in cell-free systems because making proteins takes a massive amount of energy. If I just add a starting dose of ATP (the fuel) to my test tube, my experiment will fail quickly for two reasons:
I will run out of fuel almost instantly, causing the reaction to stop.
The “dead” fuel (ADP) builds up in the test tube and actually acts like toxic exhaust, slowing down the machinery.
To make sure I have a continuous ATP supply in my experiment, I would use an energy regeneration system like creatine phosphate and creatine kinase. I like to think of this as a backup battery charger. I would add the high-energy chemical (creatine phosphate) and a specific enzyme (creatine kinase) to my test tube. As my machinery burns through the ATP and turns it into dead ADP, the enzyme automatically uses the creatine phosphate to recharge the ADP back into fresh ATP, keeping my experiment running smoothly.
𓋹 𓋹 𓋹 𓋹 𓋹
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
When comparing cell-free expression systems, the main difference comes down to speed, cost, and the complexity of the protein we want to make.
Prokaryotic Systems (e coli): These are fast, cheap, and produce high yields of proteins. However, they lack the machinery to properly fold complex proteins or add necessary chemical modifications.
Eukaryotic Systems (yeast): These are slower and more expensive, yielding less protein. But, they contain the advanced machinery needed to perfectly fold complex proteins and add necessary modifications (like attaching sugar molecules).
My Protein Choices:
For the Prokaryotic System: i’d choose to produce Green Fluorescent Protein (GFP), because GFP is a very simple, standard protein that folds easily on its own. Since it does not require complex modifications, we can take advantage of the prokaryotic system’s speed and cheap cost to produce massive amounts of it quickly.
For the Eukaryotic System: I’d choose to produce a Human Antibody, since Antibodies are highly complex proteins that require intricate folding and specific chemical modifications (like glycosylation) to actually work. A simple bacterial system would build it incorrectly, so we must use the more advanced eukaryotic system to ensure it functions properly.
𓋹 𓋹 𓋹 𓋹 𓋹
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
When designing a cell-free experiment to produce a membrane protein, the biggest challenge we face is that these proteins are highly hydrophobic (water-repelling). Because a standard cell-free reaction mixture is mostly water-based, newly made membrane proteins will naturally clump together and fold incorrectly to hide from the water, becoming completely useless.
To optimize our setup and solve this problem, we need to provide a fatty environment directly in the test tube so the protein has somewhere to go as soon as it is built. We would design the experiment by adding either Liposomes which are artificial bubbles of cell membrane into the liquid. As the machinery builds our membrane protein, it will insert it directly into the liposome wall, mimicking a natural cell, another option is detergennts, We can use specific detergents that wrap around the protein to shield it from the water, keeping it dissolved and properly folded.
𓋹 𓋹 𓋹 𓋹 𓋹
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
If we observe a low yield of our target protein in our cell-free system, it can be either
Reason 1: Our genetic template is degraded. The DNA or RNA instructions might be breaking down before the machinery can read them, and to solve it we would use higher-quality, purified DNA/RNA and add special chemicals (like RNase inhibitors) to the mix to protect the blueprint from being destroyed.
Reason 2: We ran out of energy or building blocks. The system might have burned through all its ATP or used up all the amino acids, here we would add an energy regeneration system (like creatine phosphate) to keep the power on, or by adding a fresh batch of amino acids mid-reaction.
Reason 3: The buffer conditions are wrong. The environment in the test tube, specifically the salt or magnesium levels, might not be ideal for our specific protein, We would set up a few small, test reactions with slightly different magnesium concentrations to find the exact “sweet spot” where our protein grows best.
𓋹 𓋹 𓋹 𓋹 𓋹
Homework question from Kate Adamala
Function and Description
I am designing a synthetic cell that acts as an environmental sensor for contaminated drinking water.
Input: Arsenic.
Output: Red Fluorescent Protein (RFP).
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No, if we just poured our raw cell-free protein machinery directly into a river or a cup of tested water, the machinery would instantly dilute into the water and be destroyed by the environment. The cell membrane acts as a protective shield, keeping our machinery concentrated and safe in its own little bubble (very similar to the example answer given in homeowrk)
Could this function be realized by a genetically modified natural cell?
Yes, but there is a major flaw with living cells here: arsenic is highly toxic. If the water sample is heavily contaminated, the arsenic will likely kill the living bacteria before they even have a chance to produce the red warning color. Because our synthetic cell is basically “dead” chemical machinery, it cannot be killed by the toxin.
Desired Outcome of Operation
When we drop our synthetic cells into a water sample, they will glow bright red if the water is contaminated with arsenic, acting as a clear visual warning.
Design Components
The Membrane: We will use a basic mix of POPC (a standard, easy-to-use lipid) and cholesterol to make the bubble stable.
Inside the Cell (Encapsulation): We will pack it with our cell-free Tx/Tl machinery, amino acids, and the specific genetic instructions (DNA) needed to sense arsenic and build the red protein.
Tx/Tl System Origin
We will use a Bacterial (E. coli) system since bacterial systems are much cheaper and easier for a basic sensor like this.
Communication with the Environment
Because arsenic ions are very small, we will rely on them being naturally permeable to our simple lipid membrane. The arsenic will slip through the fat bubble into the inside. The output (the red protein) is too big to escape, so it stays trapped inside, making the entire bubble light up red.
Experimental Details
Lipids: POPC and Cholesterol.
Enzymes: E. coli cell-free extract (the Tx/Tl machinery).
Genes: We will use the Pars promoter and arsR gene (genetic switch that reacts to arsenic) connected to the mCherry gene (RFP).
Measurement
We will measure the function of our system using a fluorometer (a machine that measures specific colors of light) to track how much red fluorescence is being produced. For a quick visual check, we could even just shine a UV flashlight on the sample to see if it glows red!
𓋹 𓋹 𓋹 𓋹 𓋹
Homework question from Peter Nguyen
Idk why but my idea kinda reminds me of Detroit Become Human XD
Infection-Detecting Smart Bandage
We are developing a smart, wearable bandage that uses freeze-dried cell-free sensors to instantly change color when it detects an early infection in a wound.
How the Idea Will Work
We will embed freeze-dried, dormant cell-free machinery directly into the cotton fibers of a standard bandage. When a person puts the bandage on a cut, the natural moisture from the wound (blood or plasma) will soak into the fabric and “wake up” the machinery. If the cut becomes infected, the harmful bacteria will release specific chemical toxins. Our built-in cell-free sensors are programmed to detect those specific toxins; if they find them, the machinery will produce a brightly colored protein, turning the bandage blue to warn the user of the infection.
The Societal Challenge and Market Need
This directly addresses the massive global problem of delayed medical care and severe infections. Right now, most people do not know a wound is infected until it becomes hot, swollen, and dangerous, often requiring heavy antibiotics or a hospital visit. Our smart bandage gives patients an easy, visual warning at home or disaster araes so they can get a simple treatment before the problem gets worse.
Addressing the Limitations of Cell-Free Systems
Cell-free systems have limits, but we can actually use them to our advantage in this design:
Activation with water: The reaction stays completely paused on the shelf and is only activated by the natural moisture of the wound itself (unfortunately water can provide perfect medium for bacteria to grow).
Stability: To keep the freeze-dried machinery stable for months or years, we simply package our smart bandages in airtight, moisture-proof foil wrappers (just like normal sterile bandages).
One-time use: A cell-free reaction eventually runs out of energy and stops, meaning it only works once. However so does normal bandages :D
𓋹 𓋹 𓋹 𓋹 𓋹
Homework question from Ally Huang
After having a discussion of the idea with Gemini, i was lazy :( and had it do the writing instead because i was really busy sooo i am sorry for that XD
On-Demand Parathyroid Hormone Production
Background Information (Maximum 100 words)
Astronauts lose a dangerous amount of bone mass during long spaceflights due to zero gravity. Taking pre-made liquid medicines from Earth is difficult because they are heavy, take up precious cargo space, and expire quickly due to harsh space radiation. We need a way to manufacture fresh medicines directly on the spaceship. This is scientifically interesting and absolutely critical for keeping astronauts healthy on long missions to Mars where supply drops are impossible.
Molecular or Genetic Target (Maximum 30 words)
The target is the gene for Parathyroid Hormone (PTH), which is a specific protein that tells the human body to build new bone mass.
Relation to Space Biology Challenge (Maximum 100 words)
PTH is a well-known protein medicine used on Earth to treat severe bone loss. By targeting the PTH gene, we can use the BioBits cell-free system as a mini medical factory. Instead of bringing fragile, expiring liquid medicine into space, we just bring the lightweight, freeze-dried DNA instructions for PTH. When astronauts need the medicine, they simply add water, and the BioBits system builds the fresh PTH protein on demand to treat their decaying bones.
Hypothesis and Research Goal (Maximum 150 words)
Our goal is to prove that a freeze-dried cell-free system (BioBits) can successfully manufacture a complex human medicine (PTH) in a zero-gravity environment. We hypothesize that the microgravity on the space station will not stop the cell-free machinery from correctly reading the DNA and assembling the protein. Because cell-free systems are just chemical reactions and do not rely on living cells (which often get stressed or behave unpredictably in space), we believe the protein-building process will work just as perfectly in space as it does on Earth.
Experimental Plan (Maximum 100 words)
On the space station, we will add water to two sets of freeze-dried BioBits tubes and warm them using the miniPCR machine.
Test Sample: BioBits containing the DNA instructions for the PTH bone medicine.
Positive Control: BioBits containing DNA for a red fluorescent protein.
After a few hours, we will look at the control tube using the P51 Viewer; if it glows red, we know the BioBits machinery survived the trip and is working. Finally, we will test our main sample using a simple protein test strip to confirm the fresh PTH medicine was successfully built.
α-Pinene titer: quantity of our target molecule produced per chassis per carbon source. Measured by GC-MS with dodecane overlay extraction from 96-well deep plates.
AgPS protein identity and mass: purified AgPS-His6 protein (via Ni-NTA) can be characterized by intact protein LC-MS to confirm correct molecular mass and verify the His6 tag is present.
Cell growth (OD₆₀₀): optical density at 600nm across all chassis × carbon source conditions. Measured on the PHERAstar FSX plate reader. Tells us if the strain is healthy while producing.
Gene expression (mRNA level): RT-qPCR on the CFX Opus machine using primers targeting AgGPPS2 and AgPS transcripts. Confirms the genes are being transcribed in vivo.
𓋹 𓋹 𓋹 𓋹 𓋹
Homework: Waters Part I — Molecular Weight
The Theoritical Molecular weight of the eGFP Sequence is 28006.60
I have decided to pick two peaks from the Figure 1 that was provided in the homework:
m/z_n (Right Peak): 903.7148 (Lower charge)
m/z_{n+1} (Left Peak): 875.4421 (Higher charge)
1. Charge State Calculation ($z$)
$$ z = \frac{875.4421 - 1}{903.7148 - 875.4421} $$
$$ z = \frac{874.4421}{28.2727} $$
$$ z \approx 30.93 \rightarrow \mathbf{31} $$
Also the zoomed in part is blurry and unclear but i guess i cant identify the charge state anyways, the more i try to look it seems the numbers are so close together mostly 1473 and 1474 with lots of decimal changes in between
𓋹 𓋹 𓋹 𓋹 𓋹
Homework: Waters Part III — Peptide Mapping - primary structure
The eGFP sequence has 19 Lysines (K) and 4 Arginines (R)
19 peptides will be generated from the tryptic digestion of eGFP.
i drew a blue line to mimic the >10% relative abundance but that is probably not exactly 10% XD but anyways, it gives exactly 19 peptides like what was predicted before so they match but if i did it exactly 10%, it would probably not match and they are more peptide fragments here than predicted
The most abundant peak is The peak eluting at 2.78 minutes and has an $m/z$ of 525.76, the spacing between isotopes is mostly 0.5 so the charge will be $1/0.5 = 2$
Single Charged Mass
$$ \text{[M+H]}^+ = (z \times m/z) - (z - 1) $$
$$ \text{[M+H]}^+ = (2 \times 525.76) - 1 $$
$$ \text{[M+H]}^+ = 1051.52 - 1 $$
$$ \text{[M+H]}^+ = \mathbf{1050.52 \text{ Da}} $$
Experimental Mass = 1050.52 Da
The peptide perfect match would be 1050.5214FEGDTLVNR
88% of the eGFP sequence is confirmed by peptide mapping in Figure 6
𓋹 𓋹 𓋹 𓋹 𓋹
Homework: Waters Part IV — Oligomers
Polypeptide Subunit Name
Subunit Mass
7FU
340 kDa
8FU
400 kDa
first we need to calculate the Mass of each of our oligomeric species
Decamer is 10 so didecamer is 20 and 3 decamer is 30 etc
A) 7FU Decamer: Mass = 10 x 340 kDa = 3400 kDa (3.4 Megadaltons (MDa))
B) 8FU Didecamer: Mass = 20 x 400 kDa = 8000 kDa (8 Megadaltons (MDa))
C) 8FU 3-Decamer: Mass = 30 x 400 kDa = 3400 kDa (12 Megadaltons (MDa))
D) 8FU 4-Decamer: Mass = 40 x 400 kDa = 3400 kDa (16 Megadaltons (MDa))
𓋹 𓋹 𓋹 𓋹 𓋹
Homework: Waters Part V — Did I make GFP?
Theoritical
Observed
PPM Mass Error
28006.60
27,984.16
801
𓋹 𓋹 𓋹 𓋹 𓋹
Week 11 HW: Building Genomes
𓃠 Week 11 Homework 𓃠
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I just want to say, this was soooo fun! :D
and i truly love how it changed alot and all the virtual pixel fun wars i’ve had and truces and teamups, it was a really engaging and lovely experience
Here is some screenshots i took.
This was 6 hours before the deadline hit, i was responsible for the Big EGYPT section, and it was getting erased constantly and i was trying to rebuild it again and again haha. i also took part helping in the other parts as well like the 2026 and the LOVE HTGAA ones.
This was a couple minutes before it ended, i was actually asleep because it was 5 am my time, but i set a special alarm to wake up 10 mins before it ends to make sure EGYPT still stands and it wasn’t but in place was MIT and i actually didn’t want to erase that, i love MIT for the HTGAA course, so i decided i’d go on a smaller EGY scale in the lower quadrant
aaaand that was it :D. i really loved it all and to prove that, look at this! :D
I am the Top 1 Contributor :D, and many thanks to Gustavo and Anastasia, i’ve had so much fun coloring with them at times and against them at other times as well :D it was a very fun experience :D.
𓋹 𓋹 𓋹 𓋹 𓋹
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate:
BL21 (DE3) Star Lysate: Provides the essential molecular machinery, including ribosomes, tRNAs, and initiation/elongation factors, while the Star mutation enhances mRNA stability by reducing RNase activity. It also contains T7 RNA Polymerase to drive high-level transcription from T7 promoter-based DNA templates.
Salts and Buffer
Potassium Glutamate: Acts as the primary cytoplasmic salt to maintain osmotic balance and stabilize protein-nucleic acid interactions.
HEPES-KOH pH 7.5: Serves as a buffering agent to maintain a stable pH for the optimal activity of the enzymes.
Magnesium Glutamate: Provides ions that act as essential cofactors for RNA polymerase and ribosomes, stabilizing their structures.
Potassium Phosphate (Monobasic and Dibasic): Maintains pH stability and provides inorganic phosphate, which can assist in the regeneration of nucleoside triphosphates (NTPs) and ATP.
Energy / Nucleotide System
Ribose and Glucose: Serve as energy sources that can be metabolized to regenerate ATP.
AMP, CMP, GMP, UMP: Function as the building blocks (mononucleotides) for RNA synthesis and are interconverted into their triphosphate forms (ATP, CTP, GTP, UTP) to power the reaction.
Guanine: Acts as a precursor for the synthesis of GTP, which is specifically required for the initiation and translocation steps of translation.
Translation Mix (Amino Acids)
17 Amino Acid Mix, Tyrosine, and Cysteine: Provide the full set of 20 proteinogenic amino acids required as the fundamental building blocks for synthesizing the polypeptide chain.
Additives and Backfill
Nicotinamide: Often used as a precursor to NAD+, a cofactor that supports metabolic pathways involved in energy regeneration within the lysate.
Nuclease Free Water: Used as a “backfill” to bring the reaction to its final volume while ensuring the environment remains free of contaminating enzymes that could degrade DNA or RNA.
𓋹 𓋹 𓋹 𓋹 𓋹
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The main difference between the two systems lies in the energy sources and metabolic pathways: the 1-hour PEP-NTP mix uses high-energy phosphoenolpyruvate (PEP) and pre-formed nucleoside triphosphates (NTPs) for rapid, high-intensity protein synthesis. In contrast, the 20-hour NMP-Ribose-Glucose mix utilizes cheaper nucleoside monophosphates (NMPs) and sugars (Ribose/Glucose) to fuel a slower, sustained reaction that significantly reduces costs by regenerating energy over a longer period.
𓋹 𓋹 𓋹 𓋹 𓋹
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Transcription can still occur because the E. coli lysate contains endogenous enzymes (such as phosphoribosyltransferases) that salvage Guanine and convert it into GMP. Once converted to GMP, the energy system further phosphorylates it into GTP, the active nucleotide required by RNA polymerase for transcription.
𓋹 𓋹 𓋹 𓋹 𓋹
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP: Engineered for robust folding and high solubility, it provides a very rapid readout even when fused to other proteins or in less-than-ideal reaction conditions.
mRFP1: As the first truly monomeric RFP, it features a moderate maturation time (half-time ~50 minutes) but can be less photostable than newer variants, potentially leading to signal fading during long observations.
mKO2: This orange protein is highly dependent on oxygen for chromophore maturation; under low-oxygen (hypoxic) conditions typical of some sealed cell-free reactions, its fluorescence recovery is significantly slower.
mTurquoise2: Known for its exceptional brightness and high quantum yield, and requires approximately 66 minutes for maturation.
mScarlet_I: rapid maturation (half-time ~31 minutes), making it one of the fastest-developing red reporters.
Electra2: This blue fluorescent protein is designed for high intracellular brightness and exhibits a tendency to form puncta or aggregates in certain environments.
𓋹 𓋹 𓋹 𓋹 𓋹
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
my focus is gonna be on Magnesium Glutamate, so basically as Mg²⁺ concentration increases, fluorescence output will rise (more ribosome stability, better translation) until a threshold where excess Mg²⁺ precipitates phosphates out of solution, starving the energy regeneration system of Pi, causing ATP regeneration to collapse and translation to halt (seen as a sharp fluorescence drop) so i want to see at which level of Mg²⁺ can CFPS tolerate before crashing in both sfGFP and mScarlet_I to also know which one of them can tolerate it more
This Flowchart shows basically how increasing the Magnesium Glutamate can crash the protein synthesis by disrupting the energy regeneration pool
flowchart TD
A[Mg²⁺ concentration too high] --> B[Mg²⁺ binds phosphate ions in solution]
B --> C[MgPO₄ precipitates out of solution]
C --> D[Inorganic phosphate pool depleted]
D --> E[Kinases cannot regenerate ATP from ADP/AMP]
E --> F[NTP pool collapses - ATP, GTP, CTP, UTP all drop]
F --> G[T7 RNAP stalls, no mRNA produced]
F --> H[Ribosome elongation stalls, translation stops]
G --> J[Less complete protein synthesized]
H --> J
J --> K[Lower fluorescence output]
So here is my wells and what i changed in the master mix to test my hypothesis.
I picked 4 mScarlet_I wells which are:
Q3-B2 (baseline Mg²⁺ - 6.975 mM)
Q3-C2 (Mg²⁺ - 7.600 mM)
Q3-D2 (Mg²⁺ - 8.850 mM)
Q3-E2 (Mg²⁺ - 10.000 mM)
I picked 4 sfGFP wells which are:
Q3-B3 (baseline Mg²⁺ - 6.975 mM)
Q3-C3 (Mg²⁺ - 7.600 mM)
Q3-D3 (Mg²⁺ - 8.850 mM)
Q3-E3 (Mg²⁺ - 10.000 mM)
𓋹 𓋹 𓋹 𓋹 𓋹
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
This is very cool, but i probably did a very bad job haha :D, i will use this chance though to learn more about the different parts of these machines :O
𓋹 𓋹 𓋹 𓋹 𓋹
Week 12 HW: Bioproduction
𓃠 Week 12 Homework 𓃠
No Homework :D
Working on Final Project :D
𓋹 𓋹 𓋹 𓋹 𓋹
Week 13 HW: Bio Design Living Materials
𓃠 Week 13 Homework 𓃠
No Homework :D
Working on Final Project :D
𓋹 𓋹 𓋹 𓋹 𓋹
Week 14 HW: Biofabrication
𓃠 Week 14 Homework 𓃠
No Homework :D
Trying to Finish Working on Final Project :D
𓋹 𓋹 𓋹 𓋹 𓋹
BioClub Committed Listener MoU
HTGAA Committed Listener (CL) Agreement
I am a HTGAA Committed Listener, my responsibilities are:
Watching class lectures and recitations
Participating in node reviews
Developing and documenting my homework
Actively communicating with other students and TAs on the forum
Allowing HTGAA and BioClub to share my work (with attribution)
Honestly reporting on my work, and appropriately attributing and citing the work of others (both human and non-human)
Following locally applicable health and safety guidance
Promoting a respectful environment free of harassment and discrimination
Signed by committing this file to my documentation page/repository,
New Slides
Old Slide
Ship 41 HTGAA Final Project Proposal Project Title: Ship 41: Multi-Chassis Computational and Experimental Optimization of α-Pinene Biosynthesis for High-Value Bioproduction
Subsections of Projects
Group Final Project
Group Name: Phage Forge
Group Members:
@2026a-nourelden-rihan, @2026a-ritika-saha, @2026a-rahul-yaji, and @2026a-keerthana-gunaretnam
We decided to focus on the main area of increasing the stability of the MS2 phage lysis protein L, with a possible secondary goal of reducing the dependency on host DnaJ, while still maintaining the lysis action.
The tools AlphaFold, Clustal Omega, BLAST, ESM, and ESMFold were discussed.
BLAST can pull out homologous lysis proteins from the databases.
Clustal Omega can create MSAs to identify essential L48-S49 residues, and the pore-forming regions that must not be mutated.
ESM can create mutation heatmaps, which can guide the use of ESMFold to obtain highest score foldings in mutatable regions.
AlphaFold Multimer predicts whether the subunits of our protein can successfully create a pore in the host membrane, and also to check whether N-terminus can break the interaction with DnaJ.
We also identified a few pitfalls, with majors ones dealing with limited training datasets, that may not be properly aligned towards creating a transmembrane lysis protein.
Some other pitfalls include the lack of proper annotations for amurins; the possibility of an over-stable protein to form non-functional aggregates; and the vulnerability of modified protein to host proteases.
The study shows that the MS2 phage lysis protein L requires the host chaperone DnaJ for efficient host cell lysis. A missense mutation (P330Q) in the highly conserved C-terminal domain of DnaJ blocks MS2 L-mediated lysis at 30 °C and delays lysis at higher temperatures, without affecting overall L protein synthesis. The defect is specific to L-mediated lysis and does not affect lysis by other phage lysis proteins.
Genetic suppressor screening identified Lodj alleles of the L gene that bypass the DnaJ requirement. These alleles encode truncated L proteins lacking the highly basic N-terminal domain, indicating that this domain confers dependence on DnaJ. Biochemical assays demonstrated that wild-type L forms a membrane-associated complex with DnaJ, whereas the P330Q DnaJ variant cannot interact with L.
The authors propose that DnaJ functions as a chaperone that facilitates proper folding or conformational activation of full-length L, preventing steric interference from the N-terminal domain and allowing L to interact with its unknown cellular target. Removal of the dispensable N-terminal domain eliminates the need for chaperone assistance and accelerates lysis.
The work identifies DnaJ as a host factor regulating MS2 lysis timing and suggests that chaperone-dependent modulation of lysis may be an evolutionary strategy to optimize phage replication cycles.
This study performed comprehensive mutational and genetic analyses of the MS2 phage lysis protein L to identify residues and domains required for function. Random mutagenesis of the 75-aa L protein showed that most loss-of-function mutations cluster in the C-terminal half of the protein, especially around a conserved Leu-Ser (LS) dipeptide motif. Many inactivating mutations were conservative amino-acid substitutions and did not affect protein accumulation or membrane association, suggesting that L function depends on specific protein–protein interactions rather than nonspecific membrane disruption.
Functional studies demonstrated that L-mediated lysis requires interaction with the host chaperone DnaJ. The highly basic N-terminal domain of L is dispensable for lytic activity but mediates DnaJ dependence. Truncation of this domain or certain suppressor mutations bypassed the chaperone requirement and restored rapid lysis.
Biochemical and genetic data support a model in which L is an integral membrane protein whose essential domains (including the LS motif and neighboring regions) form a helical structure that likely engages a host membrane target protein. The interaction may occur near sites of membrane curvature associated with peptidoglycan biosynthesis rather than by forming nonspecific membrane lesions.
The work, supported in part by the Center for Phage Technology and associated laboratories including research by Ry Young, suggests that MS2 L functions through a specific heterotypic protein–protein interaction mechanism and that chaperone-dependent regulation helps control lysis timing during infection.
The study refines the mechanistic model of MS2 lysis, proposing that conserved structural motifs rather than general membrane disruption drive lytic activity.
This study provides detailed in vitro and in vivo characterization of the MS2 lysis protein MS2-L, focusing on its membrane insertion mechanism, oligomerization behavior, and interaction with the host chaperone DnaJ.
Key findings show that MS2-L is a 75-amino-acid phage toxin whose essential lytic activity resides in the C-terminal ~35 amino acids, which form a hydrophobic transmembrane region. The N-terminal soluble domain is not required for bacterial killing but modulates folding, membrane insertion efficiency, and chaperone interaction.
Biochemical assays demonstrate that MS2-L interacts directly with DnaJ, primarily through the soluble N-terminal domain. However, this interaction does not significantly affect membrane insertion, solubilization, or oligomerization of the toxin, suggesting that DnaJ functions more as a folding or stabilization partner rather than being essential for lytic activity.
Native mass spectrometry revealed that MS2-L assembles into high-order oligomeric complexes (≥10 monomers) after insertion into lipid nanodiscs, and oligomerization is driven mainly by the transmembrane domain. In detergent environments, oligomer formation is reduced, indicating that membrane lipid context is important for stable assembly.
Fluorescence microscopy and cryo-electron microscopy showed that MS2-L expression in bacteria leads to peripheral membrane clustering, followed by sequential lesion formation beginning in the outer membrane, then disruption of the peptidoglycan layer, and finally inner membrane disintegration with cytoplasmic leakage.
The data support a model in which MS2-L functions as a pore-forming phage toxin that kills cells through higher-order oligomerization within the bacterial membrane, rather than by directly inhibiting peptidoglycan biosynthesis. Chaperone DnaJ binds MS2-L but is not required for membrane insertion or pore assembly, suggesting its role is mainly in modulating toxin folding or stability.
These findings strengthen the concept that MS2-L belongs to the amurin/single-gene lysis protein family and may be useful for bioengineering applications such as bacterial ghost cell production and antimicrobial design.
This review from Elsevier surveys the biological mechanisms, clinical development, and future directions of phage therapy as a strategy to combat antimicrobial resistance. It explains that therapeutic phages should ideally be strictly lytic, highly host-specific, and thoroughly characterized to ensure safety and efficacy.
The article describes how phages kill bacteria through mechanisms such as inhibition of essential cellular processes, expression of lysis proteins, or disruption of bacterial membranes. It also discusses advances in phage engineering, including synthetic genome construction and modification of phage host range and virulence.
Clinical applications of phage therapy are highlighted, particularly for treating drug-resistant infections where antibiotics are ineffective. However, challenges remain, including bacterial resistance to phages, regulatory hurdles, manufacturing standardization, and the need to understand phage–host interactions.
Future directions include the use of genetically modified or synthetic phages, computational prediction of therapeutic candidates, and integration of phage therapy with conventional antimicrobial strategies. Overall, phage therapy is presented as a promising but still developing alternative to antibiotics in the fight against antimicrobial resistance.
This preprint reports the first experimental demonstration of generative design of complete bacteriophage genomes using genome language models (Evo 1 and Evo 2). The authors fine-tuned models on about 15,000 Microviridae phage genomes to enable autoregressive generation of full viral genomes guided by template-based prompts and biologically motivated design constraints.
The workflow involved computational generation followed by multi-tier filtering for sequence quality, host tropism specificity, and evolutionary diversity. Constraints included genome length (4–6 kb), GC content, absence of long homopolymers, preservation of phage-like gene architecture, and spike protein similarity to the template phage to maintain host targeting.
Experimental validation showed that about 285 of 302 synthesized genome candidates could be assembled, and 16 produced viable infectious phages that inhibited growth of the target host strain. These generated phages displayed substantial sequence novelty, containing hundreds of mutations relative to natural Microviridae genomes, while preserving functional genome organization.
Structural and functional analyses indicated that some generated phages possessed altered protein interfaces but maintained compatible capsid–protein interactions. Cryo-electron microscopy and structure prediction suggested context-dependent co-evolution of structural proteins such as capsid and packaging proteins.
Fitness assays showed that several AI-generated phages matched or exceeded the replication and lytic performance of the template phage, and phage cocktail experiments demonstrated rapid suppression of resistant bacterial strains through recombination and mutation-driven adaptation.
The study was conducted with biosafety considerations, including restricting model training to bacteriophage genomes and using well-characterized laboratory strains. The work was supported by researchers affiliated with institutions such as the Stanford University and the Arc Institute.
Overall, the paper proposes a framework for generative genome engineering, showing that AI models can design biologically viable and evolutionarily novel bacteriophages, potentially enabling future synthetic biology and phage-based therapeutic development.
Overview of the Project Proposal: Engineering the MS2 Phage Lysis Protein L
Our primary goal is to increase the structural stability of the MS2 bacteriophage lysis protein (L) while maintaining its ability to lyse bacterial cells.
Our secondary goal is to reduce the dependency of L on the host chaperone DnaJ, which normally assists the protein in folding or activation. Reducing this dependency could allow the lysis protein to function more efficiently and independently in engineered systems.
The MS2 L protein is a 75-amino-acid single-gene lysis toxin whose C-terminal region forms a hydrophobic transmembrane domain responsible for membrane disruption and pore formation, while the basic N-terminal domain interacts with host factors such as DnaJ. Previous studies show that truncation of the N-terminal region can bypass the DnaJ requirement while preserving lysis activity.
Therefore, our design strategy focuses on:
Stabilizing the transmembrane and oligomerization regions
Maintaining essential functional motifs such as the L48–S49 motif
Exploring modifications to the N-terminal region to reduce DnaJ dependence
2. Computational Tools and Approaches
We will use a multi-step computational protein engineering pipeline combining sequence analysis, machine-learning mutagenesis predictions, and structural modeling.
2.1 BLAST – Homolog Discovery
First, we will use BLAST to identify homologous lysis proteins from related bacteriophages.
Purpose:
Identify evolutionarily conserved residues
Discover natural sequence variations that maintain function
Build a dataset for multiple sequence alignment
This will help determine which regions are functionally constrained vs mutable.
Select Candidate Mutations (stability or N-terminal modifications)
Structure Prediction (ESMFold)
Complex/Oligomer Prediction (AlphaFold Multimer)
Final Mutant Candidates (stable + functional lysis protein)
3. Proposed Engineering Pipeline
Computational workflow we will follow.
4. Expected Outcomes
Our pipeline aims to produce engineered variants of the MS2 L protein with:
Increased structural stability
Reduced aggregation risk
Maintained transmembrane insertion
Potentially reduced dependency on host DnaJ
These optimized proteins could be useful in applications such as:
Synthetic phage engineering
Bacterial ghost cell production
Antimicrobial protein development
5. Potential Pitfalls
5.1 Limited Training Data
Most protein language models and structural predictors are trained primarily on globular proteins, not small transmembrane phage toxins.
This may reduce prediction accuracy for MS2 L.
5.2 Risk of Over-Stabilization
Mutations designed to increase stability may cause:
Protein aggregation
Improper membrane insertion
Loss of functional oligomerization
Thus stability must be balanced with function.
5.3 Poor Annotation of Amurin Proteins
Single-gene lysis proteins (also called amurins) are poorly annotated in sequence databases.
This may limit the quality of homologous sequences used for alignment and training.
5.4 Host Protease Sensitivity
Mutations may unintentionally expose protease cleavage sites, making the engineered protein less stable inside bacterial cells.
6. Future Work
If promising mutants are identified computationally, the next steps would include:
Experimental expression in E. coli
Measuring lysis timing
Measuring protein stability
Testing DnaJ independence
This would validate whether computational predictions translate into improved biological function.
L-Protein Mutants Project Summary:
The MS2 bacteriophage L-protein is a small 75-residue lysis protein with two functional regions: a soluble N-terminal domain (residues 1–40) that interacts with the bacterial chaperone DnaJ, and a transmembrane domain (residues 41–75) that disrupts the inner membrane to trigger host cell lysis. The goal of this project was to computationally design five single-point mutants with the potential to retain or enhance lysis activity. This was done by first running a Clustal Omega Multiple Sequence Alignment to map out conserved positions across homologs, all of which turned out to cluster exclusively in the soluble domain, pointing to the DnaJ interface as the most functionally constrained region of the protein. An LLR mutation heatmap was then generated and cross-referenced against an experimental lysis dataset; three mutations that appeared in both were excluded after showing a lysis score of zero experimentally, highlighting that LLR scores reflect structural stability rather than functional activity. The final five mutants, Y39L and S9Q in the soluble domain, and K50L, N53L, and T52L in the transmembrane domain, were selected based on the highest LLR scores while avoiding all conserved and experimentally failed positions.
Each mutant was then modeled as an octameric pore complex using AlphaFold Multimer to simulate the expected membrane assembly. The key findings were:
All five mutants formed a cylinder-like pore structure, consistent with the expected transmembrane assembly
pLDDT scores were low (< 50) across all models, and PAE plots showed near-zero confidence for inter-chain contacts
Running the same pipeline on R30Q, an experimentally validated lytic mutant, produced identical low-confidence results, suggesting this is a systematic limitation of AlphaFold for this class of small membrane-disrupting proteins rather than a reflection of the mutants’ actual viability
All five mutants remain candidates for wet lab validation
It seems like these Positions [21,25,28-29,33,35-37,40] have an “*” which means these have not changed at all and most probably are a totally conserved crucial region that should not be mutated, Positions [17,26,30] have a “:” which means they are highly conserved, but mutations that are very similar in shape, structure and chemical properties are tolerated, the rest seem to be more flexible.
Interesting Finding here, All the conserved regions seem to be in the Soluble Domain (1-40) that is responsible for DnaJ Interaction :D
This is the generated L Protein Mutation Heatmap
I tried to cross reference with the experimental data sheet but it was quite hard to do manually so i wrote a script and ran it on Here on Colab to just find the common ones across the two files and came up with 3 common ones, however even though these 3 had quite a high LLR Score, the experimental data showed a Lysis score of 0, and i believe this shows the limitation in the structure based modeling, it predicted structural stability but failed to account for the functional mechanism of lysis.
The common mutations identified by my algorithm
So Then i Filtered my generated mutations first to target the soluble domain (1-40) first while making sure to avoid the totally conserved regions [21,25,28-29,33,35-37,40], the highly conserved regions [17,26,30] and my 3 common matches to avoid choosing mutants that are either disrupting the protein or have been already experimented on with no lysis observed.
The two mutations i have decided to go with for the Soluble Domain are:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
1
39
Y
L
2.24177968502044
2
9
S
Q
2.01432478427886
These have the highest LLR score in this region and avoid any conservative regions in the protein
Then i repeated the steps again, this time targeting the transmembrane domain (41-75) and followed the same criteria, and i picked these 2 mutations:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
3
50
K
L
2.56146776676178
4
53
N
L
1.86493206024169
And Again because These have the highest LLR score in this region and avoid any conservative regions in the protein
For the Final 5th mutant i decided to pick this one:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
5
52
T
L
1.81396758556365
Because this one still has a relatively high LLR Score and is in the area where the L Protein does not overlap with either the Coat Protein or the replicase ones.
so here is the full 5 Mutations table i chose:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
1
39
Y
L
2.24177968502044
2
9
S
Q
2.01432478427886
3
50
K
L
2.56146776676178
4
53
N
L
1.86493206024169
5
52
T
L
1.81396758556365
AlphaFold Multimer Runs
Mutant 1 (Y39L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The L Proteins do form a cylinder like shape mimicking the transmembrane pore but the piDDT score are very low (<50) so this probably won’t express well or won’t express at all if done in wet lab :(.
This is the Predicted Aligned Error (PAE), i asked AI (Gemini) how to interpret this and it said this:
So following this, it seems like each monomer folds correctly with high confidence yet their together-grouping is not reliable at all with very low confidence scores possibly hinting that the pore forming shape we saw might not actually happen (i hope i understood that correctly XD)
Mutant 2 (S9Q)
The Monomer Sequence:
METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Same exact findings for this mutant too :(.
Again, Same Here as well. (it is the same for all five mutants :(. )
Mutant 3 (K50L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
Mutant 4 (N53L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT
Mutant 5 (T52L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT
I Actually decided to pick a mutant from the Experimental Data Sheet and made sure it has been proven to have the Lysis Effect, my aim is to try to perform the AlphaFold Multimer step for it, to have a look at how different it might be, the mutant i picked was R30Q.
This is the monomer sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
This is the full Alphafold sequence i used:
METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Well i got bad news and good news, the bad news is, the structre is also of very low confidence and the Predicted Aligned Error (PAE) Plot shows the same low confidence for the monomer’s interaction with each othe, the good news though is that this is the run of a mutant that has been experimentally validated and has the lysis effect and protein level determined, so maybe this gives me hope that my five mutants might actually stand a chance in a wet lab validation regardless of the very low confidence scores it got.
We decided to focus on the main area of increasing the stability of the MS2 phage lysis protein L, with a possible secondary goal of reducing the dependency on host DnaJ, while still maintaining the lysis action.
The tools AlphaFold, Clustal Omega, BLAST, ESM, and ESMFold were discussed.
BLAST can pull out homologous lysis proteins from the databases.
Clustal Omega can create MSAs to identify essential L48-S49 residues, and the pore-forming regions that must not be mutated.
ESM can create mutation heatmaps, which can guide the use of ESMFold to obtain highest score foldings in mutatable regions.
AlphaFold Multimer predicts whether the subunits of our protein can successfully create a pore in the host membrane, and also to check whether N-terminus can break the interaction with DnaJ.
We also identified a few pitfalls, with majors ones dealing with limited training datasets, that may not be properly aligned towards creating a transmembrane lysis protein.
Some other pitfalls include the lack of proper annotations for amurins; the possibility of an over-stable protein to form non-functional aggregates; and the vulnerability of modified protein to host proteases.
The study shows that the MS2 phage lysis protein L requires the host chaperone DnaJ for efficient host cell lysis. A missense mutation (P330Q) in the highly conserved C-terminal domain of DnaJ blocks MS2 L-mediated lysis at 30 °C and delays lysis at higher temperatures, without affecting overall L protein synthesis. The defect is specific to L-mediated lysis and does not affect lysis by other phage lysis proteins.
Genetic suppressor screening identified Lodj alleles of the L gene that bypass the DnaJ requirement. These alleles encode truncated L proteins lacking the highly basic N-terminal domain, indicating that this domain confers dependence on DnaJ. Biochemical assays demonstrated that wild-type L forms a membrane-associated complex with DnaJ, whereas the P330Q DnaJ variant cannot interact with L.
The authors propose that DnaJ functions as a chaperone that facilitates proper folding or conformational activation of full-length L, preventing steric interference from the N-terminal domain and allowing L to interact with its unknown cellular target. Removal of the dispensable N-terminal domain eliminates the need for chaperone assistance and accelerates lysis.
The work identifies DnaJ as a host factor regulating MS2 lysis timing and suggests that chaperone-dependent modulation of lysis may be an evolutionary strategy to optimize phage replication cycles.
This study performed comprehensive mutational and genetic analyses of the MS2 phage lysis protein L to identify residues and domains required for function. Random mutagenesis of the 75-aa L protein showed that most loss-of-function mutations cluster in the C-terminal half of the protein, especially around a conserved Leu-Ser (LS) dipeptide motif. Many inactivating mutations were conservative amino-acid substitutions and did not affect protein accumulation or membrane association, suggesting that L function depends on specific protein–protein interactions rather than nonspecific membrane disruption.
Functional studies demonstrated that L-mediated lysis requires interaction with the host chaperone DnaJ. The highly basic N-terminal domain of L is dispensable for lytic activity but mediates DnaJ dependence. Truncation of this domain or certain suppressor mutations bypassed the chaperone requirement and restored rapid lysis.
Biochemical and genetic data support a model in which L is an integral membrane protein whose essential domains (including the LS motif and neighboring regions) form a helical structure that likely engages a host membrane target protein. The interaction may occur near sites of membrane curvature associated with peptidoglycan biosynthesis rather than by forming nonspecific membrane lesions.
The work, supported in part by the Center for Phage Technology and associated laboratories including research by Ry Young, suggests that MS2 L functions through a specific heterotypic protein–protein interaction mechanism and that chaperone-dependent regulation helps control lysis timing during infection.
The study refines the mechanistic model of MS2 lysis, proposing that conserved structural motifs rather than general membrane disruption drive lytic activity.
This study provides detailed in vitro and in vivo characterization of the MS2 lysis protein MS2-L, focusing on its membrane insertion mechanism, oligomerization behavior, and interaction with the host chaperone DnaJ.
Key findings show that MS2-L is a 75-amino-acid phage toxin whose essential lytic activity resides in the C-terminal ~35 amino acids, which form a hydrophobic transmembrane region. The N-terminal soluble domain is not required for bacterial killing but modulates folding, membrane insertion efficiency, and chaperone interaction.
Biochemical assays demonstrate that MS2-L interacts directly with DnaJ, primarily through the soluble N-terminal domain. However, this interaction does not significantly affect membrane insertion, solubilization, or oligomerization of the toxin, suggesting that DnaJ functions more as a folding or stabilization partner rather than being essential for lytic activity.
Native mass spectrometry revealed that MS2-L assembles into high-order oligomeric complexes (≥10 monomers) after insertion into lipid nanodiscs, and oligomerization is driven mainly by the transmembrane domain. In detergent environments, oligomer formation is reduced, indicating that membrane lipid context is important for stable assembly.
Fluorescence microscopy and cryo-electron microscopy showed that MS2-L expression in bacteria leads to peripheral membrane clustering, followed by sequential lesion formation beginning in the outer membrane, then disruption of the peptidoglycan layer, and finally inner membrane disintegration with cytoplasmic leakage.
The data support a model in which MS2-L functions as a pore-forming phage toxin that kills cells through higher-order oligomerization within the bacterial membrane, rather than by directly inhibiting peptidoglycan biosynthesis. Chaperone DnaJ binds MS2-L but is not required for membrane insertion or pore assembly, suggesting its role is mainly in modulating toxin folding or stability.
These findings strengthen the concept that MS2-L belongs to the amurin/single-gene lysis protein family and may be useful for bioengineering applications such as bacterial ghost cell production and antimicrobial design.
This review from Elsevier surveys the biological mechanisms, clinical development, and future directions of phage therapy as a strategy to combat antimicrobial resistance. It explains that therapeutic phages should ideally be strictly lytic, highly host-specific, and thoroughly characterized to ensure safety and efficacy.
The article describes how phages kill bacteria through mechanisms such as inhibition of essential cellular processes, expression of lysis proteins, or disruption of bacterial membranes. It also discusses advances in phage engineering, including synthetic genome construction and modification of phage host range and virulence.
Clinical applications of phage therapy are highlighted, particularly for treating drug-resistant infections where antibiotics are ineffective. However, challenges remain, including bacterial resistance to phages, regulatory hurdles, manufacturing standardization, and the need to understand phage–host interactions.
Future directions include the use of genetically modified or synthetic phages, computational prediction of therapeutic candidates, and integration of phage therapy with conventional antimicrobial strategies. Overall, phage therapy is presented as a promising but still developing alternative to antibiotics in the fight against antimicrobial resistance.
This preprint reports the first experimental demonstration of generative design of complete bacteriophage genomes using genome language models (Evo 1 and Evo 2). The authors fine-tuned models on about 15,000 Microviridae phage genomes to enable autoregressive generation of full viral genomes guided by template-based prompts and biologically motivated design constraints.
The workflow involved computational generation followed by multi-tier filtering for sequence quality, host tropism specificity, and evolutionary diversity. Constraints included genome length (4–6 kb), GC content, absence of long homopolymers, preservation of phage-like gene architecture, and spike protein similarity to the template phage to maintain host targeting.
Experimental validation showed that about 285 of 302 synthesized genome candidates could be assembled, and 16 produced viable infectious phages that inhibited growth of the target host strain. These generated phages displayed substantial sequence novelty, containing hundreds of mutations relative to natural Microviridae genomes, while preserving functional genome organization.
Structural and functional analyses indicated that some generated phages possessed altered protein interfaces but maintained compatible capsid–protein interactions. Cryo-electron microscopy and structure prediction suggested context-dependent co-evolution of structural proteins such as capsid and packaging proteins.
Fitness assays showed that several AI-generated phages matched or exceeded the replication and lytic performance of the template phage, and phage cocktail experiments demonstrated rapid suppression of resistant bacterial strains through recombination and mutation-driven adaptation.
The study was conducted with biosafety considerations, including restricting model training to bacteriophage genomes and using well-characterized laboratory strains. The work was supported by researchers affiliated with institutions such as the Stanford University and the Arc Institute.
Overall, the paper proposes a framework for generative genome engineering, showing that AI models can design biologically viable and evolutionarily novel bacteriophages, potentially enabling future synthetic biology and phage-based therapeutic development.
Overview of the Project Proposal: Engineering the MS2 Phage Lysis Protein L
Our primary goal is to increase the structural stability of the MS2 bacteriophage lysis protein (L) while maintaining its ability to lyse bacterial cells.
Our secondary goal is to reduce the dependency of L on the host chaperone DnaJ, which normally assists the protein in folding or activation. Reducing this dependency could allow the lysis protein to function more efficiently and independently in engineered systems.
The MS2 L protein is a 75-amino-acid single-gene lysis toxin whose C-terminal region forms a hydrophobic transmembrane domain responsible for membrane disruption and pore formation, while the basic N-terminal domain interacts with host factors such as DnaJ. Previous studies show that truncation of the N-terminal region can bypass the DnaJ requirement while preserving lysis activity.
Therefore, our design strategy focuses on:
Stabilizing the transmembrane and oligomerization regions
Maintaining essential functional motifs such as the L48–S49 motif
Exploring modifications to the N-terminal region to reduce DnaJ dependence
2. Computational Tools and Approaches
We will use a multi-step computational protein engineering pipeline combining sequence analysis, machine-learning mutagenesis predictions, and structural modeling.
2.1 BLAST – Homolog Discovery
First, we will use BLAST to identify homologous lysis proteins from related bacteriophages.
Purpose:
Identify evolutionarily conserved residues
Discover natural sequence variations that maintain function
Build a dataset for multiple sequence alignment
This will help determine which regions are functionally constrained vs mutable.
Select Candidate Mutations (stability or N-terminal modifications)
Structure Prediction (ESMFold)
Complex/Oligomer Prediction (AlphaFold Multimer)
Final Mutant Candidates (stable + functional lysis protein)
3. Proposed Engineering Pipeline
Computational workflow we will follow.
4. Expected Outcomes
Our pipeline aims to produce engineered variants of the MS2 L protein with:
Increased structural stability
Reduced aggregation risk
Maintained transmembrane insertion
Potentially reduced dependency on host DnaJ
These optimized proteins could be useful in applications such as:
Synthetic phage engineering
Bacterial ghost cell production
Antimicrobial protein development
5. Potential Pitfalls
5.1 Limited Training Data
Most protein language models and structural predictors are trained primarily on globular proteins, not small transmembrane phage toxins.
This may reduce prediction accuracy for MS2 L.
5.2 Risk of Over-Stabilization
Mutations designed to increase stability may cause:
Protein aggregation
Improper membrane insertion
Loss of functional oligomerization
Thus stability must be balanced with function.
5.3 Poor Annotation of Amurin Proteins
Single-gene lysis proteins (also called amurins) are poorly annotated in sequence databases.
This may limit the quality of homologous sequences used for alignment and training.
5.4 Host Protease Sensitivity
Mutations may unintentionally expose protease cleavage sites, making the engineered protein less stable inside bacterial cells.
6. Future Work
If promising mutants are identified computationally, the next steps would include:
Experimental expression in E. coli
Measuring lysis timing
Measuring protein stability
Testing DnaJ independence
This would validate whether computational predictions translate into improved biological function.
Week 5
L-Protein Mutants Project Summary:
@2026a-nourelden-rihan
The MS2 bacteriophage L-protein is a small 75-residue lysis protein with two functional regions: a soluble N-terminal domain (residues 1–40) that interacts with the bacterial chaperone DnaJ, and a transmembrane domain (residues 41–75) that disrupts the inner membrane to trigger host cell lysis. The goal of this project was to computationally design five single-point mutants with the potential to retain or enhance lysis activity. This was done by first running a Clustal Omega Multiple Sequence Alignment to map out conserved positions across homologs, all of which turned out to cluster exclusively in the soluble domain, pointing to the DnaJ interface as the most functionally constrained region of the protein. An LLR mutation heatmap was then generated and cross-referenced against an experimental lysis dataset; three mutations that appeared in both were excluded after showing a lysis score of zero experimentally, highlighting that LLR scores reflect structural stability rather than functional activity. The final five mutants, Y39L and S9Q in the soluble domain, and K50L, N53L, and T52L in the transmembrane domain, were selected based on the highest LLR scores while avoiding all conserved and experimentally failed positions.
Each mutant was then modeled as an octameric pore complex using AlphaFold Multimer to simulate the expected membrane assembly. The key findings were:
All five mutants formed a cylinder-like pore structure, consistent with the expected transmembrane assembly
pLDDT scores were low (< 50) across all models, and PAE plots showed near-zero confidence for inter-chain contacts
Running the same pipeline on R30Q, an experimentally validated lytic mutant, produced identical low-confidence results, suggesting this is a systematic limitation of AlphaFold for this class of small membrane-disrupting proteins rather than a reflection of the mutants’ actual viability
All five mutants remain candidates for wet lab validation
Full documentation, sequences, and AlphaFold outputs are available in the week 5 homework.
Ship 41 - Individual Final Project
New Slides
Old Slide
Ship 41
HTGAA Final Project Proposal
Project Title: Ship 41: Multi-Chassis Computational and Experimental Optimization of α-Pinene Biosynthesis for High-Value Bioproduction
Author: Nourelden Rihan
Course: How to Grow (Almost) Anything — HTGAA 2026
Date: April 4, 2026
Industry Partners: Ginkgo Bioworks · Twist Bioscience · Waters Corporation · New England Biolabs · Millipore Sigma
For forty ships, Egypt sailed to Byblos. Ship 41 engineers what Byblos grew.
SECTION 1: ABSTRACT
Around 2600 BCE, Pharaoh Sneferu dispatched 40 ships across the Mediterranean to the port of Byblos, Lebanon — the only place in the ancient world where cedar could be sourced in abundance. The Palermo Stone records the expedition plainly: “bringing 40 ships laden with cedar wood.” Egypt’s soil grew no conifers. For over two millennia, one of history’s greatest civilizations remained dependent on a foreign tree for its ships, its temples, and its monuments. Ship 41 does not sail to Byblos. It engineers what Byblos grew.
α-Pinene is the principal volatile monoterpene of coniferous wood — the molecule that defined the cedar of Lebanon and made it irreplaceable to the ancient world. Today, it is recognized as one of the most commercially versatile natural products in existence, with applications spanning sustainable aviation fuel (via catalytic dimerization to JP-10), fragrance and flavor industries (as a precursor to synthetic camphor, linalool, and geraniol), pharmaceuticals (anti-inflammatory, antimicrobial, and bronchodilatory properties), and polymer chemistry (as a feedstock for terpene resins). While prior work has demonstrated α-pinene biosynthesis in Escherichia coli using the heterologously expressed Abies grandis geranyl diphosphate synthase (AgGPPS2) and α-pinene synthase (AgPS), no systematic multi-chassis, multi-carbon-source benchmarking study has been conducted to identify the optimal production host and feedstock for high-yield industrial bioproduction. This project — Ship 41 — addresses that gap by integrating computational flux balance analysis (FBA) using COBRApy genome-scale metabolic models across three industrially relevant chassis — E. coli BL21(DE3), Saccharomyces cerevisiae BY4741, and Yarrowia lipolytica CLIB89 — with a high-throughput automated experimental screening pipeline executed on Ginkgo Bioworks’ robotics infrastructure. Constructs encoding bicistronic AgGPPS2-AgPS-sfGFP-His6 cassettes will be synthesized as whole plasmids by Twist Bioscience and validated in cell-free expression systems prior to in vivo transformation. α-Pinene titers will be quantified by GC-MS using dodecane overlay extraction from 96-well deep-well plates, with growth monitored by OD₆₀₀ on the PHERAstar FSX. The best-performing chassis and carbon source combination identified in Aim 1 will inform strain optimization and pathway intensification strategies in subsequent aims, ultimately establishing a rational, scalable biological platform for α-pinene bioproduction across the full spectrum of its industrial applications.
SECTION 2: PROJECT AIMS
Aim 1 — Experimental Aim
The first aim of this project is to computationally predict and experimentally validate the optimal microbial chassis and carbon source for α-pinene biosynthesis by utilizing COBRApy flux balance analysis across three genome-scale metabolic models, followed by high-throughput automated screening of engineered E. coli, S. cerevisiae, and Y. lipolytica strains expressing Twist Bioscience-synthesized AgGPPS2-AgPS constructs, with α-pinene titers quantified by GC-MS — establishing a rational production baseline applicable across all high-value α-pinene markets.
This aim encompasses:
COBRApy FBA, FVA, and gene knockout analysis across iJO1366 (E. coli), Yeast9 (S. cerevisiae), and the latest Y. lipolytica GEM
Whole plasmid DNA synthesis ordered from Twist Bioscience (2 constructs)
Cell-free construct validation using Ginkgo Bioworks BL21 DE3 lysate + GFP fluorescence on the Spark Plate Reader
α-Pinene quantification by GC-MS (Waters Corporation) from dodecane overlay extractions
Aim 2 — Medium-Term Aim
Having identified the best-performing chassis and carbon source combination in Aim 1, the second aim is to intensify α-pinene production through chassis-specific pathway engineering and promoter optimization. This will involve redesigning the genetic construct with chassis-matched regulatory elements (e.g., T7 for E. coli, TEF1-intron for Y. lipolytica) and overexpressing MEP or MVA pathway bottleneck genes identified by FVA in Aim 1. Additionally, computationally identified beneficial gene knockouts (e.g., disruption of competing farnesyl diphosphate branches) will be introduced using CRISPR-Cas9 or homologous recombination, and the resulting strains will be subjected to fed-batch fermentation optimization to achieve titers relevant for downstream industrial application — whether in fragrance, pharmaceuticals, polymer chemistry, or fuel. This aim will leverage the Asimov Kernel Platform for genetic part design and the Cultivarium’s expertise in non-model organism strain engineering, particularly for Y. lipolytica metabolic rewiring.
Aim 3 — Visionary Aim
Engineer a living biofoundry-on-a-chip: a fully autonomous, closed-loop biosynthetic system where AI-guided metabolic models continuously retrain on fermentation data to evolve self-optimizing microbial strains capable of converting atmospheric CO₂ and waste feedstocks directly into α-pinene — the ancient molecule of cedar — and routing it on demand toward any of its industrial destinies: fuel, fragrance, medicine, or material.
In the long term, Ship 41 envisions a carbon-negative, AI-driven α-pinene biorefinery where genome-scale metabolic modeling, automated strain construction, and real-time fermentation analytics are deeply integrated in a Design-Build-Test-Learn (DBTL) cycle operating without human intervention. Because α-pinene sits at the entry point of an extraordinarily diverse downstream chemical space — it can be dimerized into JP-10 jet fuel, isomerized into camphor, oxidized into verbenone, rearranged into limonene or linalool precursors, or polymerized into terpene resins — a high-yield microbial production platform for this single molecule effectively unlocks a portfolio of sustainable industrial chemicals simultaneously. The broader impact extends to establishing a generalizable computational-experimental framework for rapid monoterpene pathway optimization in any chassis, democratizing access to high-value terpene bioproduction across the developing world using locally available feedstocks — and finally, permanently, solving the problem that sent Pharaoh Sneferu’s 40 ships north across the sea.
SECTION 3: BACKGROUND
The Lore of Ship 41
Around 2600 BCE, the Palermo Stone records one of the most consequential supply chain decisions in ancient history: Pharaoh Sneferu, founder of the Fourth Dynasty and builder of the first true pyramids, dispatched a fleet of 40 ships northward across the Mediterranean to the Phoenician port of Byblos — present-day Lebanon — to obtain cedar wood. The inscription reads plainly: “bringing 40 ships laden with cedar wood.” The following year, he had three further 52-meter ships built from the same imported timber, and cedar doors installed in his palace at Dahshur. Remnants of that very wood survive inside the Bent Pyramid to this day.
Egypt was timber-poor by geography. The Nile Valley produced date palms and sycamore figs — serviceable for furniture, inadequate for ships, insufficient for the monumental ambitions of a civilization that was simultaneously inventing the state. Cedar of Lebanon (Cedrus libani) was in a different category entirely: dense, resinous, aromatic, resistant to rot and insects, straight-grained enough for ship planks and long enough for obelisk sledges. It was the structural material of the ancient world’s greatest projects. And Egypt could not grow it.
For over two thousand years — through the Old Kingdom, the Middle Kingdom, the New Kingdom, through Sneferu and Khufu and Thutmose and Ramesses — Egypt continued to sail for cedar. The Wenamun Papyrus, written around 1075 BCE, documents an Egyptian official’s fraught journey to Byblos to negotiate cedar logs for a ceremonial barge, some 1,500 years after Sneferu’s fleet. The dependency never ended. The ships kept sailing.
α-Pinene is the primary monoterpene constituent of cedar heartwood resin — the molecule responsible for the wood’s legendary resistance to decay, its distinctive scent, and much of its structural character. It is, in a molecular sense, what Egypt was sailing for. Today, α-pinene is understood as one of the most industrially versatile natural products known: a precursor to sustainable aviation fuel (JP-10), a feedstock for fragrance and flavor chemistry, a pharmaceutical candidate with documented anti-inflammatory and antimicrobial properties, and a building block for terpene-based polymers.
Ship 41 does not sail to Byblos. We are the 41st ship — except we carry no cargo hold and need no harbor. We carry a genome-scale metabolic model, a codon-optimized gene construct, and a 96-well deep plate. We are not imitators of that civilization. We are Egyptians, and we are its continuation. The cedar Egypt could never grow on its own soil, we now produce in a flask — on glucose, on glycerol, on fatty acids, from microbes of our design. The loop that opened with Sneferu’s fleet closes here.
Literature Context
Sarria et al. (2014) demonstrated the first microbial synthesis of pinene in E. coli by co-expressing a codon-optimized A. grandis GPPS and pinene synthase alongside an enhanced MEP pathway, achieving titers of approximately 32 mg/L of total pinene — establishing proof-of-concept for monoterpene biofuel production in a bacterial chassis. Subsequent work by Cao et al. (2016) expanded this effort by systematically engineering the E. coli MEP pathway through overexpression of rate-limiting enzymes (DXS, IspG, IspH) and achieved improved α-pinene titers, demonstrating that carbon flux toward the isoprenoid precursor pool is a critical bottleneck in monoterpene production. In parallel, the oleaginous yeast Yarrowia lipolytica has emerged as a compelling alternative chassis for terpenoid production due to its naturally high acetyl-CoA flux, tolerance to hydrophobic compounds, and compatibility with lipid-rich feedstocks, with several studies documenting its superior performance for sesquiterpene and diterpene accumulation relative to S. cerevisiae. Together, these bodies of work reveal a critical knowledge gap: no systematic, quantitative comparison of α-pinene production across these three industrially relevant chassis under matched genetic designs and multiple carbon sources has been performed, leaving the field without a rational basis for chassis selection during scale-up.
Innovation
This project is novel in three key dimensions. First, it is the first study to apply genome-scale metabolic modeling via COBRApy — including FBA, FVA, and systematic gene knockout screening — across all three chassis (E. coli, S. cerevisiae, Y. lipolytica) simultaneously, providing a computational-first rational framework for chassis selection that goes beyond empirical trial-and-error. Second, by using a single standardized genetic insert (AgGPPS2-AgPS-sfGFP-His6, synthesized by Twist Bioscience) expressed from host-compatible promoters, this work controls for genetic design variables and isolates the contribution of chassis metabolism and carbon source to α-pinene yield — a controlled comparison that has not previously been reported. Third, the integration of cell-free expression-based construct validation prior to in vivo transformation, combined with fully automated GC-MS-coupled high-throughput screening on Ginkgo Bioworks’ liquid handling robotics, represents a methodological advance in how monoterpene pathway screening is conducted at the course and early research stage.
Significance
α-Pinene is not a single-market molecule — it is a gateway compound whose downstream chemical space spans at least four major industrial sectors simultaneously. In sustainable fuels, it can be catalytically dimerized and hydrogenated into JP-10 (volumetric energy density ~40 MJ/L), a high-density jet fuel of particular value for military and advanced air mobility applications where energy density per volume is critical. In fragrance and flavor, α-pinene is a direct precursor to synthetic camphor, α-terpineol, linalool, and geraniol — globally traded aroma chemicals with a combined market exceeding $6 billion annually. In pharmaceuticals, α-pinene and its oxidized derivatives exhibit documented anti-inflammatory, bronchodilatory, and antimicrobial activities, with active research programs in respiratory and oncology indications. In polymer chemistry, terpene resins derived from pinene are used as tackifiers in pressure-sensitive adhesives, a market valued at over $3 billion. Building a high-yield microbial production platform for α-pinene therefore does not serve a single industry — it serves all of them. The significance of this project lies precisely in that breadth: by identifying the optimal chassis and carbon source through a rigorous computational-experimental framework, this work creates a foundational production baseline from which any downstream application can be pursued. Furthermore, the multi-chassis, multi-carbon-source comparative approach developed here is immediately generalizable to other high-value monoterpenes — limonene, linalool, geraniol — making Ship 41 not just a project about α-pinene, but a template for rational monoterpene bioproduction at large.
Bioethical Considerations
Paragraph 1 — Ethical Considerations:
The engineering of microorganisms for high-value chemical production raises important biosafety and environmental ethics questions that must be considered from the earliest stages of project design. The three chassis used in this project (E. coli BL21(DE3), S. cerevisiae BY4741, and Y. lipolytica CLIB89) are all well-characterized biosafety level 1 organisms with long histories of safe use in industrial biotechnology, and none possess pathogenic traits. The synthetic constructs encoding AgGPPS2 and AgPS do not confer selective advantages in natural environments, as monoterpene overproduction is metabolically costly and α-pinene itself is a volatile compound that does not accumulate in soil or water in toxic concentrations under normal conditions. Nevertheless, the intentional design of organisms with enhanced metabolic capabilities — even benign ones — raises broader questions about genetic sovereignty, access to engineered production strains, and the economic impact on existing natural α-pinene supply chains, including the global turpentine and pine resin industries that employ thousands of workers across developing economies. Any project advancing toward commercial bioproduction must engage with these displacement effects transparently and consider benefit-sharing frameworks with communities whose livelihoods depend on natural terpene harvesting.
Paragraph 2 — Responsible Implementation and Risk Mitigation:
All experimental work in this project will be conducted under BSL-1 containment at Ginkgo Bioworks’ certified laboratory facility, with all engineered strains handled, stored, and disposed of in accordance with institutional biosafety committee (IBC) protocols. All DNA constructs will be screened through SecureDNA prior to synthesis to ensure that no sequences of concern are inadvertently introduced during construct design. Engineered strains will be maintained with auxotrophic markers or antibiotic selection dependencies that prevent survival outside controlled laboratory conditions, and no environmental release is contemplated at any stage of this project. Longer-term, should this work advance toward pilot-scale fermentation, engagement with regulatory agencies (EPA, FDA, USDA) under the Coordinated Framework for Regulation of Biotechnology will be initiated proactively, and open publication of all metabolic modeling code and construct sequences on platforms such as Addgene and GitHub will ensure that the scientific community can build on, scrutinize, and improve this work without proprietary barriers.
SECTION 4: EXPERIMENTAL DESIGN
Overview
The experimental workflow proceeds in three logical phases: (1) Computational prediction, (2) Construct design and cell-free validation, and (3) Automated in vivo screening. All liquid handling automation is performed at Ginkgo Bioworks unless otherwise noted. Opentrons OT-2 may be used for preparatory steps at the bench.
Detailed Workflow (17 Steps)
Step 1 — COBRApy Flux Balance Analysis (FBA)
Method: Download the latest genome-scale metabolic models — iJO1366 (E. coli), Yeast9 (S. cerevisiae), and iYLI647 or the latest curated model for Y. lipolytica — from BiGG or the respective repositories. Add a heterologous α-pinene exchange reaction and set the objective function to maximize α-pinene flux. Simulate FBA under four carbon sources (glucose, glycerol, acetate, fatty acids/oleate) at matched carbon equivalents.
Expected Result: Predicted maximum theoretical α-pinene yield (mmol/g DCW/h) per chassis per carbon source; ranking of chassis–feedstock combinations
Timeline: Days 1–4
Step 2 — COBRApy Flux Variability Analysis (FVA)
Method: Run FVA on the top two chassis–feedstock combinations identified in Step 1. Identify reactions with high flux variability intersecting the MEP/MVA pathway and GPP branch point (IDI, GPPS). These are metabolic bottlenecks for experimental engineering targets.
Automation: Python/Jupyter notebook
Plate: N/A
Expected Result: List of bottleneck reactions; identification of gene overexpression targets (e.g., dxs, idi, ispG)
Timeline: Days 3–5
Step 3 — COBRApy Gene Knockout Screening
Method: Perform single and double gene knockout simulations (using cobra.flux_analysis.single_gene_deletion) targeting competing isoprenoid branches (e.g., ispA farnesyl diphosphate synthase competition, erg9 squalene synthase in yeast). Identify knockouts that improve GPP/α-pinene flux without abolishing growth.
Automation: Python/Jupyter notebook
Plate: N/A
Expected Result: Ranked list of gene knockouts predicted to increase α-pinene yield ≥ 1.5× relative to wild-type flux; shortlist for Aim 2 CRISPR targeting
Timeline: Days 4–6
Step 4 — DNA Construct Design
Method: Design two whole plasmid constructs using Benchling or SnapGene. Construct 1 (E. coli): pET28a backbone with bicistronic T7→RBS→AgGPPS2→RBS→AgPS-sfGFP-His6→T7term. Construct 2 (S. cerevisiae / Y. lipolytica shuttle): p426TEF backbone with TEF1→AgGPPS2→CYC1term; TEF2→AgPS-His6→ADH1term; URA3; 2μ ori. All coding sequences are E. coli or yeast codon-optimized using the Integrated DNA Technologies (IDT) codon optimization tool.
Expected Result: Two complete annotated plasmid maps ready for Twist submission; GenBank files exported (see Appendix A)
Timeline: Days 5–7
Step 5 — Twist Bioscience DNA Order
Method: Submit both plasmid designs (GenBank format) to Twist Bioscience as whole plasmid synthesis orders via the Twist online portal. Select clonal gene backbone option. Specify kanamycin resistance for Construct 1 and uracil prototrophy for Construct 2. Expected delivery: 10–14 business days.
Partner: Twist Bioscience
Plate: N/A (shipping)
Expected Result: Two lyophilized whole plasmids, sequence-verified by Twist, received at Ginkgo Bioworks
Timeline: Days 7–21 (concurrent with Steps 6–7)
Step 6 — Cell-Free System Preparation
Method: Prepare cell-free expression (CFE) reactions using Ginkgo Bioworks BL21 DE3 lysate + master mix (pre-prepared). Rehydrate Construct 1 plasmid (Twist-supplied) to 50 ng/μL in nuclease-free water. Design a CFE plate with titrated plasmid concentrations (0, 1, 5, 10, 25, 50 ng/μL) in triplicate to determine optimal DNA input for GFP expression.
Automation (Liquid Transfer): Echo525 acoustic liquid handler — transfers plasmid DNA and master mix into 384 Greiner black-well clear-bottom plate (384-well black-well clear-bottom)
Plate: 384 Greiner black-well clear-bottom
Expected Result: CFE reaction volumes of 10 μL per well, zero dead volume, accurate nanoliter-scale transfers
Method: Seal the 384-well CFE plate with breathable A4s seal (A4S seal applied by Plateloc). Incubate at 29°C for 3 hours in the Inheco Plate Incubator. Read GFP fluorescence (Ex: 488 nm / Em: 520 nm) on the Spark Plate Reader at t=0, t=1h, t=2h, t=3h.
Expected Result: GFP fluorescence signal ≥ 5× background in wells containing Construct 1, confirming successful transcription/translation of AgPS-sfGFP fusion; dose-response curve identifies optimal DNA input concentration
Timeline: Day 22–23
Step 8 — E. coli Transformation
Method: Transform Construct 1 (pET28a-AgGPPS2-AgPS-sfGFP-His6) into chemically competent E. coli BL21(DE3) cells (NEB C2527I) by heat shock (42°C, 30 sec). Recover in SOC medium for 1 hour at 37°C. Plate on LB + kanamycin (50 μg/mL). Pick 6 colonies; verify by colony PCR using T7 promoter and T7 terminator primers on the ATC Thermal Cycler.
Expected Result: ≥4/6 colonies confirmed correct insert by PCR band size (~3.4 kb); one verified colony inoculated into overnight LB + Kan culture
Timeline: Days 22–24
Step 9 — S. cerevisiae and Y. lipolytica Transformation
Method: Transform Construct 2 (p426TEF-AgGPPS2-AgPS-His6) into S. cerevisiae BY4741 (URA3Δ) by lithium acetate/PEG/heat shock protocol. Separately, adapt the construct backbone and transform into Y. lipolytica CLIB89 using electroporation (Gene Pulser, external step). Select on SC-Ura dropout plates (yeast) and YPD + hygromycin plates (Yarrowia). Verify transformants by colony PCR.
Machines: ATC Thermal Cycler (colony PCR)
Plate: 96-Armadillo-PCR-AB2396X
Expected Result: Verified transformants for both yeast species; starter cultures established in YPD or SC-Ura medium
Timeline: Days 23–26
Step 10 — Overnight Seed Culture Preparation
Method: Inoculate verified E. coli (LB + Kan), S. cerevisiae (SC-Ura), and Y. lipolytica (YPD) transformants into 5 mL liquid cultures. Grow overnight at 37°C (E. coli), 30°C (S. cerevisiae), 28°C (Y. lipolytica) with 200 rpm shaking. Measure OD₆₀₀ to normalize inoculation density.
Machines: Cytomat (30°C shaking incubator for yeast/Yarrowia); standard bench shaker for E. coli overnight
Plate: N/A (tube culture)
Expected Result: OD₆₀₀ of 1.5–4.0 for all three seed cultures; normalize to OD₆₀₀ = 0.05 for plate inoculation
Method: Prepare the multi-chassis × multi-carbon-source screening plate using a 96-well deep plate (96-v-eppendorf-951033502-deep). Add 500 μL of appropriate defined media + carbon source per well (see plate layout below). Dispense media using the Multiflo Automated Microplate Dispenser. Add dodecane overlay (100 μL/well) to trap volatile α-pinene. Inoculate each well with normalized seed culture using the Bravo-96 plate stamp from a source plate.
Expected Result: 96-well plate fully inoculated with 12 chassis-carbon source combinations in triplicate, plus negative controls and blanks; dodecane overlay in place
Timeline: Day 27
Step 12 — Induction and Incubation
Method: For E. coli wells, induce with IPTG (0.5 mM final, dispensed by Echo525 from 100 mM IPTG stock in DMSO) at OD₆₀₀ ≈ 0.4–0.6. Transfer sealed plate to Cytomat shaking incubator (30°C, 300 rpm) for 48 hours. For yeast and Yarrowia wells (no induction required; constitutive TEF1 promoter), incubate directly in Cytomat at 28–30°C, 300 rpm, 48 hours.
Expected Result: All strains growing under respective conditions; dodecane overlay capturing emitted α-pinene over 48 hours
Timeline: Days 27–29
Step 13 — OD₆₀₀ Growth Monitoring
Method: At t=0, t=12h, t=24h, t=48h, briefly remove plate from Cytomat, peel A4s seal (XPeel plate peeler), read OD₆₀₀ on the PHERAstar FSX (absorbance module, 600 nm). Reseal with A4s (Plateloc) and return to Cytomat.
Expected Result: Growth curves for all 12 conditions; identification of chassis-carbon source pairs with optimal growth without product toxicity; expected OD₆₀₀ range: 1.5–6.0 at 48h
Timeline: Days 27–29 (concurrent with Step 12)
Step 14 — Dodecane Layer Extraction and GC-MS Sample Preparation
Method: After 48h incubation, transfer 80 μL of the dodecane overlay from each well into a fresh 384-well Echo PP plate using the Bravo-384 plate stamp or liquid handler. Dilute samples 1:5 in dodecane. Prepare an α-pinene standard curve in dodecane (0, 1, 5, 10, 25, 50, 100, 200 mg/L) in the remaining wells of the Echo PP plate. Submit plate to GC-MS analysis (Waters GC-MS system, external analytical service via Waters Corporation collaboration).
Expected Result: Collected dodecane samples for 36 experimental wells + 12 control wells + 8-point standard curve; samples ready for GC-MS injection
Timeline: Day 29–30
Step 15 — GC-MS α-Pinene Quantification
Method: Inject dodecane samples onto GC-MS (Waters Xevo TQ-S or equivalent; column: HP-5MS, 30m × 0.25mm × 0.25μm; oven program: 40°C → 200°C at 10°C/min; MS detection: SIM mode m/z 136 for α-pinene, retention time ~5.8 min). Quantify α-pinene titers from standard curve. Calculate volumetric titer (mg/L culture) and specific yield (mg/g DCW).
Partner: Waters Corporation (GC-MS instrumentation and method)
Plate: N/A (GC-MS vials)
Expected Result: α-Pinene titers for all 12 chassis-carbon source combinations; identification of best-performing condition
Timeline: Days 30–32
Step 16 — qPCR Expression Validation
Method: Collect 1 mL cell pellets from each well at t=48h. Extract RNA using RNeasy Mini Kit (Qiagen). Perform RT-qPCR on the CFX Opus qPCR machine using primers targeting AgGPPS2, AgPS, and reference genes (16S rRNA for E. coli; ACT1 for yeasts). Confirm that mRNA expression levels of both transgenes correlate with α-pinene titer.
Machines: HiG Centrifuge (pellet) → CFX Opus (qPCR)
Plate: 96-Armadillo-PCR-AB2396X (qPCR reactions)
Expected Result: ΔΔCt values showing 10–100-fold overexpression of AgGPPS2 and AgPS relative to empty vector controls; correlation between transcript level and titer confirms pathway expression is functional
Timeline: Days 30–33
Step 17 — IPTG Dose-Response Optimization (E. coli)
Method: For the best-performing E. coli condition identified in Step 15, perform a fine-grained IPTG dose-response experiment (0, 0.05, 0.1, 0.25, 0.5, 1.0, 2.0 mM) in a 384-well format using the Echo525 for IPTG titration. Monitor GFP fluorescence (sfGFP fusion as proxy for AgPS expression) on the Spark Plate Reader and collect dodecane samples for GC-MS at 48h.
Plate: 384 Greiner black-well clear-bottom (fluorescence) + 96-v-eppendorf deep (culture)
Expected Result: Optimal IPTG concentration identified (expected: 0.1–0.5 mM); GFP signal correlates with α-pinene titer; induction condition carried forward to Aim 2
Timeline: Days 33–37
Assay Plate Layout — 96-Well Deep Plate (Step 11–13)
The following layout illustrates the experimental design for the multi-chassis carbon source screen. All volumes: 500 μL defined media + carbon source + 100 μL dodecane overlay.
Cell-free protein expression (CFPE) is an in vitro transcription-translation system in which a cell lysate — containing ribosomes, RNA polymerases, tRNAs, and associated cofactors — is combined with an energy regeneration system and a DNA or RNA template to produce functional proteins outside of a living cell. In this project, CFPE using Ginkgo Bioworks’ pre-prepared BL21 DE3 lysate serves as a rapid validation gateway for Construct 1, allowing confirmation that the AgPS-sfGFP fusion protein is correctly transcribed and translated before committing to the more time-intensive in vivo transformation, selection, and growth experiments. The sfGFP reporter fused to AgPS provides a direct, plate-reader-detectable proxy for construct functionality: if GFP fluorescence is observed in the CFE reaction above background, it confirms that the T7 promoter, RBS, and coding sequence architecture are intact and functional, significantly reducing the risk of propagating non-functional constructs into downstream experiments. CFPE is also advantageous because it eliminates concerns about cell viability, metabolic burden, or plasmid instability that can confound in vivo expression, making it an ideal screening tool for rapidly evaluating construct variants before committing to in vivo work.
Technique 2: Genome-Scale Metabolic Modeling with COBRApy
Genome-scale metabolic modeling (GSMM) is a constraint-based computational approach that uses a mathematical reconstruction of all known metabolic reactions in an organism — encoded as a stoichiometric matrix — to predict metabolic fluxes under defined growth conditions using linear programming. COBRApy (COnstraint-Based Reconstruction and Analysis for Python) is the leading open-source software package for GSMM, supporting FBA, FVA, gene knockout simulation, and phenotype phase plane analysis on genome-scale metabolic models (GEMs) containing thousands of reactions and metabolites. In this project, COBRApy is used in three distinct modes: first, FBA maximizes a synthetic α-pinene exchange flux under each chassis–carbon source combination to predict theoretical production yields; second, FVA identifies reactions whose flux ranges overlap with MEP/MVA pathway intermediates, pinpointing bottleneck steps that constrain GPP availability for pinene synthesis; and third, single and double gene knockout simulations identify genetic modifications that redirect carbon flux away from competing terpenoid branches (e.g., farnesyl diphosphate and squalene synthesis) toward the desired α-pinene product without lethality. The computational predictions generated by COBRApy directly inform both the experimental design of this project — guiding which chassis and carbon source to prioritize in the automated screen — and the strain engineering targets for Aim 2, creating a tightly integrated computational-experimental DBTL cycle.
SECTION 6: PROJECT VALIDATION
10a — Validation Choice
The primary validation experiment for Aim 1 is cell-free expression of Construct 1 (pET28a-AgGPPS2-AgPS-sfGFP-His6) with GFP fluorescence readout on the Spark Plate Reader, chosen because it directly confirms that the DNA construct synthesized by Twist Bioscience produces a functional, detectable protein product before any in vivo transformation is attempted, and because sfGFP fluorescence serves as a quantitative, plate-reader-compatible proxy for AgPS expression that can be measured in a single automated 3-hour experiment with no specialized equipment beyond the Ginkgo Bioworks automation stack.
Construct 1 plasmid DNA (Twist Bioscience, resuspended to 50 ng/μL in nuclease-free water)
384 Greiner black-well clear-bottom plate
A4s breathable seal + Plateloc sealer
Echo525 acoustic liquid handler
Inheco Plate Incubator (pre-equilibrated to 29°C)
Spark Plate Reader
Protocol:
Thaw BL21 DE3 lysate and master mix on ice (15 min). Gently mix by inversion; do not vortex.
Prepare source plate: In a 384-well Echo PP plate, prepare Construct 1 DNA at 6 concentrations: 0, 1, 5, 10, 25, 50 ng/μL (20 μL each) in columns 1–6. Prepare water-only negative control in column 7.
Using Echo525, transfer 20 nL of each DNA concentration into the destination 384 Greiner black-well plate (4 replicates per concentration = 24 wells total).
Using Multiflo, dispense 10 μL of cell-free master mix + lysate (pre-mixed 7:3 v/v on ice) into each well of the 384 destination plate.
Apply A4s breathable seal using Plateloc to prevent evaporation while allowing gas exchange.
Transfer sealed plate to Inheco Plate Incubator (29°C). Begin incubation timer (3 hours total).
At t=0, t=60 min, t=120 min, t=180 min: transfer plate to Spark Plate Reader for GFP fluorescence measurement (Ex 488 nm / Em 520 nm / gain 60 / 25 flashes per well). Return to Inheco after each read.
Export raw fluorescence data to CSV. Subtract background (water-only control well average from each timepoint).
Plot background-corrected fluorescence vs. DNA concentration vs. time. Identify optimal DNA input (expected: 5–25 ng/μL) for maximum GFP signal.
Confirm: ≥ 5-fold fluorescence increase over background at optimal DNA concentration indicates successful construct expression. Flag for in vivo transformation.
10c — Techniques Used
This validation protocol employs four distinct techniques in an integrated pipeline. First, cell-free protein expression leverages the BL21 DE3 transcription-translation machinery to produce the AgPS-sfGFP fusion protein directly from plasmid DNA without any cellular growth, transformation, or selection steps, making it the fastest possible route to functional protein validation. Second, acoustic liquid handling via the Echo525 enables nanoliter-precision transfer of DNA samples across a concentration gradient into the 384-well plate without tip contamination or carryover, which is critical for generating a clean dose-response curve with minimal reagent consumption. Third, fluorescence plate reader detection on the Spark provides a quantitative, kinetic readout of sfGFP accumulation over the 3-hour incubation period, allowing observation of the expression time course and identification of the plateau phase that indicates maximal translation of the AgPS fusion. Fourth, automated plate sealing and incubation management via the Plateloc (A4s seal), Inheco incubator, and programmed Spark time-course reading ensures that all replicates experience identical environmental conditions throughout the experiment, which is essential for reliable, reproducible kinetic fluorescence data that can be compared across DNA concentrations.
The following table represents hypothetical cell-free expression results for Construct 1 at t=180 min, showing GFP fluorescence (relative fluorescence units, RFU) as a function of plasmid DNA input concentration:
DNA Input (ng/μL)
Rep 1 (RFU)
Rep 2 (RFU)
Rep 3 (RFU)
Rep 4 (RFU)
Mean RFU
Std Dev
0 (water control)
312
298
321
305
309
±9.7
1
1,847
1,923
1,788
1,902
1,865
±59.1
5
7,412
7,088
7,521
7,203
7,306
±191
10
14,832
15,201
14,677
15,088
14,950
±237
25
21,403
22,107
21,889
21,644
21,761
±295
50
21,982
22,391
21,677
22,103
22,038
±289
Interpretation: GFP signal saturates between 25–50 ng/μL DNA input, indicating the cell-free system is limiting (not DNA), consistent with published CFE saturation kinetics. The 47-fold signal-to-background at 10 ng/μL confirms robust expression of the AgPS-sfGFP fusion. Optimal DNA concentration for downstream cell-free assays: 10–25 ng/μL.
Hypothetical α-Pinene Titers — In Vivo Multi-Chassis Screen (Aim 1, Step 15)
The following table shows hypothetical GC-MS α-pinene titers (mg/L) at t=48h across all chassis and carbon source combinations:
Chassis
Carbon Source
α-Pinene Titer (mg/L)
Std Dev
OD₆₀₀ at 48h
E. coli BL21(DE3)
Glucose
85.3
±8.2
3.2
E. coli BL21(DE3)
Glycerol
112.7
±11.4
2.8
E. coli BL21(DE3)
Acetate
43.2
±6.8
1.9
E. coli BL21(DE3)
Fatty acids
28.4
±4.1
1.4
S. cerevisiae BY4741
Glucose
67.8
±9.3
4.1
S. cerevisiae BY4741
Glycerol
78.2
±8.7
3.6
S. cerevisiae BY4741
Acetate
31.5
±5.2
2.3
S. cerevisiae BY4741
Fatty acids
45.3
±7.1
3.8
Y. lipolytica CLIB89
Glucose
58.4
±7.8
5.2
Y. lipolytica CLIB89
Glycerol
89.1
±10.2
4.8
Y. lipolytica CLIB89
Acetate
22.7
±4.3
2.1
Y. lipolytica CLIB89
Fatty acids
134.6
±15.8
5.9
Negative controls (all)
All
<2.1
±0.4
varied
Interpretation: Two conditions emerge as clear leaders: E. coli on glycerol (112.7 mg/L) and Y. lipolytica on fatty acids (134.6 mg/L). This is consistent with COBRApy predictions — glycerol feeds the MEP pathway efficiently in E. coli, while Y. lipolytica’s native high-flux acetyl-CoA metabolism is amplified by fatty acid substrates. Acetate performs poorly across all chassis, suggesting it imposes pH stress or insufficient flux. These two winning conditions advance to Aim 2 strain optimization.
Challenge 1 — Low or absent GFP signal in cell-free validation. If GFP fluorescence fails to rise above background in the CFPE experiment, the most likely causes are plasmid damage during shipping/resuspension, an incorrect construct architecture (e.g., frame shift in the sfGFP fusion junction), or degraded lysate; troubleshooting steps include running the Twist-supplied plasmid on an agarose gel to confirm supercoiled band integrity, re-sequencing the construct junction region with Sanger sequencing, and testing a positive control plasmid (e.g., a known GFP-expressing plasmid) in parallel with the same lysate batch.
Challenge 2 — No detectable α-pinene in GC-MS despite confirmed GFP expression. α-Pinene is a highly volatile monoterpene (boiling point: 155°C) that can escape from culture wells if the dodecane overlay evaporates or if the plate seal is compromised; if GC-MS returns below-detection titers from experimental wells that showed GFP expression, the dodecane overlay volume should be increased to 200 μL, the seal integrity should be verified, and a second extraction with fresh dodecane should be performed to confirm the volatile product is being captured.
Challenge 3 — Poor yeast transformation efficiency. If S. cerevisiae or Y. lipolytica transformants are not recovered at sufficient frequency, an alternative strategy is to clone the insert into a linearized integrating vector (e.g., pRS306 for yeast) for genomic integration at a defined locus (e.g., HIS3), which typically gives more stable expression and avoids the metabolic burden of maintaining a high-copy episomal plasmid.
Challenge 4 — Growth inhibition by α-pinene accumulation. Monoterpenes including α-pinene can be toxic to microbial membranes at concentrations above ~200 mg/L; if growth curves (OD₆₀₀) plateau abnormally early in the highest-producing wells, a two-phase fermentation strategy using an increased dodecane overlay ratio (1:1 v/v culture:dodecane) can be implemented to continuously extract α-pinene from the aqueous phase, relieving product toxicity and potentially improving total titer by 2–5× as demonstrated in the literature for similar volatile terpenoids.
SECTION 7: ADDITIONAL INFORMATION
References
Sarria, S., Wong, B., Martín, H. G., Keasling, J. D., & Peralta-Yahya, P. (2014). Microbial synthesis of pinene. ACS Synthetic Biology, 3(7), 466–475.
Cao, X., Wei, L.-J., Lin, J.-Y., & Hua, Q. (2016). Enhancing linalool production by engineering oleaginous yeast Yarrowia lipolytica. Bioresource Technology, 245, 1641–1644. (verify DOI before submission)
Jongedijk, E., Cankar, K., Ranzijn, J., van der Krol, S., Bouwmeester, H., & Beekwilder, J. (2016). Capturing of the monoterpene olefin limonene produced in Saccharomyces cerevisiae. Yeast, 33(8), 331–343.
Meylemans, H. A., Quintana, R. L., Goldsmith, B. R., & Harvey, B. G. (2011). Solvent-free conversion of linalool to methylcyclopentadiene dimers and tricyclodecanes: selective synthesis of high-density fuels. ChemSusChem, 4(4), 465–469.
Ebrahim, A., Lerman, J. A., Palsson, B. O., & Hyduke, D. R. (2013). COBRApy: Constraints-based reconstruction and analysis for Python. BMC Systems Biology, 7(1), 74.
Gu, Y., et al. (2021). Advances and prospects of Yarrowia lipolytica as a typical oleaginous yeast. Microbial Biotechnology, 14(4), 1335–1342.
Lu, X., et al. (2019). Constructing a synthetic pathway for acetyl-coenzyme A from one-carbon through enzyme design. Nature Communications, 10, 1378. (background for acetate metabolism)
Noor, E., et al. (2016). The protein cost of metabolic fluxes: prediction from enzymatic rate laws and cost minimization. PLOS Computational Biology, 12(11), e1005167.
King, Z. A., et al. (2016). BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Research, 44(D1), D515–D522.
Note: All DOIs and publication details should be verified via PubMed or Google Scholar before final submission. Some citations above are approximated from training knowledge and require confirmation.
APPENDIX A: DNA CONSTRUCT DESIGNS (GenBank Format)
These GenBank files are formatted for direct import into Benchling or SnapGene, and for submission to Twist Bioscience as whole plasmid synthesis orders. Select “Clonal Gene — Plasmid” on the Twist portal and upload the respective .gb file. All coding sequences are synthetic and codon-optimized; verify codon optimization using the IDT Codon Optimization Tool (https://www.idtdna.com/CodonOpt) before ordering.
Construct 1: pET28a-AgGPPS2-AgPS-sfGFP-His6 (E. coli)
Note: The ORIGIN section above shows the first ~2,800 bp of the ~8,923 bp plasmid. The complete sequence should be generated by assembling the codon-optimized insert sequences (generated via IDT Codon Optimization Tool from AgGPPS2 and AgPS native protein sequences) into the pET28a backbone. Export the final annotated map as .gb from Benchling and upload directly to the Twist Bioscience portal.
Construct 2: p426TEF-AgGPPS2-AgPS-His6 (S. cerevisiae / Y. lipolytica Shuttle)
Note: The ORIGIN section above is a representative excerpt. Generate the full annotated plasmid in Benchling by inserting yeast-codon-optimized AgGPPS2 and AgPS sequences (generated from the IDT Codon Optimization Tool, organism: Saccharomyces cerevisiae) into the p426TEF backbone (Addgene #26362). Export as .gb and upload to Twist Bioscience portal as a whole plasmid synthesis order.
End of HTGAA Final Project Proposal
Document assembled with assistance from the HTGAA Project Design AI AssistantPhase 1 design dialogue completed: April 4, 2026
𓋹 𓋹 𓋹 𓋹 𓋹
BioClub Committed Listener MoU
HTGAA Committed Listener (CL) Agreement
I am a HTGAA Committed Listener, my responsibilities are:
Watching class lectures and recitations
Participating in node reviews
Developing and documenting my homework
Actively communicating with other students and TAs on the forum
Allowing HTGAA and BioClub to share my work (with attribution)
Honestly reporting on my work, and appropriately attributing and citing the work of others (both human and non-human)
Following locally applicable health and safety guidance
Promoting a respectful environment free of harassment and discrimination
Signed by committing this file to my documentation page/repository,