New Slides
Old Slide
Ship 41 HTGAA Final Project Proposal Project Title: Ship 41: Multi-Chassis Computational and Experimental Optimization of α-Pinene Biosynthesis for High-Value Bioproduction
Subsections of Projects
Group Final Project
Group Name: Phage Forge
Group Members:
@2026a-nourelden-rihan, @2026a-ritika-saha, @2026a-rahul-yaji, and @2026a-keerthana-gunaretnam
We decided to focus on the main area of increasing the stability of the MS2 phage lysis protein L, with a possible secondary goal of reducing the dependency on host DnaJ, while still maintaining the lysis action.
The tools AlphaFold, Clustal Omega, BLAST, ESM, and ESMFold were discussed.
BLAST can pull out homologous lysis proteins from the databases.
Clustal Omega can create MSAs to identify essential L48-S49 residues, and the pore-forming regions that must not be mutated.
ESM can create mutation heatmaps, which can guide the use of ESMFold to obtain highest score foldings in mutatable regions.
AlphaFold Multimer predicts whether the subunits of our protein can successfully create a pore in the host membrane, and also to check whether N-terminus can break the interaction with DnaJ.
We also identified a few pitfalls, with majors ones dealing with limited training datasets, that may not be properly aligned towards creating a transmembrane lysis protein.
Some other pitfalls include the lack of proper annotations for amurins; the possibility of an over-stable protein to form non-functional aggregates; and the vulnerability of modified protein to host proteases.
The study shows that the MS2 phage lysis protein L requires the host chaperone DnaJ for efficient host cell lysis. A missense mutation (P330Q) in the highly conserved C-terminal domain of DnaJ blocks MS2 L-mediated lysis at 30 °C and delays lysis at higher temperatures, without affecting overall L protein synthesis. The defect is specific to L-mediated lysis and does not affect lysis by other phage lysis proteins.
Genetic suppressor screening identified Lodj alleles of the L gene that bypass the DnaJ requirement. These alleles encode truncated L proteins lacking the highly basic N-terminal domain, indicating that this domain confers dependence on DnaJ. Biochemical assays demonstrated that wild-type L forms a membrane-associated complex with DnaJ, whereas the P330Q DnaJ variant cannot interact with L.
The authors propose that DnaJ functions as a chaperone that facilitates proper folding or conformational activation of full-length L, preventing steric interference from the N-terminal domain and allowing L to interact with its unknown cellular target. Removal of the dispensable N-terminal domain eliminates the need for chaperone assistance and accelerates lysis.
The work identifies DnaJ as a host factor regulating MS2 lysis timing and suggests that chaperone-dependent modulation of lysis may be an evolutionary strategy to optimize phage replication cycles.
This study performed comprehensive mutational and genetic analyses of the MS2 phage lysis protein L to identify residues and domains required for function. Random mutagenesis of the 75-aa L protein showed that most loss-of-function mutations cluster in the C-terminal half of the protein, especially around a conserved Leu-Ser (LS) dipeptide motif. Many inactivating mutations were conservative amino-acid substitutions and did not affect protein accumulation or membrane association, suggesting that L function depends on specific protein–protein interactions rather than nonspecific membrane disruption.
Functional studies demonstrated that L-mediated lysis requires interaction with the host chaperone DnaJ. The highly basic N-terminal domain of L is dispensable for lytic activity but mediates DnaJ dependence. Truncation of this domain or certain suppressor mutations bypassed the chaperone requirement and restored rapid lysis.
Biochemical and genetic data support a model in which L is an integral membrane protein whose essential domains (including the LS motif and neighboring regions) form a helical structure that likely engages a host membrane target protein. The interaction may occur near sites of membrane curvature associated with peptidoglycan biosynthesis rather than by forming nonspecific membrane lesions.
The work, supported in part by the Center for Phage Technology and associated laboratories including research by Ry Young, suggests that MS2 L functions through a specific heterotypic protein–protein interaction mechanism and that chaperone-dependent regulation helps control lysis timing during infection.
The study refines the mechanistic model of MS2 lysis, proposing that conserved structural motifs rather than general membrane disruption drive lytic activity.
This study provides detailed in vitro and in vivo characterization of the MS2 lysis protein MS2-L, focusing on its membrane insertion mechanism, oligomerization behavior, and interaction with the host chaperone DnaJ.
Key findings show that MS2-L is a 75-amino-acid phage toxin whose essential lytic activity resides in the C-terminal ~35 amino acids, which form a hydrophobic transmembrane region. The N-terminal soluble domain is not required for bacterial killing but modulates folding, membrane insertion efficiency, and chaperone interaction.
Biochemical assays demonstrate that MS2-L interacts directly with DnaJ, primarily through the soluble N-terminal domain. However, this interaction does not significantly affect membrane insertion, solubilization, or oligomerization of the toxin, suggesting that DnaJ functions more as a folding or stabilization partner rather than being essential for lytic activity.
Native mass spectrometry revealed that MS2-L assembles into high-order oligomeric complexes (≥10 monomers) after insertion into lipid nanodiscs, and oligomerization is driven mainly by the transmembrane domain. In detergent environments, oligomer formation is reduced, indicating that membrane lipid context is important for stable assembly.
Fluorescence microscopy and cryo-electron microscopy showed that MS2-L expression in bacteria leads to peripheral membrane clustering, followed by sequential lesion formation beginning in the outer membrane, then disruption of the peptidoglycan layer, and finally inner membrane disintegration with cytoplasmic leakage.
The data support a model in which MS2-L functions as a pore-forming phage toxin that kills cells through higher-order oligomerization within the bacterial membrane, rather than by directly inhibiting peptidoglycan biosynthesis. Chaperone DnaJ binds MS2-L but is not required for membrane insertion or pore assembly, suggesting its role is mainly in modulating toxin folding or stability.
These findings strengthen the concept that MS2-L belongs to the amurin/single-gene lysis protein family and may be useful for bioengineering applications such as bacterial ghost cell production and antimicrobial design.
This review from Elsevier surveys the biological mechanisms, clinical development, and future directions of phage therapy as a strategy to combat antimicrobial resistance. It explains that therapeutic phages should ideally be strictly lytic, highly host-specific, and thoroughly characterized to ensure safety and efficacy.
The article describes how phages kill bacteria through mechanisms such as inhibition of essential cellular processes, expression of lysis proteins, or disruption of bacterial membranes. It also discusses advances in phage engineering, including synthetic genome construction and modification of phage host range and virulence.
Clinical applications of phage therapy are highlighted, particularly for treating drug-resistant infections where antibiotics are ineffective. However, challenges remain, including bacterial resistance to phages, regulatory hurdles, manufacturing standardization, and the need to understand phage–host interactions.
Future directions include the use of genetically modified or synthetic phages, computational prediction of therapeutic candidates, and integration of phage therapy with conventional antimicrobial strategies. Overall, phage therapy is presented as a promising but still developing alternative to antibiotics in the fight against antimicrobial resistance.
This preprint reports the first experimental demonstration of generative design of complete bacteriophage genomes using genome language models (Evo 1 and Evo 2). The authors fine-tuned models on about 15,000 Microviridae phage genomes to enable autoregressive generation of full viral genomes guided by template-based prompts and biologically motivated design constraints.
The workflow involved computational generation followed by multi-tier filtering for sequence quality, host tropism specificity, and evolutionary diversity. Constraints included genome length (4–6 kb), GC content, absence of long homopolymers, preservation of phage-like gene architecture, and spike protein similarity to the template phage to maintain host targeting.
Experimental validation showed that about 285 of 302 synthesized genome candidates could be assembled, and 16 produced viable infectious phages that inhibited growth of the target host strain. These generated phages displayed substantial sequence novelty, containing hundreds of mutations relative to natural Microviridae genomes, while preserving functional genome organization.
Structural and functional analyses indicated that some generated phages possessed altered protein interfaces but maintained compatible capsid–protein interactions. Cryo-electron microscopy and structure prediction suggested context-dependent co-evolution of structural proteins such as capsid and packaging proteins.
Fitness assays showed that several AI-generated phages matched or exceeded the replication and lytic performance of the template phage, and phage cocktail experiments demonstrated rapid suppression of resistant bacterial strains through recombination and mutation-driven adaptation.
The study was conducted with biosafety considerations, including restricting model training to bacteriophage genomes and using well-characterized laboratory strains. The work was supported by researchers affiliated with institutions such as the Stanford University and the Arc Institute.
Overall, the paper proposes a framework for generative genome engineering, showing that AI models can design biologically viable and evolutionarily novel bacteriophages, potentially enabling future synthetic biology and phage-based therapeutic development.
Overview of the Project Proposal: Engineering the MS2 Phage Lysis Protein L
Our primary goal is to increase the structural stability of the MS2 bacteriophage lysis protein (L) while maintaining its ability to lyse bacterial cells.
Our secondary goal is to reduce the dependency of L on the host chaperone DnaJ, which normally assists the protein in folding or activation. Reducing this dependency could allow the lysis protein to function more efficiently and independently in engineered systems.
The MS2 L protein is a 75-amino-acid single-gene lysis toxin whose C-terminal region forms a hydrophobic transmembrane domain responsible for membrane disruption and pore formation, while the basic N-terminal domain interacts with host factors such as DnaJ. Previous studies show that truncation of the N-terminal region can bypass the DnaJ requirement while preserving lysis activity.
Therefore, our design strategy focuses on:
Stabilizing the transmembrane and oligomerization regions
Maintaining essential functional motifs such as the L48–S49 motif
Exploring modifications to the N-terminal region to reduce DnaJ dependence
2. Computational Tools and Approaches
We will use a multi-step computational protein engineering pipeline combining sequence analysis, machine-learning mutagenesis predictions, and structural modeling.
2.1 BLAST – Homolog Discovery
First, we will use BLAST to identify homologous lysis proteins from related bacteriophages.
Purpose:
Identify evolutionarily conserved residues
Discover natural sequence variations that maintain function
Build a dataset for multiple sequence alignment
This will help determine which regions are functionally constrained vs mutable.
Select Candidate Mutations (stability or N-terminal modifications)
Structure Prediction (ESMFold)
Complex/Oligomer Prediction (AlphaFold Multimer)
Final Mutant Candidates (stable + functional lysis protein)
3. Proposed Engineering Pipeline
Computational workflow we will follow.
4. Expected Outcomes
Our pipeline aims to produce engineered variants of the MS2 L protein with:
Increased structural stability
Reduced aggregation risk
Maintained transmembrane insertion
Potentially reduced dependency on host DnaJ
These optimized proteins could be useful in applications such as:
Synthetic phage engineering
Bacterial ghost cell production
Antimicrobial protein development
5. Potential Pitfalls
5.1 Limited Training Data
Most protein language models and structural predictors are trained primarily on globular proteins, not small transmembrane phage toxins.
This may reduce prediction accuracy for MS2 L.
5.2 Risk of Over-Stabilization
Mutations designed to increase stability may cause:
Protein aggregation
Improper membrane insertion
Loss of functional oligomerization
Thus stability must be balanced with function.
5.3 Poor Annotation of Amurin Proteins
Single-gene lysis proteins (also called amurins) are poorly annotated in sequence databases.
This may limit the quality of homologous sequences used for alignment and training.
5.4 Host Protease Sensitivity
Mutations may unintentionally expose protease cleavage sites, making the engineered protein less stable inside bacterial cells.
6. Future Work
If promising mutants are identified computationally, the next steps would include:
Experimental expression in E. coli
Measuring lysis timing
Measuring protein stability
Testing DnaJ independence
This would validate whether computational predictions translate into improved biological function.
L-Protein Mutants Project Summary:
The MS2 bacteriophage L-protein is a small 75-residue lysis protein with two functional regions: a soluble N-terminal domain (residues 1–40) that interacts with the bacterial chaperone DnaJ, and a transmembrane domain (residues 41–75) that disrupts the inner membrane to trigger host cell lysis. The goal of this project was to computationally design five single-point mutants with the potential to retain or enhance lysis activity. This was done by first running a Clustal Omega Multiple Sequence Alignment to map out conserved positions across homologs, all of which turned out to cluster exclusively in the soluble domain, pointing to the DnaJ interface as the most functionally constrained region of the protein. An LLR mutation heatmap was then generated and cross-referenced against an experimental lysis dataset; three mutations that appeared in both were excluded after showing a lysis score of zero experimentally, highlighting that LLR scores reflect structural stability rather than functional activity. The final five mutants, Y39L and S9Q in the soluble domain, and K50L, N53L, and T52L in the transmembrane domain, were selected based on the highest LLR scores while avoiding all conserved and experimentally failed positions.
Each mutant was then modeled as an octameric pore complex using AlphaFold Multimer to simulate the expected membrane assembly. The key findings were:
All five mutants formed a cylinder-like pore structure, consistent with the expected transmembrane assembly
pLDDT scores were low (< 50) across all models, and PAE plots showed near-zero confidence for inter-chain contacts
Running the same pipeline on R30Q, an experimentally validated lytic mutant, produced identical low-confidence results, suggesting this is a systematic limitation of AlphaFold for this class of small membrane-disrupting proteins rather than a reflection of the mutants’ actual viability
All five mutants remain candidates for wet lab validation
It seems like these Positions [21,25,28-29,33,35-37,40] have an “*” which means these have not changed at all and most probably are a totally conserved crucial region that should not be mutated, Positions [17,26,30] have a “:” which means they are highly conserved, but mutations that are very similar in shape, structure and chemical properties are tolerated, the rest seem to be more flexible.
Interesting Finding here, All the conserved regions seem to be in the Soluble Domain (1-40) that is responsible for DnaJ Interaction :D
This is the generated L Protein Mutation Heatmap
I tried to cross reference with the experimental data sheet but it was quite hard to do manually so i wrote a script and ran it on Here on Colab to just find the common ones across the two files and came up with 3 common ones, however even though these 3 had quite a high LLR Score, the experimental data showed a Lysis score of 0, and i believe this shows the limitation in the structure based modeling, it predicted structural stability but failed to account for the functional mechanism of lysis.
The common mutations identified by my algorithm
So Then i Filtered my generated mutations first to target the soluble domain (1-40) first while making sure to avoid the totally conserved regions [21,25,28-29,33,35-37,40], the highly conserved regions [17,26,30] and my 3 common matches to avoid choosing mutants that are either disrupting the protein or have been already experimented on with no lysis observed.
The two mutations i have decided to go with for the Soluble Domain are:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
1
39
Y
L
2.24177968502044
2
9
S
Q
2.01432478427886
These have the highest LLR score in this region and avoid any conservative regions in the protein
Then i repeated the steps again, this time targeting the transmembrane domain (41-75) and followed the same criteria, and i picked these 2 mutations:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
3
50
K
L
2.56146776676178
4
53
N
L
1.86493206024169
And Again because These have the highest LLR score in this region and avoid any conservative regions in the protein
For the Final 5th mutant i decided to pick this one:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
5
52
T
L
1.81396758556365
Because this one still has a relatively high LLR Score and is in the area where the L Protein does not overlap with either the Coat Protein or the replicase ones.
so here is the full 5 Mutations table i chose:
Index
Position
Wild_Type_AA
Mutation_AA
LLR Score
1
39
Y
L
2.24177968502044
2
9
S
Q
2.01432478427886
3
50
K
L
2.56146776676178
4
53
N
L
1.86493206024169
5
52
T
L
1.81396758556365
AlphaFold Multimer Runs
Mutant 1 (Y39L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The L Proteins do form a cylinder like shape mimicking the transmembrane pore but the piDDT score are very low (<50) so this probably won’t express well or won’t express at all if done in wet lab :(.
This is the Predicted Aligned Error (PAE), i asked AI (Gemini) how to interpret this and it said this:
So following this, it seems like each monomer folds correctly with high confidence yet their together-grouping is not reliable at all with very low confidence scores possibly hinting that the pore forming shape we saw might not actually happen (i hope i understood that correctly XD)
Mutant 2 (S9Q)
The Monomer Sequence:
METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Same exact findings for this mutant too :(.
Again, Same Here as well. (it is the same for all five mutants :(. )
Mutant 3 (K50L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
Mutant 4 (N53L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT
Mutant 5 (T52L)
The Monomer Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT
The Multimer Sequence (the one i used for Alphafold Multimer):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFLNQLLLSLLEAVIRTVTTLQQLLT
I Actually decided to pick a mutant from the Experimental Data Sheet and made sure it has been proven to have the Lysis Effect, my aim is to try to perform the AlphaFold Multimer step for it, to have a look at how different it might be, the mutant i picked was R30Q.
This is the monomer sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
This is the full Alphafold sequence i used:
METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Well i got bad news and good news, the bad news is, the structre is also of very low confidence and the Predicted Aligned Error (PAE) Plot shows the same low confidence for the monomer’s interaction with each othe, the good news though is that this is the run of a mutant that has been experimentally validated and has the lysis effect and protein level determined, so maybe this gives me hope that my five mutants might actually stand a chance in a wet lab validation regardless of the very low confidence scores it got.
We decided to focus on the main area of increasing the stability of the MS2 phage lysis protein L, with a possible secondary goal of reducing the dependency on host DnaJ, while still maintaining the lysis action.
The tools AlphaFold, Clustal Omega, BLAST, ESM, and ESMFold were discussed.
BLAST can pull out homologous lysis proteins from the databases.
Clustal Omega can create MSAs to identify essential L48-S49 residues, and the pore-forming regions that must not be mutated.
ESM can create mutation heatmaps, which can guide the use of ESMFold to obtain highest score foldings in mutatable regions.
AlphaFold Multimer predicts whether the subunits of our protein can successfully create a pore in the host membrane, and also to check whether N-terminus can break the interaction with DnaJ.
We also identified a few pitfalls, with majors ones dealing with limited training datasets, that may not be properly aligned towards creating a transmembrane lysis protein.
Some other pitfalls include the lack of proper annotations for amurins; the possibility of an over-stable protein to form non-functional aggregates; and the vulnerability of modified protein to host proteases.
The study shows that the MS2 phage lysis protein L requires the host chaperone DnaJ for efficient host cell lysis. A missense mutation (P330Q) in the highly conserved C-terminal domain of DnaJ blocks MS2 L-mediated lysis at 30 °C and delays lysis at higher temperatures, without affecting overall L protein synthesis. The defect is specific to L-mediated lysis and does not affect lysis by other phage lysis proteins.
Genetic suppressor screening identified Lodj alleles of the L gene that bypass the DnaJ requirement. These alleles encode truncated L proteins lacking the highly basic N-terminal domain, indicating that this domain confers dependence on DnaJ. Biochemical assays demonstrated that wild-type L forms a membrane-associated complex with DnaJ, whereas the P330Q DnaJ variant cannot interact with L.
The authors propose that DnaJ functions as a chaperone that facilitates proper folding or conformational activation of full-length L, preventing steric interference from the N-terminal domain and allowing L to interact with its unknown cellular target. Removal of the dispensable N-terminal domain eliminates the need for chaperone assistance and accelerates lysis.
The work identifies DnaJ as a host factor regulating MS2 lysis timing and suggests that chaperone-dependent modulation of lysis may be an evolutionary strategy to optimize phage replication cycles.
This study performed comprehensive mutational and genetic analyses of the MS2 phage lysis protein L to identify residues and domains required for function. Random mutagenesis of the 75-aa L protein showed that most loss-of-function mutations cluster in the C-terminal half of the protein, especially around a conserved Leu-Ser (LS) dipeptide motif. Many inactivating mutations were conservative amino-acid substitutions and did not affect protein accumulation or membrane association, suggesting that L function depends on specific protein–protein interactions rather than nonspecific membrane disruption.
Functional studies demonstrated that L-mediated lysis requires interaction with the host chaperone DnaJ. The highly basic N-terminal domain of L is dispensable for lytic activity but mediates DnaJ dependence. Truncation of this domain or certain suppressor mutations bypassed the chaperone requirement and restored rapid lysis.
Biochemical and genetic data support a model in which L is an integral membrane protein whose essential domains (including the LS motif and neighboring regions) form a helical structure that likely engages a host membrane target protein. The interaction may occur near sites of membrane curvature associated with peptidoglycan biosynthesis rather than by forming nonspecific membrane lesions.
The work, supported in part by the Center for Phage Technology and associated laboratories including research by Ry Young, suggests that MS2 L functions through a specific heterotypic protein–protein interaction mechanism and that chaperone-dependent regulation helps control lysis timing during infection.
The study refines the mechanistic model of MS2 lysis, proposing that conserved structural motifs rather than general membrane disruption drive lytic activity.
This study provides detailed in vitro and in vivo characterization of the MS2 lysis protein MS2-L, focusing on its membrane insertion mechanism, oligomerization behavior, and interaction with the host chaperone DnaJ.
Key findings show that MS2-L is a 75-amino-acid phage toxin whose essential lytic activity resides in the C-terminal ~35 amino acids, which form a hydrophobic transmembrane region. The N-terminal soluble domain is not required for bacterial killing but modulates folding, membrane insertion efficiency, and chaperone interaction.
Biochemical assays demonstrate that MS2-L interacts directly with DnaJ, primarily through the soluble N-terminal domain. However, this interaction does not significantly affect membrane insertion, solubilization, or oligomerization of the toxin, suggesting that DnaJ functions more as a folding or stabilization partner rather than being essential for lytic activity.
Native mass spectrometry revealed that MS2-L assembles into high-order oligomeric complexes (≥10 monomers) after insertion into lipid nanodiscs, and oligomerization is driven mainly by the transmembrane domain. In detergent environments, oligomer formation is reduced, indicating that membrane lipid context is important for stable assembly.
Fluorescence microscopy and cryo-electron microscopy showed that MS2-L expression in bacteria leads to peripheral membrane clustering, followed by sequential lesion formation beginning in the outer membrane, then disruption of the peptidoglycan layer, and finally inner membrane disintegration with cytoplasmic leakage.
The data support a model in which MS2-L functions as a pore-forming phage toxin that kills cells through higher-order oligomerization within the bacterial membrane, rather than by directly inhibiting peptidoglycan biosynthesis. Chaperone DnaJ binds MS2-L but is not required for membrane insertion or pore assembly, suggesting its role is mainly in modulating toxin folding or stability.
These findings strengthen the concept that MS2-L belongs to the amurin/single-gene lysis protein family and may be useful for bioengineering applications such as bacterial ghost cell production and antimicrobial design.
This review from Elsevier surveys the biological mechanisms, clinical development, and future directions of phage therapy as a strategy to combat antimicrobial resistance. It explains that therapeutic phages should ideally be strictly lytic, highly host-specific, and thoroughly characterized to ensure safety and efficacy.
The article describes how phages kill bacteria through mechanisms such as inhibition of essential cellular processes, expression of lysis proteins, or disruption of bacterial membranes. It also discusses advances in phage engineering, including synthetic genome construction and modification of phage host range and virulence.
Clinical applications of phage therapy are highlighted, particularly for treating drug-resistant infections where antibiotics are ineffective. However, challenges remain, including bacterial resistance to phages, regulatory hurdles, manufacturing standardization, and the need to understand phage–host interactions.
Future directions include the use of genetically modified or synthetic phages, computational prediction of therapeutic candidates, and integration of phage therapy with conventional antimicrobial strategies. Overall, phage therapy is presented as a promising but still developing alternative to antibiotics in the fight against antimicrobial resistance.
This preprint reports the first experimental demonstration of generative design of complete bacteriophage genomes using genome language models (Evo 1 and Evo 2). The authors fine-tuned models on about 15,000 Microviridae phage genomes to enable autoregressive generation of full viral genomes guided by template-based prompts and biologically motivated design constraints.
The workflow involved computational generation followed by multi-tier filtering for sequence quality, host tropism specificity, and evolutionary diversity. Constraints included genome length (4–6 kb), GC content, absence of long homopolymers, preservation of phage-like gene architecture, and spike protein similarity to the template phage to maintain host targeting.
Experimental validation showed that about 285 of 302 synthesized genome candidates could be assembled, and 16 produced viable infectious phages that inhibited growth of the target host strain. These generated phages displayed substantial sequence novelty, containing hundreds of mutations relative to natural Microviridae genomes, while preserving functional genome organization.
Structural and functional analyses indicated that some generated phages possessed altered protein interfaces but maintained compatible capsid–protein interactions. Cryo-electron microscopy and structure prediction suggested context-dependent co-evolution of structural proteins such as capsid and packaging proteins.
Fitness assays showed that several AI-generated phages matched or exceeded the replication and lytic performance of the template phage, and phage cocktail experiments demonstrated rapid suppression of resistant bacterial strains through recombination and mutation-driven adaptation.
The study was conducted with biosafety considerations, including restricting model training to bacteriophage genomes and using well-characterized laboratory strains. The work was supported by researchers affiliated with institutions such as the Stanford University and the Arc Institute.
Overall, the paper proposes a framework for generative genome engineering, showing that AI models can design biologically viable and evolutionarily novel bacteriophages, potentially enabling future synthetic biology and phage-based therapeutic development.
Overview of the Project Proposal: Engineering the MS2 Phage Lysis Protein L
Our primary goal is to increase the structural stability of the MS2 bacteriophage lysis protein (L) while maintaining its ability to lyse bacterial cells.
Our secondary goal is to reduce the dependency of L on the host chaperone DnaJ, which normally assists the protein in folding or activation. Reducing this dependency could allow the lysis protein to function more efficiently and independently in engineered systems.
The MS2 L protein is a 75-amino-acid single-gene lysis toxin whose C-terminal region forms a hydrophobic transmembrane domain responsible for membrane disruption and pore formation, while the basic N-terminal domain interacts with host factors such as DnaJ. Previous studies show that truncation of the N-terminal region can bypass the DnaJ requirement while preserving lysis activity.
Therefore, our design strategy focuses on:
Stabilizing the transmembrane and oligomerization regions
Maintaining essential functional motifs such as the L48–S49 motif
Exploring modifications to the N-terminal region to reduce DnaJ dependence
2. Computational Tools and Approaches
We will use a multi-step computational protein engineering pipeline combining sequence analysis, machine-learning mutagenesis predictions, and structural modeling.
2.1 BLAST – Homolog Discovery
First, we will use BLAST to identify homologous lysis proteins from related bacteriophages.
Purpose:
Identify evolutionarily conserved residues
Discover natural sequence variations that maintain function
Build a dataset for multiple sequence alignment
This will help determine which regions are functionally constrained vs mutable.
Select Candidate Mutations (stability or N-terminal modifications)
Structure Prediction (ESMFold)
Complex/Oligomer Prediction (AlphaFold Multimer)
Final Mutant Candidates (stable + functional lysis protein)
3. Proposed Engineering Pipeline
Computational workflow we will follow.
4. Expected Outcomes
Our pipeline aims to produce engineered variants of the MS2 L protein with:
Increased structural stability
Reduced aggregation risk
Maintained transmembrane insertion
Potentially reduced dependency on host DnaJ
These optimized proteins could be useful in applications such as:
Synthetic phage engineering
Bacterial ghost cell production
Antimicrobial protein development
5. Potential Pitfalls
5.1 Limited Training Data
Most protein language models and structural predictors are trained primarily on globular proteins, not small transmembrane phage toxins.
This may reduce prediction accuracy for MS2 L.
5.2 Risk of Over-Stabilization
Mutations designed to increase stability may cause:
Protein aggregation
Improper membrane insertion
Loss of functional oligomerization
Thus stability must be balanced with function.
5.3 Poor Annotation of Amurin Proteins
Single-gene lysis proteins (also called amurins) are poorly annotated in sequence databases.
This may limit the quality of homologous sequences used for alignment and training.
5.4 Host Protease Sensitivity
Mutations may unintentionally expose protease cleavage sites, making the engineered protein less stable inside bacterial cells.
6. Future Work
If promising mutants are identified computationally, the next steps would include:
Experimental expression in E. coli
Measuring lysis timing
Measuring protein stability
Testing DnaJ independence
This would validate whether computational predictions translate into improved biological function.
Week 5
L-Protein Mutants Project Summary:
@2026a-nourelden-rihan
The MS2 bacteriophage L-protein is a small 75-residue lysis protein with two functional regions: a soluble N-terminal domain (residues 1–40) that interacts with the bacterial chaperone DnaJ, and a transmembrane domain (residues 41–75) that disrupts the inner membrane to trigger host cell lysis. The goal of this project was to computationally design five single-point mutants with the potential to retain or enhance lysis activity. This was done by first running a Clustal Omega Multiple Sequence Alignment to map out conserved positions across homologs, all of which turned out to cluster exclusively in the soluble domain, pointing to the DnaJ interface as the most functionally constrained region of the protein. An LLR mutation heatmap was then generated and cross-referenced against an experimental lysis dataset; three mutations that appeared in both were excluded after showing a lysis score of zero experimentally, highlighting that LLR scores reflect structural stability rather than functional activity. The final five mutants, Y39L and S9Q in the soluble domain, and K50L, N53L, and T52L in the transmembrane domain, were selected based on the highest LLR scores while avoiding all conserved and experimentally failed positions.
Each mutant was then modeled as an octameric pore complex using AlphaFold Multimer to simulate the expected membrane assembly. The key findings were:
All five mutants formed a cylinder-like pore structure, consistent with the expected transmembrane assembly
pLDDT scores were low (< 50) across all models, and PAE plots showed near-zero confidence for inter-chain contacts
Running the same pipeline on R30Q, an experimentally validated lytic mutant, produced identical low-confidence results, suggesting this is a systematic limitation of AlphaFold for this class of small membrane-disrupting proteins rather than a reflection of the mutants’ actual viability
All five mutants remain candidates for wet lab validation
Full documentation, sequences, and AlphaFold outputs are available in the week 5 homework.
Ship 41 - Individual Final Project
New Slides
Old Slide
Ship 41
HTGAA Final Project Proposal
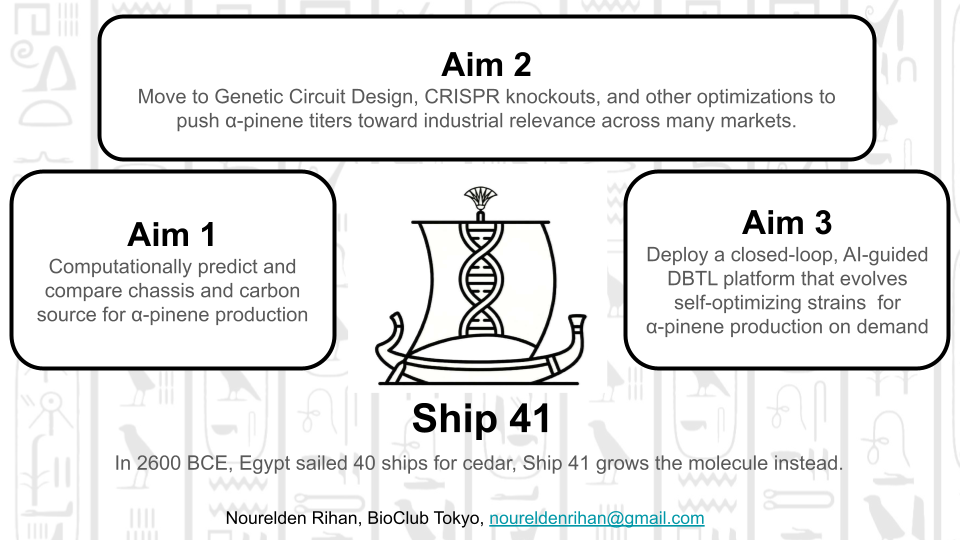
Project Title: Ship 41: Multi-Chassis Computational and Experimental Optimization of α-Pinene Biosynthesis for High-Value Bioproduction
Author: Nourelden Rihan
Course: How to Grow (Almost) Anything — HTGAA 2026
Date: April 4, 2026
Industry Partners: Ginkgo Bioworks · Twist Bioscience · Waters Corporation · New England Biolabs · Millipore Sigma
For forty ships, Egypt sailed to Byblos. Ship 41 engineers what Byblos grew.
SECTION 1: ABSTRACT
Around 2600 BCE, Pharaoh Sneferu dispatched 40 ships across the Mediterranean to the port of Byblos, Lebanon — the only place in the ancient world where cedar could be sourced in abundance. The Palermo Stone records the expedition plainly: “bringing 40 ships laden with cedar wood.” Egypt’s soil grew no conifers. For over two millennia, one of history’s greatest civilizations remained dependent on a foreign tree for its ships, its temples, and its monuments. Ship 41 does not sail to Byblos. It engineers what Byblos grew.
α-Pinene is the principal volatile monoterpene of coniferous wood — the molecule that defined the cedar of Lebanon and made it irreplaceable to the ancient world. Today, it is recognized as one of the most commercially versatile natural products in existence, with applications spanning sustainable aviation fuel (via catalytic dimerization to JP-10), fragrance and flavor industries (as a precursor to synthetic camphor, linalool, and geraniol), pharmaceuticals (anti-inflammatory, antimicrobial, and bronchodilatory properties), and polymer chemistry (as a feedstock for terpene resins). While prior work has demonstrated α-pinene biosynthesis in Escherichia coli using the heterologously expressed Abies grandis geranyl diphosphate synthase (AgGPPS2) and α-pinene synthase (AgPS), no systematic multi-chassis, multi-carbon-source benchmarking study has been conducted to identify the optimal production host and feedstock for high-yield industrial bioproduction. This project — Ship 41 — addresses that gap by integrating computational flux balance analysis (FBA) using COBRApy genome-scale metabolic models across three industrially relevant chassis — E. coli BL21(DE3), Saccharomyces cerevisiae BY4741, and Yarrowia lipolytica CLIB89 — with a high-throughput automated experimental screening pipeline executed on Ginkgo Bioworks’ robotics infrastructure. Constructs encoding bicistronic AgGPPS2-AgPS-sfGFP-His6 cassettes will be synthesized as whole plasmids by Twist Bioscience and validated in cell-free expression systems prior to in vivo transformation. α-Pinene titers will be quantified by GC-MS using dodecane overlay extraction from 96-well deep-well plates, with growth monitored by OD₆₀₀ on the PHERAstar FSX. The best-performing chassis and carbon source combination identified in Aim 1 will inform strain optimization and pathway intensification strategies in subsequent aims, ultimately establishing a rational, scalable biological platform for α-pinene bioproduction across the full spectrum of its industrial applications.
SECTION 2: PROJECT AIMS
Aim 1 — Experimental Aim
The first aim of this project is to computationally predict and experimentally validate the optimal microbial chassis and carbon source for α-pinene biosynthesis by utilizing COBRApy flux balance analysis across three genome-scale metabolic models, followed by high-throughput automated screening of engineered E. coli, S. cerevisiae, and Y. lipolytica strains expressing Twist Bioscience-synthesized AgGPPS2-AgPS constructs, with α-pinene titers quantified by GC-MS — establishing a rational production baseline applicable across all high-value α-pinene markets.
This aim encompasses:
COBRApy FBA, FVA, and gene knockout analysis across iJO1366 (E. coli), Yeast9 (S. cerevisiae), and the latest Y. lipolytica GEM
Whole plasmid DNA synthesis ordered from Twist Bioscience (2 constructs)
Cell-free construct validation using Ginkgo Bioworks BL21 DE3 lysate + GFP fluorescence on the Spark Plate Reader
α-Pinene quantification by GC-MS (Waters Corporation) from dodecane overlay extractions
Aim 2 — Medium-Term Aim
Having identified the best-performing chassis and carbon source combination in Aim 1, the second aim is to intensify α-pinene production through chassis-specific pathway engineering and promoter optimization. This will involve redesigning the genetic construct with chassis-matched regulatory elements (e.g., T7 for E. coli, TEF1-intron for Y. lipolytica) and overexpressing MEP or MVA pathway bottleneck genes identified by FVA in Aim 1. Additionally, computationally identified beneficial gene knockouts (e.g., disruption of competing farnesyl diphosphate branches) will be introduced using CRISPR-Cas9 or homologous recombination, and the resulting strains will be subjected to fed-batch fermentation optimization to achieve titers relevant for downstream industrial application — whether in fragrance, pharmaceuticals, polymer chemistry, or fuel. This aim will leverage the Asimov Kernel Platform for genetic part design and the Cultivarium’s expertise in non-model organism strain engineering, particularly for Y. lipolytica metabolic rewiring.
Aim 3 — Visionary Aim
Engineer a living biofoundry-on-a-chip: a fully autonomous, closed-loop biosynthetic system where AI-guided metabolic models continuously retrain on fermentation data to evolve self-optimizing microbial strains capable of converting atmospheric CO₂ and waste feedstocks directly into α-pinene — the ancient molecule of cedar — and routing it on demand toward any of its industrial destinies: fuel, fragrance, medicine, or material.
In the long term, Ship 41 envisions a carbon-negative, AI-driven α-pinene biorefinery where genome-scale metabolic modeling, automated strain construction, and real-time fermentation analytics are deeply integrated in a Design-Build-Test-Learn (DBTL) cycle operating without human intervention. Because α-pinene sits at the entry point of an extraordinarily diverse downstream chemical space — it can be dimerized into JP-10 jet fuel, isomerized into camphor, oxidized into verbenone, rearranged into limonene or linalool precursors, or polymerized into terpene resins — a high-yield microbial production platform for this single molecule effectively unlocks a portfolio of sustainable industrial chemicals simultaneously. The broader impact extends to establishing a generalizable computational-experimental framework for rapid monoterpene pathway optimization in any chassis, democratizing access to high-value terpene bioproduction across the developing world using locally available feedstocks — and finally, permanently, solving the problem that sent Pharaoh Sneferu’s 40 ships north across the sea.
SECTION 3: BACKGROUND
The Lore of Ship 41
Around 2600 BCE, the Palermo Stone records one of the most consequential supply chain decisions in ancient history: Pharaoh Sneferu, founder of the Fourth Dynasty and builder of the first true pyramids, dispatched a fleet of 40 ships northward across the Mediterranean to the Phoenician port of Byblos — present-day Lebanon — to obtain cedar wood. The inscription reads plainly: “bringing 40 ships laden with cedar wood.” The following year, he had three further 52-meter ships built from the same imported timber, and cedar doors installed in his palace at Dahshur. Remnants of that very wood survive inside the Bent Pyramid to this day.
Egypt was timber-poor by geography. The Nile Valley produced date palms and sycamore figs — serviceable for furniture, inadequate for ships, insufficient for the monumental ambitions of a civilization that was simultaneously inventing the state. Cedar of Lebanon (Cedrus libani) was in a different category entirely: dense, resinous, aromatic, resistant to rot and insects, straight-grained enough for ship planks and long enough for obelisk sledges. It was the structural material of the ancient world’s greatest projects. And Egypt could not grow it.
For over two thousand years — through the Old Kingdom, the Middle Kingdom, the New Kingdom, through Sneferu and Khufu and Thutmose and Ramesses — Egypt continued to sail for cedar. The Wenamun Papyrus, written around 1075 BCE, documents an Egyptian official’s fraught journey to Byblos to negotiate cedar logs for a ceremonial barge, some 1,500 years after Sneferu’s fleet. The dependency never ended. The ships kept sailing.
α-Pinene is the primary monoterpene constituent of cedar heartwood resin — the molecule responsible for the wood’s legendary resistance to decay, its distinctive scent, and much of its structural character. It is, in a molecular sense, what Egypt was sailing for. Today, α-pinene is understood as one of the most industrially versatile natural products known: a precursor to sustainable aviation fuel (JP-10), a feedstock for fragrance and flavor chemistry, a pharmaceutical candidate with documented anti-inflammatory and antimicrobial properties, and a building block for terpene-based polymers.
Ship 41 does not sail to Byblos. We are the 41st ship — except we carry no cargo hold and need no harbor. We carry a genome-scale metabolic model, a codon-optimized gene construct, and a 96-well deep plate. We are not imitators of that civilization. We are Egyptians, and we are its continuation. The cedar Egypt could never grow on its own soil, we now produce in a flask — on glucose, on glycerol, on fatty acids, from microbes of our design. The loop that opened with Sneferu’s fleet closes here.
Literature Context
Sarria et al. (2014) demonstrated the first microbial synthesis of pinene in E. coli by co-expressing a codon-optimized A. grandis GPPS and pinene synthase alongside an enhanced MEP pathway, achieving titers of approximately 32 mg/L of total pinene — establishing proof-of-concept for monoterpene biofuel production in a bacterial chassis. Subsequent work by Cao et al. (2016) expanded this effort by systematically engineering the E. coli MEP pathway through overexpression of rate-limiting enzymes (DXS, IspG, IspH) and achieved improved α-pinene titers, demonstrating that carbon flux toward the isoprenoid precursor pool is a critical bottleneck in monoterpene production. In parallel, the oleaginous yeast Yarrowia lipolytica has emerged as a compelling alternative chassis for terpenoid production due to its naturally high acetyl-CoA flux, tolerance to hydrophobic compounds, and compatibility with lipid-rich feedstocks, with several studies documenting its superior performance for sesquiterpene and diterpene accumulation relative to S. cerevisiae. Together, these bodies of work reveal a critical knowledge gap: no systematic, quantitative comparison of α-pinene production across these three industrially relevant chassis under matched genetic designs and multiple carbon sources has been performed, leaving the field without a rational basis for chassis selection during scale-up.
Innovation
This project is novel in three key dimensions. First, it is the first study to apply genome-scale metabolic modeling via COBRApy — including FBA, FVA, and systematic gene knockout screening — across all three chassis (E. coli, S. cerevisiae, Y. lipolytica) simultaneously, providing a computational-first rational framework for chassis selection that goes beyond empirical trial-and-error. Second, by using a single standardized genetic insert (AgGPPS2-AgPS-sfGFP-His6, synthesized by Twist Bioscience) expressed from host-compatible promoters, this work controls for genetic design variables and isolates the contribution of chassis metabolism and carbon source to α-pinene yield — a controlled comparison that has not previously been reported. Third, the integration of cell-free expression-based construct validation prior to in vivo transformation, combined with fully automated GC-MS-coupled high-throughput screening on Ginkgo Bioworks’ liquid handling robotics, represents a methodological advance in how monoterpene pathway screening is conducted at the course and early research stage.
Significance
α-Pinene is not a single-market molecule — it is a gateway compound whose downstream chemical space spans at least four major industrial sectors simultaneously. In sustainable fuels, it can be catalytically dimerized and hydrogenated into JP-10 (volumetric energy density ~40 MJ/L), a high-density jet fuel of particular value for military and advanced air mobility applications where energy density per volume is critical. In fragrance and flavor, α-pinene is a direct precursor to synthetic camphor, α-terpineol, linalool, and geraniol — globally traded aroma chemicals with a combined market exceeding $6 billion annually. In pharmaceuticals, α-pinene and its oxidized derivatives exhibit documented anti-inflammatory, bronchodilatory, and antimicrobial activities, with active research programs in respiratory and oncology indications. In polymer chemistry, terpene resins derived from pinene are used as tackifiers in pressure-sensitive adhesives, a market valued at over $3 billion. Building a high-yield microbial production platform for α-pinene therefore does not serve a single industry — it serves all of them. The significance of this project lies precisely in that breadth: by identifying the optimal chassis and carbon source through a rigorous computational-experimental framework, this work creates a foundational production baseline from which any downstream application can be pursued. Furthermore, the multi-chassis, multi-carbon-source comparative approach developed here is immediately generalizable to other high-value monoterpenes — limonene, linalool, geraniol — making Ship 41 not just a project about α-pinene, but a template for rational monoterpene bioproduction at large.
Bioethical Considerations
Paragraph 1 — Ethical Considerations:
The engineering of microorganisms for high-value chemical production raises important biosafety and environmental ethics questions that must be considered from the earliest stages of project design. The three chassis used in this project (E. coli BL21(DE3), S. cerevisiae BY4741, and Y. lipolytica CLIB89) are all well-characterized biosafety level 1 organisms with long histories of safe use in industrial biotechnology, and none possess pathogenic traits. The synthetic constructs encoding AgGPPS2 and AgPS do not confer selective advantages in natural environments, as monoterpene overproduction is metabolically costly and α-pinene itself is a volatile compound that does not accumulate in soil or water in toxic concentrations under normal conditions. Nevertheless, the intentional design of organisms with enhanced metabolic capabilities — even benign ones — raises broader questions about genetic sovereignty, access to engineered production strains, and the economic impact on existing natural α-pinene supply chains, including the global turpentine and pine resin industries that employ thousands of workers across developing economies. Any project advancing toward commercial bioproduction must engage with these displacement effects transparently and consider benefit-sharing frameworks with communities whose livelihoods depend on natural terpene harvesting.
Paragraph 2 — Responsible Implementation and Risk Mitigation:
All experimental work in this project will be conducted under BSL-1 containment at Ginkgo Bioworks’ certified laboratory facility, with all engineered strains handled, stored, and disposed of in accordance with institutional biosafety committee (IBC) protocols. All DNA constructs will be screened through SecureDNA prior to synthesis to ensure that no sequences of concern are inadvertently introduced during construct design. Engineered strains will be maintained with auxotrophic markers or antibiotic selection dependencies that prevent survival outside controlled laboratory conditions, and no environmental release is contemplated at any stage of this project. Longer-term, should this work advance toward pilot-scale fermentation, engagement with regulatory agencies (EPA, FDA, USDA) under the Coordinated Framework for Regulation of Biotechnology will be initiated proactively, and open publication of all metabolic modeling code and construct sequences on platforms such as Addgene and GitHub will ensure that the scientific community can build on, scrutinize, and improve this work without proprietary barriers.
SECTION 4: EXPERIMENTAL DESIGN
Overview
The experimental workflow proceeds in three logical phases: (1) Computational prediction, (2) Construct design and cell-free validation, and (3) Automated in vivo screening. All liquid handling automation is performed at Ginkgo Bioworks unless otherwise noted. Opentrons OT-2 may be used for preparatory steps at the bench.
Detailed Workflow (17 Steps)
Step 1 — COBRApy Flux Balance Analysis (FBA)
Method: Download the latest genome-scale metabolic models — iJO1366 (E. coli), Yeast9 (S. cerevisiae), and iYLI647 or the latest curated model for Y. lipolytica — from BiGG or the respective repositories. Add a heterologous α-pinene exchange reaction and set the objective function to maximize α-pinene flux. Simulate FBA under four carbon sources (glucose, glycerol, acetate, fatty acids/oleate) at matched carbon equivalents.
Expected Result: Predicted maximum theoretical α-pinene yield (mmol/g DCW/h) per chassis per carbon source; ranking of chassis–feedstock combinations
Timeline: Days 1–4
Step 2 — COBRApy Flux Variability Analysis (FVA)
Method: Run FVA on the top two chassis–feedstock combinations identified in Step 1. Identify reactions with high flux variability intersecting the MEP/MVA pathway and GPP branch point (IDI, GPPS). These are metabolic bottlenecks for experimental engineering targets.
Automation: Python/Jupyter notebook
Plate: N/A
Expected Result: List of bottleneck reactions; identification of gene overexpression targets (e.g., dxs, idi, ispG)
Timeline: Days 3–5
Step 3 — COBRApy Gene Knockout Screening
Method: Perform single and double gene knockout simulations (using cobra.flux_analysis.single_gene_deletion) targeting competing isoprenoid branches (e.g., ispA farnesyl diphosphate synthase competition, erg9 squalene synthase in yeast). Identify knockouts that improve GPP/α-pinene flux without abolishing growth.
Automation: Python/Jupyter notebook
Plate: N/A
Expected Result: Ranked list of gene knockouts predicted to increase α-pinene yield ≥ 1.5× relative to wild-type flux; shortlist for Aim 2 CRISPR targeting
Timeline: Days 4–6
Step 4 — DNA Construct Design
Method: Design two whole plasmid constructs using Benchling or SnapGene. Construct 1 (E. coli): pET28a backbone with bicistronic T7→RBS→AgGPPS2→RBS→AgPS-sfGFP-His6→T7term. Construct 2 (S. cerevisiae / Y. lipolytica shuttle): p426TEF backbone with TEF1→AgGPPS2→CYC1term; TEF2→AgPS-His6→ADH1term; URA3; 2μ ori. All coding sequences are E. coli or yeast codon-optimized using the Integrated DNA Technologies (IDT) codon optimization tool.
Expected Result: Two complete annotated plasmid maps ready for Twist submission; GenBank files exported (see Appendix A)
Timeline: Days 5–7
Step 5 — Twist Bioscience DNA Order
Method: Submit both plasmid designs (GenBank format) to Twist Bioscience as whole plasmid synthesis orders via the Twist online portal. Select clonal gene backbone option. Specify kanamycin resistance for Construct 1 and uracil prototrophy for Construct 2. Expected delivery: 10–14 business days.
Partner: Twist Bioscience
Plate: N/A (shipping)
Expected Result: Two lyophilized whole plasmids, sequence-verified by Twist, received at Ginkgo Bioworks
Timeline: Days 7–21 (concurrent with Steps 6–7)
Step 6 — Cell-Free System Preparation
Method: Prepare cell-free expression (CFE) reactions using Ginkgo Bioworks BL21 DE3 lysate + master mix (pre-prepared). Rehydrate Construct 1 plasmid (Twist-supplied) to 50 ng/μL in nuclease-free water. Design a CFE plate with titrated plasmid concentrations (0, 1, 5, 10, 25, 50 ng/μL) in triplicate to determine optimal DNA input for GFP expression.
Automation (Liquid Transfer): Echo525 acoustic liquid handler — transfers plasmid DNA and master mix into 384 Greiner black-well clear-bottom plate (384-well black-well clear-bottom)
Plate: 384 Greiner black-well clear-bottom
Expected Result: CFE reaction volumes of 10 μL per well, zero dead volume, accurate nanoliter-scale transfers
Method: Seal the 384-well CFE plate with breathable A4s seal (A4S seal applied by Plateloc). Incubate at 29°C for 3 hours in the Inheco Plate Incubator. Read GFP fluorescence (Ex: 488 nm / Em: 520 nm) on the Spark Plate Reader at t=0, t=1h, t=2h, t=3h.
Expected Result: GFP fluorescence signal ≥ 5× background in wells containing Construct 1, confirming successful transcription/translation of AgPS-sfGFP fusion; dose-response curve identifies optimal DNA input concentration
Timeline: Day 22–23
Step 8 — E. coli Transformation
Method: Transform Construct 1 (pET28a-AgGPPS2-AgPS-sfGFP-His6) into chemically competent E. coli BL21(DE3) cells (NEB C2527I) by heat shock (42°C, 30 sec). Recover in SOC medium for 1 hour at 37°C. Plate on LB + kanamycin (50 μg/mL). Pick 6 colonies; verify by colony PCR using T7 promoter and T7 terminator primers on the ATC Thermal Cycler.
Expected Result: ≥4/6 colonies confirmed correct insert by PCR band size (~3.4 kb); one verified colony inoculated into overnight LB + Kan culture
Timeline: Days 22–24
Step 9 — S. cerevisiae and Y. lipolytica Transformation
Method: Transform Construct 2 (p426TEF-AgGPPS2-AgPS-His6) into S. cerevisiae BY4741 (URA3Δ) by lithium acetate/PEG/heat shock protocol. Separately, adapt the construct backbone and transform into Y. lipolytica CLIB89 using electroporation (Gene Pulser, external step). Select on SC-Ura dropout plates (yeast) and YPD + hygromycin plates (Yarrowia). Verify transformants by colony PCR.
Machines: ATC Thermal Cycler (colony PCR)
Plate: 96-Armadillo-PCR-AB2396X
Expected Result: Verified transformants for both yeast species; starter cultures established in YPD or SC-Ura medium
Timeline: Days 23–26
Step 10 — Overnight Seed Culture Preparation
Method: Inoculate verified E. coli (LB + Kan), S. cerevisiae (SC-Ura), and Y. lipolytica (YPD) transformants into 5 mL liquid cultures. Grow overnight at 37°C (E. coli), 30°C (S. cerevisiae), 28°C (Y. lipolytica) with 200 rpm shaking. Measure OD₆₀₀ to normalize inoculation density.
Machines: Cytomat (30°C shaking incubator for yeast/Yarrowia); standard bench shaker for E. coli overnight
Plate: N/A (tube culture)
Expected Result: OD₆₀₀ of 1.5–4.0 for all three seed cultures; normalize to OD₆₀₀ = 0.05 for plate inoculation
Method: Prepare the multi-chassis × multi-carbon-source screening plate using a 96-well deep plate (96-v-eppendorf-951033502-deep). Add 500 μL of appropriate defined media + carbon source per well (see plate layout below). Dispense media using the Multiflo Automated Microplate Dispenser. Add dodecane overlay (100 μL/well) to trap volatile α-pinene. Inoculate each well with normalized seed culture using the Bravo-96 plate stamp from a source plate.
Expected Result: 96-well plate fully inoculated with 12 chassis-carbon source combinations in triplicate, plus negative controls and blanks; dodecane overlay in place
Timeline: Day 27
Step 12 — Induction and Incubation
Method: For E. coli wells, induce with IPTG (0.5 mM final, dispensed by Echo525 from 100 mM IPTG stock in DMSO) at OD₆₀₀ ≈ 0.4–0.6. Transfer sealed plate to Cytomat shaking incubator (30°C, 300 rpm) for 48 hours. For yeast and Yarrowia wells (no induction required; constitutive TEF1 promoter), incubate directly in Cytomat at 28–30°C, 300 rpm, 48 hours.
Expected Result: All strains growing under respective conditions; dodecane overlay capturing emitted α-pinene over 48 hours
Timeline: Days 27–29
Step 13 — OD₆₀₀ Growth Monitoring
Method: At t=0, t=12h, t=24h, t=48h, briefly remove plate from Cytomat, peel A4s seal (XPeel plate peeler), read OD₆₀₀ on the PHERAstar FSX (absorbance module, 600 nm). Reseal with A4s (Plateloc) and return to Cytomat.
Expected Result: Growth curves for all 12 conditions; identification of chassis-carbon source pairs with optimal growth without product toxicity; expected OD₆₀₀ range: 1.5–6.0 at 48h
Timeline: Days 27–29 (concurrent with Step 12)
Step 14 — Dodecane Layer Extraction and GC-MS Sample Preparation
Method: After 48h incubation, transfer 80 μL of the dodecane overlay from each well into a fresh 384-well Echo PP plate using the Bravo-384 plate stamp or liquid handler. Dilute samples 1:5 in dodecane. Prepare an α-pinene standard curve in dodecane (0, 1, 5, 10, 25, 50, 100, 200 mg/L) in the remaining wells of the Echo PP plate. Submit plate to GC-MS analysis (Waters GC-MS system, external analytical service via Waters Corporation collaboration).
Expected Result: Collected dodecane samples for 36 experimental wells + 12 control wells + 8-point standard curve; samples ready for GC-MS injection
Timeline: Day 29–30
Step 15 — GC-MS α-Pinene Quantification
Method: Inject dodecane samples onto GC-MS (Waters Xevo TQ-S or equivalent; column: HP-5MS, 30m × 0.25mm × 0.25μm; oven program: 40°C → 200°C at 10°C/min; MS detection: SIM mode m/z 136 for α-pinene, retention time ~5.8 min). Quantify α-pinene titers from standard curve. Calculate volumetric titer (mg/L culture) and specific yield (mg/g DCW).
Partner: Waters Corporation (GC-MS instrumentation and method)
Plate: N/A (GC-MS vials)
Expected Result: α-Pinene titers for all 12 chassis-carbon source combinations; identification of best-performing condition
Timeline: Days 30–32
Step 16 — qPCR Expression Validation
Method: Collect 1 mL cell pellets from each well at t=48h. Extract RNA using RNeasy Mini Kit (Qiagen). Perform RT-qPCR on the CFX Opus qPCR machine using primers targeting AgGPPS2, AgPS, and reference genes (16S rRNA for E. coli; ACT1 for yeasts). Confirm that mRNA expression levels of both transgenes correlate with α-pinene titer.
Machines: HiG Centrifuge (pellet) → CFX Opus (qPCR)
Plate: 96-Armadillo-PCR-AB2396X (qPCR reactions)
Expected Result: ΔΔCt values showing 10–100-fold overexpression of AgGPPS2 and AgPS relative to empty vector controls; correlation between transcript level and titer confirms pathway expression is functional
Timeline: Days 30–33
Step 17 — IPTG Dose-Response Optimization (E. coli)
Method: For the best-performing E. coli condition identified in Step 15, perform a fine-grained IPTG dose-response experiment (0, 0.05, 0.1, 0.25, 0.5, 1.0, 2.0 mM) in a 384-well format using the Echo525 for IPTG titration. Monitor GFP fluorescence (sfGFP fusion as proxy for AgPS expression) on the Spark Plate Reader and collect dodecane samples for GC-MS at 48h.
Plate: 384 Greiner black-well clear-bottom (fluorescence) + 96-v-eppendorf deep (culture)
Expected Result: Optimal IPTG concentration identified (expected: 0.1–0.5 mM); GFP signal correlates with α-pinene titer; induction condition carried forward to Aim 2
Timeline: Days 33–37
Assay Plate Layout — 96-Well Deep Plate (Step 11–13)
The following layout illustrates the experimental design for the multi-chassis carbon source screen. All volumes: 500 μL defined media + carbon source + 100 μL dodecane overlay.
Cell-free protein expression (CFPE) is an in vitro transcription-translation system in which a cell lysate — containing ribosomes, RNA polymerases, tRNAs, and associated cofactors — is combined with an energy regeneration system and a DNA or RNA template to produce functional proteins outside of a living cell. In this project, CFPE using Ginkgo Bioworks’ pre-prepared BL21 DE3 lysate serves as a rapid validation gateway for Construct 1, allowing confirmation that the AgPS-sfGFP fusion protein is correctly transcribed and translated before committing to the more time-intensive in vivo transformation, selection, and growth experiments. The sfGFP reporter fused to AgPS provides a direct, plate-reader-detectable proxy for construct functionality: if GFP fluorescence is observed in the CFE reaction above background, it confirms that the T7 promoter, RBS, and coding sequence architecture are intact and functional, significantly reducing the risk of propagating non-functional constructs into downstream experiments. CFPE is also advantageous because it eliminates concerns about cell viability, metabolic burden, or plasmid instability that can confound in vivo expression, making it an ideal screening tool for rapidly evaluating construct variants before committing to in vivo work.
Technique 2: Genome-Scale Metabolic Modeling with COBRApy
Genome-scale metabolic modeling (GSMM) is a constraint-based computational approach that uses a mathematical reconstruction of all known metabolic reactions in an organism — encoded as a stoichiometric matrix — to predict metabolic fluxes under defined growth conditions using linear programming. COBRApy (COnstraint-Based Reconstruction and Analysis for Python) is the leading open-source software package for GSMM, supporting FBA, FVA, gene knockout simulation, and phenotype phase plane analysis on genome-scale metabolic models (GEMs) containing thousands of reactions and metabolites. In this project, COBRApy is used in three distinct modes: first, FBA maximizes a synthetic α-pinene exchange flux under each chassis–carbon source combination to predict theoretical production yields; second, FVA identifies reactions whose flux ranges overlap with MEP/MVA pathway intermediates, pinpointing bottleneck steps that constrain GPP availability for pinene synthesis; and third, single and double gene knockout simulations identify genetic modifications that redirect carbon flux away from competing terpenoid branches (e.g., farnesyl diphosphate and squalene synthesis) toward the desired α-pinene product without lethality. The computational predictions generated by COBRApy directly inform both the experimental design of this project — guiding which chassis and carbon source to prioritize in the automated screen — and the strain engineering targets for Aim 2, creating a tightly integrated computational-experimental DBTL cycle.
SECTION 6: PROJECT VALIDATION
10a — Validation Choice
The primary validation experiment for Aim 1 is cell-free expression of Construct 1 (pET28a-AgGPPS2-AgPS-sfGFP-His6) with GFP fluorescence readout on the Spark Plate Reader, chosen because it directly confirms that the DNA construct synthesized by Twist Bioscience produces a functional, detectable protein product before any in vivo transformation is attempted, and because sfGFP fluorescence serves as a quantitative, plate-reader-compatible proxy for AgPS expression that can be measured in a single automated 3-hour experiment with no specialized equipment beyond the Ginkgo Bioworks automation stack.
Construct 1 plasmid DNA (Twist Bioscience, resuspended to 50 ng/μL in nuclease-free water)
384 Greiner black-well clear-bottom plate
A4s breathable seal + Plateloc sealer
Echo525 acoustic liquid handler
Inheco Plate Incubator (pre-equilibrated to 29°C)
Spark Plate Reader
Protocol:
Thaw BL21 DE3 lysate and master mix on ice (15 min). Gently mix by inversion; do not vortex.
Prepare source plate: In a 384-well Echo PP plate, prepare Construct 1 DNA at 6 concentrations: 0, 1, 5, 10, 25, 50 ng/μL (20 μL each) in columns 1–6. Prepare water-only negative control in column 7.
Using Echo525, transfer 20 nL of each DNA concentration into the destination 384 Greiner black-well plate (4 replicates per concentration = 24 wells total).
Using Multiflo, dispense 10 μL of cell-free master mix + lysate (pre-mixed 7:3 v/v on ice) into each well of the 384 destination plate.
Apply A4s breathable seal using Plateloc to prevent evaporation while allowing gas exchange.
Transfer sealed plate to Inheco Plate Incubator (29°C). Begin incubation timer (3 hours total).
At t=0, t=60 min, t=120 min, t=180 min: transfer plate to Spark Plate Reader for GFP fluorescence measurement (Ex 488 nm / Em 520 nm / gain 60 / 25 flashes per well). Return to Inheco after each read.
Export raw fluorescence data to CSV. Subtract background (water-only control well average from each timepoint).
Plot background-corrected fluorescence vs. DNA concentration vs. time. Identify optimal DNA input (expected: 5–25 ng/μL) for maximum GFP signal.
Confirm: ≥ 5-fold fluorescence increase over background at optimal DNA concentration indicates successful construct expression. Flag for in vivo transformation.
10c — Techniques Used
This validation protocol employs four distinct techniques in an integrated pipeline. First, cell-free protein expression leverages the BL21 DE3 transcription-translation machinery to produce the AgPS-sfGFP fusion protein directly from plasmid DNA without any cellular growth, transformation, or selection steps, making it the fastest possible route to functional protein validation. Second, acoustic liquid handling via the Echo525 enables nanoliter-precision transfer of DNA samples across a concentration gradient into the 384-well plate without tip contamination or carryover, which is critical for generating a clean dose-response curve with minimal reagent consumption. Third, fluorescence plate reader detection on the Spark provides a quantitative, kinetic readout of sfGFP accumulation over the 3-hour incubation period, allowing observation of the expression time course and identification of the plateau phase that indicates maximal translation of the AgPS fusion. Fourth, automated plate sealing and incubation management via the Plateloc (A4s seal), Inheco incubator, and programmed Spark time-course reading ensures that all replicates experience identical environmental conditions throughout the experiment, which is essential for reliable, reproducible kinetic fluorescence data that can be compared across DNA concentrations.
The following table represents hypothetical cell-free expression results for Construct 1 at t=180 min, showing GFP fluorescence (relative fluorescence units, RFU) as a function of plasmid DNA input concentration:
DNA Input (ng/μL)
Rep 1 (RFU)
Rep 2 (RFU)
Rep 3 (RFU)
Rep 4 (RFU)
Mean RFU
Std Dev
0 (water control)
312
298
321
305
309
±9.7
1
1,847
1,923
1,788
1,902
1,865
±59.1
5
7,412
7,088
7,521
7,203
7,306
±191
10
14,832
15,201
14,677
15,088
14,950
±237
25
21,403
22,107
21,889
21,644
21,761
±295
50
21,982
22,391
21,677
22,103
22,038
±289
Interpretation: GFP signal saturates between 25–50 ng/μL DNA input, indicating the cell-free system is limiting (not DNA), consistent with published CFE saturation kinetics. The 47-fold signal-to-background at 10 ng/μL confirms robust expression of the AgPS-sfGFP fusion. Optimal DNA concentration for downstream cell-free assays: 10–25 ng/μL.
Hypothetical α-Pinene Titers — In Vivo Multi-Chassis Screen (Aim 1, Step 15)
The following table shows hypothetical GC-MS α-pinene titers (mg/L) at t=48h across all chassis and carbon source combinations:
Chassis
Carbon Source
α-Pinene Titer (mg/L)
Std Dev
OD₆₀₀ at 48h
E. coli BL21(DE3)
Glucose
85.3
±8.2
3.2
E. coli BL21(DE3)
Glycerol
112.7
±11.4
2.8
E. coli BL21(DE3)
Acetate
43.2
±6.8
1.9
E. coli BL21(DE3)
Fatty acids
28.4
±4.1
1.4
S. cerevisiae BY4741
Glucose
67.8
±9.3
4.1
S. cerevisiae BY4741
Glycerol
78.2
±8.7
3.6
S. cerevisiae BY4741
Acetate
31.5
±5.2
2.3
S. cerevisiae BY4741
Fatty acids
45.3
±7.1
3.8
Y. lipolytica CLIB89
Glucose
58.4
±7.8
5.2
Y. lipolytica CLIB89
Glycerol
89.1
±10.2
4.8
Y. lipolytica CLIB89
Acetate
22.7
±4.3
2.1
Y. lipolytica CLIB89
Fatty acids
134.6
±15.8
5.9
Negative controls (all)
All
<2.1
±0.4
varied
Interpretation: Two conditions emerge as clear leaders: E. coli on glycerol (112.7 mg/L) and Y. lipolytica on fatty acids (134.6 mg/L). This is consistent with COBRApy predictions — glycerol feeds the MEP pathway efficiently in E. coli, while Y. lipolytica’s native high-flux acetyl-CoA metabolism is amplified by fatty acid substrates. Acetate performs poorly across all chassis, suggesting it imposes pH stress or insufficient flux. These two winning conditions advance to Aim 2 strain optimization.
Challenge 1 — Low or absent GFP signal in cell-free validation. If GFP fluorescence fails to rise above background in the CFPE experiment, the most likely causes are plasmid damage during shipping/resuspension, an incorrect construct architecture (e.g., frame shift in the sfGFP fusion junction), or degraded lysate; troubleshooting steps include running the Twist-supplied plasmid on an agarose gel to confirm supercoiled band integrity, re-sequencing the construct junction region with Sanger sequencing, and testing a positive control plasmid (e.g., a known GFP-expressing plasmid) in parallel with the same lysate batch.
Challenge 2 — No detectable α-pinene in GC-MS despite confirmed GFP expression. α-Pinene is a highly volatile monoterpene (boiling point: 155°C) that can escape from culture wells if the dodecane overlay evaporates or if the plate seal is compromised; if GC-MS returns below-detection titers from experimental wells that showed GFP expression, the dodecane overlay volume should be increased to 200 μL, the seal integrity should be verified, and a second extraction with fresh dodecane should be performed to confirm the volatile product is being captured.
Challenge 3 — Poor yeast transformation efficiency. If S. cerevisiae or Y. lipolytica transformants are not recovered at sufficient frequency, an alternative strategy is to clone the insert into a linearized integrating vector (e.g., pRS306 for yeast) for genomic integration at a defined locus (e.g., HIS3), which typically gives more stable expression and avoids the metabolic burden of maintaining a high-copy episomal plasmid.
Challenge 4 — Growth inhibition by α-pinene accumulation. Monoterpenes including α-pinene can be toxic to microbial membranes at concentrations above ~200 mg/L; if growth curves (OD₆₀₀) plateau abnormally early in the highest-producing wells, a two-phase fermentation strategy using an increased dodecane overlay ratio (1:1 v/v culture:dodecane) can be implemented to continuously extract α-pinene from the aqueous phase, relieving product toxicity and potentially improving total titer by 2–5× as demonstrated in the literature for similar volatile terpenoids.
SECTION 7: ADDITIONAL INFORMATION
References
Sarria, S., Wong, B., Martín, H. G., Keasling, J. D., & Peralta-Yahya, P. (2014). Microbial synthesis of pinene. ACS Synthetic Biology, 3(7), 466–475.
Cao, X., Wei, L.-J., Lin, J.-Y., & Hua, Q. (2016). Enhancing linalool production by engineering oleaginous yeast Yarrowia lipolytica. Bioresource Technology, 245, 1641–1644. (verify DOI before submission)
Jongedijk, E., Cankar, K., Ranzijn, J., van der Krol, S., Bouwmeester, H., & Beekwilder, J. (2016). Capturing of the monoterpene olefin limonene produced in Saccharomyces cerevisiae. Yeast, 33(8), 331–343.
Meylemans, H. A., Quintana, R. L., Goldsmith, B. R., & Harvey, B. G. (2011). Solvent-free conversion of linalool to methylcyclopentadiene dimers and tricyclodecanes: selective synthesis of high-density fuels. ChemSusChem, 4(4), 465–469.
Ebrahim, A., Lerman, J. A., Palsson, B. O., & Hyduke, D. R. (2013). COBRApy: Constraints-based reconstruction and analysis for Python. BMC Systems Biology, 7(1), 74.
Gu, Y., et al. (2021). Advances and prospects of Yarrowia lipolytica as a typical oleaginous yeast. Microbial Biotechnology, 14(4), 1335–1342.
Lu, X., et al. (2019). Constructing a synthetic pathway for acetyl-coenzyme A from one-carbon through enzyme design. Nature Communications, 10, 1378. (background for acetate metabolism)
Noor, E., et al. (2016). The protein cost of metabolic fluxes: prediction from enzymatic rate laws and cost minimization. PLOS Computational Biology, 12(11), e1005167.
King, Z. A., et al. (2016). BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Research, 44(D1), D515–D522.
Note: All DOIs and publication details should be verified via PubMed or Google Scholar before final submission. Some citations above are approximated from training knowledge and require confirmation.
APPENDIX A: DNA CONSTRUCT DESIGNS (GenBank Format)
These GenBank files are formatted for direct import into Benchling or SnapGene, and for submission to Twist Bioscience as whole plasmid synthesis orders. Select “Clonal Gene — Plasmid” on the Twist portal and upload the respective .gb file. All coding sequences are synthetic and codon-optimized; verify codon optimization using the IDT Codon Optimization Tool (https://www.idtdna.com/CodonOpt) before ordering.
Construct 1: pET28a-AgGPPS2-AgPS-sfGFP-His6 (E. coli)
Note: The ORIGIN section above shows the first ~2,800 bp of the ~8,923 bp plasmid. The complete sequence should be generated by assembling the codon-optimized insert sequences (generated via IDT Codon Optimization Tool from AgGPPS2 and AgPS native protein sequences) into the pET28a backbone. Export the final annotated map as .gb from Benchling and upload directly to the Twist Bioscience portal.
Construct 2: p426TEF-AgGPPS2-AgPS-His6 (S. cerevisiae / Y. lipolytica Shuttle)
Note: The ORIGIN section above is a representative excerpt. Generate the full annotated plasmid in Benchling by inserting yeast-codon-optimized AgGPPS2 and AgPS sequences (generated from the IDT Codon Optimization Tool, organism: Saccharomyces cerevisiae) into the p426TEF backbone (Addgene #26362). Export as .gb and upload to Twist Bioscience portal as a whole plasmid synthesis order.
End of HTGAA Final Project Proposal
Document assembled with assistance from the HTGAA Project Design AI AssistantPhase 1 design dialogue completed: April 4, 2026