Week 5: Protein design, part II
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
UniProt ID: P37840
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQA4V mutation introduced:
MATK**V**VCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ2. Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
| index | Binder | Pseudo Perplexity |
|---|---|---|
| 1 | WRYYVVGLRHKX | 13.80 |
| 2 | HRYPVVVVALGX | 17.15 |
| 3 | WRVYAVGLALWX | 11.84 |
| 4 | WLYPATVLEWKX | 12.49 |
More peptides were generated to screen the ones with higher scores (above 30%):
| index | Binder | Pseudo Perplexity |
|---|---|---|
| 1 | WRYYVVVVELKK | 33.87 |
| 2 | HLYYVAGVRWKK | 30.94 |
| 3 | HHYYVAVLELWK | 31.88 |
| 4 | HLYYVTVVRLGK | 48.23 |
| known | FLYRWLPSRRGG | - |
Part 2: Evaluation of Binders with AlphaFold3
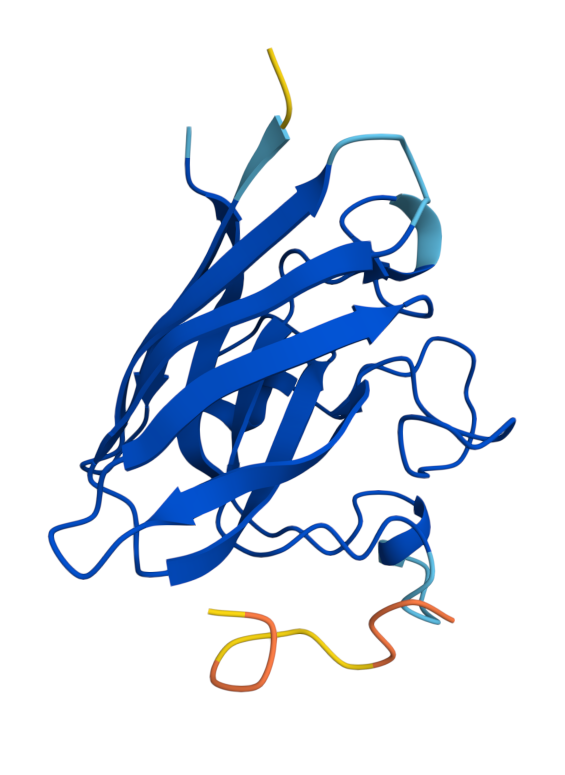 Known (FLYRWLPSRRGG) ipTM = 0.38, pTM = 0.78, Seed 1074152779 | 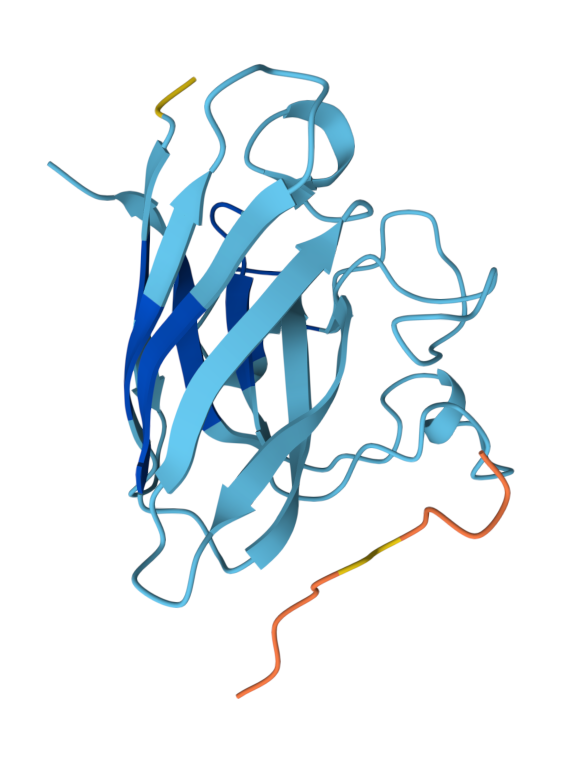 Peptide 1 (WRYYVVVVELKK) ipTM = 0.24 , pTM = 0.76, Seed 1561902944 | 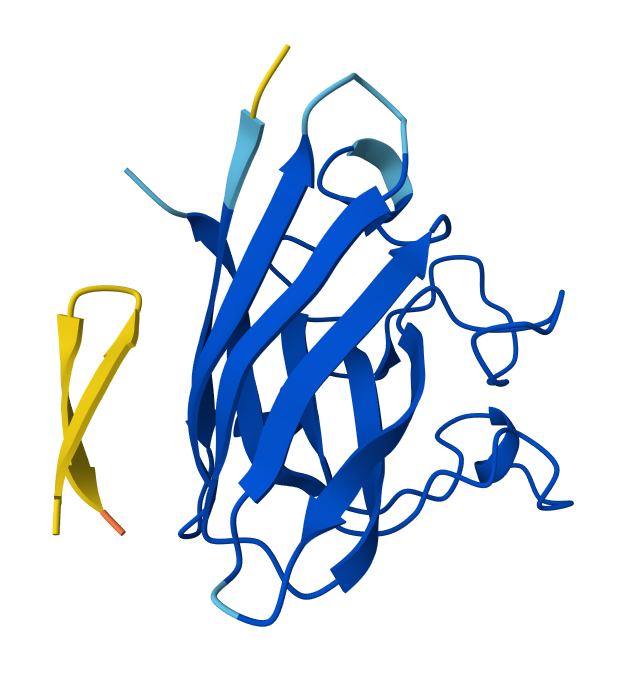 Peptide 2 (HLYYVAGVRWKK) ipTM = 0.39, pTM = 0.86 , Seed 1713765441 |
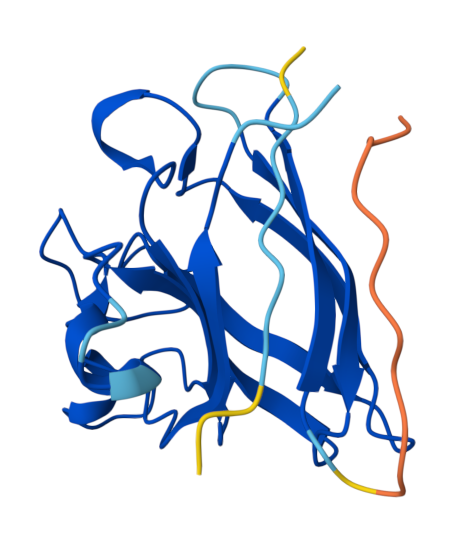 Peptide 3 (HHYYVAVLELWK) ipTM = 0.74, pTM = 0.86, Seed 1794605497 | 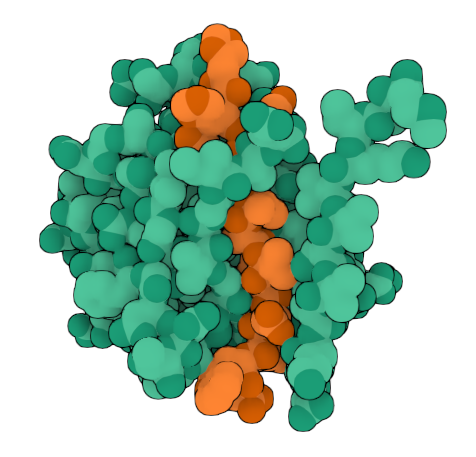 Peptide 3 (HHYYVAVLELWK) ipTM = 0.74, pTM = 0.86, Seed 1794605497 | 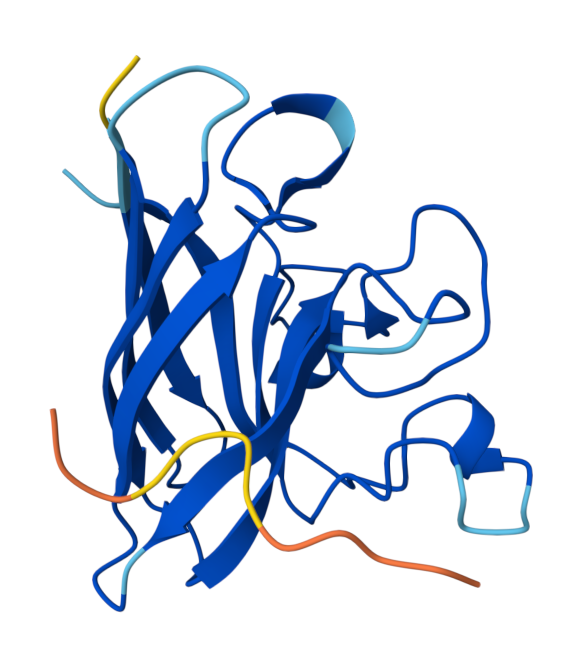 Peptide 4 (HLYYVTVVRLGK) ipTM = 0.45, pTM = 0.86, Seed 1556305056 |
The ipTM scores and peptide localization/binding were recorded:
| Binder | near A4V | engage the β-barrel region | dimer interface | surface-bound | partially buried |
|---|---|---|---|---|---|
| 1 | WRYYVVVVELKK | no | a bit | no | yes |
| 2 | HLYYVAGVRWKK | no | yes | no | yes |
| 3 | HHYYVAVLELWK | yes | yes | yes | |
| 4 | HLYYVTVVRLGK | no | yes | no | |
| known | FLYRWLPSRRGG | no | no | no |
Based on the predicted template modeling scores, all of the overall predicted folds for the complexes might be similar to the true structure (pTM scores>0.5), and the model for Peptide 3 (HHYYVAVLELWK) has exceptionally high accuracy of the predicted relative positions of the ligand and SOD1 within the complex. However, according to Alphafold3 instructions, values between 0.6 and 0.8 are a gray zone, where predictions could still be either correct or incorrect.
Peptide 3 and Peptide 4 ipTM values (Peptide 3: ipTM = 0.74; Peptide 4: ipTM = 0.45) exceed the ipTM value of the known binder (ipTM = 0.38), and Peptide 1 and Peptide 2 ipTM values are close to that of the known binder.
Part 3: Evaluation of the Properties of Generated Peptides in the PeptiVerse
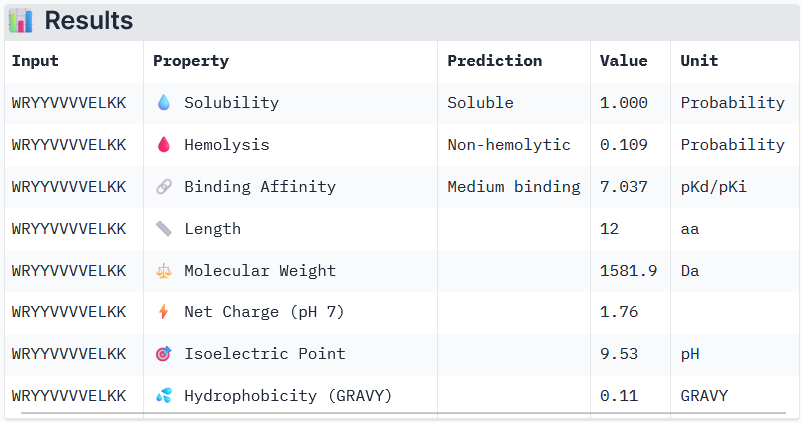
Binder 1: WRYYVVVVELKK
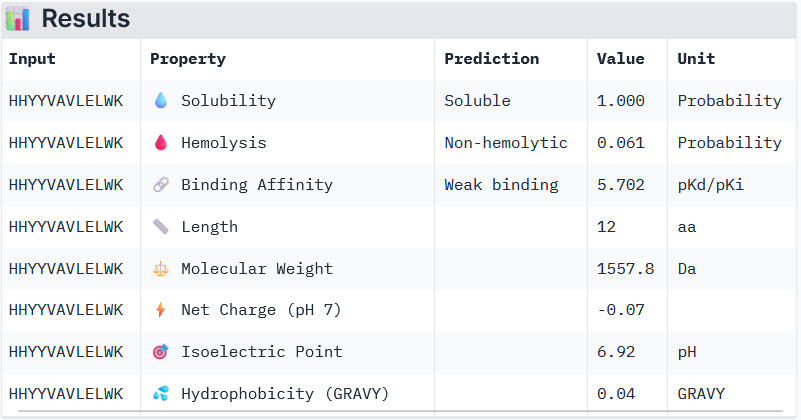
Binder 2: HHYYVAVLELWK
Binder 3: HLYYVAGVRWKK
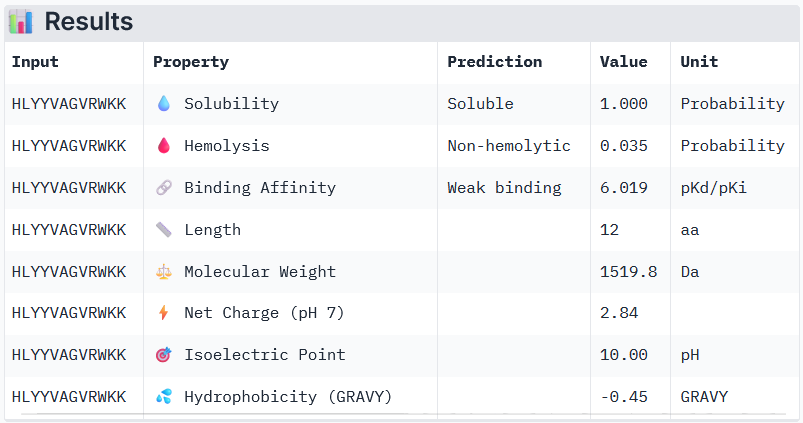
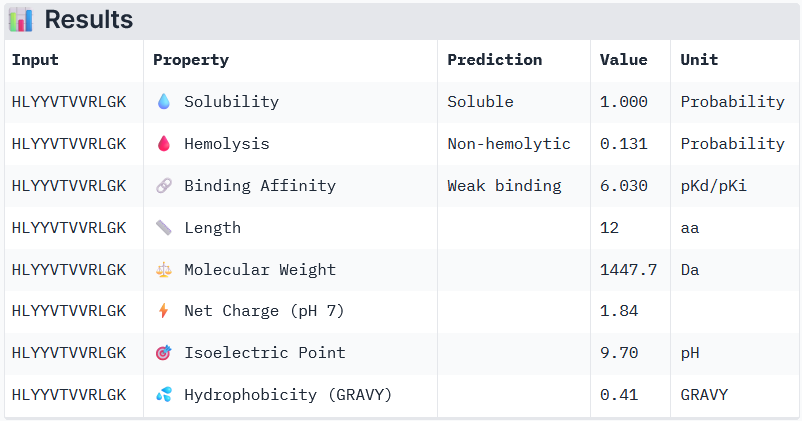
Binder 3: HLYYVTVVRLGK
Known Binder: FLYRWLPSRRGG
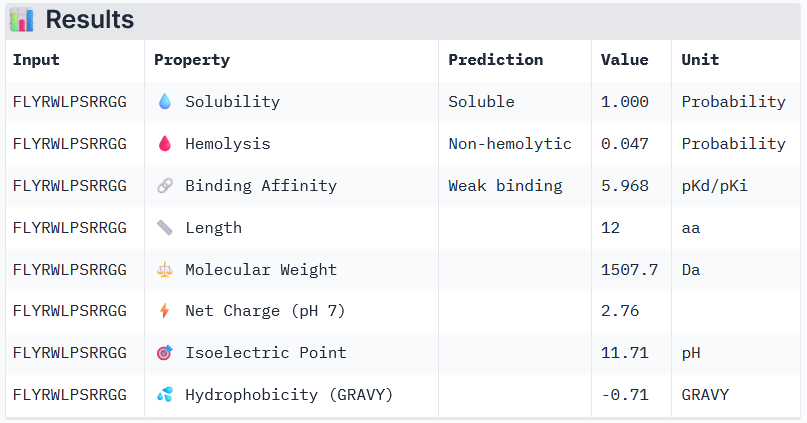
Peptide 3 and Peptide 4, with higher ipTM, both show weak affinity, whereas Peptide 1, having an ipTM score lower than that of the known peptide, shows medium affinity.
None of the peptides shows poor solubility or is hemolytic.
Therapeutic properties in this case are related to where a peptide binds. Essentially, only Peptide 3 with the highest ipTM was selective to the mutant SOD1, but it showed weak binding, although it is partially buried and binds the dimer interface, whereas Peptide 1, showing medium binding, is not selective and may inhibit the native SOD1. So, Peptide 3, although weak-binding, may be best at balancing predicted binding and therapeutic properties.
Interestingly, Peptide 4 also interacts with a trimer interface, which is more important than interaction with just the dimer interface alone. Trimers were found to be toxic and produced off-pathway, whereas dimers and fibrils are protective forms (Hnath and Dokholyan, 2022). Peptide 3 interacts with Lys23, Glu21, Phe22, Ala152, Gly14, Ile18, Gly16, Cys146, 3 of which are ALS mutation sites (14, 16, and 23) and 100% of the contact residues (all 8) are trimer interface residues (with one of them experimentally validated (Hnath, Dokholyan, 2022) and all 8 being predicted (Proctor et al., 2016)). Therefore, Peptide 3 is by far the more promising candidate overall, and in particular, for disrupting toxic SOD1 trimerization.
Part 4: Generation of Optimized Peptides with moPPIt; Evaluation of the Properties of Generated Peptides in the PeptiVerse
Parameters for binders generated in moPPIt: Peptide length: 12 amino acids SELECTED_OBJECTIVES = [‘Hemolysis’, ‘Solubility’, ‘Affinity’, ‘Motif’] OBJECTIVE_WEIGHTS_DICT = {‘Hemolysis’: 1.0, ‘Solubility’: 1.0, ‘Affinity’: 1.0, ‘Motif’: 1.0} OBJECTIVE_WEIGHTS_LIST = [1.0, 1.0, 1.0, 1.0] motif_positions = 4-7
Generated Binders
| index | Binder | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|---|
| 1 | RHIARGYRYYTP | 0.952 | 0.75 | 6.442 | 0.0289 |
| 2 | GEKKTQRSKQCG | 0.965 | 1.00 | 6.469 | 0.8644 |
| 3 | EEPDKTGDKTPF | 0.966 | 0.75 | 5.228 | 0.8947 |
The controlled design generated binders with greater affinity overall, but all of them are hemolytic. These peptides are not useful for further. Overall, Peptide 3 generated with PepMLM could be further tested experimentally if it indeed prevents trimer formation (there is also a chance that it would stabilize trimers).
The peptide first needs to be validated with mass spectrometry to assess if it matches the designed structure and is pure. Hi-res mass spectrometry will confirm the sequence and the mass, and LC-MS will assess purity and detect contaminating byproducts. Additionally, NMR would be needed to confirm atom connectivity and their stereochemistry, and HPLC is needed for quantitative purity. Then, binding needs to be confirmed, and after that, biochemical assays are needed to aggregation. After that, crystallography could confirm whether the ligand is in the interface, and the selectivity of binding to the mutated SOD1 in neurons could be confirmed. Lastly, solubility, metabolic stability, plasma protein binding, and cytochrome P450 inhibition could be assessed with LC-MS/MS, cytotoxicity, in vivo pharmacokinetics, efficacy in a rodent and primate model can be measured, and GLP toxicology studies are needed. Overall: confirm the molecule -> confirm binding -> confirm function -> confirm binding mode -> confirm selectivity and cellular activity -> confirm drug-like properties and safety, with computational analysis run in between stages.
Part C: Final Project: L-Protein Mutants
Original Sequence
Soluble N-terminal domain C-terminal domain METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT
Variable sites identified aligning BLAST results in ClustalOmega (8 in the N-terminus and 4 in the transmembrane domain highlighted):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT
Mutated Sequence 1
METRFPRQSQETLESTNRRRPRKHEDYPCRRQQRSST LYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
For this mutant, I modified the N-terminal domain, aiming to stabilize the disordered domain. I introduced as many charged pairs as possible in the variable sites (changed 4 out of 8 in the N-terminal domain), and additionally changed one conserved site on the left side of the 2nd pair.
Summary of mutations
Conserved site changed: 13P->L
Variable sites changed: (7Q->R, 11Q->E, 14A->E, 22F->R)
Pairs introduced by changing the 4 variable sites: Pair 1 (R7–E11), Pair 2 (E14–R18), Pair 3 (R22–D26)
Soluble N-terminal domain C-terminal domain
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Original Sequence)
R---E LE--- R--- (Mutated Sites)
V V CV V (Conserved / Variable)
METRFPRQSQETLESTNRRRPRKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Mutated Sequence 1)
Mutated Sequence 2
METRFPRQSQETLRSTNERRPRKHEDYPCRRQQRSST LYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
For this mutant, I modified the previous sequence (Mutated Sequence 1), aiming to further stabilize the disordered domain.
I introduced 1 more mutation to a variable site to invert the second pair.
Summary of mutations
Conserved site changed: 13P->L
Variable sites changed: (7Q->R, 11Q->E, 14A->R, 18A->E, 22R->E)
Pairs introduced by changing the 5 variable sites: Pair 1 (R7–E11), Pair 2 (R14–E18), Pair 3 (R22–D26)
Soluble N-terminal domain C-terminal domain
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Original Sequence)
R---E LR---E R--- (Mutated Sites)
V V CV V V (Conserved / Variable)
METRFPRQSQETLRSTNERRPRKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Mutated Sequence 2)
AlphafoldServer was used to fold the monomers of Mutated Sequence 1 and Mutated Sequence 2. alfafold2_multimer_v2 was used to fold the multimers. alfafold2_multimer_v2 parameters used:
num_relax: 0
template_mode: none
msa_mode: mmseqs2_uniref_env
Pair mode: paired
num_recycles: 3
recycle_early_stop_tolerance: auto
relax_max_iterations: 200
pairing_strategy: greedy
max_msa: auto
num_seeds: 1
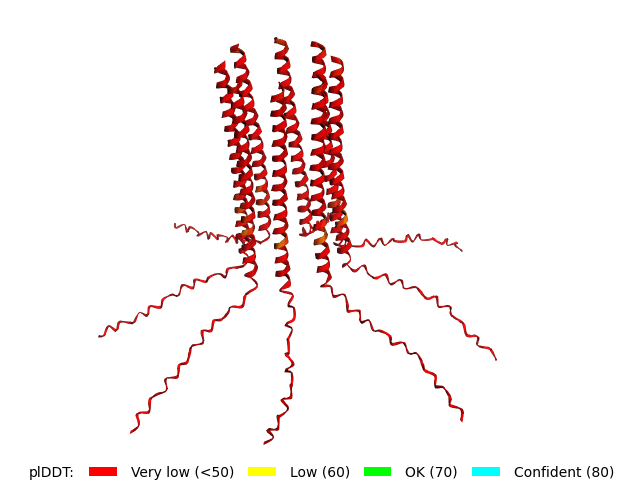 Original Sequence Multimer | 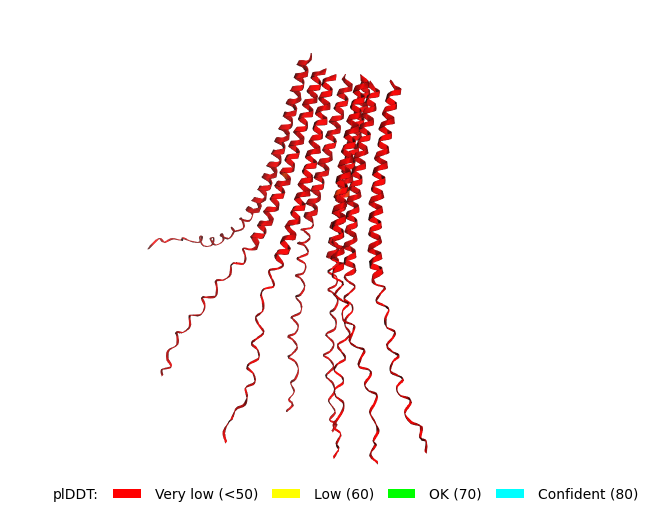 Mutant 1 pLDDT=37.6, pTM-0.189, ipTM = 0.127. 3 pairs/bridges introduced, 1 conserved site changed (13P->L), RRR site kept, (1 conserved and 4 variable sites changed) | 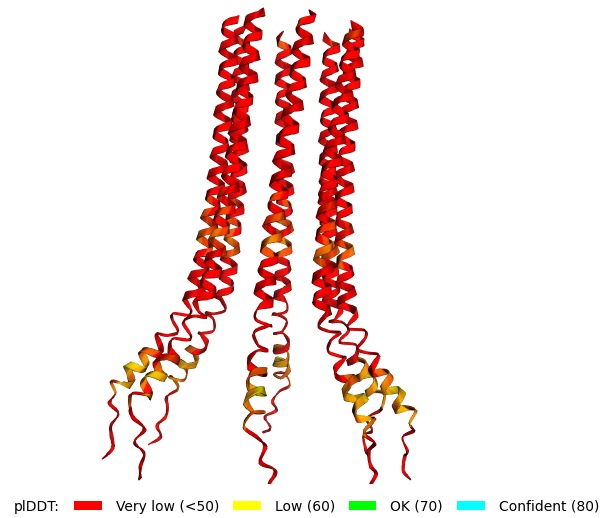 Mutant 2 pLDDT=45.8, pTM-0.187, ipTM = 0.126. 3 pairs/bridges introduced, 1 conserved site changed (13P->L), 2nd pair inverted, no RRR site (1 conserved and 5 variable sites changed) |
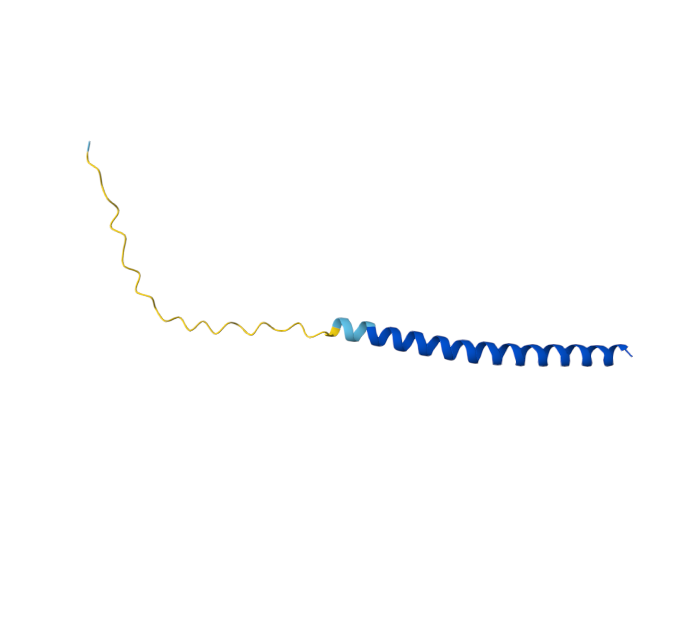 Original Sequence AlphaFold ipTM = -pTM = 0.44 | 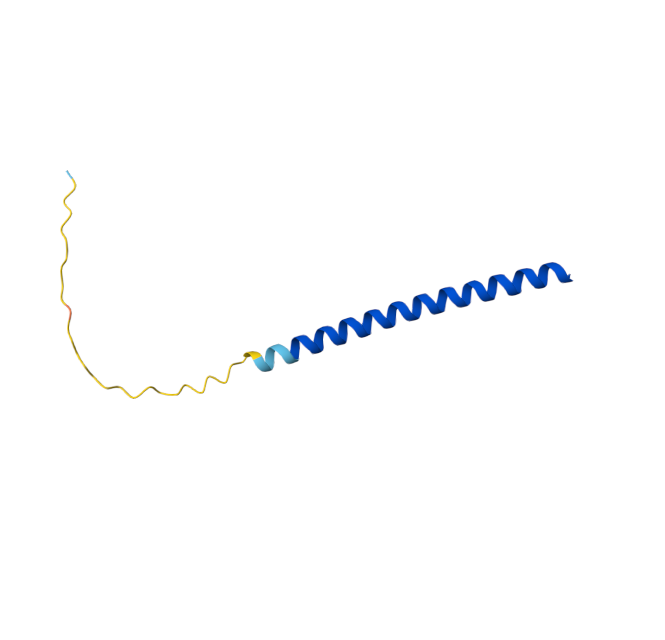 Mutant 1 AlphaFold ipTM = -pTM = 0.43 |  Mutant 2 AlphaFold ipTM = - , pTM = 0.44 |
Mutated Sequence 3
METRFPRQSQETLESTNRRRPRKHEDYPCRRQQRSST LYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
This sequence was designed to explore whether changing the conserved site (13P->L) was required to achieve the same structure as that of the Mutated Sequence 1. For that, the mutated conserved site of the Sequence 1 was changed back to the original (13L->P).
Summary of mutations
Conserved site changed: None
Variable sites changed (as in Mutated Sequence 1): (7Q->R, 11Q->E, 14A->E, 22F->R)
Pairs introduced by changing the 4 variable sites (as in Mutated Sequence 1): Pair 1 (R7–E11), Pair 2 (E14–R18), Pair 3 (R22–D26)
Soluble N-terminal domain C-terminal domain
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Original Sequence)
R---E LE--- R--- (Mutated Sites)
V V CV V (Conserved / Variable)
METRFPRQSQETLESTNRRRPRKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Mutated Sequence 1)
L (Reverted site)
C (Conserved / Variable)
METRFPRQSQETLESTNRRRPRKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Mutated Sequence 3)
Mutated Sequence 4
METRFPRQSQETLESTNRRRPRKHEDYPCRRQQRSST LYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
This sequence was designed to explore whether changing the conserved site (13P->L) was required to achieve the helix as in the Mutated Sequence 2. For that, the mutated conserved site of the Sequence 2 was changed back to the original (13L->P).
Summary of mutations
Conserved site changed: None
Variable sites changed (as in Mutated Sequence 1): (7Q->R, 11Q->E, 14A->E, 22F->R)
Pairs introduced by changing the 4 variable sites (as in Mutated Sequence 2): Pair 1 (R7–E11), Pair 2 (E14–R18), Pair 3 (R22–D26)
Soluble N-terminal domain C-terminal domain
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Original Sequence)
R---E LR---E R--- (Mutated Sites)
V V CV V V (Conserved / Variable)
METRFPRQSQETLRSTNERRPRKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Mutated Sequence 2)
L (Reverted site)
C (Conserved / Variable)
METRFPRQSQETLRSTNERRPRKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Mutated Sequence 4)
alfafold2_multimer_v2 was used to fold the multimers of Mutated Sequence 3 and Mutated Sequence 4. alfafold2_multimer_v2 parameters used:
num_relax: 0
template_mode: none
msa_mode: mmseqs2_uniref_env
Pair mode: paired
num_recycles: 3
recycle_early_stop_tolerance: auto
relax_max_iterations: 200
pairing_strategy: greedy
max_msa: auto
num_seeds: 1
Mutant 1 pLDDT=37.6, pTM-0.189, ipTM = 0.127. 3 pairs/bridges introduced, 1 conserved site changed (13P->L), RRR site kept, (1 conserved and 4 variable sites changed) | 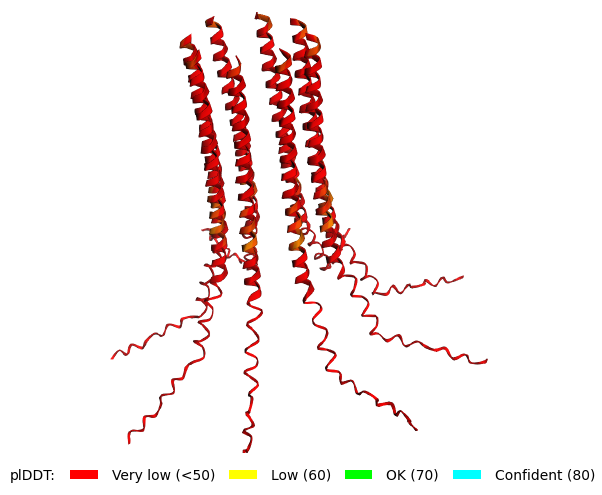 Mutant 3 pLDDT=43.3, pTM-0.188, ipTM = 0.127. Mutant 1 -> the conserved site mutation reverted (13L->P) (4 variable sites of the Original Sequence changed) |
Mutant 2 pLDDT=45.8, pTM-0.187, ipTM = 0.126. 3 pairs/bridges introduced, 1 conserved site changed (13P->L), 2nd pair inverted, no RRR site (1 conserved and 5 variable sites changed) | 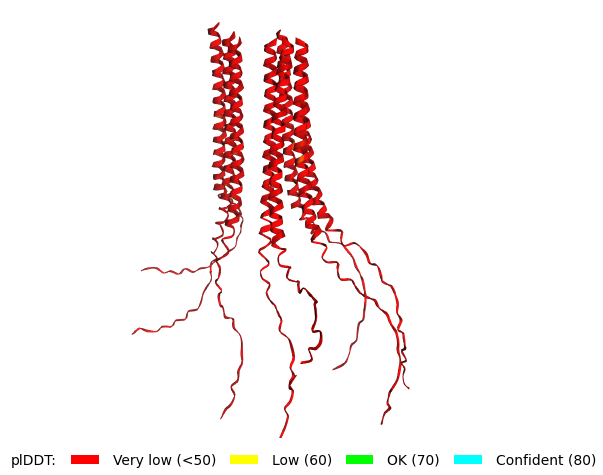 Mutant 4 pLDDT=37, pTM-0.189, ipTM = 0.127. Mutant 2 -> the conserved site mutation reverted (13L->P) (5 variable sites of the Original Sequence changed) |