Week 6 HW: Genetic Circuits Part I: Assembly Technologies
DNA Assembly
Components in the Phusion High-Fidelity PCR Master Mix and their purpose*
The mix contains:
A thermostable high-fidelity and high-speed DNA polymerase with 5´→ 3´ polymerase activity and 3´→ 5´exonuclease activity for proofreading and correcting (the enzyme was engineered so that a polymerase is fused to a small DNA-binding domain, and the domain gives the polymerase high processivity and low error rate; the exonuclease activity, unlike that of Taq polymerase, gives blunt-ended products);
Deoxynucleotides used by DNA polymerase to synthesize a new DNA strand;
Reaction buffer to maintain the optimal pH and ion concentration, with MgCl2 for Mg2+ ions (cofactors) that coordinate the active site of the DNA-polymerase and the phosphate groups of the dNTP, for the phosphodiester bonds that extend the growing DNA strand to form.
There can also be stabilizers added.
Factors that determine primer annealing temperature during PCR
Primer melting temperature. According to the protocol, the annealing temperature is typically set a few degrees below it (2-5 degrees). Primer melting temperature or Tm depends on primer length (longer primers require higher temperature), GC content (GC-richer primers require higher temperature), and nearest-neighbour thermodynamics (nearest neighbour interactions affect duplex stability and the temperature needed).
Primer pair matching. If two primers have very different melting temperatures, then it’s harder to find the optimum. According to the protocol, the two primers (Color Forward and Color Reverse) are advised to keep within 5 degrees. In this case, the temperature needs to be set to be a few degrees below the lower one.
Mismatches between a primer and a template. Mismatches destabilise the two strands and lower the annealing temperature.
Ion concentration. Mg and K neutralize the negatively charged phosphate backbone and stabilize the duplex, and so a higher annealing temperature is needed.
Primer concentration. The higher the concentration of a primer, the higher the annealing temperature can be used.
DMSO concentration. DMSO lowers the temperature.
PCR vs restriction enzyme digests
In this lab, PCR and restriction digestion were used for different purposes. PCR built the inserts and added the overhangs and mutations, and restriction digestion linearized the backbone and verified the final product, and the restriction enzyme DpnI cleaned the template.
The main difference between the methods is that PCR is a DNA synthesis method, and through it, the product is amplified, whereas restriction digestion does not synthesize and does not use primers or nucleotides, but instead cuts the DNA that is supplied. Also, PCR products have blunt ends, and restriction enzymes produce sticky or blunt ends. In terms of protocols, PCR requires primers, nucleotides, polymerase, Mg, and cycling temperature for the primers to melt, anneal, and extend. Restriction digestion is performed at the constant physiological temperature, within approximately 15 min (instead of 1-2 hrs). The cut positions are set by specific sequences included in the DNA supplied and the specificity of the enzymes.
PCR is useful when more DNA needs to be produced, when the DNA needs to be changed. Restriction digestion is useful when DNA linearization is needed and cuts at specific sequences, in general, when sticky ends are needed, when errors that polymerases produce are not allowed, and when a diagnostic test is performed to run on a gel and verify the construct.
How to ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning
Proper overhangs need to be designed. The lab used 20 nt overlap to create adjacent fragments sharing identical sequences. The documents also mentioned 15-25 range, and up to 40 nt are also used. The whole plasmid should first be designed as assembled, and the primers need to be designed so that every junction has a matched overlap. Each overlap must be relatively unique so that fragments assemble in the correct orientation.
Check that each fragment covers its desired region (promoter + partial gene, mutation + terminators, etc.).
Overlaps should have similar melting temperatures so that they all anneal during the assembly.
PCR products must be verified using gel electrophoresis, and the concentration of each product needs to be quantified using Nanodrop or similar. According to the protocol, the expected band size can be calculated in Benchling and then checked on a gel. The concentrations are measured because appropriate ratios of PCR products are needed for the assembly (concentrations above 30 µg/mL were used in the lab to check that PCR worked).
High-fidelity polymerase must be used to generate blunt ends, so that the end of a fragment is the sequence that has been designed.
The original template must be removed (the protocol used DpnI)
The products must be purified to remove primers, nucleotides, polymerase, and primer dimers (the protocol used silica-column).
How does the plasmid DNA enter the E. coli cells during transformation
Plasmids are forced to pass a cell barrier. Cells are first made competent with the addition of calcium chloride, which neutralizes negative charges on DNA’s phosphate backbone and on the cell surface (the treatment is done on ice). Either heat shock is used (fast temperature increase) or electroporation (brief electrical pulse) to create transient pores for the plasmids to diffuse into cells, and then the cells are left to recover. Cells are then selected with culturing on antibiotic plates, and antibiotic resistance genes are switched on due to transfection.
Golden Gate Assembly
- Description
Like Gibson Assembly method, Golden Gate is a cloning method that joins DNA fragments, with the reaction performed in a single tube. It does not use homology overlaps but uses a special class of restriction enzymes (Type IIS enzymes) to build overhangs (Figure 1). The enzymes recognize an asymmetric site GGTCTC and cut at a certain distance near this site, producing a custom overhang of several bases long. The sequence where the same enzyme cuts is designed, in order to arrange different parts of the plasmid in order; assembly standards exist that define the rules for overhangs corresponding to any part (Modular Cloning or MoClo) (Figure 2, Figure 3). The reaction requires DNA fragments, Type IIS enzyme, and T4 DNA ligase, and cycling temperature, and recognition sites are removed during the process (Figure 4).
 Figure 1. Common types of restriction enzymes used for Golden Gate Assembly Source: iGEM.
Figure 1. Common types of restriction enzymes used for Golden Gate Assembly Source: iGEM.
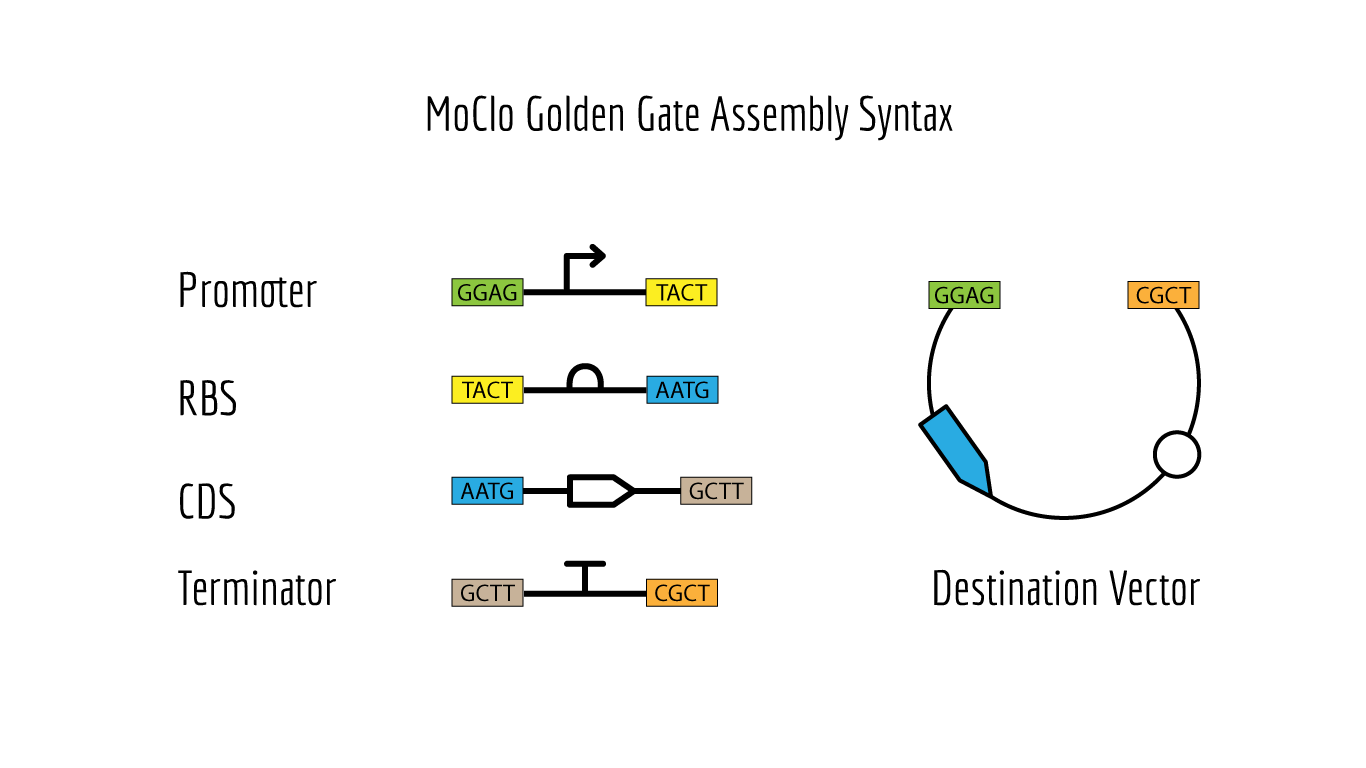 Figure 2. MoClo Sybtax Standards Source: iGEM.
Figure 2. MoClo Sybtax Standards Source: iGEM.
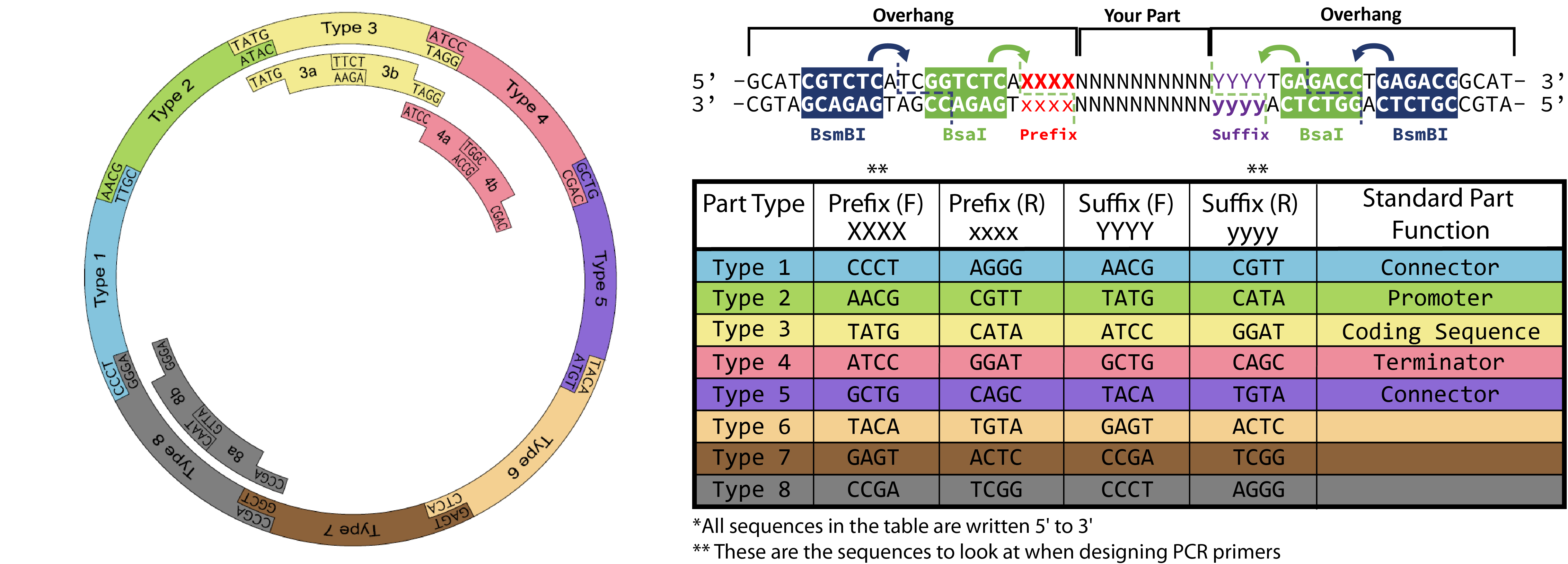 Figure 3. Reaction Source: iGEM.
Figure 3. Reaction Source: iGEM.
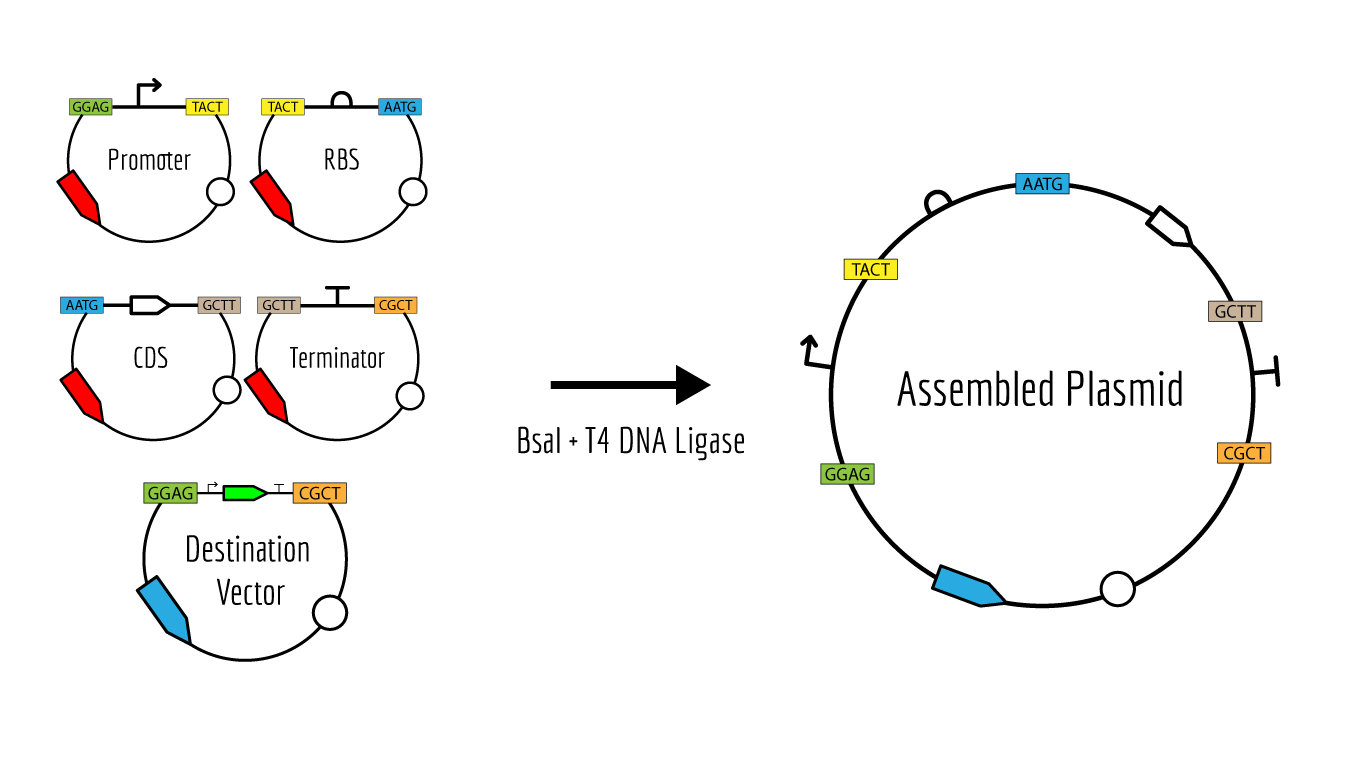 Figure 4. Reaction Source: iGEM.
Figure 4. Reaction Source: iGEM.
- Modeling in Benchling
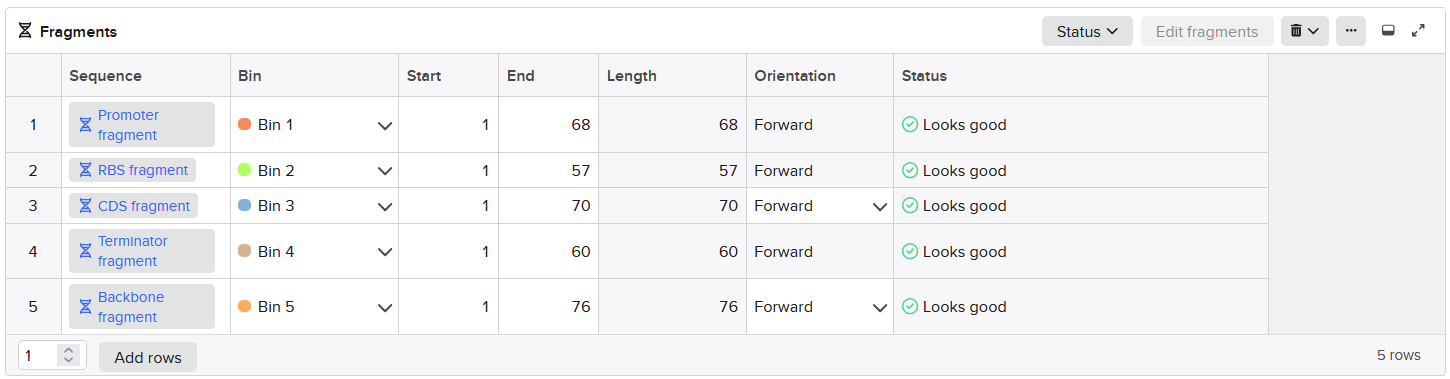
Annotated linear fragments were first created with the corresponding overhang sequences and random sequences for the parts (promoter, RBS, CDS, terminator, origin):
| index | Fragment Name | Overhang (Forward 1) | Overhang (Forward 2) |
|---|---|---|---|
| 1 | Promoter fragment | CCCT | AACG |
| 2 | RBS fragment | AACG | TATG |
| 3 | CDS fragment | TATG | ATCC |
| 4 | Terminator fragment | ATCC | GCTG |
| 5 | Bakcbone fragment | GCTG | TTGC |
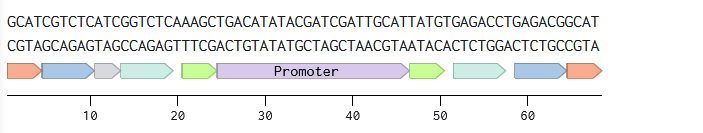
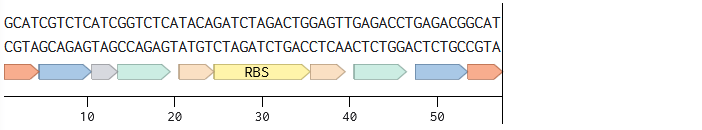
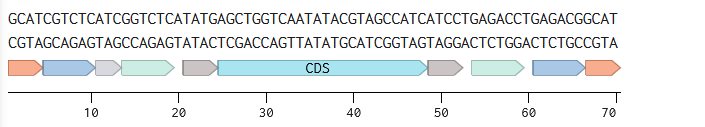
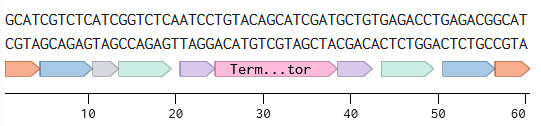
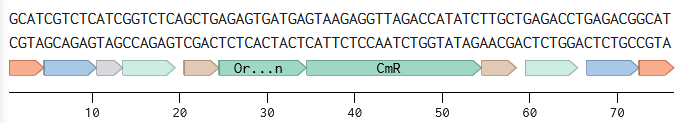
Two BsmBI sites were verified on each fragment, and virtual digests were performed on each fragment to confirm 2 pieces and 1 dropout as products of a BsmBI digest.
The assembly modeling wizard was run (DNA concatenation)
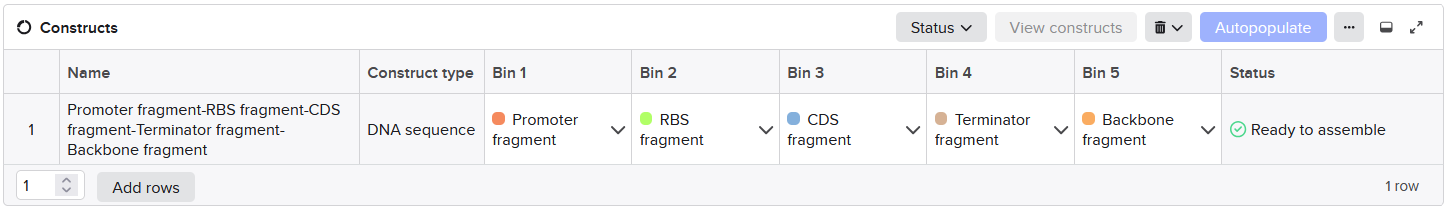
The assembly product confirmed successful concatenations (outer BsmBI sites were consumed in Stage 1 to build the part plasmid, and inner BsaI sites are preserved through Stage 1 and then consumed in Stage 2 to build the final construct).
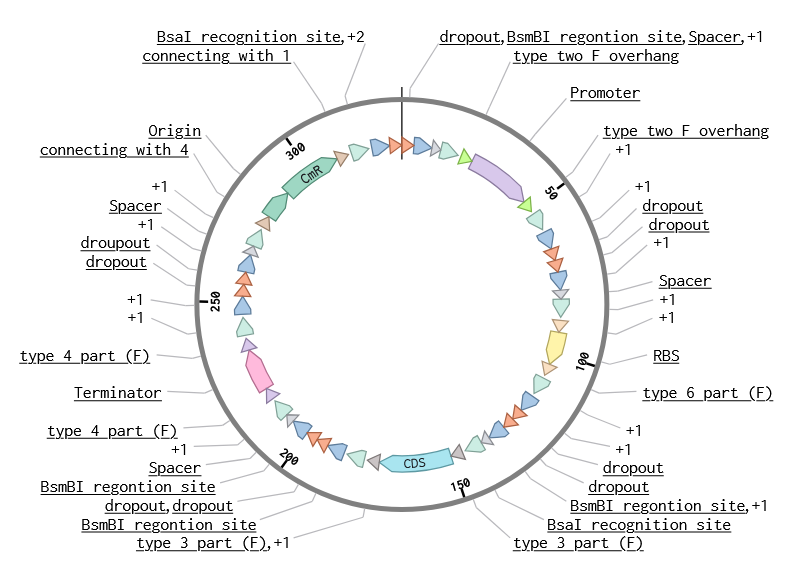
Asimov Kernel
A repository ‘Olga Mineyeva’ was created in Kernel (containing the constructs and a notebook).
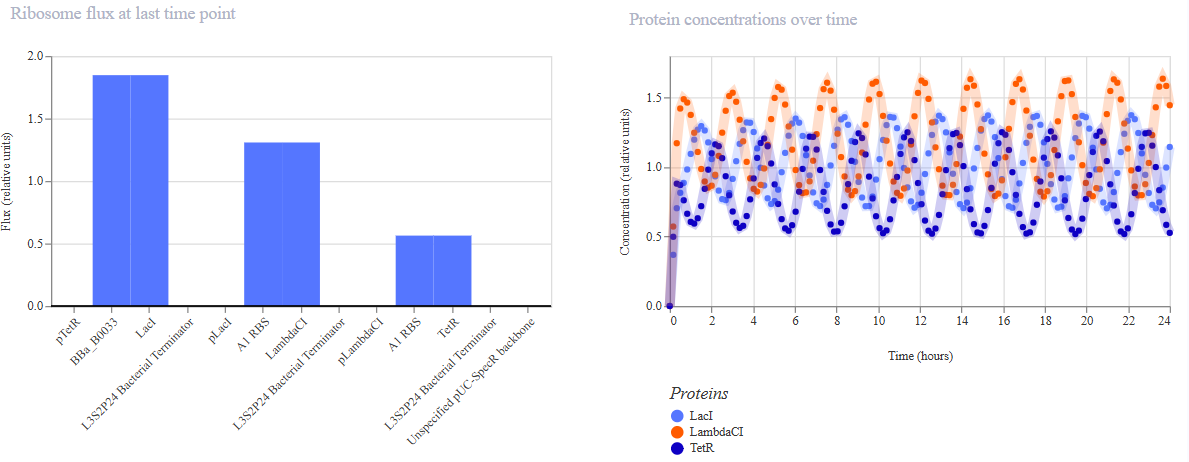
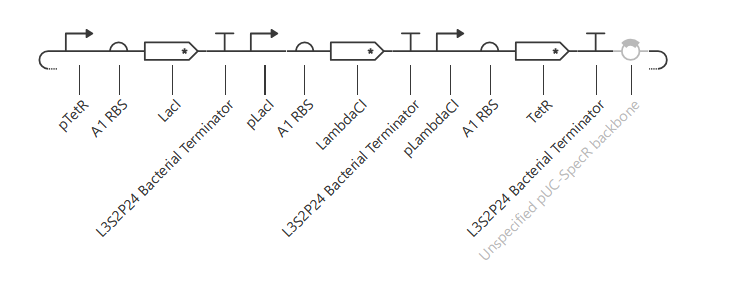
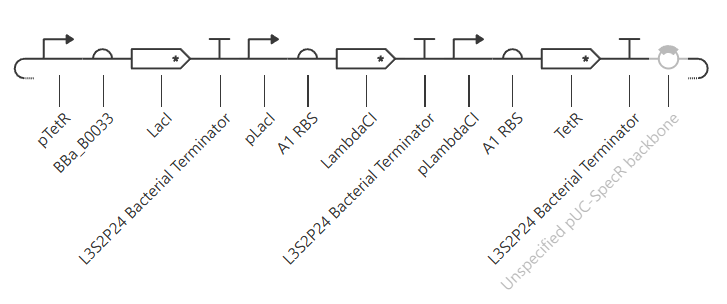
The following 13 parts were assembled to recreate the original repressilator (construct ‘repressilator version’):
- pTetR
- A1 RBS
- LacI CDC
- L3S2P24 Bacterial Terminator
- pLacI
- A1RBS
- LambdaCI
- L3S2P24 Bacterial Terminator
- pLambdaCI
- A1RBS
- TetR
- L3S2P24 Bacterial Terminator
- Unspecified pIC-SpecR backbone
The resulting construct:
Simulations were performed to confirm the reconstructed repressilator behaviour:
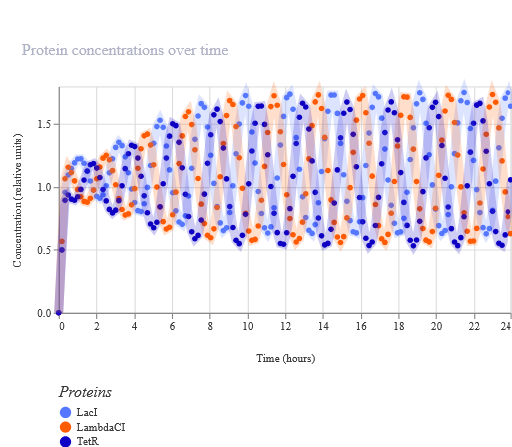
The simulations confirmed that the recreated repressilator behaves the same way as the original.
The first variation of the circuit (construct ‘rep v1’) targeted the circuit topology: pLacI was deleted, so pLac should never be repressed, and so LambdaCI should go high and stay high, and TetR should not be produced, and no oscillations should be possible.
The resulting construct:
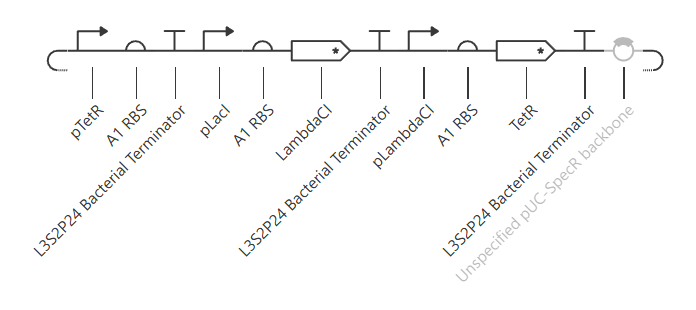
A simulation (with the same parameters as before) was performed to confirm the modified repressilator (rep v1) behaviour:
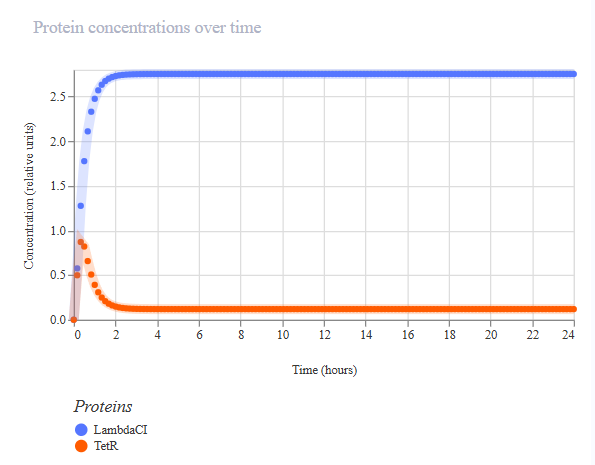
The simulation confirmed LacI is not produced, LambdaCI stays high, but TetR stays near zero, and no oscillations occur.
The second variation of the circuit (construct ‘rep v2’) targeted the production rate of LacI. To see a lower production of LacI, the first RBS was changed to the weakest B0033. Ribosomes should initiate mRNA synthesis less frequently, and so less LacI protein should be produced more slowly. A slower-rising LacI peak is expected, and shifted phases of oscillations.
A1: AATGTTCCCTAATAATCAGCAAAGAGGTTACTAG -> B0033: tcacacaggac
The resulting construct:
A simulation (with the same parameters as before) was performed to confirm the modified repressilator (rep v2) behaviour:
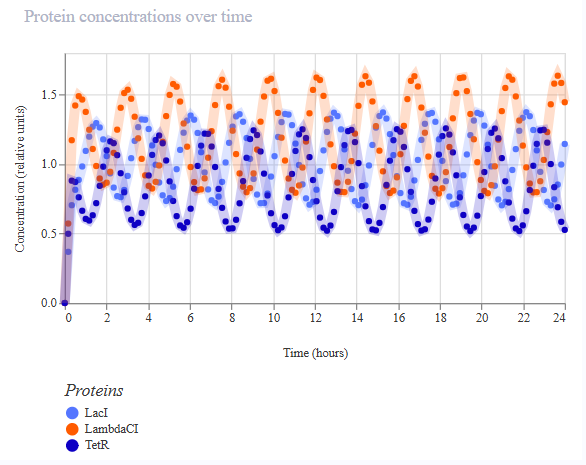
The simulation confirmed LacI is accumulating more slowly, and so LambdaCI is more intensely produced and also stays high, and the phases are shifted.
The third variation of the circuit (construct ‘rep v3’) targeted production rate of LacI again. To see a higher production of LacI, the first RBS was changed to the weakest B0033. Ribosomes should initiate mRNA synthesis more frequently and so more LacI protein should be produced faster. A taller LacI peak is expected, and shifted amplitude and phases of the other two repressors are expected.
A1: AATGTTCCCTAATAATCAGCAAAGAGGTTACTAG -> B0030: ttaaagaggagaaa
The resulting construct:

A simulation (with the same parameters as before) was performed to confirm the modified repressilator (rep v3) behaviour:
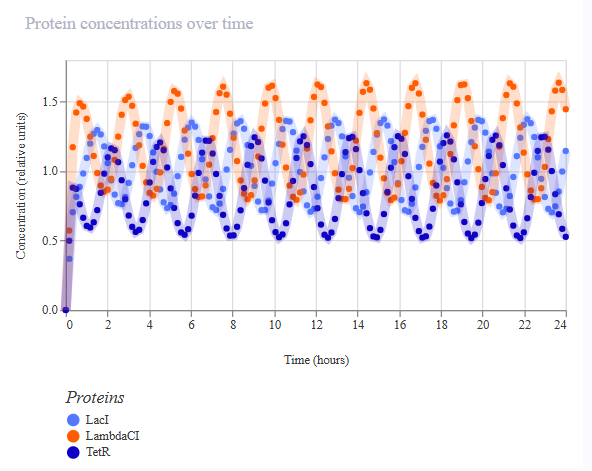
The simulation, however, did not confirm the expectations and was identical to the previous one (with the weaker RBS).
The changes should be observed immediately, so changing the parameters to a longer simulation wouldn’t change the result.
The same behaviour observed for both B0030 and B0033 (instead of A1) can be related to the structure of A1 (spacers, special motifs, or length) that is not present in either B0030 or B0033, so does not allow to make LacI production more efficient than with A.
The following screenshots confirm that different RBS parts were used in the simulation, although the simulation results are identical: