I previously did some studies in neurophysiology addressing neural stem-progenitor cell lifecycle, traumatic brain injury, neuromodulation, and behavioural flexibility in rodent models. I am now interested in neurodevelopmental and neurodegenerative disorders, optimizing disease signature extraction in complex tissue-culture models, culture platform optimization, workflow automation.
Class Assignment Describe a biological engineering application or tool you want to develop and why. The project aims to develop a tool to promote Parkinson’s disease phenotype manifestation in human brain organoids by controllable induction of alpha-synuclein protein expression in dopaminergic neurons. The tool is a genetic construct containing switches and regulators to produce alpha-synuclein beyond normal levels in a subpopulation of cells in patient-derived brain organoids for the investigation of patient-specific pathogenic mechanisms, pathways, and phenotypes.
Waters Part I — Molecular Weight Theoretical molecular weight Based on the 247 aa sequence (including the initiator methionine, linker, and His-tag), the calculated average molecular weight is 28006.60 Da. Chromophore maturation (an autocatalytic post-translational modification where residues T66-Y67-G68 undergo cyclization resulting in −18.02) Da and oxidation (resulting in −2.02 Da) contribute to a 20.04 Da mass loss. The corrected predicted mass would therefore be 27,986.56 Da.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Artwork link
I contributed a pixel in the middle of the letter O in the word LOVE, but the pixel was later replaced.
What I did like was that there is a chance to contribute to the automation protocol design.
What I guess could be better is if we had access to the past year’s experiments (at least for the baseline concentrations) in order to estimate the variability, better plan their 3 wells, and see what changes worked for specific fluorescent proteins. Overall, I think for the problem presented as is (identify a fluorescent protein to work with and present experiments using just 3 wells, with no prior knowledge available), the nature of experiments each student proposes could reveal some interesting personality traits that characterise the HTGAA community; that would be interesting to collect and analyse this information involving those students who have a psychology background.
Part 1: Benchling & In-silico Gel Art This virtual digest of phage Lambda DNA was performed in Benchling. The enzymes used to process the DNA are listed below each column.
Part 3: DNA Design Challenge I chose to practice designing a fluorescent-tagged human tyrosine hydroxylase (TH), relevant for my project on Parkinson’s disease. Tyrosine Hydroxylase converts tyrosine to dopamine and is an essential marker for my target cell population, dopaminergic neurons. Although in real applications, GFP under the TH promoter is used to trace dopaminergic neurons, and GFP fused to large (~56 kDa) TH can disrupt tetramerization, enzymatic activity, and folding, I chose to design a TH-GFP construct for training purposes.
Part A. Conceptual Questions Q1: How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
1 Da equals 1.66053906892(52)×10−27 kg, so 1 aa is 1.66053906892(52)×10−25 kg. The average protein fraction in meat is ~20%. Therefore, the total amount of protein is 100g. The number of aa in 100g = 0.1kg of protein is ~6×10²³ or Avogadro number, 1 mole.
DNA Assembly Components in the Phusion High-Fidelity PCR Master Mix and their purpose*
The mix contains:
A thermostable high-fidelity and high-speed DNA polymerase with 5´→ 3´ polymerase activity and 3´→ 5´exonuclease activity for proofreading and correcting (the enzyme was engineered so that a polymerase is fused to a small DNA-binding domain, and the domain gives the polymerase high processivity and low error rate; the exonuclease activity, unlike that of Taq polymerase, gives blunt-ended products);
Part 1: Intracellular Artificial Neural Networks (IANNs) Advantages IANNs have over traditional genetic circuits There are several advantages: 1) IANN processing uses graded values and the output is analog, just like continuous biochemical signals being graded and variable, not digital on/off; 2) adaptability of the circuit design as any function can be designed and simulated with changed weights instead of rebuilding circuit topology; 3) weights in IANN correspond directly to a real biological interactions (activation/repression of a gene); 4) IANNs more efficiently use limited cellular resources (weights are tuned instead of gates chained).
Part A: General and Lecturer-Specific Questions general questions Advantages of cell-free protein synthesis (CFPS) over in vivo methods
Mainly, there is no living cell and no cell membrane involved, and so any component of the reaction can be added or removed in the course of the controlled reaction. There are fewer variables to control compared to an experiment and synthesis in a living cell. No transformation process is involved, and so the reaction is faster. Cell-free is more beneficial when a toxic protein is synthesized or a protein that incorporates non-canonical amino acids.
Subsections of Homework
Week 1 HW: Principles and Practices
Class Assignment
Describe a biological engineering application or tool you want to develop and why.
The project aims to develop a tool to promote Parkinson’s disease phenotype manifestation in human brain organoids by controllable induction of alpha-synuclein protein expression in dopaminergic neurons. The tool is a genetic construct containing switches and regulators to produce alpha-synuclein beyond normal levels in a subpopulation of cells in patient-derived brain organoids for the investigation of patient-specific pathogenic mechanisms, pathways, and phenotypes.
Patient-derived brain organoids naturally recapitulate some neurodegenerative and neurodevelopmental disease features and are used as models to study human-specific pathology and test potential therapeutics. One of the significant and costly problems of these models is the time needed (months) for organoid maturation and manifestation of pathological phenotypes, involving protein accumulation, mitochondrial dysfunction, and neuronal death. Therefore, approaches to speed up growth, maturation, and phenotype development are currently needed and being devised. In Parkinson’s disease in particular, death of dopaminergic neurons causing movement deficiencies is caused by alpha-synuclein protein misfolding and accumulation, triggered by failures in different interconnected processes (mitochondrial dysfunction, dopamine metabolism, inflammation, autophagy dysfunction) and various mutations (SNCA, LRRK2, PINK1, Parkin, DJ-1, GBA). The tool for premature controllable production of alpha-synuclein will allow standardized promotion of Parkinson’s disease phenotype to be used in studies of individual and patient-specific factors leading to the disease (specific dysfunctions leading to inefficient protein degradation and alpha-synuclein protein accumulation) and devise personalized treatment strategies within platforms working with patient-derived brain organoids/assembloids.
Describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
One goal can be related to environmental and resource considerations. The tool may substantially reduce the time and resources for Parkinson’s research, so these benefits need to be documented and promoted. Therefore, systematic quantification, validation, and dissemination of resource efficiency of this method for accelerated Parkinson’s phenotype modeling is needed to promote adoption of sustainable research practices and resource allocation decisions in neurodegenerative research and drug discovery.
Describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g., 3D printing, drones, financial systems, etc.).
Action 1: Conduct and share the analysis of the process
Purpose: measure costs and environmental footprint of the tool versus old approaches; the results need to be published.
Design: data needs to be collected by a lab; environmental specialists, including institutional ones, need to be involved to share their expertise on the methodology and the analysis, as well as consider both relative and absolute environmental impact; additional funding can be requested from the Parkinson’s Foundation.
What could be wrong, incorrect assumptions: The environmental and resource benefits may not be sufficient or comparable to old protocols like fibril seeding; there could be hidden costs for monitoring or equipment that cancel out the saved time; when these organoids are produced at scale, the dynamics and the resources needed may be different from those for lab scales.
Risks: initial monitoring, as well as monitoring in the labs that acquire this method, may require considerable investments that can be hard to acquire, and therefore, this tool may lack monitoring, be less preferred; the results of monitoring may show that the method is not saving time or resources but rather is more resource-intensive
Action 2: Develop and promote a framework for resource efficiency reporting
Purpose: create a standardized method for reporting the use of resources that can become a standard for the field and include environmental measures into research workflow. This could be a template or checklist to increase transparency and improve reproducibility across studies and labs.
Design: tool developers need to agree on metrics, sharing the standard with agencies, societies, and databases that could adopt and promote it (SFN, CAN, Stem Cell Research foundations, etc.); software developers can create tools for efficient and easy reporting of data across the tool users
What could be wrong, incorrect assumptions: the reporting burden might be too large for researchers to comply with; standardization may not be possible across different research environments; the chosen metrics fail in reporting real efficiency gains
Risks: users may refuse to adopt or ignore the standard because it’s too complex, expensive, or not enforced
Action 3: Develop an open-access resource optimization database and tools
Purpose: create a community-maintained platform where researchers working with organoids and the developed tool share their protocols, techniques that save resources, troubleshooting, quality control, and cost-benefit analysis.
Design: this database can be part of an already existing open-access platform for organoid research, such as the one maintained by the Early Drug Discovery Unit at McGill University. Tool developers need to develop the initial database and report their content there. Contributing researchers need to volunteer time to share their data and protocols; the university needs to approve sharing potentially patentable information; moderators are required to guide and encourage participation.
What could be wrong, incorrect assumptions: publications can be sufficient to report protocols and sustainability gains, and no new resource is needed; efficiency improvements in different contexts may not translate to universal strategies.
Risks: the resource can become outdated and not used; companies might take advantage to promote their reagents; funding may not be sufficient; researchers may choose not to share the most valuable information; bad protocols might be propagated.
Score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
3
3
2
• By helping respond
3
3
2
Foster Lab Safety
• By preventing incident
3
2
1
• By helping respond
3
3
1
Protect the environment
• By preventing incidents
2
1
3
• By helping respond
3
2
1
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
1
• Feasibility?
1
3
2
• Not impede research
1
3
2
• Promote constructive applications
2
1
3
Drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Among the proposed 3, Action 1, i.e., the monitoring, needs to be prioritized because it will generate and assess evidence for the tool and for whether the other two actions are needed. The data obtained through Action 1 will provide the base for protocol optimization. Action 1 also mainly depends on the tool developers rather than on other contributors, and so it’s easier to lead. The main risk of this action is choosing the wrong metrics. Also, the efficiency of the tool in terms of the environment may not be accepted as a priority by the community of researchers in the field (Parkinson’s or neurodegenerative disorders). Also, if in fact, there is no environmental benefit of the tool, some lessons on the disease-accelerating approach, metrics approach, and costs, need to nevertheless be learned and published. An uncertainty is what agencies would be willing to fund this.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
I’ve learned about governance for synthetic biology projects in general and about the boundary between technical practice in science and governance. Regarding the latter, the design of controls is part of scientific methodology. But control design can actually become governance if new and specific standards for controls are disseminated, when control requirements are institutionalized, standardized control frameworks are proposed or built onto a tool, minimal control standards are created, or when expectations about reporting controls are established to foster transparency. This is applicable to my project on accelerated disease phenotype manifestation as well, and some related governance actions can be developed on how to ensure proper controls are used. These actions can include 1) establishing minimum control standards both for within-organoid controls (the organoids are intended to be chimeras) and parallel controls with natural manifestation (normally aging organoids); 2) creating a standardized control protocol repository; 3) providing training on the protocols and certifications; 4) other (to be added).
Assignment (Week 2 Lecture Prep)
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate of DNA polymerase depends on the organism and on the type of polymerase. The error rate of replicative human polymerases δ and ε before proofreading with their endonuclease activity is estimated as about one wrong nucleotide in every hundred thousand to million nucleotides added. Compared to diploid human genome 6 billion base pairs long, this error rate means that replication mediated by polymerase alone will produce about 610-910-5 = ~60,000 errors per genome with 10-5 rate, which is why proofreading is essential. To deal with that discrepancy, errors are corrected through immediate proofreading, reducing mistakes to about 1-2 orders and through mismatch repair reducing mistakes to about another 1-2 orders, eventually reducing the mistakes to ~10⁻9–10⁻10 rate. To immediately correct the mistakes of Pol ε, Pols δ and ε conduct proofreading themselves by shifting the DNA strand to the exonuclease site. Right after replication, post-replication errors are corrected in the process called mismatch repair correcting patches of mismatching pairs and involving proteins recognising a mistake on a new strand, endonucleases and free exonucleases to remove the mismatch, the polymerase delta to fill the gap with the correct sequence, and ligase to seal the strand.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The number of ways to code an average human protein is equal to the average number of codons per amino acid (~3) to the power of the average protein size (~400 amino acids) = 3400. Some of the reasons all these different codes don’t work and codons are not equally likely include: sequence proofreading correcting mistakes in codons; evolution leading to codon selection removing sequences that caused problems in mRNA folding, splicing, were slow in translation and affect protein folding making the protein non-functional; the polymerase being prone to some errors but not others; certain mutations accruing non-randomly, with pyrimidine-purine less likely than purine-purine or pyrimidine- pyrimidine.
Homework Questions from Professor Proust:
What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite synthesis is the most common.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Its difficult to synthesise oligos longer than 200 nucleotides because phosphoramidite synthesis efficiency decreases with length due to errors accumulation (as more synthesis steps are needed and more opportunities for mistakes arise, nucleotides may fail to attach, chemical errors upon nucleotide attachment may occur, and inefficient capping may allow for next synthesis cycles while truncated sequences are produced that compromise purification), enzymatic synthesis capable of making longer oligos expensive, less standardized, and still under development, and microarray-based synthesis produces shorter sequences.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Such a long synthesis will mainly produce incorrect sequences and a tiny fraction of correct ones due to the accumulation of errors, and extracting the correct ones will be expensive, time-consuming, impossible, and overall impractical.
Homework Questions from Professor Church:
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids in all animals are: Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, Valine, and Arginine. ‘Lysine Contingency’ was intended as a method to make dinosaurs unable to synthesize Lysine, which makes no sense, as lysine is not produced by animals, regardless of the species they used for cloning.
Week 10 HW: Imaging and Measurement (Mass Spectrometry)
Waters Part I — Molecular Weight
Theoretical molecular weight
Based on the 247 aa sequence (including the initiator methionine, linker, and His-tag), the calculated average molecular weight is 28006.60 Da. Chromophore maturation (an autocatalytic post-translational modification where residues T66-Y67-G68 undergo cyclization resulting in −18.02) Da and oxidation (resulting in −2.02 Da) contribute to a 20.04 Da mass loss. The corrected predicted mass would therefore be 27,986.56 Da.
Experimental molecular weight
z for adjacent peaks: z = m/z(n+1) / (m/z(n) - m/z(n+1)). Two peaks were chosen, m/z(n+1)=875.4421 and m/z(n)=903.7148, therefore, z = 875.4421/(903.7148-875.4421) = 30.9642 ~ 31 charges the 28kDa eGFP protein carries. z(n) = 31; z(n+1) = 32.
MW = z × (m/z(n) − 1.00728): MW = 31*(903.7148-1.00728)= 31*902.70752 = 27983.93312 Da
Individual peaks cannot be resolved in the zoomed-in peak as the difference between those peaks is too low and cannot represent a charge. The zoomed-in peaks are isotope peaks within the charge state envelope.
The mass spectrum shifts toward lower m/z when a denatured protein is evaluated, and shifts toward higher m/z when a native protein is evaluated. In the case of denatured protein, charge envelopes become broader as many charge states are observed.
The charge state of the peak at ~2800 ….
Waters Part III — Peptide Mapping - primary structure
Lysine (K) and Arginine (R) in the eGFP: 20 Lysines and 6 Arginines.
Theoretical digest produced 19 peptides (Expasy output):
Selected enzyme:Trypsin
Maximum number of missed cleavages (MC): 0
Cysteines modifications: All cysteines in reduced form
Methionines modifications: Methionines have not been oxidized.
Mass of displayed peptides: > 500 Dalton
Mass calculation: Using monoisotopic masses of the occurring amino acid residues and giving peptide masses as [M+H]+.
If peaks of >10% abundance are considered, ~ 19 peaks can be counted, about the same number as the peptides:
Chromotogram with peaks that are >10% relative abundance highlighted. Red line marks the 10% cutoff.
The mass-to-charge (m/z) of the peptide shown in Figure 5b.
The isotope spacing is= 1/z; the peaks are spaced ~0.5 m/z apart (525.76712, 526.25918), therefore z = +2.
The mass of the singly charged form of the peptide ([M+H]+) based on m/z=525.76712 and z=2: M neutral =(m/z x z)-(z x 1.0073)= 525.76712 x 2 – 2 x 1.0073 = 1,051.53424 - 2.0146 = 1,049.51964 m/z, the singly charged form [M+H]⁺= = M+1.0073 = 1,049.51964 + 1.0073 = 1,050.52694 Da
Identification of the peptide
The peptide is FEGDTLVNR, with the mass of 1050.5214 Da.
Accuracy = |MWexp - MWtheor|/MWtheor: Accuracy = (1,050.52694 – 1050.5214)/ 1050.5214 = 5.27ppm, which is within the 10 ppm mass error tolerance for peptide evaluation.
The percentage of the sequence that is confirmed by peptide mapping is 88% (see Figure 6).
Based on the ion masses, 7 y ions can be identified in the spectrum:
Figure 5c. Fragmentation spectrum of the peptide eluting at retention time 2.78 minutes in Figure 5a.
No b ions were identified on the spectrum.
Claude explains why:
b and y ions are not produced equally — the chemistry of fragmentation strongly favors one series over the other depending on the peptide
Arginine (R) at the C-terminus — your peptide ends in R, which is highly basic and strongly attracts the charge. This means the C-terminal fragments (y-ions) tend to retain the charge and be detected, while the complementary b-ions lose the charge and become invisible. This is actually a well-known rule: tryptic peptides ending in R or K almost always show dominant y-ion series with weak or absent b-ions
Several peaks do not match with any b or y ions.
Claude explains what these peaks might refer to:
536.74844 and 525.76712 - these are spaced ~11 m/z apart and are in the 500s range, which strongly suggests these are doubly charged ions (z=+2) of larger fragments or possibly the precursor isotope peaks.
214.09165 - The most likely explanation is an internal fragment ion. These form when two bond breaks happen in the same fragmentation event, releasing a small piece from the middle of the peptide. They are named by the residues they contain. T + L = 101.05 + 113.08 = 214.13 (very close ✅)
86.09155, 122.08946 - immonium ions, which are small diagnostic ions produced as side products of fragmentation. They are specific to individual amino acids. 86.09155 — immonium ion of Leucine (L), which is indeed in the sequence. 122.08946 — immonium ion of Phenylalanine (F), the first residue in the sequence.
The y ions found in the fragmentation spectrum are consistent with the predicted sequence (FEGDTLVNR).
Figure 6 depicting the % of aa coverage of peptides positively identified using their calculated mass and fragmentation, confirms that the sequence of this peptide (FEGDTLVNR) matched.
Waters Part IV — Oligomers
Waters Part V — Did I make GFP?
Week 11 HW: Bioproduction and Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I contributed a pixel in the middle of the letter O in the word LOVE, but the pixel was later replaced.
What I did like was that there is a chance to contribute to the automation protocol design.
What I guess could be better is if we had access to the past year’s experiments (at least for the baseline concentrations) in order to estimate the variability, better plan their 3 wells, and see what changes worked for specific fluorescent proteins. Overall, I think for the problem presented as is (identify a fluorescent protein to work with and present experiments using just 3 wells, with no prior knowledge available), the nature of experiments each student proposes could reveal some interesting personality traits that characterise the HTGAA community; that would be interesting to collect and analyse this information involving those students who have a psychology background.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Components and their role
E. coli Lysate
• BL21 (DE3) Star Lysate (includes T7 RNA Polymerase) – The lysate is an unfractionated content of the cytoplasm, it supplies ribosomes, tRNAs, aminoacyl-tRNA synthetases, elongation and initiation factors, and the metabolic enzymes. T7 RNA polymerase is the most important enzyme in the reaction; it’s a bacteriophage polymerase but has been integrated into the E.coli strain because it’s faster than the native polymerase and does not require transcription machinery, which I guess is important for the transcription not to be a limiting factor.
Salts/Buffer
• Potassium Glutamate – Potassium is the main cation cells are adapted to, and glutamate is E.coli’s major cytoplasmic anion modulating osmolarity, and so both are recreating the native ionic environment. Potassium is needed for ribosomes to fold and assemble (by neutralizing negative phosphate charges on rRNA and allowing ribosomal subunits to not repel) and for translation itself, but too high concentration inhibits translation (promotes dissociation of the ribosomal subunits and initiation complex destabilize) as many of the interactions involved in the process are reversible electrostatic; glutamate unlike chloride is large and it does not interact with protein surfaces with positively charged regions and functional interfaces.
• HEPES-KOH pH 7.5 – It’s a buffer needed to keep pH for the optimal work of transcription and translation enzymes by counteracting accumulating acidic byproducts of energy metabolism. Buffer titration experiments may be beneficial.
• Magnesium Glutamate – Magnesium is the major cofactor for T7 polymerase (uses two ions at the active site to form phosphodiester bonds), it defines structure and function of ribosomes (neutralizes negative phosphate charges of rRNA), it stabilises nucleotides and RNA (binds to phosphates of nucleotides, mRNA, tRNA); and glutamate is a safe (large) counter-ion maintaining osmolarity as in potassium glutamate. Magnesium is the most nonlinear parameter in the cell-free reaction system; at too low a concentration, transcription and translation efficiency decrease, and upon too high concentration, RNA molecules crosslink, and nucleotides precipitate as magnesium phosphate salts.
• Potassium phosphate monobasic/dibasic – another buffer system, but for a different pH range than HEPES, and so may add the capacity to counteract pH drift from accumulating acetate. Phosphate serves as an energy metabolism substrate and regenerates ATP and nucleotide triphosphates, but too much phosphate can diminish magnesium (as the magnesium salt is insoluble) and inhibit the reaction.
Energy / Nucleotide System
• Ribose – replenishes the NTP pool, in the system where nucleoside monophosphates and free guanine are supplied instead of ready-made triphosphates. It is converted to the sugar-phosphate backbone (phosphoribosyl pyrophosphate) that is needed to build nucleotides.
• Glucose – primary energy source, substrate for glycolysis to generate ATP for transcription and translation. Acetate produced as a byproduct shifts the pH to acidic.
• AMP, CMP, GMP, UMP – ribonucleoside monophosphates phosphorylated by kinases in the lysate, precursors to ATP, GTP, CTP, UTP for mRNA synthesis.
• Guanine – converts to guanine nucleotides through the purine salvage pathway. Replenishes GTP pool that is consumed in transcription and translation.
Translation Mix (Amino Acids)
• 17 Amino Acid Mix, Tyrosine, Cysteine – building blocks for proteins, fluorescent proteins in our case. Tyrosine is added separately because of its different solubility (in high pH), and cysteine oxidizes quickly.
Additives
• Nicotinamide – precursor for NAD⁺/NADH indispensable for glycolysis; these are not consumed in transcription or translation.
Backfill
• Nuclease Free Water – dissolves all the components, is deprived of RNases or DNases that would degrade the template and mRNA.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix.
The 1-hr master mix is optimized for speed and contains triphosphates and a high-energy phosphate donor (PEP, to quickly recharge ATP) as well as NTPs to quickly supply transcription and translation, higher concentration HEPES as the only buffer, and a number of additives including energy equivalents and redox cofactors, and stabilizers for nucleic acids.
The 20-hour mix is optimized to sustain the long reaction and for the cost. It contains nucleoside monophosphates and free guanine phosphorylated by lysate enzymes, ribose and glucose a phosphate buffer, in addition to HEPES, but also as a substrate for energy-regeneration to recharge nucleotides, higher concentrations of amino acids as more total protein is synthesized through a longer reaction, and only nicotinamide as an additive needed as a precursor for regenerating NAD.
How can transcription occur if GMP is not included but Guanine is?
Guanine converts to guanine nucleotides through purine salvage pathway. Replenishes GTP pool that is consumed in transcription and translation.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc):
sfGFP – is optimized for efficient folding (a very small fraction of molecules fail to fold) and fast maturation (in minutes), making sfGFP the most reliable reporter. It’s also very stable as its barrel is quite resistant to proteases in E.Coli lysate, and so the fluorescent signal mostly accumulates.
mRFP1 – slow chromophore maturation (~ tens of minutes, closer to 1 hr) and relatively low brightness (lower than that of mScarlet-I), so no fast kinetics can be observed. More synthesized protein would help to increase brightness. It’s historically interesting as mRFP1 opened the palette for monomer red and orange fluorescent proteins; mRFP1 was developed with a large number of mutations, and in its slow maturation, the fluorophore passes a stage with green fluorescence.
mKO2 - maturating faster than its parent mKO, but with pH of 5.5, which can be reached with accumulating acetate if HEPES is not buffering it, half of the molecules lose fluorescence. The latter feature was actually part of the mKO2 design that was developed for cell cycle monitoring timer/indicator in the Fucci system, where green and red proteins are fluorescent in different cell cycle phases; the name of the protein is actually Kusabira-Orange, which is probably the name of the coral, and “Kusabira” means mushroom in old Japanese, as the coral reminded one of a mushroom.
mTurquoise2 – very high resistance to acid, fast maturation, and high quantum yield for a cyan protein. Its pKA is much lower than that of mKO2 (3.1 vs 5.5), which can be useful in reactions where pH is predicted to shift to acidic; however, the dynamic range is narrow because of the high autofluorescence of lysate components, having excitation and emission overlapping with that of mTurquoise2.
mScarlet_I – matures faster than mRFP and mScarlet (~40 min vs ~100 min of mScarlet) and has slightly lower brightness than mScarlet. It can capture fast dynamics, and its pKa is ~5.4 (moderate acid resistance).
Electra2 – it’s a blue fluorescent protein engineered from mRuby3, and it can form aggregates, which affects the reproducibility of the signal in cell-free systems. Also, the dynamic range is narrow as well (lower than that of green and red reporters) for the same reason of high autofluorescence in near UV.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Higher concentration of HEPES buffer may make the reaction more resistant to pH drift due to acetate accumulation through the long reaction and protect sfGFP signal (decrease chromophore protonation). Some HEPEs concentration optimum (a plateau in fluorescent signal) is expected upon increasing HEPES concentration because the ionic strength also increases.
Additionally, sfGFP expression and green fluorescence could be increased by adding magnesium (but the effect is not specific to sfGFP), and reagents modulating oxygen availability could improve maturation and fluorescence of sfGFP over the long incubation time.
Define the precise reagent concentrations for a cell-free experiment.
8 wells were registered with the following parameters:
This virtual digest of phage Lambda DNA was performed in Benchling. The enzymes used to process the DNA are listed below each column.
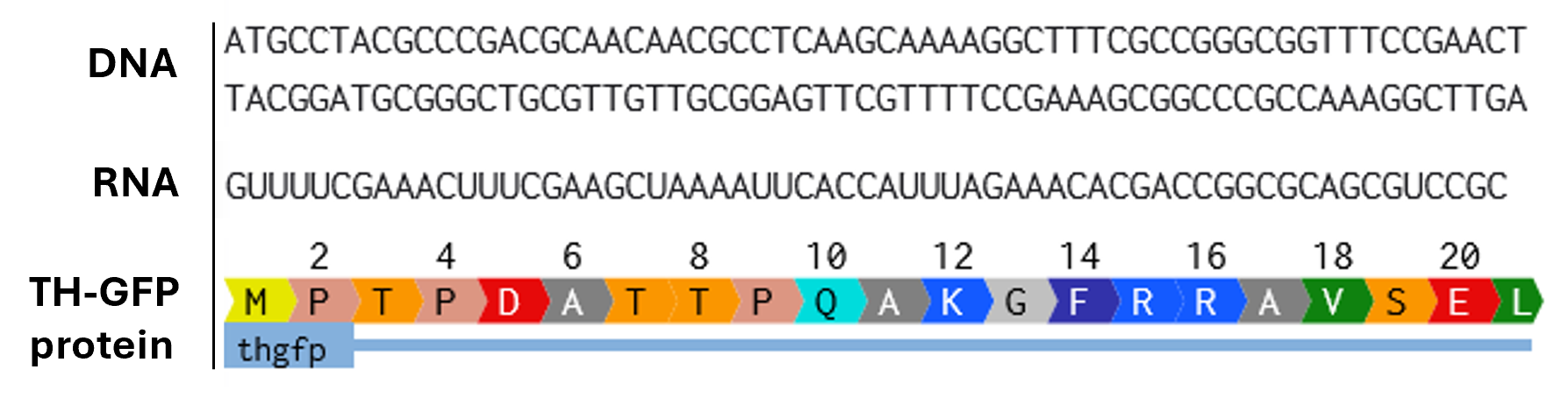
Part 3: DNA Design Challenge
I chose to practice designing a fluorescent-tagged human tyrosine hydroxylase (TH), relevant for my project on Parkinson’s disease. Tyrosine Hydroxylase converts tyrosine to dopamine and is an essential marker for my target cell population, dopaminergic neurons. Although in real applications, GFP under the TH promoter is used to trace dopaminergic neurons, and GFP fused to large (~56 kDa) TH can disrupt tetramerization, enzymatic activity, and folding, I chose to design a TH-GFP construct for training purposes.
3.1. Choose your protein.
For TH, UniProt P07101 (Tyrosine 3-monooxygenase, Tyrosine 3-hydroxylase (TH)), I chose TH isoform 1 of 528 amino acids as it’s the canonical and most common isoform in the brain.
As a linker, I chose a flexible (GGGGS)3 linker of 15 aa, as this type of linker is used in design in recombinant fusion proteins to increase spatial separation between domains, which could be useful for fusing GFP with a large protein. Since both N and C domains of TH are functional, with the N-terminal domain containing a phosphorylation site needed for the enzyme activation and the C-terminal domain allowing tetramerization, and the fused domain functionality can only be tested empirically, I chose to add a flexible linker to the C-terminal domain, as this linker can allow some spatial freedom for tetramerization.
Codon optimization is needed to adapt the DNA sequence to the different frequencies of tRNAs in the target organism in which expression is desired. It’s necessary because the same amino acid can be encoded by multiple synonymous codons, while different organisms have different preferences for which codons they use most frequently. By choosing codons that match the most abundant tRNAs in that organism, translation becomes more efficient.
Protein synthesis in cells. A double-stranded DNA first needs to be resolved by helicases to obtain a single strand accessible for RNA polymerase. RNA polymerase binds to a promoter on a 3’ to 5’ ‘template’ strand and produces a 5 to 3 strand of mRNA. In eukaryotic cells, mRNA gets modified right during transcription and just after that. It gets stabilised, protected from endonucleases, prepared for export to the cytoplasm, recognition by ribosomes, and translation initiation, i.e., a cap (a triphosphate link and a modified G) is added on its 5’ end, mRNA is spliced, and a poly-A tail sequence is added on its 3’ end. Mature mRNA then leaves the nucleus for translation in the cytoplasm. mRNA is read in 5’ to 3’ direction, in codons, within a ribosome, a protein-RNA complex or ribozyme, with a catalytic domain in ribosomal RNA. In a ribosome, protein synthesis is a series of processes (initiation, elongation, and termination) with ribosomal proteins acting as initiation factors, terminating factors, and mediating mRNA and tRNA meeting and release between ribosomal subunits. tRNAs recognise codons with their anti-codon and carry the corresponding amino acid, which binds with the previous one with the catalytic activity of the ribosome. If a protein is produced in bacteria, no preprocessing of mRNA occurs (no splicing like in eukaryotes as bacteria lack introns, but there is 5’end protection and addition of poly-A tail for degradation), and translation is coupled with transcription within the cytoplasm, in ribosomes that are smaller than eukaryotic (70S vs 80S). After synthesis, proteins undergo modifications, which are much more complex in eukaryotic cells than in bacteria. Post-translational modifications in eucaryotes occur in membrane organelles, endoplasmic reticulum, and Golgi apparatus, and include a variety of modifications (including phosphorylation, acetylation, formation of disulfide bonds, glycosylation, ubiquitination, most well-studied). In bacteria, these modifications are much simpler and happen in the cytoplasm.
Cell-free protein synthesis. A cell extract (eucaryotic or bacterial) or a PURE system is added to a DNA, and the extract/mix contains all enzymes and factors needed for transcription and translation, including energy sources and helicases.
3.5. How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level.
During transcription in eukaryotes, RNA (pre-mRNA) gets spliced, and introns, which are non-coding sequences, are cut out while exons are joined. mRNA goes through either regular or alternative splicing in a spliceosome. In regular splicing, all introns are removed, and all exons are joined together, while in alternative splicing, some exons are removed and some are included. Either one or another splicing occurs, depending on the gene, cell type, and conditions (including developmental stage), instructing specific regulatory proteins mediating splicing. There are variations in how alternative splicing occurs (either an exon is skipped, an intron is left in the sequence, or a specific site is selected in the sequence). The splicing process is regulated by proteins that bind to pre-mRNA sites near exons and introns and either enhance or repress splicing (their relative concentration can also be a regulating factor, not just absence or presence). These proteins regulate the assembly of spliceosomes (complexes of specialized proteins and small RNA) on pre-mRNA and instruct the sites of spliceosome assembly and the efficiency of its binding. A spliceosome cuts out and joins fragments of pre-mRNA. The process overall allows for a variety of proteins that are synthesized from a single gene.
During transcription in bacteria, almost no splicing occurs, but still, different proteins can be produced from a single gene. RNA polymerase can be directed to different sequences related to a single gene by transcription factors, and so different transcripts are produced. Also, a ribosome can shift a reading frame due to specific patterns in mRNA sequence.
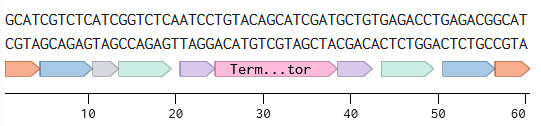
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
Rearranged snapshot of TH-GFP protein information flow from DNA to RNA to Protein. Captured from my Benchling and stitched together in a ppt.
4.1. Create a Twist account and a Benchling account
4.2. Build Your DNA Insert Sequence
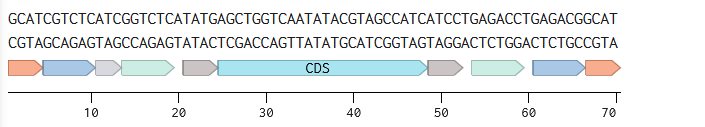
A sequence for expression in E.coli:
Promoter (e.g. BBa_J23106): TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC
RBS (e.g. BBa_B0034 with spacers for optimal expression): CATTAAAGAGGAGAAAGGTACC
Start Codon: ATG
Coding Sequence (codon optimized HT_GFP DNA): ATGCCTACGCCCGACGCAACAACGCCTCAAGCAAAAGGCTTTCGCCGGGCGGTTTCCGAACTGGATGCCAAGCAGGCGGAAGCCATTATGGTTCGTGGACAAGGCGCACCGGGTCCCAGCCTTACGGGTAGCCCTTGGCCGGGTACTGCGGCACCTGCTGCTAGCTACACGCCTACTCCTCGCTCACCCCGTTTTATAGGACGTCGTCAATCTCTCATAGAAGATGCTCGCAAAGAACGCGAAGCAGCAGTTGCAGCAGCGGCAGCAGCGGTACCTTCCGAGCCCGGAGACCCTTTAGAGGCTGTTGCATTTGAGGAAAAAGAAGGTAAAGCAGTTCTGAATTTGCTTTTCTCTCCTCGTGCGACAAAACCTTCGGCACTGTCACGGGCTGTCAAGGTTTTCGAAACTTTCGAAGCTAAAATTCACCATTTAGAAACACGACCGGCGCAGCGTCCGCGTGCCGGGGGGCCTCACTTGGAGTACTTCGTGCGTCTGGAGGTTCGACGTGGCGACCTTGCTGCTCTGTTGAGCGGTGTGCGCCAGGTTTCCGAAGATGTTCGTAGTCCTGCCGGACCTAAAGTACCATGGTTTCCGCGCAAAGTTTCCGAATTGGATAAGTGTCATCATCTTGTGACGAAATTTGATCCGGATCTTGACCTCGACCATCCGGGGTTCTCTGATCAGGTGTATCGTCAGCGTCGCAAACTCATTGCAGAGATTGCTTTTCAATATCGCCATGGCGACCCGATTCCCCGCGTAGAGTATACCGCTGAAGAAATAGCTACTTGGAAAGAAGTGTACACAACCCTGAAGGGCTTATATGCTACACACGCGTGTGGCGAACATTTAGAAGCCTTTGCTCTTCTCGAACGTTTCTCAGGTTATAGAGAGGACAACATTCCACAGTTAGAGGACGTTTCCCGATTTCTCAAAGAACGTACCGGCTTTCAGCTGAGACCCGTGGCCGGTTTATTGTCTGCTCGTGATTTCCTGGCATCACTGGCCTTTAGAGTATTCCAGTGTACTCAGTATATTCGCCATGCTTCCTCGCCAATGCACTCACCCGAACCAGATTGTTGCCATGAGTTACTTGGACATGTACCAATGCTCGCAGATCGAACATTTGCGCAATTCTCTCAAGATATCGGCCTGGCTAGTTTAGGCGCTTCAGATGAAGAAATTGAAAAGCTGTCCACACTGTACTGGTTCACCGTAGAATTTGGACTGTGCAAACAGAATGGCGAGGTTAAAGCGTACGGTGCCGGGCTTCTGTCCAGCTATGGTGAATTACTGCACTGTCTGTCAGAGGAGCCGGAGATTCGCGCATTTGATCCTGAAGCAGCCGCCGTCCAGCCATATCAAGATCAGACGTACCAGTCTGTGTATTTTGTTTCCGAAAGCTTTTCAGATGCCAAGGATAAGTTGCGCTCTTACGCTTCACGTATCCAACGCCCGTTTTCTGTAAAGTTCGACCCGTATACGCTGGCCATTGACGTCCTGGATAGCCCACAGGCAGTGCGCAGAAGTCTTGAAGGGGTTCAAGATGAGCTCGATACACTTGCCCATGCCCTTTCCGCTATAGGCGGGGGTGGTGGCTCTGGCGGTGGAGGTAGTGGAGGGGGTGGGAGCATGGTTTCAAAAGGGGAGGAGTTGTTTACTGGCGTGGTCCCAATCCTGGTAGAGTTAGACGGAGATGTTAACGGGCACAAATTCAGCGTTAGTGGTGAAGGGGAAGGCGACGCTACATATGGTAAACTGACACTGAAATTTATTTGTACCACCGGTAAGCTCCCAGTGCCCTGGCCGACTTTGGTTACCACGTTGACATATGGTGTACAATGTTTCTCCCGCTATCCTGACCACATGAAACAACATGATTTTTTCAAATCTGCTATGCCGGAAGGATATGTACAGGAACGTACGATCTTCTTCAAAGATGATGGCAACTATAAAACACGTGCCGAGGTTAAATTTGAGGGTGATACGCTGGTGAATCGCATTGAGTTAAAAGGAATAGACTTTAAGGAGGATGGGAATATTCTTGGCCACAAACTGGAGTACAATTACAATTCTCATAATGTGTATATCATGGCTGATAAACAGAAAAATGGTATCAAGGTTAACTTCAAAATCCGTCATAATATCGAGGATGGTTCTGTTCAGCTTGCTGATCATTATCAGCAAAATACGCCAATCGGTGATGGACCAGTCCTGTTGCCTGATAATCATTACCTCTCTACACAGTCAGCGCTGTCCAAAGACCCAAATGAGAAACGAGATCATATGGTATTGCTGGAATTCGTTACCGCTGCCGGAATTACACTTGGCATGGATGAATTATACAAATAA
7x His Tag (Let’s add a 7×His tag at the C-terminus of the protein to enable protein purification from E. coli): CATCACCATCACCATCATCAC
Stop Codon: TAA
Terminator (e.g. BBa_B0015): CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
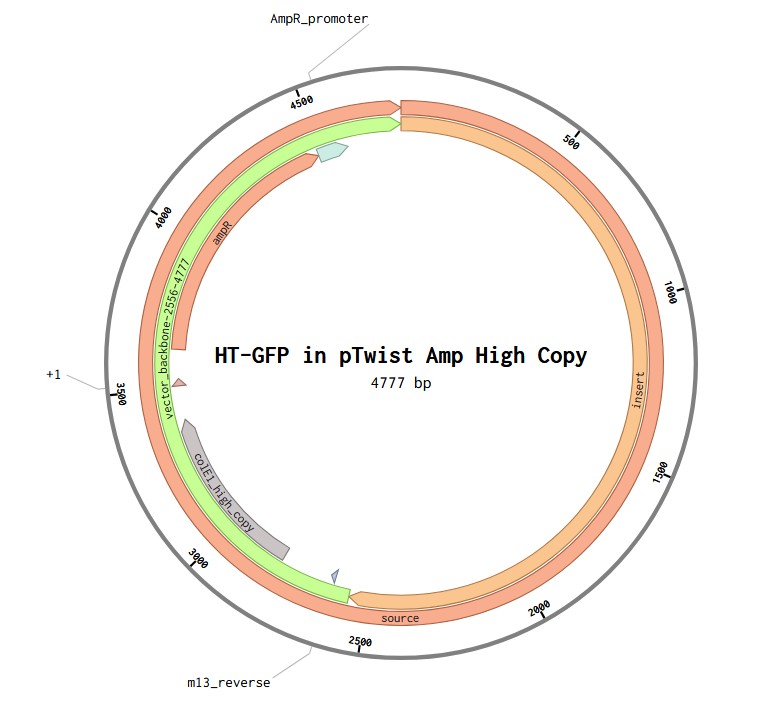
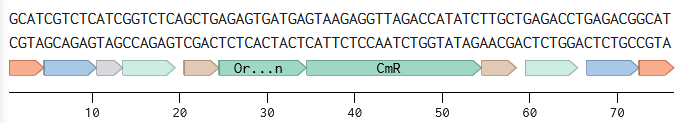
4.6. Choose Your Vector
Vector: pTwist Amp High Copy
TH-GFP Plasmid
Part 5: DNA Read/Write/Edit
5.1 DNA Read
What DNA would you want to sequence (e.g., read) and why?
It could be useful to sequence mitochondrial DNA of dopaminergic neurons and other cells of PD patients. As mutant mtDNA accumulates with age, mitochondrial dysfunction is a common phenotype observed in brain tissue, and the link between mtDNA maintenance and neurodegeneration is fairly recognised. Sequencing may reveal patterns of mitochondrial dysfunction and mt genome instability useful for predicting vulnerability to PD, neurodegenerative, and non-neurodegenerative conditions, as well as investigating fundamental aspects of aging in general. Concentrating on mtDNA involves a large patient population. However, to be a practical diagnostic tool (mtDNA biomarker), more accessible mtDNA in CSF or blood, or peripheral mtDNA has to be included, as tissue decline during aging is heterogeneous, and peripheral mtDNA may also contain information on the vulnerability of neuronal mtDNA to PD. It could be useful to aim at sequencing all 37 mitochondrial genes.
DNA Write
That would be useful to synthesize a construct for inducible and controllable induction of an amyloid protein alpha-synuclein that would be used for modeling Parkinson’s disease in patient-derived cells. This construct would be an upgrade to currently used ones (fibril seeding and constitutive alpha-synuclein overexpression) and can be used along with fibril seeding as well to produce more specific and more controllable models of the disease.
As the construct would be quite large, it could be synthesized in pieces, with either phosphoramidite chip synthesis (being the most cost-efficient) or enzymatic synthesis (to avoid the error-prone polymerase chain assembly step). The essential steps of phosphoramidite chip synthesis (Twit order) are: 1) designing the construct (colonogenes or gene fragments) in silico with overhangs for Gibson or Golden Gate assembly, and codon optimization, 2) synthesis, 3) polymerase chain assembly, 4) verification of clonogenes with NGS, 5) PCR amplification, 6) Gibson or Golden Gate assembly, 7) sequence verification of the assembled plasmid. Overall, the limitation of phosphoramidite chip synthesis would be mostly accuracy, and errors would be caught at the step of colony sequencing, when the assembled plasmid is analyzed.
DNA Edit
Editing human mitochondrial protease ClpP so that it does not bind alpha-synuclein would be useful to prevent proteostatic failure and unload dopaminergic neurons from the misfolded amyloid protein (Parkinson’s disease). DNA editing can be done with site-directed mutagenesis, prime editing, base editing, Cas9 TDR system. Base editing only works with transition mutations; prime editing does not have this constraint and can also add small insertions or deletions; CRISPR-Cas9 with HDR can also change, but it’s cell cycle dependent and is error-prone (which can be improved by using TALENs and Zinc fingers requiring specific fragments included in the construct).
As for preparation, the interface between ClpP and alpha-synuclein and candidate substitutions must be defined. Based on the nature of edits required, either prime or base editing can be chosen. If more complex editing is chosen, the essential steps of CRISPR-Cas9 and HDR are designing the donor template, checking the specificity of the guide, collecting the reagents (Cas9, sgRNA, and the donor template), delivery as RNP into iPSCs cells (Cas9 protein assembled with its guide RNA), increasing the proportion of cells that carry the precise homology-directed DNA repair, isolation of clones, genotyping and sequencing, validation of the clones, differentiation of the cells for functional validation.
Although the paper is still unpublished, the paper describes a unit for drug development using brain organoids and patient-derived cells, which hasn’t been achieved before. Mainly, three automation tools are used: automated high-content imaging, automated image and PCR analysis pipelines. High-content imaging is achieved with a screening platform using 96-well plates; this way, throughput bottleneck s removed and the platform deals with variability of standard imaging. The data is processed with complementary custom image data analysis tools, created to trace specific disease hallmarks for the disease. For PCR, an automated outlier removal and relative-expression calculations are achieved through a custom Python-based algorithms. The tools allowed to have both acquisition and analysis automated and run dose-response experiments over time (identified myricetin and resveratrol as dose-dependent inhibitors of aggregate formation in a model of Parkinson’s). This is essentially the core of the drug discovery screening platform with an opportunity to scale a Parkinson’s model in human iPSC-derived neurons (typically done in rodent cells, on a smaller scale).
Han, C., Nguyen-Renou, E., Benaliouad, F., Luo, W., Chen, C. X., Alluli, A., Lorenza Villegas, Lenore K Beitel, Irina Shlaifer, Wolfgang E Reintsch, Andrea I Krahn, Esther
Del Cid Pellitero2, Edward A Fon2 & Durcan, T. M. (2025). An automated workflow for quantifying the formation of synuclein aggregates in human dopaminergic neurons. bioRxiv, 2025-12. Link
For my final projects on an inducible system (as opposed to the fibril-driven pathology used in the paper), all of these automation tools (and disease hallmarks the authors are measuring) as well as the unit infrastructure, are directly transferable at the starting point and validation. However, live cell imaging will be primarily needed to trace the resulting oscillatory alpha-synuclein expression.
Part 3: Final Project Ideas
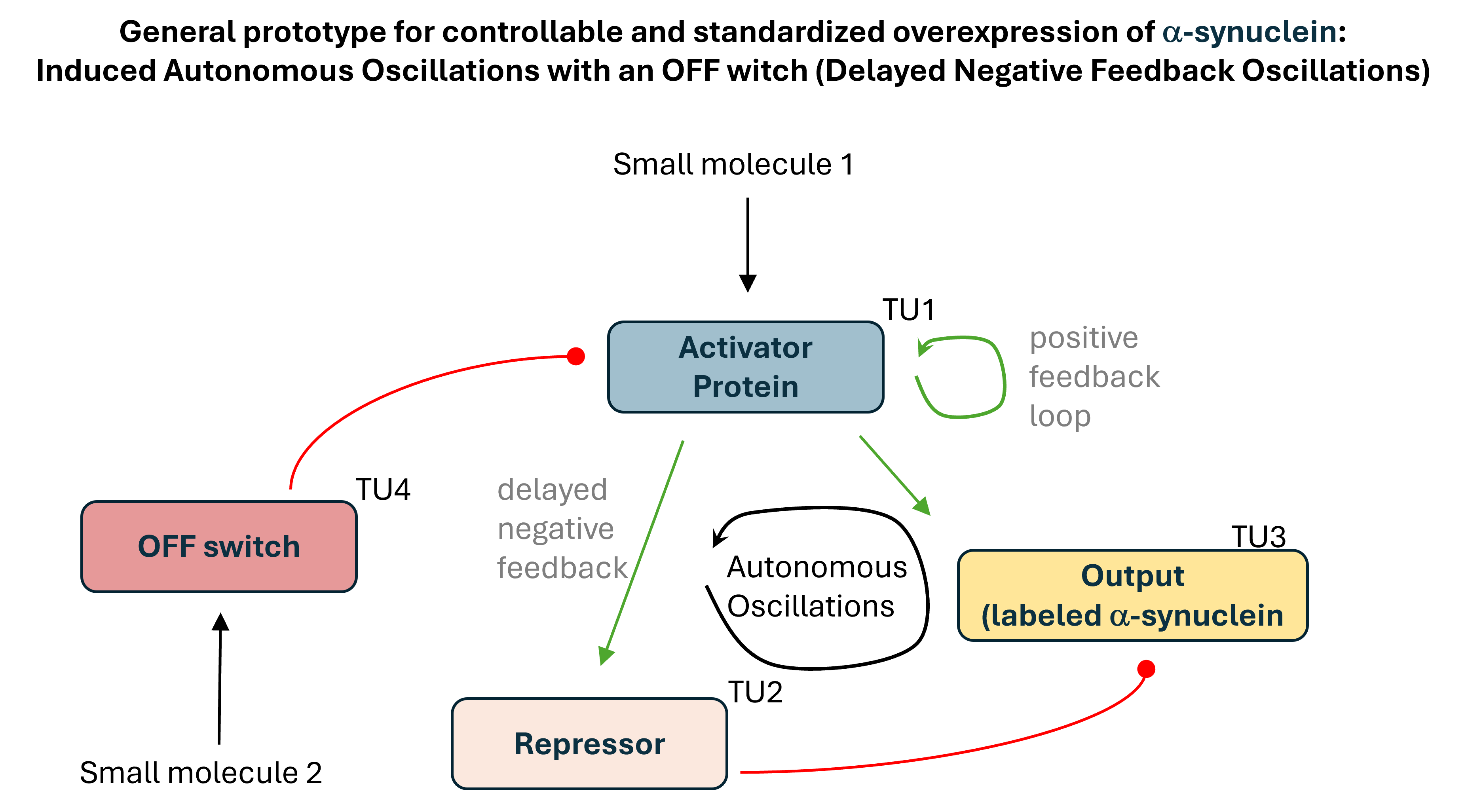
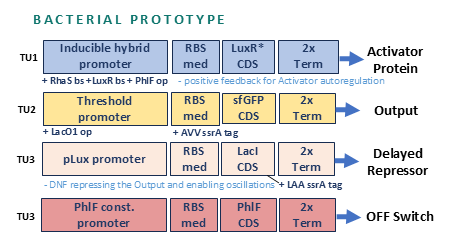
Project 1: Tunable Induction of Alpha-Synuclein Expression for Modeling Parkinson’s Disease
Aim:
This project aims to develop a tool to promote Parkinson’s disease phenotype manifestation by controllable induction of alpha-synuclein expression in dopaminergic neurons within patient-derived brain organoids.
Background:
Parkinson’s disease (PD) is driven by alpha-synuclein misfolding and accumulation in dopaminergic neurons, triggered by interconnected failures in multiple cellular processes. Current PD models using AAV-mediated alpha-synuclein overexpression and exogenous fibril seeding are effective at replicating key features of sporadic PD but lack controllability, limiting their value for investigating which cellular systems fail under pathological alpha-synuclein load in individual patients.
Patient-derived brain organoids naturally recapitulate human-specific neurodegenerative features, but their use for studying PD is constrained by the months required for PD phenotype manifestation. Tools that accelerate and standardize pathological phenotype induction in human tissue culture models are therefore needed.
Tool Description:
This project aims to develop a tool to promote Parkinson’s disease phenotype manifestation by controllable induction of alpha-synuclein expression in dopaminergic neurons within patient-derived brain organoids. The tool uses a genetic circuit for controllable oscillatory overexpression of alpha-synuclein. The circuit will employ a small molecule-activated sensor-promoter to initiate alpha-synuclein expression, a delayed negative feedback loop with a repressor to generate self-limiting oscillatory expression, and an external OFF switch to terminate the expression.
Significance:
The tool will hopefully:
enable standardized and accelerated induction of PD phenotypes and
allow probing patient-specific vulnerabilities and
allow testing personalized therapeutic strategies in organoid platforms.
Project 2: Sensing α-Synuclein-Driven Mitochondrial Proteostatic Failure in Parkinson’s Disease with an RNA Toehold Switch
Aim:
This project aims to design a sensor for mitochondrial dysfunction in models of Parkinson’s disease (PD). An RNA toehold switch sensor will target mitochondrial protease (ClpP) mRNA, which is expected to rise in Parkinson’s disease models.
Background:
ClpP is a mitochondrial matrix protease that degrades misfolded or damaged proteins and recently shown to be inhibited by α-synuclein (through direct binding at the NAC domain), representing a novel mechanistic link between α-synuclein pathology and mitochondrial proteostatic failure in Parkinson’s disease.
When ClpP activity is chronically suppressed by accumulating α-synuclein, the cell is expected to sense the resulting proteostatic stress through the mitochondrial unfolded protein response, which in mammals involves a nuclear transcriptional response involving ATF5-driven upregulation of ClpP as a compensatory mechanism. This creates a scenario where ClpP mRNA levels is expected to rise despite and because of functional ClpP insufficiency at the protein level.
Sensor Description:
An RNA sensor targeting ClpP mRNA would therefore report not on ClpP activity directly, but on the cell’s transcriptional response to its own proteostatic failure, serving as an indirect proxy for the α-synuclein-driven mitochondrial dysfunction that precedes late neurodegeneration and neuronal death.
Significance:
If 1) ATF5-driven ClpP upregulation in dopaminergic neurons is confirmed and 2) the sensor construct is validated in dopaminergic neurons, this sensor could provide an early, mitochondria-specific readout of PD-relevant stress in brain organoid models and become a drug screening tool to identify small molecules that restore the activity of the protease and reduce pathological α-synuclein accumulation (shift the tetrameric:monomeric α-synuclein balance), with the sensor itself as the readout.
Week 4: Protein Design Part I
Part A. Conceptual Questions
Q1: How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
1 Da equals 1.66053906892(52)×10−27 kg, so 1 aa is 1.66053906892(52)×10−25 kg. The average protein fraction in meat is ~20%. Therefore, the total amount of protein is 100g. The number of aa in 100g = 0.1kg of protein is ~6×10²³ or Avogadro number, 1 mole.
Q2: Why do humans eat beef but do not become a cow, eat fish but do not become fish?
To become another organism, we need to use its DNA to produce proteins within us. However, DNA, RNA, and proteins, we consume are broken down and absorbed, and so only their building blocks and not the information their sequences carry is used in our body.
Q3: Why are there only 20 natural amino acids?
The set of 20 amino acids has been established through evolution, and it would be impossible to change already fixed codons because a change would corrupt thousands of proteins.
Q4: Can you make other non-natural amino acids? Design some new amino acids.
Yes. Some design strategies can include adding an azide to a sidechain for the aa to react in click chemistry reactions or an isotope.
Q5: Where did amino acids come from before enzymes that make them, and before life started?
The production of amino acids does not require enzymes. Enzyme-independent production of amino acids was possible in the early Earth atmosphere, in hydrothermal vents, and through the Strecher Synthesis. Additionally, amino acids reach Earth with meteorite material.
Q6: If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
The resulting helix will be left-handed.
Q7: Can you discover additional helices in proteins?
Yes. Several other types (4+) of helices have been identified beyond α-helices.
Q8: Why are most molecular helices right-handed?
Most polymer molecules are right-handed because they are comprised from left-handed monomers. In helices, left-handed monomers would occupy more space near the backbone, which is energetically inefficient.
Q9: Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
They aggregate due to exposed NH group being hydrogen donors and C=O groups being acceptors on the edge of a sheet, so edges form hydrogen bonds (the driving force); due to hydrophobic sides associating with each other; due to steric compatibility of identical sheets; due to matching electrostatic periodicity in sheets; due to low configurational entropy cost of aggregations.
Q10: Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Yes, β-sheets are used as materials (Kevlar, peptide hydrogels as tissue engineering scaffolds).
Due to stress, mutations, and aging, proteins partially unfold.
β-sheets with an alternation of hydrophobic and hydrophilic sites ease aggregation and then the growth of stable aggregates. Sequences with alternating hydrophobic and hydrophilic residues get exposed and form β-sheet conformation due to thermodynamic stability. These sheets bind and stack due to hydrogen bonds and hydrophobic interactions, and the oligomers then grow to exceptionally stable fibers.
Q11: Design a β-sheet motif that forms a well-ordered structure.
In the week 5 homework, one of the generated binders produced a beta sheet structure upon co-folding it with the target (SODI), and so while a well-ordered structure is more of an empirical claim, I chose to analyze the binder I previously got and potentially improve it in silico.
PepMLM-generated binder to human SOD1: HLYYVAGVRWKK (Peptide 2, Psudo Perplexity 30.9)
To find out whether it is stably folded into beta sheets even when no target template is present, the peptide was folded separately (without SOD1) in AlphaFold3. Three folds (the highest-ranked model considered per seed) confirmed that the hairpin structure is reproducible. In all the folds, average pLDDT was uniformly high (>90%), and the Expected Position Error graphs showed uniformly low Error (Figure 1).
To explore this hairpin structure further and predict its intermolecular behavior, adding this to the confidence metrics observed in AlphaFold3, the peptide was analyzed in the AGGRESCAN web interface (Conchillo-Solé, O., de Groot, N. S., Avilés, F. X., Vendrell, J., Daura, X., & Ventura, S. (2007). AGGRESCAN: a server for the prediction and evaluation of" hot spots" of aggregation in polypeptides. BMC bioinformatics, 8(1), 65). This analysis would inform whether the strands are prone to intermolecular β-aggregation.
AGGRESCAN results highlighted 1 hot-spot consisting of 9 residues, peak aggregation propensity at Y4 (1.051) and Y3 (0.852), and a3vSA = 0.268: net aggregation-prone on average (Fig 1, last insert, and Figure 2).
To find out whether changing Ala to Asn (to make an Asn-Gly turn instead of Ala-Gly turn (Griffiths-Jones et al. 1998)) improves net aggregation propensity, the modified sequence was analyzed in AlphaFold3:
The folding results are identical, and the two sequences can both fold to the same hairpin. Thermodynamic stability can be more informative to confirm if the modification influenced the stability of the structure. Other methods (e.g. adding cross-strand pairs, cation or aromatic) may further reinforce the structure.
(Ref.
Griffiths-Jones, S., Maynard, A., & Sharman, G. (1998). NMR evidence for the nucleation of a β-hairpin peptide conformation in water by an Asn-Gly type I′ β-turn sequence. Chemical Communications, (7), 789-790.)
Part B: Protein Analysis and Visualization
1. Protein choice
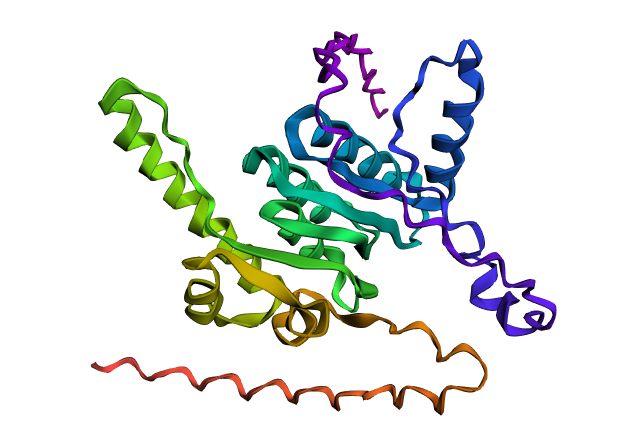
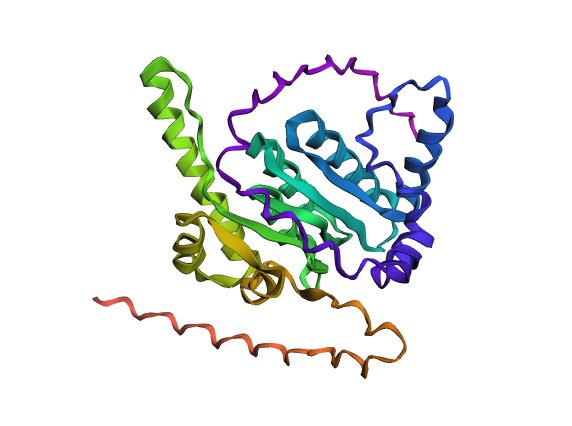
I chose alpha-synuclein (SNCA) Uniprot P37840 because its toxic form accumulates in Parkinson’s disease, and the mechanisms of alpha-synuclein aggregation are still being investigated.
The protein is comprised of 3 domains: an amphipathic N-terminal domain (1-60), a hydrophobic NAC domain (61-95), and an acidic C-terminus (96-140). Three isoforms produced by alternative splicing are: isoform 1 of 140 aa canonical, isoform 2 of 112 aa, and isoform 3 of 126 aa. The protein is normally a monomer, with the highest concentration in the brain and concentrated in the presynaptic terminals of nerve cells. The ’non A-beta component of Alzheimer disease amyloid plaque’ domain (NAC domain) is hydrophobic and is involved in fibril formation. The C-terminus may regulate aggregation and determine the diameter of the filaments.
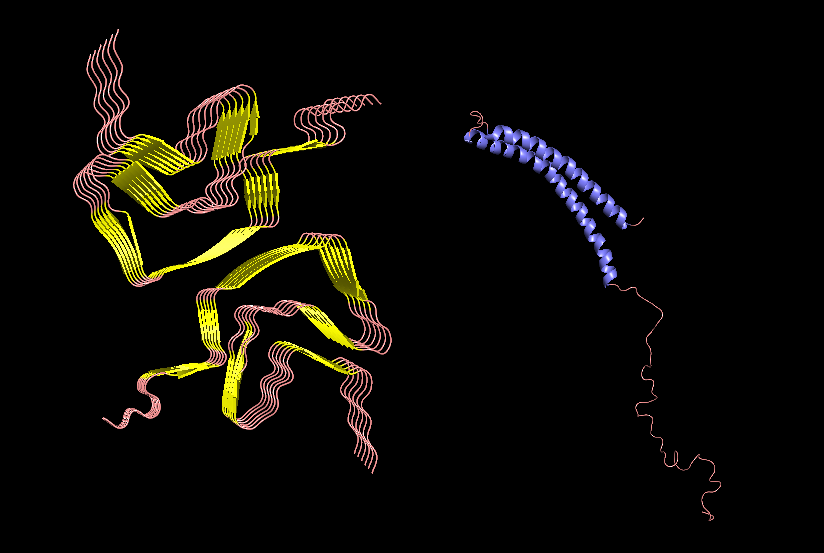
The structure of the monomer was solved with NMR and published in 2005. The structure of the fiber was solved with EM, with a resolution of 3.07 Å and published in 2018. No other molecules other than SNCA are present in the structures.
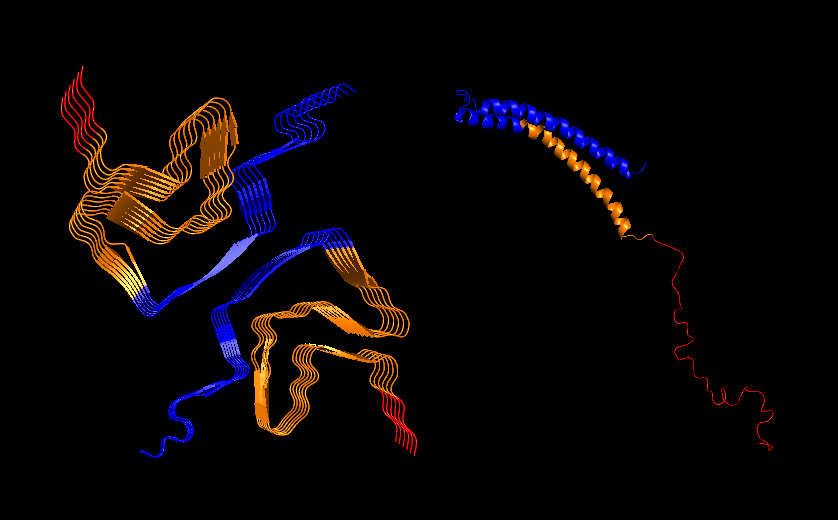
PyMol Cartoon visualization of an SCNA aggregate (6A6B) EM structure and of an SNCA monomer (1XQ8) NMR structure. Colours by secondary structure.
PyMol Cartoon visualization of the same structures. Blue (37–60) - N-terminal region, Orange (61–95) - NAC region, Red (96–99)- C-terminal region.
Part C. Using ML-Based Protein Design Tools
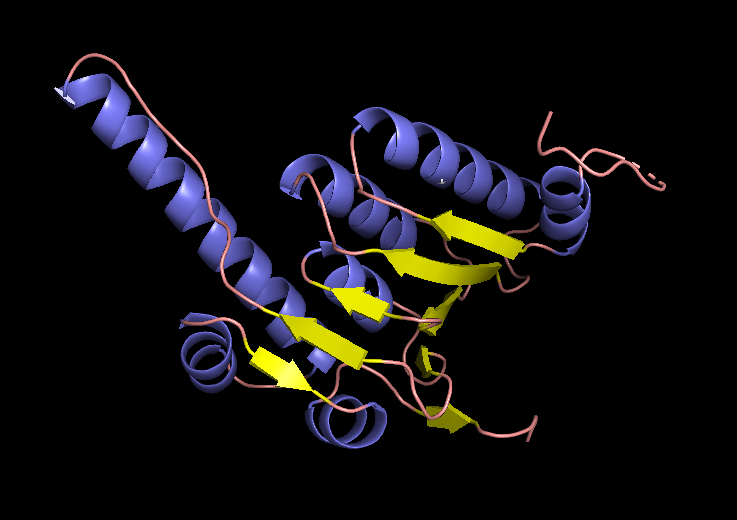
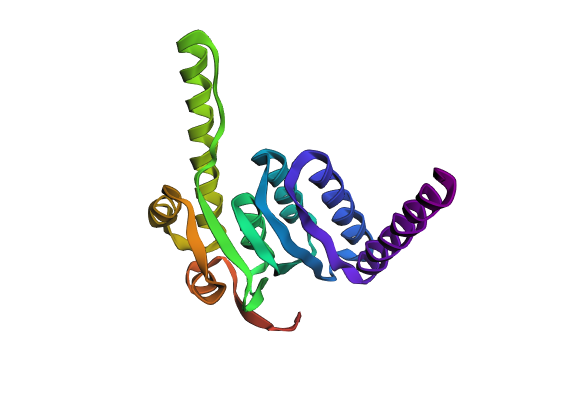
I chose human mitochondrial protease ClpP, the protease component of the ClpXP complex that cleaves peptides and various proteins in an ATP-dependent process. ClpP binds the NAC domain of SCNA, and this binding inhibits the protease.
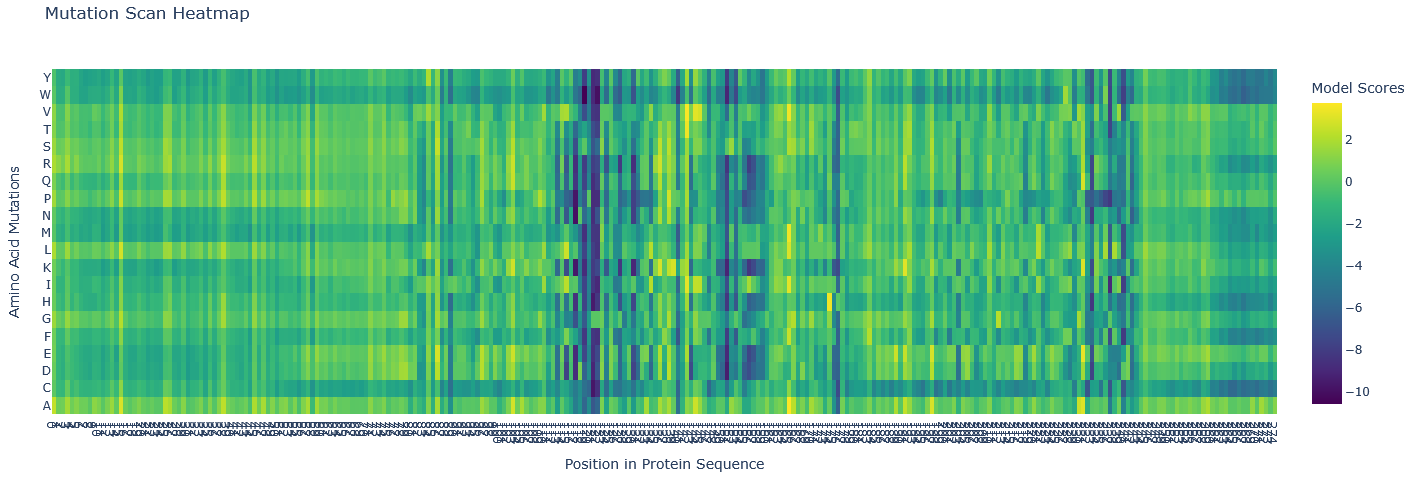
Esm2_t6_8M_UR50D (the shallowest one and using fewer parameters) model was used to generate an unsupervised deep mutational scan of ClpP protein based on language model likelihoods.
Most mutations are neutral (green/as likely as wildtype). There are several blue regions (121-122, 151,153, and 176) in which all mutations score highly unlikely, so these are structurally and functionally important, not likely to mutate, and these are probably part of active centres of the protease.
The mutation scan mapped the active site neighbourhood. The active sites mentioned in UniPort are TYR 153 and GLU 178. The sites with almost all aa unlikely are: HIS 122, TYR 153, PRO 176, ILE 121, ASN 151.
2. Latent Space Analysis
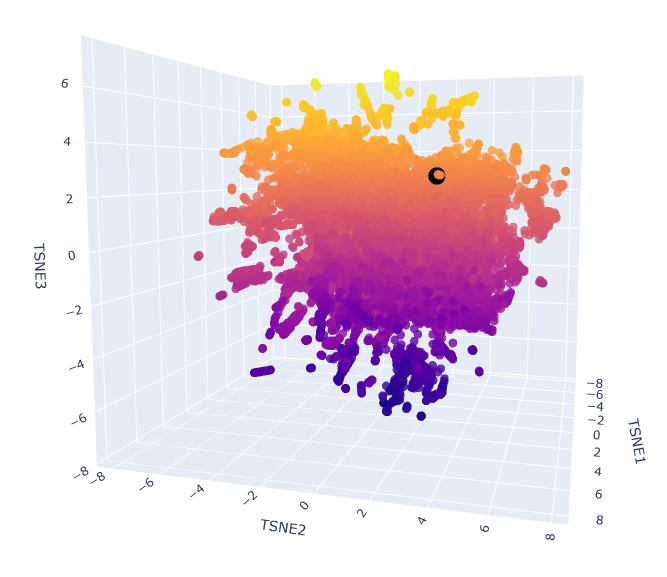
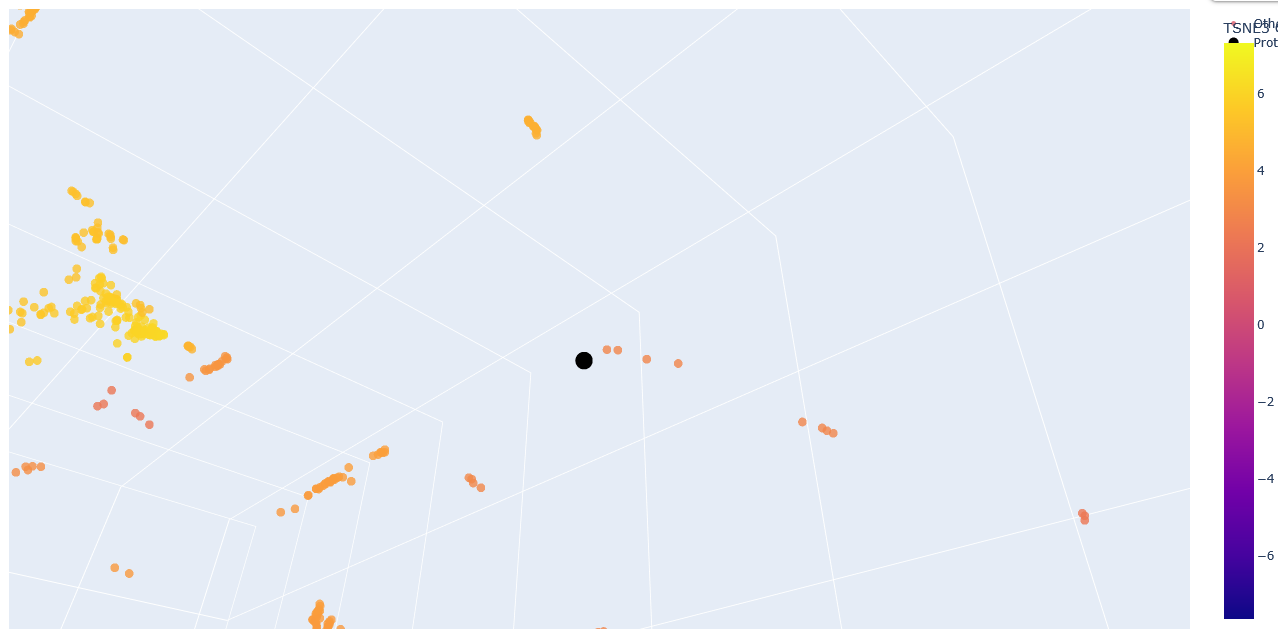
The provided sequence dataset was used to embed proteins in a reduced dimensionality space.
To place ClpP into the resulting latent space map, its ESM2 embedding was first generated. Then, this new embedding was combined with the existing dataset’s embeddings, and the t-SNE algorithm on this combined set was re-run.
Esm2_t6_8M_UR50D latent space with embedded human ClpP Q16740 (black).
Zoom-in into the Esm2_t6_8M_UR50D latent space with embedded human ClpP Q16740 (black).
The neighbours are all ClpP homologs; therefore, the 6-layer model is correctly identifying sequence homology. Larger models like esm2_t33_650M or esm2_t36_3B are needed to capture more nuanced functional information.
C2. Protein Folding
Folding the original sequence
ClpP folded with ESMFold.
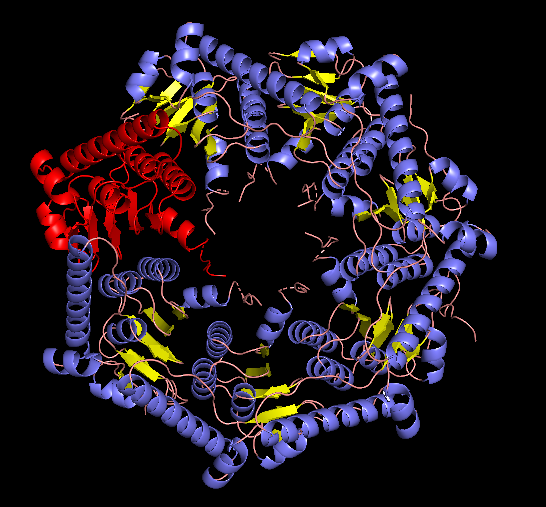
EM structure of the ClpP subunit visualized with Pymol (1TG6 | pdb_00001tg6).
EM structure of the ClpP with the subunit highlighted, visualized with Pymol (1TG6 | pdb_00001tg6).
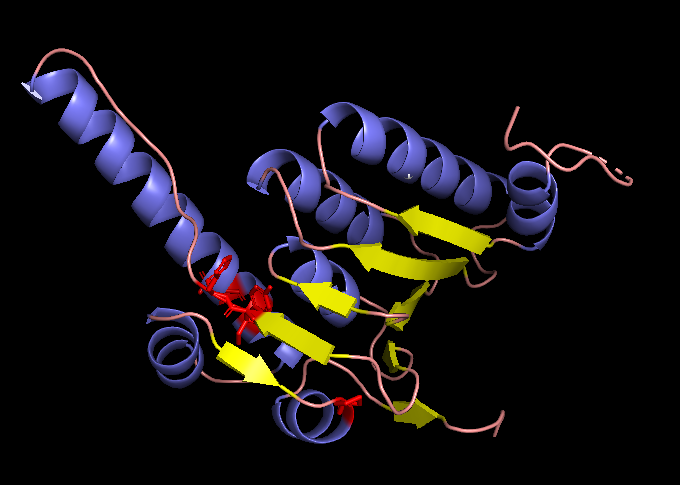
Resilience of the structure to changes in the original sequence
To test the resilience of the predicted fold, I changed 5 positions (121, 122, 151, 153, 176) that are highly unlikely to mutate to Tryptophane.
121 Ser (S)→ Trp (W)
122 Pro (P)→ Trp (W)
151 Ala (A)→ Trp (W)
153 Ser (S)→ Trp (W)
176 Met (M)→ Trp (W)
The generated sequence (with the Xs changed to As) was then folded with EMS fold to compare with the fold predicted from the original sequence and with the published ClpP structure.
A fold of the ProteinMPNN-generated ClpP sequence predicted by ESMFold.
ESMFold inference for a sequence with length 193. ptm: 0.861 plddt: 90.378.
The original ClpP subunit structure, Q16740.
A fold of the original ClpP sequence predicted with ESMFold.
Part D. Group Brainstorm on Bacteriophage Engineering
Our group (Abhishek Udawat, Tammy Sisodiya, Nour Abdelrahman, Nurlenden Rihan, and myself) focused on targeting increased stability as a goal for engineering the L Protein.
Protein Language Models (ESM2) and the analysis of sequence alignment (BLAST/ ClustalOmega) will identify conserved and variable sites and therefore inform a mutagenesis strategy.
Analyses of mutated sequences Alphafold-Multimer will reveal a change in pLDDT, ipTM, which may indicate higher stability of the tail.
Engineering Plan
Review the guidelines.
Isolate the soluble N-terminus and the middle part of the protein non-overlapping with the coat or the replicase sequence.
Evaluate the mutational scan in ESM2 to identify candidate substitutions.
Check BLAST results of sequence alignment (the layout of conserved and variable sites).
Define a specific strategy (e.g., conservation, creating salt bridges to create a helix, etc.).
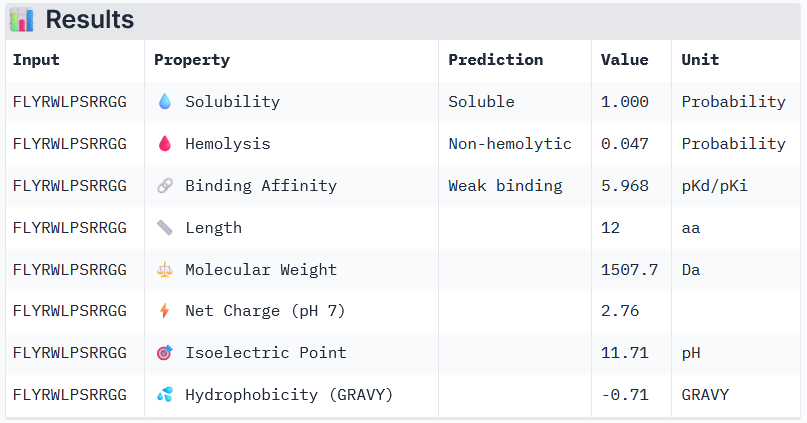
2. Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
index
Binder
Pseudo Perplexity
1
WRYYVVGLRHKX
13.80
2
HRYPVVVVALGX
17.15
3
WRVYAVGLALWX
11.84
4
WLYPATVLEWKX
12.49
More peptides were generated to screen the ones with higher scores (above 30%):
index
Binder
Pseudo Perplexity
1
WRYYVVVVELKK
33.87
2
HLYYVAGVRWKK
30.94
3
HHYYVAVLELWK
31.88
4
HLYYVTVVRLGK
48.23
known
FLYRWLPSRRGG
-
Part 2: Evaluation of Binders with AlphaFold3
Known (FLYRWLPSRRGG) ipTM = 0.38, pTM = 0.78, Seed 1074152779
The ipTM scores and peptide localization/binding were recorded:
Binder
near A4V
engage the β-barrel region
dimer interface
surface-bound
partially buried
1
WRYYVVVVELKK
no
a bit
no
yes
2
HLYYVAGVRWKK
no
yes
no
yes
3
HHYYVAVLELWK
yes
yes
yes
4
HLYYVTVVRLGK
no
yes
no
known
FLYRWLPSRRGG
no
no
no
Based on the predicted template modeling scores, all of the overall predicted folds for the complexes might be similar to the true structure (pTM scores>0.5), and the model for Peptide 3 (HHYYVAVLELWK) has exceptionally high accuracy of the predicted relative positions of the ligand and SOD1 within the complex. However, according to Alphafold3 instructions, values between 0.6 and 0.8 are a gray zone, where predictions could still be either correct or incorrect.
Peptide 3 and Peptide 4 ipTM values (Peptide 3: ipTM = 0.74; Peptide 4: ipTM = 0.45) exceed the ipTM value of the known binder (ipTM = 0.38), and Peptide 1 and Peptide 2 ipTM values are close to that of the known binder.
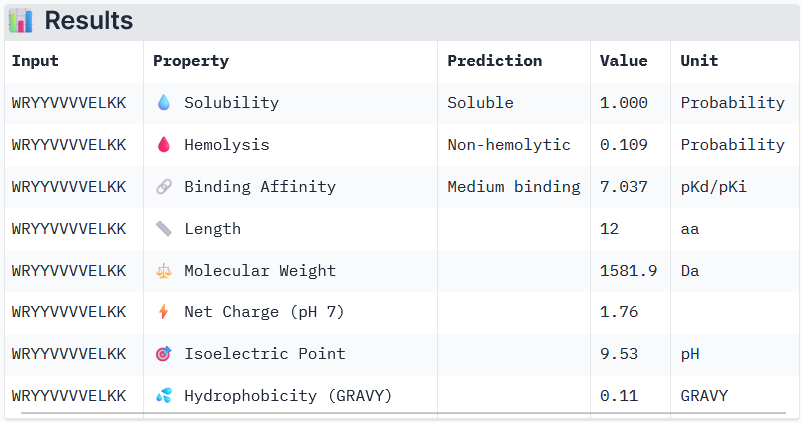
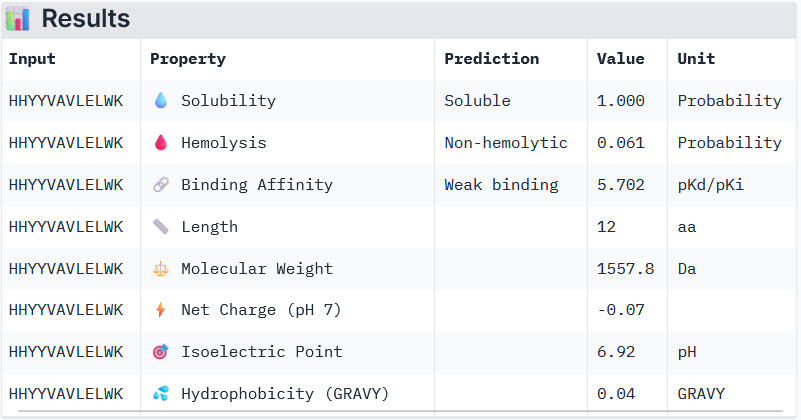
Part 3: Evaluation of the Properties of Generated Peptides in the PeptiVerse
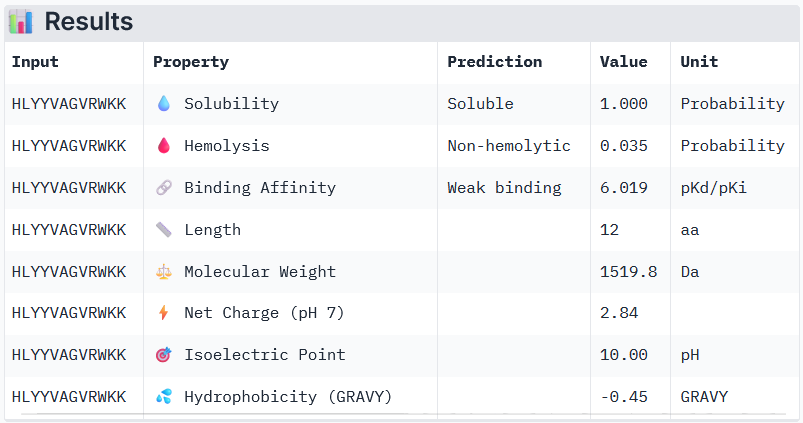
Binder 1: WRYYVVVVELKK
Binder 2: HHYYVAVLELWK
Binder 3: HLYYVAGVRWKK
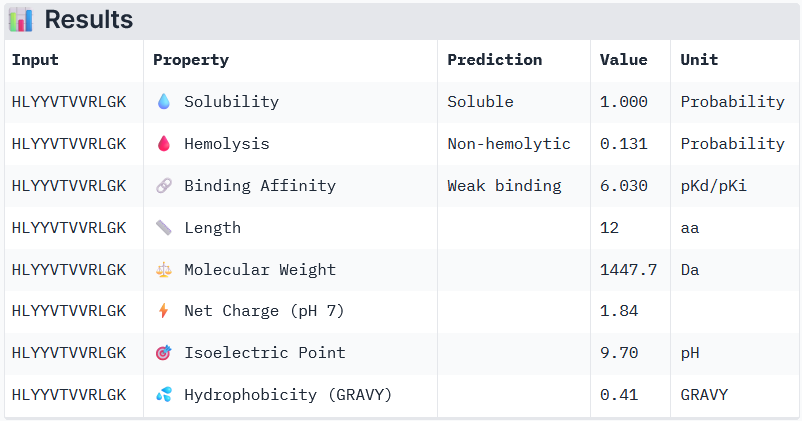
Binder 3: HLYYVTVVRLGK
Known Binder: FLYRWLPSRRGG
Peptide 3 and Peptide 4, with higher ipTM, both show weak affinity, whereas Peptide 1, having an ipTM score lower than that of the known peptide, shows medium affinity.
None of the peptides shows poor solubility or is hemolytic.
Therapeutic properties in this case are related to where a peptide binds. Essentially, only Peptide 3 with the highest ipTM was selective to the mutant SOD1, but it showed weak binding, although it is partially buried and binds the dimer interface, whereas Peptide 1, showing medium binding, is not selective and may inhibit the native SOD1. So, Peptide 3, although weak-binding, may be best at balancing predicted binding and therapeutic properties.
Interestingly, Peptide 4 also interacts with a trimer interface, which is more important than interaction with just the dimer interface alone. Trimers were found to be toxic and produced off-pathway, whereas dimers and fibrils are protective forms (Hnath and Dokholyan, 2022). Peptide 3 interacts with Lys23, Glu21, Phe22, Ala152, Gly14, Ile18, Gly16, Cys146, 3 of which are ALS mutation sites (14, 16, and 23) and 100% of the contact residues (all 8) are trimer interface residues (with one of them experimentally validated (Hnath, Dokholyan, 2022) and all 8 being predicted (Proctor et al., 2016)). Therefore, Peptide 3 is by far the more promising candidate overall, and in particular, for disrupting toxic SOD1 trimerization.
Part 4: Generation of Optimized Peptides with moPPIt; Evaluation of the Properties of Generated Peptides in the PeptiVerse
The controlled design generated binders with greater affinity overall, but all of them are hemolytic. These peptides are not useful for further. Overall, Peptide 3 generated with PepMLM could be further tested experimentally if it indeed prevents trimer formation (there is also a chance that it would stabilize trimers).
The peptide first needs to be validated with mass spectrometry to assess if it matches the designed structure and is pure. Hi-res mass spectrometry will confirm the sequence and the mass, and LC-MS will assess purity and detect contaminating byproducts. Additionally, NMR would be needed to confirm atom connectivity and their stereochemistry, and HPLC is needed for quantitative purity. Then, binding needs to be confirmed, and after that, biochemical assays are needed to aggregation. After that, crystallography could confirm whether the ligand is in the interface, and the selectivity of binding to the mutated SOD1 in neurons could be confirmed. Lastly, solubility, metabolic stability, plasma protein binding, and cytochrome P450 inhibition could be assessed with LC-MS/MS, cytotoxicity, in vivo pharmacokinetics, efficacy in a rodent and primate model can be measured, and GLP toxicology studies are needed. Overall: confirm the molecule -> confirm binding -> confirm function -> confirm binding mode -> confirm selectivity and cellular activity -> confirm drug-like properties and safety, with computational analysis run in between stages.
For this mutant, I modified the N-terminal domain, aiming to stabilize the disordered domain. I introduced as many charged pairs as possible in the variable sites (changed 4 out of 8 in the N-terminal domain), and additionally changed one conserved site on the left side of the 2nd pair.
For this mutant, I modified the previous sequence (Mutated Sequence 1), aiming to further stabilize the disordered domain.
I introduced 1 more mutation to a variable site to invert the second pair.
Pairs introduced by changing the 5 variable sites: Pair 1 (R7–E11), Pair 2 (R14–E18), Pair 3 (R22–D26)
Soluble N-terminal domain C-terminal domain
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Original Sequence)
R---E LR---E R--- (Mutated Sites)
V V CV V V (Conserved / Variable)
METRFPRQSQETLRSTNERRPRKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Mutated Sequence 2)
AlphafoldServer was used to fold the monomers of Mutated Sequence 1 and Mutated Sequence 2. alfafold2_multimer_v2 was used to fold the multimers. alfafold2_multimer_v2 parameters used:
This sequence was designed to explore whether changing the conserved site (13P->L) was required to achieve the same structure as that of the Mutated Sequence 1. For that, the mutated conserved site of the Sequence 1 was changed back to the original (13L->P).
This sequence was designed to explore whether changing the conserved site (13P->L) was required to achieve the helix as in the Mutated Sequence 2. For that, the mutated conserved site of the Sequence 2 was changed back to the original (13L->P).
Mutant 1 pLDDT=37.6, pTM-0.189, ipTM = 0.127. 3 pairs/bridges introduced, 1 conserved site changed (13P->L), RRR site kept, (1 conserved and 4 variable sites changed)
Mutant 3 pLDDT=43.3, pTM-0.188, ipTM = 0.127. Mutant 1 -> the conserved site mutation reverted (13L->P) (4 variable sites of the Original Sequence changed)
Mutant 2 pLDDT=45.8, pTM-0.187, ipTM = 0.126. 3 pairs/bridges introduced, 1 conserved site changed (13P->L), 2nd pair inverted, no RRR site (1 conserved and 5 variable sites changed)
Mutant 4 pLDDT=37, pTM-0.189, ipTM = 0.127. Mutant 2 -> the conserved site mutation reverted (13L->P) (5 variable sites of the Original Sequence changed)
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
DNA Assembly
Components in the Phusion High-Fidelity PCR Master Mix and their purpose*
The mix contains:
A thermostable high-fidelity and high-speed DNA polymerase with 5´→ 3´ polymerase activity and 3´→ 5´exonuclease activity for proofreading and correcting (the enzyme was engineered so that a polymerase is fused to a small DNA-binding domain, and the domain gives the polymerase high processivity and low error rate; the exonuclease activity, unlike that of Taq polymerase, gives blunt-ended products);
Deoxynucleotides used by DNA polymerase to synthesize a new DNA strand;
Reaction buffer to maintain the optimal pH and ion concentration, with MgCl2 for Mg2+ ions (cofactors) that coordinate the active site of the DNA-polymerase and the phosphate groups of the dNTP, for the phosphodiester bonds that extend the growing DNA strand to form.
There can also be stabilizers added.
Factors that determine primer annealing temperature during PCR
Primer melting temperature. According to the protocol, the annealing temperature is typically set a few degrees below it (2-5 degrees). Primer melting temperature or Tm depends on primer length (longer primers require higher temperature), GC content (GC-richer primers require higher temperature), and nearest-neighbour thermodynamics (nearest neighbour interactions affect duplex stability and the temperature needed).
Primer pair matching. If two primers have very different melting temperatures, then it’s harder to find the optimum. According to the protocol, the two primers (Color Forward and Color Reverse) are advised to keep within 5 degrees. In this case, the temperature needs to be set to be a few degrees below the lower one.
Mismatches between a primer and a template. Mismatches destabilise the two strands and lower the annealing temperature.
Ion concentration. Mg and K neutralize the negatively charged phosphate backbone and stabilize the duplex, and so a higher annealing temperature is needed.
Primer concentration. The higher the concentration of a primer, the higher the annealing temperature can be used.
DMSO concentration. DMSO lowers the temperature.
PCR vs restriction enzyme digests
In this lab, PCR and restriction digestion were used for different purposes. PCR built the inserts and added the overhangs and mutations, and restriction digestion linearized the backbone and verified the final product, and the restriction enzyme DpnI cleaned the template.
The main difference between the methods is that PCR is a DNA synthesis method, and through it, the product is amplified, whereas restriction digestion does not synthesize and does not use primers or nucleotides, but instead cuts the DNA that is supplied. Also, PCR products have blunt ends, and restriction enzymes produce sticky or blunt ends.
In terms of protocols, PCR requires primers, nucleotides, polymerase, Mg, and cycling temperature for the primers to melt, anneal, and extend. Restriction digestion is performed at the constant physiological temperature, within approximately 15 min (instead of 1-2 hrs). The cut positions are set by specific sequences included in the DNA supplied and the specificity of the enzymes.
PCR is useful when more DNA needs to be produced, when the DNA needs to be changed. Restriction digestion is useful when DNA linearization is needed and cuts at specific sequences, in general, when sticky ends are needed, when errors that polymerases produce are not allowed, and when a diagnostic test is performed to run on a gel and verify the construct.
How to ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning
Proper overhangs need to be designed. The lab used 20 nt overlap to create adjacent fragments sharing identical sequences. The documents also mentioned 15-25 range, and up to 40 nt are also used. The whole plasmid should first be designed as assembled, and the primers need to be designed so that every junction has a matched overlap. Each overlap must be relatively unique so that fragments assemble in the correct orientation.
Check that each fragment covers its desired region (promoter + partial gene, mutation + terminators, etc.).
Overlaps should have similar melting temperatures so that they all anneal during the assembly.
PCR products must be verified using gel electrophoresis, and the concentration of each product needs to be quantified using Nanodrop or similar. According to the protocol, the expected band size can be calculated in Benchling and then checked on a gel. The concentrations are measured because appropriate ratios of PCR products are needed for the assembly (concentrations above 30 µg/mL were used in the lab to check that PCR worked).
High-fidelity polymerase must be used to generate blunt ends, so that the end of a fragment is the sequence that has been designed.
The original template must be removed (the protocol used DpnI)
The products must be purified to remove primers, nucleotides, polymerase, and primer dimers (the protocol used silica-column).
How does the plasmid DNA enter the E. coli cells during transformation
Plasmids are forced to pass a cell barrier. Cells are first made competent with the addition of calcium chloride, which neutralizes negative charges on DNA’s phosphate backbone and on the cell surface (the treatment is done on ice). Either heat shock is used (fast temperature increase) or electroporation (brief electrical pulse) to create transient pores for the plasmids to diffuse into cells, and then the cells are left to recover. Cells are then selected with culturing on antibiotic plates, and antibiotic resistance genes are switched on due to transfection.
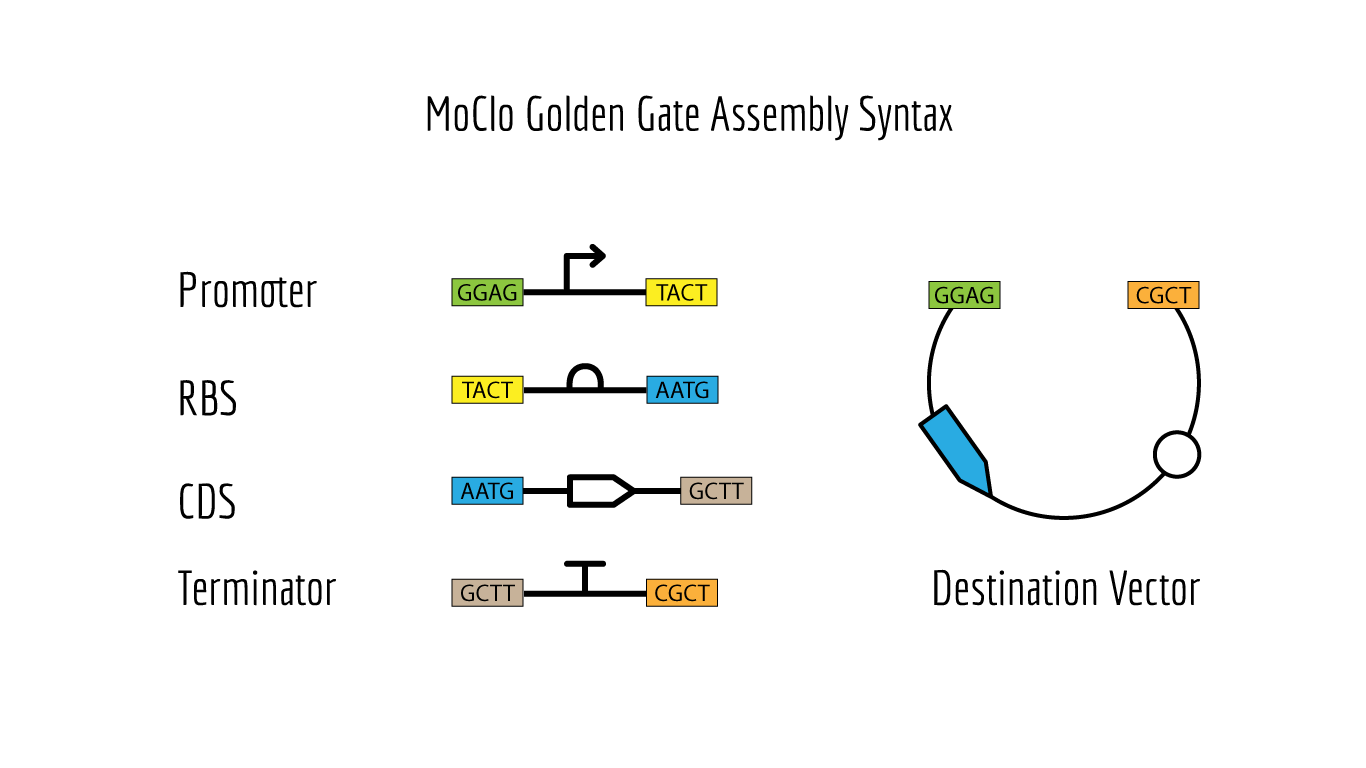
Golden Gate Assembly
Description
Like Gibson Assembly method, Golden Gate is a cloning method that joins DNA fragments, with the reaction performed in a single tube. It does not use homology overlaps but uses a special class of restriction enzymes (Type IIS enzymes) to build overhangs (Figure 1). The enzymes recognize an asymmetric site GGTCTC and cut at a certain distance near this site, producing a custom overhang of several bases long. The sequence where the same enzyme cuts is designed, in order to arrange different parts of the plasmid in order; assembly standards exist that define the rules for overhangs corresponding to any part (Modular Cloning or MoClo) (Figure 2, Figure 3). The reaction requires DNA fragments, Type IIS enzyme, and T4 DNA ligase, and cycling temperature, and recognition sites are removed during the process (Figure 4).
Figure 1. Common types of restriction enzymes used for Golden Gate Assembly Source: iGEM.
Annotated linear fragments were first created with the corresponding overhang sequences and random sequences for the parts (promoter, RBS, CDS, terminator, origin):
index
Fragment Name
Overhang (Forward 1)
Overhang (Forward 2)
1
Promoter fragment
CCCT
AACG
2
RBS fragment
AACG
TATG
3
CDS fragment
TATG
ATCC
4
Terminator fragment
ATCC
GCTG
5
Bakcbone fragment
GCTG
TTGC
Two BsmBI sites were verified on each fragment, and virtual digests were performed on each fragment to confirm 2 pieces and 1 dropout as products of a BsmBI digest.
The assembly modeling wizard was run (DNA concatenation)
The assembly product confirmed successful concatenations (outer BsmBI sites were consumed in Stage 1 to build the part plasmid, and inner BsaI sites are preserved through Stage 1 and then consumed in Stage 2 to build the final construct).
Asimov Kernel
A repository ‘Olga Mineyeva’ was created in Kernel (containing the constructs and a notebook).
The following 13 parts were assembled to recreate the original repressilator (construct ‘repressilator version’):
pTetR
A1 RBS
LacI CDC
L3S2P24 Bacterial Terminator
pLacI
A1RBS
LambdaCI
L3S2P24 Bacterial Terminator
pLambdaCI
A1RBS
TetR
L3S2P24 Bacterial Terminator
Unspecified pIC-SpecR backbone
The resulting construct:
Simulations were performed to confirm the reconstructed repressilator behaviour:
The simulations confirmed that the recreated repressilator behaves the same way as the original.
The first variation of the circuit (construct ‘rep v1’) targeted the circuit topology: pLacI was deleted, so pLac should never be repressed, and so LambdaCI should go high and stay high, and TetR should not be produced, and no oscillations should be possible.
The resulting construct:
A simulation (with the same parameters as before) was performed to confirm the modified repressilator (rep v1) behaviour:
The simulation confirmed LacI is not produced, LambdaCI stays high, but TetR stays near zero, and no oscillations occur.
The second variation of the circuit (construct ‘rep v2’) targeted the production rate of LacI. To see a lower production of LacI, the first RBS was changed to the weakest B0033. Ribosomes should initiate mRNA synthesis less frequently, and so less LacI protein should be produced more slowly. A slower-rising LacI peak is expected, and shifted phases of oscillations.
A simulation (with the same parameters as before) was performed to confirm the modified repressilator (rep v2) behaviour:
The simulation confirmed LacI is accumulating more slowly, and so LambdaCI is more intensely produced and also stays high, and the phases are shifted.
The third variation of the circuit (construct ‘rep v3’) targeted production rate of LacI again. To see a higher production of LacI, the first RBS was changed to the weakest B0033. Ribosomes should initiate mRNA synthesis more frequently and so more LacI protein should be produced faster. A taller LacI peak is expected, and shifted amplitude and phases of the other two repressors are expected.
A simulation (with the same parameters as before) was performed to confirm the modified repressilator (rep v3) behaviour:
The simulation, however, did not confirm the expectations and was identical to the previous one (with the weaker RBS).
The changes should be observed immediately, so changing the parameters to a longer simulation wouldn’t change the result.
The same behaviour observed for both B0030 and B0033 (instead of A1) can be related to the structure of A1 (spacers, special motifs, or length) that is not present in either B0030 or B0033, so does not allow to make LacI production more efficient than with A.
The following screenshots confirm that different RBS parts were used in the simulation, although the simulation results are identical:
Week 7 HW: Genetic Circuit Part II: Neuromorphic Circuits
Part 1: Intracellular Artificial Neural Networks (IANNs)
Advantages IANNs have over traditional genetic circuits
There are several advantages: 1) IANN processing uses graded values and the output is analog, just like continuous biochemical signals being graded and variable, not digital on/off; 2) adaptability of the circuit design as any function can be designed and simulated with changed weights instead of rebuilding circuit topology; 3) weights in IANN correspond directly to a real biological interactions (activation/repression of a gene); 4) IANNs more efficiently use limited cellular resources (weights are tuned instead of gates chained).
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
There is a recent study (Hu, Sun, Qi, 2025) that reveals a mechanistic link between α-Synuclein and mitochondrial stress in Parkinson’s disease and a self-reinforcing pathogenic loop that is involved in the disease progression, and so an IANN could be useful to build on their results.
The paper showed that αSyn (its special NAC domain that targets αSyn to mitochondria) binds the mitochondrial matrix protease ClpP, which normally counteracts α-Syn pathology by stabilizing the native tetramer of α-Syn. This interaction blocks ClpP activity and causes mitochondrial proteotoxic stress, and the loss of ClpP function in turn leads to further αSyn aggregation, which forms a self-reinforcing pathogenic loop. The authors also developed a decoy peptide (CS2) that binds the NAC domain of α-Syn, prevents the interaction between α-Syn and ClpP, and restores ClpP function. Importantly, CS2 reduced α-Syn pathology and neurotoxicity in primary neurons inoculated with αSyn fibrils, PD patient iPSC-derived dopaminergic neurons, and an A53T α-Syn transgenic mouse model.
An IANN can be designed to sense pathogenic processes and produce the decoy peptide as therapy on demand. The circuit is integrates three analog inputs: 1) a proximity sensor (positive weight) that reports interaction between αSyn and ClpP (could be built with one split-reporter fragment at the N- or C-terminus of α-Syn, leaving the NAC domain free, and the other on ClpP; 2) a ClpP-activity reporter (negative weight) that signals functional protease and the system that could still be self-correcting; 3) a mitochondrial-stress reporter (positive weight). The weighted sum passed through a nonlinear activation function drives the expression of the decoy peptide CS2. A functional ClpP freed from α-Syn would restabilize the αSyn tetramer and lower the interaction signal; CS2 output increases with pathology and falls as the decoy peptide frees the ClpP protease.
Perceptron form: y= w₁x₁ - w₂x₂ + w₃x₃ + b; output drives expression of CS2. Low severity of the pathology -> y low -> the output σ(y) low -> low CS2. Rising αSyn–ClpP interaction (x1 increases), poor ClpP activity (x2 decreases), and increasing mitochondrial stress (x3 increases) increase the y and the output -> CS2 expression increases proportionally -> CS2 binds the NAC domain of α-Syn -> α-Syn cannot bind to ClpP -> ClpP disinhibited -> native α-Syn tetramer restabilized -> the proximity sensor signal (x1) decreases -> output decreases.
The limitations include sensor sensitivity and proper organization of the circuit in the correct compartment. Although it’s relatively clear how the sensor should be designed, the sensor would be complex and may not be specific enough to the pathological α-Syn interaction with ClpP. As for compartmentalization, the output must work in the mitochondrial matrix, where the input event occurs, while it would be synthesized outside mitochondria, and so careful engineering is required.
A diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
packaging alternatives: custom-shaped protective packaging for agricultural waste as a replacement for expanded polystyrene foam, Styrofoam (the advantage is energy efficiency because the packaging is produced at room temperature and the packaging is compostable)
leather alternatives: used to produce fashion products (the advantage is no animal-welfare and ecological concerns)
sound-absorbing alternatives: used as acoustic insulation and decorative elements (the advantage is natural appearance, that density and thickness can be tuned during mycelium growth to filter particular frequencies).
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I’d engineer grown-to-shape, biodegradable, single-trip mycelium cradles for museum-to-museum loans and travelling exhibitions. The art or historical objects inside the cradles would be transported as usual, in climate-controlled vehicles. Fingi could be engineered to over-express hydrophobins, uniformly through the material, and to be a poor host for mold. The cradles can compete with the currently used carved Ethafoam because of their custom fit and environmental sustainability. The fungi could also be engineered to pass archival-safety testing.
The cradles could be produced ahead of a planned shipment of art objects; for that, they would be grown against a 3D-printed or cast replica. Also, mycelium can be grown for an artifact-supporting and moving assisting object. Cradles would be custom fit, low cost, low carbon compared to petroleum foam, and compostable.
The advantage would be that fungi already make the material that is fibrous, self-assembling, and has an architecture resistant to load, while bacteria do not. Also, the grown-to-shape feature is native to fungal growth.
First DNA Twist Order
Week 9 HW: Cell-Free Systems
Part A: General and Lecturer-Specific Questions
general questions
Advantages of cell-free protein synthesis (CFPS) over in vivo methods
Mainly, there is no living cell and no cell membrane involved, and so any component of the reaction can be added or removed in the course of the controlled reaction. There are fewer variables to control compared to an experiment and synthesis in a living cell. No transformation process is involved, and so the reaction is faster. Cell-free is more beneficial when a toxic protein is synthesized or a protein that incorporates non-canonical amino acids.
Main components of a cell-free expression system
Main components: cell extract, ribosomes, tRNAs, aminoacyl-tRNA synthetases, factors for elongation, initiation, and release, a template, NTPs, amino acids, energy regeneration system, and a buffer.
Lysate: a lysate is an unfractionated content of the cytoplasm, it supplies ribosomes, tRNAs, aminoacyl-tRNA synthetases, elongation and initiation factors, and the metabolic enzymes.
Buffer: a buffer needed to keep pH for the optimal work of transcription and translation enzymes by counteracting accumulating acidic byproducts of energy metabolism. Potassium Glutamate provides potassium as main cation ions cells are adapted to, and glutamate is E.coli’s major cytoplasmic anion modulating osmolarity, and so both are recreating the native ionic environment. Potassium is needed for ribosomes to fold and assemble (by neutralizing negative phosphate charges on rRNA and allowing ribosomal subunits to not repel) and for translation itself, but too high concentration inhibits translation (promotes dissociation of the ribosomal subunits and initiation complex destabilize) as many of the interactions involved in the process are reversible electrostatic; glutamate unlike chloride is large and it does not interact with protein surfaces with positively charged regions and functional interfaces. Magnesium Glutamate provides magnesium ions as the major cofactors for polymerases, it defines the structure and function of ribosomes (neutralizes negative phosphate charges of rRNA), it stabilises nucleotides and RNA (binds to phosphates of nucleotides, mRNA, tRNA); and glutamate is a safe (large) counter-ion maintaining osmolarity as in potassium glutamate. Potassium phosphate monobasic/dibasic provides an additional buffer system, and so may add the capacity to counteract pH drift from accumulating acetate. Phosphate serves as an energy metabolism substrate and regenerates ATP and nucleotide triphosphates, but too much phosphate can diminish magnesium (as the magnesium salt is insoluble) and inhibit the reaction.
Energy and nucleotides: Ribose replenishes the NTP pool in the system, where nucleoside monophosphates and free guanine are supplied instead of ready-made triphosphates. Glucose is the primary energy source and substrate for glycolysis to generate ATP for transcription and translation. Acetate produced as a byproduct shifts the pH to acidic. AMP, CMP, GMP, UMP are ribonucleoside monophosphates phosphorylated by kinases in the lysate, precursors to ATP, GTP, CTP, UTP for mRNA synthesis. Guanine converts to guanine nucleotides through the purine salvage pathway, it replenishes GTP pool that is consumed in transcription and translation.
Amino Acids: 17 Amino Acid Mix, Tyrosine, and Cysteine are building blocks for proteins. Tyrosine is added separately because of its different solubility (in high pH), and cysteine oxidizes quickly.
Additives: Nicotinamide is a precursor for NAD⁺/NADH indispensable for glycolysis; these are not consumed in transcription or translation. Nuclease Free Water dissolves all the components, is deprived of RNases or DNases that would degrade the template and mRNA.
The importance of energy regeneration and a method for continuous ATP supply
Energy equivalents are indispensable for chemical bonds to form and transcription and translation to occur. A secondary energy sources (phosphoenolpyruvate, creatine phosphate) along with the corresponding kinases can be added to regenerate ATP.
Prokaryotic vs. eukaryotic cell-free systems
Prokaryotic systems give high yields, they are cheaper and more scalable; eukaryotic are slower in their kinetics, more expensive and give lower yields. An antibody fragment or a protein that requires posttranslational modifications of multi-domain folding would require the eukaryotic systems; a small protein not requiring modifications would fit into a prokaryotic system.
An experiment for a membrane protein
Micelles or liposomes can be added into the mix to provide a hydrophobic support to hydrophobic domains of transmembrane proteins so that they don’t aggregate and misfold. An experiment would require selecting (screening) the best hydrophobic supplement, optimizing temperature and cation concentrations to achieve the optimal time for proper folding, optimizing concentrations and membrane insertion factors of chaperons if necessary. Folding needs to be verified with downstream assays involving binding analysis, activity assays, and aggregation assays.
Addressing the low protein yield problem
One reason could be a bad template design, a degraded template, or a non-optimized template; the template can be verified on a gel. Another reason could be not enough energy equivalents present in the mix; energy substrates can be added or the reaction can be supplied with a system that replenishes the substrates and removes byproducts that inhibit the reaction (continuous-exchange). Another reason can be too fast synthesis kinetics that does not allow proper folding or an environment that favors aggregation or degradation; reaction temperature can be lowered, chaperons and protease inhibitors can be added.
question from Kate Adamala
Function: sensing cortisol in wearable cortisol patch for hours-long continuous monitoring. A microfluidic sweat collector built with lipid-vesicle-encapsulated TX-TL with a glucocorticoid receptor would collect sweat and output a reporter fluorescent protein as the result of cortisol binding to its receptor.
Could cell-free Tx/Tl alone do this? Yes, with riboswitches described below, but a patch with a minimal cell system would be more selective and pharmacologically relevant.
Could a genetically modified natural cell do this? Yes
Desired outcome: sweat passively crosses the vesicle bilayer and cortisol binds to the receptor, and reporter synthesis starts.
Components include a membrane and the encapsulated contents. The latter includes a eukaryotic cell-free Tx-Tl system (HeLa lysate) with chaperones, human glucocorticoid receptor, a template for a chromoprotein under a specific promoter, energy equivalents, and kinases.
Source of the Tx/Tl system: HeLa cells.
Communication with the environment: cortisol will passively cross the membrane as it’s hydrophilic.
Measuring the function: reading mNeonGreen fluorescence with single-vesicle imaging, in dose-response experiments addressing the sensor’s dynamic range covering physiological levels of cortisol. Affinity and selectivity measurements are required.
questions from Peter Nguyen
Disposable nitrile gloves with freeze-dried cell-free biosensors that react to cortisol levels by changing colour and supply the information about animal’s physiological state at the time of handling.
How would it work: a glove material contains micro wells with lyophilized cell-free transcription-translation machinery and genetic circuits. A riboswitch responsive to cortisol would trigger the expression of a chromoprotein. Rodent urine or sweat released during animal handling would activate the reaction reservoirs and a reporter in it, causing the colour to change on the glove surface within minutes.
Societal challenge or market need: animal handling that is part of drug testing or neuroscience studies is frequently unstandardized, although it defines animal physiology and behaviour, and efficient techniques to handle animals gently, without triggering a stress response or for quick mitigation of a stress response are available. These gloves would feel a demand for basic science, conservation biologists, and animal welfare auditors for a non-invasive and real-time stress and health assay.
The limitations: 1) a liquid activating the reaction may not be sufficient or optimal; 2) reaction reagents may lose stability over time; 3) the sensitivity for sweat cortisol and urine cortisol needs to be very different, so a large dynamic range has to be factored into the design.
question from Ally Huang
Hippocampal neural stem cells are exceptionally radiation-sensitive; their loss during spaceflight underlies loss of new neurons that is tied to behavioural flexibility, persistent cognitive decline, memory, and mood deficits documented in rodents exposed to X-rays, gamma-rays, and heavy particles. As Mars class missions are planned by multiple companies and governments, and the missions exceed current chronic exposure limits, understanding how cosmic radiation damages neurogenic gene templates is critical for the health of astronauts. Radiation damage to DNA templates encoding neurogenic genes directly reduces protein output, providing an adequate model of the molecular vulnerability driving astronaut cognitive decline, and cell-free TX-TL systems serve as faithful molecular biosensors.
The target: A linear or plasmid template with the human BDNF coding sequence fused to sfGFP.
The target and the challenge: BDNF controls neuronal maturation dependent on synaptic plasticity and neural stem cell proliferation, and its levels are decreased in irradiated hippocampus; however, the integrity of its template has not been studied sufficiently yet. Radiation-induced lesions on the BDNF template in a cell-free model may impair transcription and translation.
Hypothesis: Space radiation will produce dose-dependent damage to the BDNF-sfGFP template that significantly reduces cell-free protein output relative to ground controls, with the longer BDNF–sfGFP construct showing greater vulnerability per reaction than the shorter sfGFP-only control.
Reasoning: Damage probability increases with template length, radiation exposure time, and the mode of pre-exposures, and so longer transcripts are more likely to carry lesions or mutations that block transcription and translation. Demonstrating time and dose-dependent vulnerability of the BDNF template under real space flight conditions would provide evidence that BDNF is affected in space.
Experimental plan: Samples would include the BDNF-sfGFP template, an sfGFP-only template with a length and GC-content matching the first template. Both will be aliquoted as dried DNA. Samples pre-irradiated with low and moderate doses of gamma-rays and samples left on Earth must be included. MiniPCR® thermal cycler can be used to detect template degradation. sfGFP can be quantified with imaging (P51® viewer).
Part B: Individual Final Project
The final project slide was put in the slide deck.
A mock DNA order was placed.
Our group (Abhishek Udawat, Tammy Sisodiya, Nour Abdelrahman, Nurlenden Rihan, and I) focused on targeting increased stability as a goal for engineering the L Protein.
Protein Language Models (ESM2) and the analysis of sequence alignment (BLAST/ ClustalOmega) will identify conserved and variable sites and therefore inform a mutagenesis strategy. Analyses of mutated sequences Alphafold-Multimer will reveal a change in pLDDT, ipTM, which may indicate higher stability of the tail. Engineering Plan
Controllable Induction of Alpha-Synuclein Expression for Modeling Parkinson’s Disease When we model a disease, we introduce pathological changes through genetic manipulations. These changes produce a pathological phenotype, but frequently non-uniformly or too fast to assess possible compensatory mechanisms normally developed in patients. Parkinson’s disease is modeled in patient-derived cells cultured in 3D organoids. As in patients, an amyloid protein, a-syn, misfolds and accumulates in cells. However, months of culture are needed for pathological processes to naturally emerge, and methods that stimulate a-synuclein accumulation have two main problems: they don’t allow temporal control of α-syn load, and they don’t standardize the dose of a-syn per cell.
Subsections of Projects
Group Final Project
Our group (Abhishek Udawat, Tammy Sisodiya, Nour Abdelrahman, Nurlenden Rihan, and I) focused on targeting increased stability as a goal for engineering the L Protein.
Protein Language Models (ESM2) and the analysis of sequence alignment (BLAST/ ClustalOmega) will identify conserved and variable sites and therefore inform a mutagenesis strategy.
Analyses of mutated sequences Alphafold-Multimer will reveal a change in pLDDT, ipTM, which may indicate higher stability of the tail.
Engineering Plan
Review the guidelines.
Isolate the soluble N-terminus as well as the middle part of the protein, non-overlapping with the coat or the replicase sequence.
Evaluate the mutational scan in ESM2 to identify candidate substitutions.
Check BLAST results of sequence alignment (the layout of conserved and variable sites).
Define a specific strategy (e.g., conservation, creating salt bridges to create a helix, etc.).
The strategy that was chosen targeted stabilizing the disordered N-terminal domain by creating salt bridges to introduce an alpha-helix into the disordered domain structure.
For this mutant, I modified the N-terminal domain, aiming to stabilize the disordered domain. I introduced as many charged pairs as possible in the variable sites (changed 4 out of 8 in the N-terminal domain), and additionally changed one conserved site on the left side of the 2nd pair.
For this mutant, I modified the previous sequence (Mutated Sequence 1), aiming to further stabilize the disordered domain.
I introduced 1 more mutation to a variable site to invert the second pair.
Pairs introduced by changing the 5 variable sites: Pair 1 (R7–E11), Pair 2 (R14–E18), Pair 3 (R22–D26)
Soluble N-terminal domain C-terminal domain
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Original Sequence)
R---E LR---E R--- (Mutated Sites)
V V CV V V (Conserved / Variable)
METRFPRQSQETLRSTNERRPRKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Mutated Sequence 2)
AlphafoldServer was used to fold the monomers of Mutated Sequence 1 and Mutated Sequence 2. alfafold2_multimer_v2 was used to fold the multimers. alfafold2_multimer_v2 parameters used:
This sequence was designed to explore whether changing the conserved site (13P->L) was required to achieve the same structure as that of the Mutated Sequence 1. For that, the mutated conserved site of the Sequence 1 was changed back to the original (13L->P).
This sequence was designed to explore whether changing the conserved site (13P->L) was required to achieve the helix as in the Mutated Sequence 2. For that, the mutated conserved site of the Sequence 2 was changed back to the original (13L->P).
Mutant 1 pLDDT=37.6, pTM-0.189, ipTM = 0.127. 3 pairs/bridges introduced, 1 conserved site changed (13P->L), RRR site kept, (1 conserved and 4 variable sites changed)
Mutant 3 pLDDT=43.3, pTM-0.188, ipTM = 0.127. Mutant 1 -> the conserved site mutation reverted (13L->P) (4 variable sites of the Original Sequence changed)
Mutant 2 pLDDT=45.8, pTM-0.187, ipTM = 0.126. 3 pairs/bridges introduced, 1 conserved site changed (13P->L), 2nd pair inverted, no RRR site (1 conserved and 5 variable sites changed)
Mutant 4 pLDDT=37, pTM-0.189, ipTM = 0.127. Mutant 2 -> the conserved site mutation reverted (13L->P) (5 variable sites of the Original Sequence changed)
Further analysis is needed to evaluate whether the mutations affect conserved sites and the folding of the coat protein and the replicase.
Individual Final Project:
Controllable Induction of Alpha-Synuclein Expression for Modeling Parkinson’s Disease
When we model a disease, we introduce pathological changes through genetic manipulations. These changes produce a pathological phenotype, but frequently non-uniformly or too fast to assess possible compensatory mechanisms normally developed in patients.
Parkinson’s disease is modeled in patient-derived cells cultured in 3D organoids. As in patients, an amyloid protein, a-syn, misfolds and accumulates in cells. However, months of culture are needed for pathological processes to naturally emerge, and methods that stimulate a-synuclein accumulation have two main problems: they don’t allow temporal control of α-syn load, and they don’t standardize the dose of a-syn per cell.
In particular, AAV-mediated SNCA overexpression rapidly elevates α-syn levels, or preformed fibril (PFF) seeding introduces exogenous aggregates that template endogenous α-syn misfolding. None of these allows fine-grained temporal control of α-syn load after induction, and none standardizes the per-cell dose across a population, leaving how much α-syn does each cell actually contain and for how long uncontrolled. This limits quantitative phenotype-dose mapping and makes it difficult to isolate which cellular systems fail at which protein loads in individual patient backgrounds.
Overall, it’s currently difficult to model Parkinson’s disease efficiently, i.e., to induce the disease gradually and dynamically isolate which cellular systems become vulnerable. And therefore, the goal of this project was to design a tool that promotes Parkinson’s disease phenotype manifestation in dopaminergic neurons within patient-derived brain organoids by controllable and standardized induction of α-synuclein expression. The tool will enable standardized and accelerated induction of PD phenotypes, allowing probing patient-specific vulnerabilities and testing personalized therapeutic strategies.
Aim 1. Rational circuit design
Design a genetic circuit that drives oscillatory, standardized α-synuclein expression with a built-in OFF switch, from prokaryotic logic to a mammalian-ready construct.
1.1. Design a bacterial prototype of an oscillatory circuit for induced autonomous oscillations with a delayed negative feedback loop (see a prototype on the scheme).
1.2. Redesign the construct with mammalian-compatible parts and predict its dynamics for expression in HEK293 cells.
1.3. Define a delivery and promoter strategy to carry forward the validated construct into iPSC-derived dopaminergic neurons.
Results
The starting point was a principal design of a genetic circuit for autonomous oscillations of α-Synuclein (Figure 1). Once it’s switched on, it would deliver repeated pulses of α-Synuclein with no further intervention needed, and uninterrupted imaging would let drug microscopy platforms watch how cells cope with dosed cyclical protein loads.
A bacterial prototype was then developed as a proof of concept. The prototype circuit included 4 transcription units.
Transcription unit 1 (TU1) initiates the circuit. A small molecule (L-rhamnose, added to media) triggers an activator protein, encoded by TU1, for the oscillations to begin and for positive autoregulation. The promoter carries three operators. The first operator sequence would bind the circuit initiator (a complex consisting of L-rhamnose and endogenously expressed Ras protein). The second operator is designated for the activator protein to sustain it’s own expression after L-rhamnose is gone. The third operator is for the OFF-switch complex (a second small molecule, DAPG, and an OFF-switch protein PhiF expressed with TU4) to bind and terminate the expression of the activator protein.
TU2 produces a simplified but graded output, sfGFP, through a threshold promoter, the target of the activator protein.
TU3 produces delayed negative feedback. It encodes a repressor for the activator protein, a simplified output (just sfGFP) under a threshold promoter that would allow standardization of the output.
TU4 provides an OFF switch. It encodes PhilF protein that binds the second added small molecule DAPG, and the complex binds the corresponding operator in the promoter sequence of TU1 and terminates oscillations by repressing the transcription of the activator protein.
Through the subsequent design, TU1 was split into two, because of a conflict between the three operators that wasn’t factored in at the earlier stage.
The sequences for the units were designed in Kernel and Benchling and codon-optimized.
The DNA design for the two units was attempted as a Twist order. It wasn’t uploaded to the form for a real order as it’s only partial design that was developed at a late stage.
This design, however, couldn’t be carried forward to mammalian cells because the parts depend on bacterial machinery. A simplified inducible transcriptionally-controlled mammalian construct was therefore approached.
Control levels were considered for the mammalian inducible system. Protein-level (switch on the protein with a degron) was ruled out because the mechanism depends on the ubiquitin-proteasome system, which α-syn directly inhibits in synucleinopathy. RNA-level (switch on the mRNA, riboswitches, aptazymes) were also ruled out because α-syn has documented interactions with ribosomes and translation factors. Therefore, transcription-level (switch on the promoter) was chosen.
An inducible construct concept was compared with the existing constructs and with the more advanced mammalian oscillator, yet not designed.
Table 1. Comparison of existing constructs (Constitutive AAV-α-syn, Tet-AAV-α-syn) and more advanced inducible ones, not yet designed (Transcription-level inducible construct, Mammalian oscillator)
Feature
Constitutive AAV-α-syn
Tet-AAV-α-syn
Transcription-level inducible construct
Mammalian oscillator
Developed
Yes
Yes
No
No
Turn on/off
No
Yes (doxycycline)
Yes (ABA/Mandi)
Yes (autonomous)
Dose tunability
No
Yes
Yes
pulse amplitude, set by circuit
Doxycycline confounds
No
Yes
No
No
Per-cell dose standardization
No
No
Yes (miRNA-IFFL)
Yes (built-in via threshold motif)
Turn on/off
Yes
Yes
No
No
Hands-off operation after start
No
No: scheduled dosing
No: scheduled dosing
Yes: runs autonomously
Validated in organoids
Yes
Limited validation
Not yet
Not yet
Engineering risk
Low
Moderate
Moderate
High
A prototype of the simplified inducible circuit was designed.
Specifically, a phytohormone (ABA) triggers the first module, which serves as a sensor. The second module driving the output (alpha-synuclein) also drives miRNA that standardizes the output. The small initiating molecule is then washed away. The two modules are gene fragments to be assembled into a single plasmid with a lentiviral backbone.
Aim 2 Validation in mammalian cells
Experimentally validate the construct in HEK293 cells and build the imaging and delivery infrastructure required for organoid deployment.
2.1. Measure oscillation dynamics, OFF-switch efficiency, and cell-to-cell variability of the construct.
2.2. Refine the neuronal delivery strategy and establish a high-throughput imaging pipeline for organoid-scale experiments.
Aim 3 Integration in human brain organoids
Integrate the validated tool in human brain organoids to model PD and probe patient-specific vulnerabilities at scale.
3.1 Confirm emergence of PD phenotypes.
3.2 Map patient-specific thresholds and pilot therapeutic screening.
A downstream pipeline outline was designed to plan construct delivery into HeLa cells, to modify the construct for neuronal cells, and deliver into IPS cells, as well as measure dose-response, kinetics, and standardization efficiency.
Preliminary governance strategies were discussed in the week 1 homework.
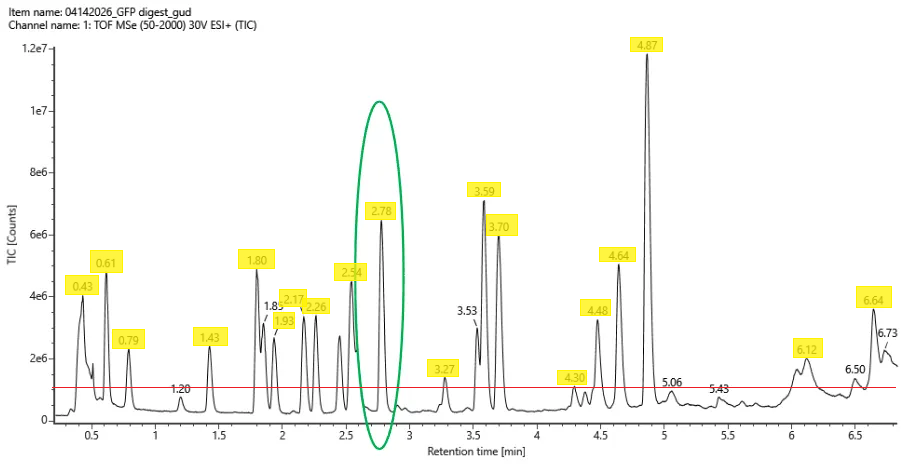
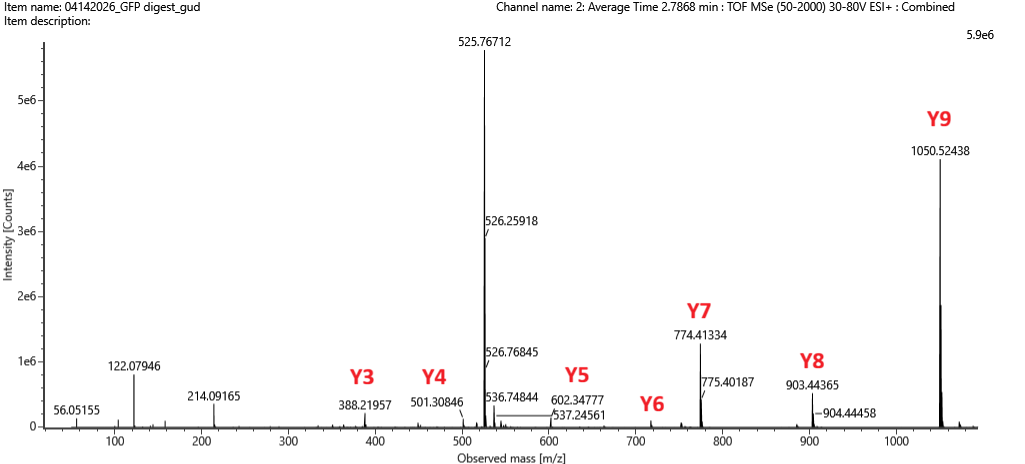
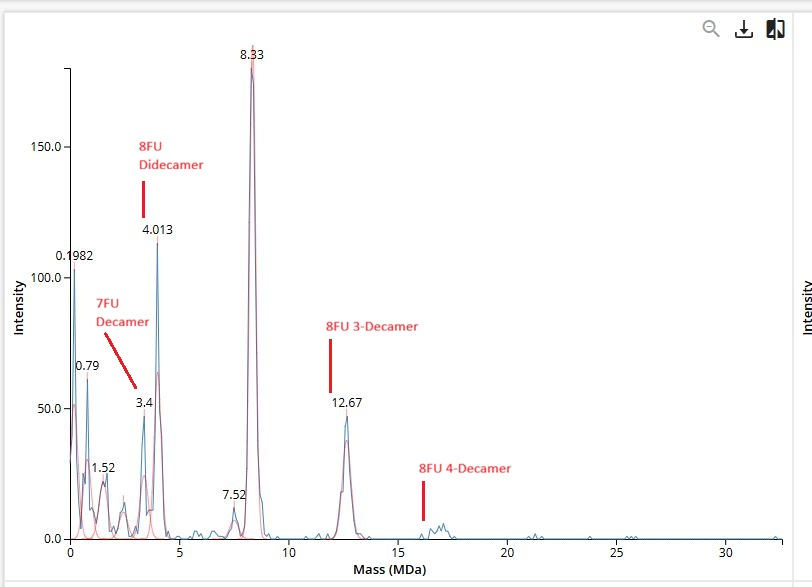
.png)