Our group (Abhishek Udawat, Tammy Sisodiya, Nour Abdelrahman, Nurlenden Rihan, and I) focused on targeting increased stability as a goal for engineering the L Protein.
Protein Language Models (ESM2) and the analysis of sequence alignment (BLAST/ ClustalOmega) will identify conserved and variable sites and therefore inform a mutagenesis strategy. Analyses of mutated sequences Alphafold-Multimer will reveal a change in pLDDT, ipTM, which may indicate higher stability of the tail. Engineering Plan
Controllable Induction of Alpha-Synuclein Expression for Modeling Parkinson’s Disease When we model a disease, we introduce pathological changes through genetic manipulations. These changes produce a pathological phenotype, but frequently non-uniformly or too fast to assess possible compensatory mechanisms normally developed in patients. Parkinson’s disease is modeled in patient-derived cells cultured in 3D organoids. As in patients, an amyloid protein, a-syn, misfolds and accumulates in cells. However, months of culture are needed for pathological processes to naturally emerge, and methods that stimulate a-synuclein accumulation have two main problems: they don’t allow temporal control of α-syn load, and they don’t standardize the dose of a-syn per cell.
Subsections of Projects
Group Final Project
Our group (Abhishek Udawat, Tammy Sisodiya, Nour Abdelrahman, Nurlenden Rihan, and I) focused on targeting increased stability as a goal for engineering the L Protein.
Protein Language Models (ESM2) and the analysis of sequence alignment (BLAST/ ClustalOmega) will identify conserved and variable sites and therefore inform a mutagenesis strategy.
Analyses of mutated sequences Alphafold-Multimer will reveal a change in pLDDT, ipTM, which may indicate higher stability of the tail.
Engineering Plan
Review the guidelines.
Isolate the soluble N-terminus as well as the middle part of the protein, non-overlapping with the coat or the replicase sequence.
Evaluate the mutational scan in ESM2 to identify candidate substitutions.
Check BLAST results of sequence alignment (the layout of conserved and variable sites).
Define a specific strategy (e.g., conservation, creating salt bridges to create a helix, etc.).
The strategy that was chosen targeted stabilizing the disordered N-terminal domain by creating salt bridges to introduce an alpha-helix into the disordered domain structure.
For this mutant, I modified the N-terminal domain, aiming to stabilize the disordered domain. I introduced as many charged pairs as possible in the variable sites (changed 4 out of 8 in the N-terminal domain), and additionally changed one conserved site on the left side of the 2nd pair.
For this mutant, I modified the previous sequence (Mutated Sequence 1), aiming to further stabilize the disordered domain.
I introduced 1 more mutation to a variable site to invert the second pair.
Pairs introduced by changing the 5 variable sites: Pair 1 (R7–E11), Pair 2 (R14–E18), Pair 3 (R22–D26)
Soluble N-terminal domain C-terminal domain
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Original Sequence)
R---E LR---E R--- (Mutated Sites)
V V CV V V (Conserved / Variable)
METRFPRQSQETLRSTNERRPRKHEDYPCRRQQRSST LYVLIFLAIFLS KFTNQLLLSLLEAVIRTVTTLQQLLT (Mutated Sequence 2)
AlphafoldServer was used to fold the monomers of Mutated Sequence 1 and Mutated Sequence 2. alfafold2_multimer_v2 was used to fold the multimers. alfafold2_multimer_v2 parameters used:
This sequence was designed to explore whether changing the conserved site (13P->L) was required to achieve the same structure as that of the Mutated Sequence 1. For that, the mutated conserved site of the Sequence 1 was changed back to the original (13L->P).
This sequence was designed to explore whether changing the conserved site (13P->L) was required to achieve the helix as in the Mutated Sequence 2. For that, the mutated conserved site of the Sequence 2 was changed back to the original (13L->P).
Mutant 1 pLDDT=37.6, pTM-0.189, ipTM = 0.127. 3 pairs/bridges introduced, 1 conserved site changed (13P->L), RRR site kept, (1 conserved and 4 variable sites changed)
Mutant 3 pLDDT=43.3, pTM-0.188, ipTM = 0.127. Mutant 1 -> the conserved site mutation reverted (13L->P) (4 variable sites of the Original Sequence changed)
Mutant 2 pLDDT=45.8, pTM-0.187, ipTM = 0.126. 3 pairs/bridges introduced, 1 conserved site changed (13P->L), 2nd pair inverted, no RRR site (1 conserved and 5 variable sites changed)
Mutant 4 pLDDT=37, pTM-0.189, ipTM = 0.127. Mutant 2 -> the conserved site mutation reverted (13L->P) (5 variable sites of the Original Sequence changed)
Further analysis is needed to evaluate whether the mutations affect conserved sites and the folding of the coat protein and the replicase.
Individual Final Project:
Controllable Induction of Alpha-Synuclein Expression for Modeling Parkinson’s Disease
When we model a disease, we introduce pathological changes through genetic manipulations. These changes produce a pathological phenotype, but frequently non-uniformly or too fast to assess possible compensatory mechanisms normally developed in patients.
Parkinson’s disease is modeled in patient-derived cells cultured in 3D organoids. As in patients, an amyloid protein, a-syn, misfolds and accumulates in cells. However, months of culture are needed for pathological processes to naturally emerge, and methods that stimulate a-synuclein accumulation have two main problems: they don’t allow temporal control of α-syn load, and they don’t standardize the dose of a-syn per cell.
In particular, AAV-mediated SNCA overexpression rapidly elevates α-syn levels, or preformed fibril (PFF) seeding introduces exogenous aggregates that template endogenous α-syn misfolding. None of these allows fine-grained temporal control of α-syn load after induction, and none standardizes the per-cell dose across a population, leaving how much α-syn does each cell actually contain and for how long uncontrolled. This limits quantitative phenotype-dose mapping and makes it difficult to isolate which cellular systems fail at which protein loads in individual patient backgrounds.
Overall, it’s currently difficult to model Parkinson’s disease efficiently, i.e., to induce the disease gradually and dynamically isolate which cellular systems become vulnerable. And therefore, the goal of this project was to design a tool that promotes Parkinson’s disease phenotype manifestation in dopaminergic neurons within patient-derived brain organoids by controllable and standardized induction of α-synuclein expression. The tool will enable standardized and accelerated induction of PD phenotypes, allowing probing patient-specific vulnerabilities and testing personalized therapeutic strategies.
Aim 1. Rational circuit design
Design a genetic circuit that drives oscillatory, standardized α-synuclein expression with a built-in OFF switch, from prokaryotic logic to a mammalian-ready construct.
1.1. Design a bacterial prototype of an oscillatory circuit for induced autonomous oscillations with a delayed negative feedback loop (see a prototype on the scheme).
1.2. Redesign the construct with mammalian-compatible parts and predict its dynamics for expression in HEK293 cells.
1.3. Define a delivery and promoter strategy to carry forward the validated construct into iPSC-derived dopaminergic neurons.
Results
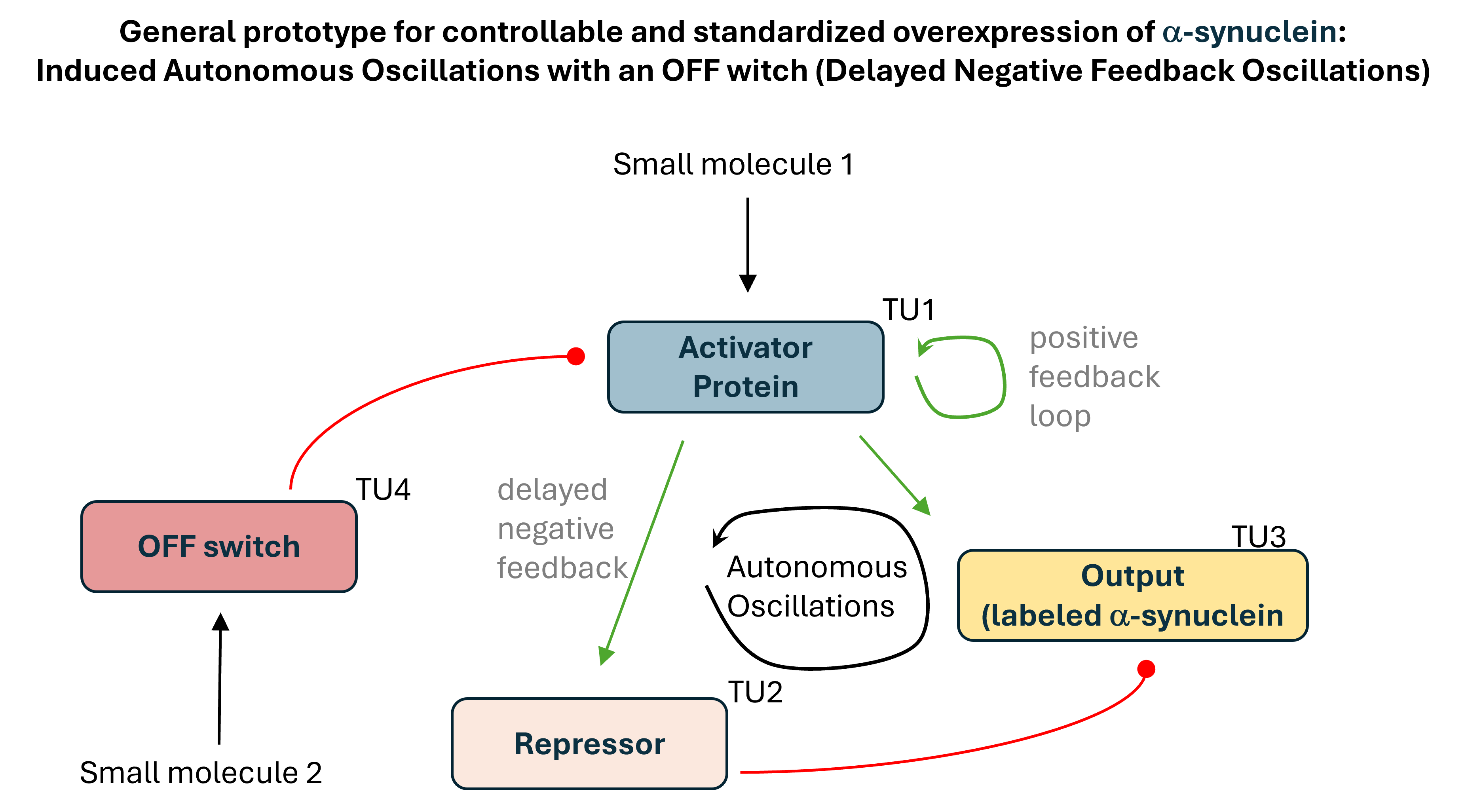
The starting point was a principal design of a genetic circuit for autonomous oscillations of α-Synuclein (Figure 1). Once it’s switched on, it would deliver repeated pulses of α-Synuclein with no further intervention needed, and uninterrupted imaging would let drug microscopy platforms watch how cells cope with dosed cyclical protein loads.
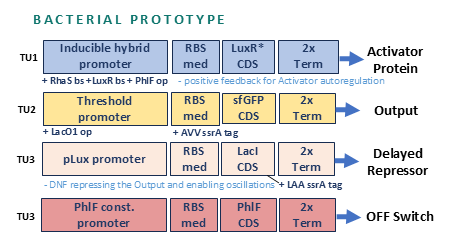
A bacterial prototype was then developed as a proof of concept. The prototype circuit included 4 transcription units.
Transcription unit 1 (TU1) initiates the circuit. A small molecule (L-rhamnose, added to media) triggers an activator protein, encoded by TU1, for the oscillations to begin and for positive autoregulation. The promoter carries three operators. The first operator sequence would bind the circuit initiator (a complex consisting of L-rhamnose and endogenously expressed Ras protein). The second operator is designated for the activator protein to sustain it’s own expression after L-rhamnose is gone. The third operator is for the OFF-switch complex (a second small molecule, DAPG, and an OFF-switch protein PhiF expressed with TU4) to bind and terminate the expression of the activator protein.
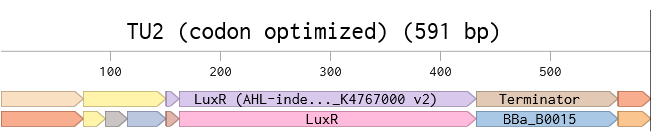
TU2 produces a simplified but graded output, sfGFP, through a threshold promoter, the target of the activator protein.
TU3 produces delayed negative feedback. It encodes a repressor for the activator protein, a simplified output (just sfGFP) under a threshold promoter that would allow standardization of the output.
TU4 provides an OFF switch. It encodes PhilF protein that binds the second added small molecule DAPG, and the complex binds the corresponding operator in the promoter sequence of TU1 and terminates oscillations by repressing the transcription of the activator protein.
Through the subsequent design, TU1 was split into two, because of a conflict between the three operators that wasn’t factored in at the earlier stage.
The sequences for the units were designed in Kernel and Benchling and codon-optimized.
The DNA design for the two units was attempted as a Twist order. It wasn’t uploaded to the form for a real order as it’s only partial design that was developed at a late stage.
This design, however, couldn’t be carried forward to mammalian cells because the parts depend on bacterial machinery. A simplified inducible transcriptionally-controlled mammalian construct was therefore approached.
Control levels were considered for the mammalian inducible system. Protein-level (switch on the protein with a degron) was ruled out because the mechanism depends on the ubiquitin-proteasome system, which α-syn directly inhibits in synucleinopathy. RNA-level (switch on the mRNA, riboswitches, aptazymes) were also ruled out because α-syn has documented interactions with ribosomes and translation factors. Therefore, transcription-level (switch on the promoter) was chosen.
An inducible construct concept was compared with the existing constructs and with the more advanced mammalian oscillator, yet not designed.
Table 1. Comparison of existing constructs (Constitutive AAV-α-syn, Tet-AAV-α-syn) and more advanced inducible ones, not yet designed (Transcription-level inducible construct, Mammalian oscillator)
Feature
Constitutive AAV-α-syn
Tet-AAV-α-syn
Transcription-level inducible construct
Mammalian oscillator
Developed
Yes
Yes
No
No
Turn on/off
No
Yes (doxycycline)
Yes (ABA/Mandi)
Yes (autonomous)
Dose tunability
No
Yes
Yes
pulse amplitude, set by circuit
Doxycycline confounds
No
Yes
No
No
Per-cell dose standardization
No
No
Yes (miRNA-IFFL)
Yes (built-in via threshold motif)
Turn on/off
Yes
Yes
No
No
Hands-off operation after start
No
No: scheduled dosing
No: scheduled dosing
Yes: runs autonomously
Validated in organoids
Yes
Limited validation
Not yet
Not yet
Engineering risk
Low
Moderate
Moderate
High
A prototype of the simplified inducible circuit was designed.
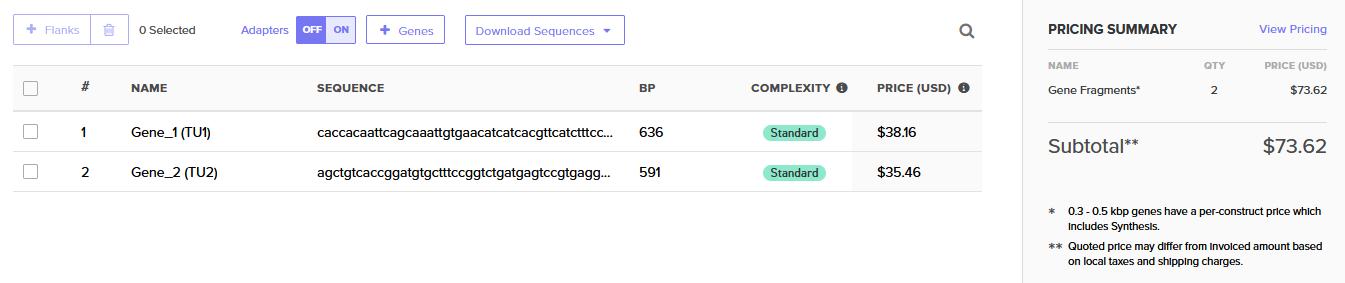
Specifically, a phytohormone (ABA) triggers the first module, which serves as a sensor. The second module driving the output (alpha-synuclein) also drives miRNA that standardizes the output. The small initiating molecule is then washed away. The two modules are gene fragments to be assembled into a single plasmid with a lentiviral backbone.
Aim 2 Validation in mammalian cells
Experimentally validate the construct in HEK293 cells and build the imaging and delivery infrastructure required for organoid deployment.
2.1. Measure oscillation dynamics, OFF-switch efficiency, and cell-to-cell variability of the construct.
2.2. Refine the neuronal delivery strategy and establish a high-throughput imaging pipeline for organoid-scale experiments.
Aim 3 Integration in human brain organoids
Integrate the validated tool in human brain organoids to model PD and probe patient-specific vulnerabilities at scale.
3.1 Confirm emergence of PD phenotypes.
3.2 Map patient-specific thresholds and pilot therapeutic screening.
A downstream pipeline outline was designed to plan construct delivery into HeLa cells, to modify the construct for neuronal cells, and deliver into IPS cells, as well as measure dose-response, kinetics, and standardization efficiency.
Preliminary governance strategies were discussed in the week 1 homework.