Week 2 HW: DNA Read/Write/Edit
Part 1: Benchling & In-silico Gel Art
For this part, I created an account on Benchling –> imported the Lambda DNA –> played around with 7 restriction enzymes to get the gel patterns from the digests. Here’s the enzymes I used and the resulting gel pattern of digests:
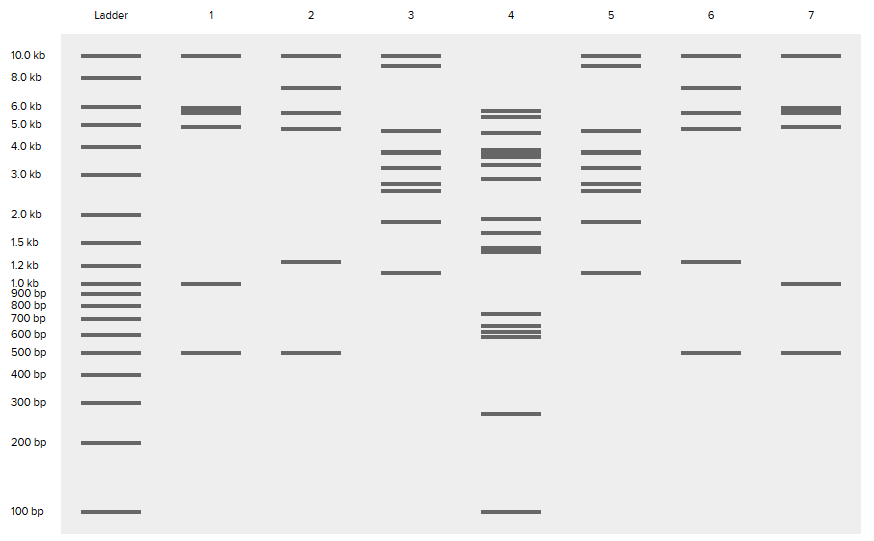
(I envisioned creating a dragonfly, so if you look closely I hope you can see it 😄)
Part 3: DNA Design Challenge
3.1. Choose your protein
The protein I chose for this assignment is the Tardigrade Dsup (Damage Suppressor), which is a unique DNA-binding protein produced only by tardigrades (Phylum Tardgrada) that enables them to survive ROS and radiation-induced DNA damage (Hashimoto et.al 2016). It’s one of the key reasons these tiny organisms can tolerate the extreme conditions of space (Jonsson et.al 2008). What I found especially fascinating is that when Dsup was expressed in human cultured cells (HEK293), it also reduced X-ray- and hydrogen-peroxide-induced DNA damage by lowering DNA breaks and fragmentation. Furthermore, a lot of exciting studies on Dsup-based gene and mRNA delivery approaches as potential radioprotective therapies, which are especially relevant to cancer treatment (Cui et. al. 2025). The (UniProt sequence)[https://www.uniprot.org/uniprotkb/P0DOW4/entry] of Dsup is pasted below:
MASTHQSSTEPSSTGKSEETKKDASQGSGQDSKNVTVTKGTGSSATSAAIVKTGGSQGKDSSTTAGSSSTQGQKFSTTPTDPKTFSSDQKEKSKSPAKEVPSGGDSKSQGDTKSQSDAKSSGQSQGQSKDSGKSSSDSSKSHSVIGAVKDVVAGAKDVAGKAVEDAPSIMHTAVDAVKNAATTVKDVASSAASTVAEKVVDAYHSVVGDKTDDKKEGEHSGDKKDDSKAGSGSGQGGDNKKSEGETSGQAESSSGNEGAAPAKGRGRGRPPAAAKGVAKGAAKGAAASKGAKSGAESSKGGEQSSGDIEMADASSKGGSDQRDSAATVGEGGASGSEGGAKKGRGRGAGKKADAGDTSAEPPRRSSRLTSSGTGAGSAPAAAKGGAKRAASSSSTPSNAKKQATGGAGKAAATKATAAKSAASKAPQNGAGAKKKGGKAGGRKRK
References
- Hashimoto, T., Horikawa, D. D., Saito, Y., Kuwahara, H., Kozuka-Hata, H., Shin-I, T., Minakuchi, Y., Ohishi, K., Motoyama, A., Aizu, T., Enomoto, A., Kondo, K., Tanaka, S., Hara, Y., Koshikawa, S., Sagara, H., Miura, T., Yokobori, S. I., Miyagawa, K., Suzuki, Y., … Kunieda, T. (2016). Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nature communications, 7, 12808. https://doi.org/10.1038/ncomms12808
- Jönsson, K. I., Rabbow, E., Schill, R. O., Harms-Ringdahl, M., & Rettberg, P. (2008). Tardigrades survive exposure to space in low Earth orbit. Current Biology, 18(17), R729–R731. https://doi.org/10.1016/j.cub.2008.06.048
- Cui, Z., Lin, C., Zhao, H., & Wang, X. (2025). Radioprotection redefined: drug discovery at the intersection of tardigrade biology and translational pharmacology. Frontiers in pharmacology, 16, 1713914. https://doi.org/10.3389/fphar.2025.1713914
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Using the “Reverse Translate” tool of the Sequence Manipulation Suite (https://www.bioinformatics.org/sms2/rev_trans.html)
atggcgagcacccatcagagcagcaccgaaccgagcagcaccggcaaaagcgaagaaacc aaaaaagatgcgagccagggcagcggccaggatagcaaaaacgtgaccgtgaccaaaggc accggcagcagcgcgaccagcgcggcgattgtgaaaaccggcggcagccagggcaaagat agcagcaccaccgcgggcagcagcagcacccagggccagaaatttagcaccaccccgacc gatccgaaaacctttagcagcgatcagaaagaaaaaagcaaaagcccggcgaaagaagtg ccgagcggcggcgatagcaaaagccagggcgataccaaaagccagagcgatgcgaaaagc agcggccagagccagggccagagcaaagatagcggcaaaagcagcagcgatagcagcaaa agccatagcgtgattggcgcggtgaaagatgtggtggcgggcgcgaaagatgtggcgggc aaagcggtggaagatgcgccgagcattatgcataccgcggtggatgcggtgaaaaacgcg gcgaccaccgtgaaagatgtggcgagcagcgcggcgagcaccgtggcggaaaaagtggtg gatgcgtatcatagcgtggtgggcgataaaaccgatgataaaaaagaaggcgaacatagc ggcgataaaaaagatgatagcaaagcgggcagcggcagcggccagggcggcgataacaaa aaaagcgaaggcgaaaccagcggccaggcggaaagcagcagcggcaacgaaggcgcggcg ccggcgaaaggccgcggccgcggccgcccgccggcggcggcgaaaggcgtggcgaaaggc gcggcgaaaggcgcggcggcgagcaaaggcgcgaaaagcggcgcggaaagcagcaaaggc ggcgaacagagcagcggcgatattgaaatggcggatgcgagcagcaaaggcggcagcgat cagcgcgatagcgcggcgaccgtgggcgaaggcggcgcgagcggcagcgaaggcggcgcg aaaaaaggccgcggccgcggcgcgggcaaaaaagcggatgcgggcgataccagcgcggaa ccgccgcgccgcagcagccgcctgaccagcagcggcaccggcgcgggcagcgcgccggcg gcggcgaaaggcggcgcgaaacgcgcggcgagcagcagcagcaccccgagcaacgcgaaa aaacaggcgaccggcggcgcgggcaaagcggcggcgaccaaagcgaccgcggcgaaaagc gcggcgagcaaagcgccgcagaacggcgcgggcgcgaaaaaaaaaggcggcaaagcgggc ggccgcaaacgcaaa
3.3. Codon optimization
When expressing a foreign gene in a host organism (chassis), it’s important to consider codon usage bias, which refers to the host’s preference of translating a synonymous codon for an amino acid over one that is in the genetic sequence, which is not frequently translated by the host. This is where codon optimization comes into play, by replacing some codons with those the host prefers to translate (but not changing the amino acid sequence), to ensure that the host organism optimally translates our protein of interest.
Supposing I want to acquire an engineered Dsup protein for therapeutic use, then I would choose [E.coli] as the chassis/delivery system. E.coli is one of the standard and most reliable chassis in synthetic biology - thoroughly studied, well characterized, and it grows rapidly in large volumes inexpensively.
Using the Codon Optimization Tool of [VectorBuilder]:
I get the following improved sequence: 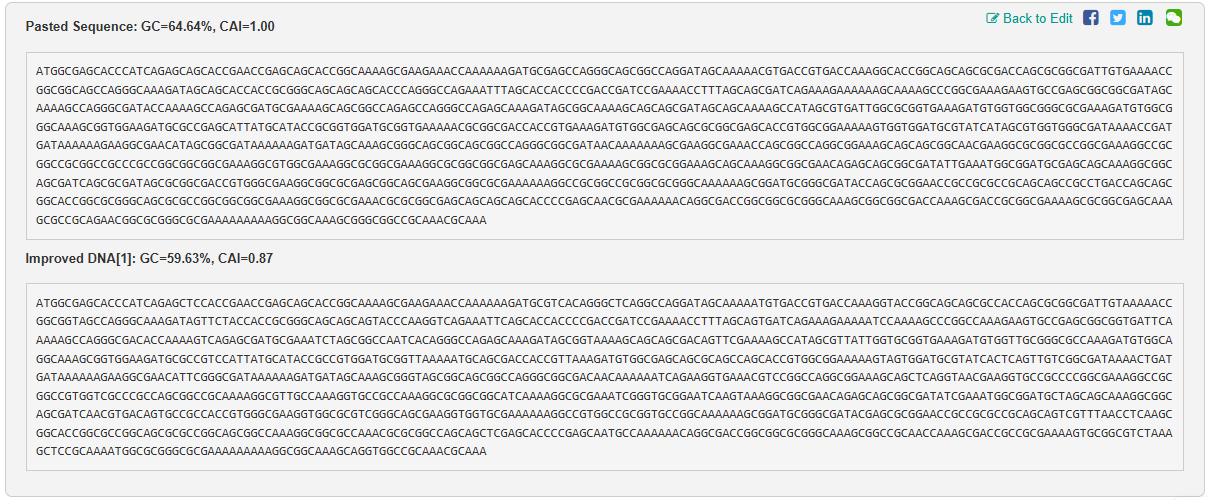
ATGGCGAGCACCCATCAGAGCTCCACCGAACCGAGCAGCACCGGCAAAAGCGAAGAAACCAAAAAAGATGCGTCACAGGGCTCAGGCCAGGATAGCAAAAATGTGACCGTGACCAAAGGTACCGGCAGCAGCGCCACCAGCGCGGCGATTGTAAAAACCGGCGGTAGCCAGGGCAAAGATAGTTCTACCACCGCGGGCAGCAGCAGTACCCAAGGTCAGAAATTCAGCACCACCCCGACCGATCCGAAAACCTTTAGCAGTGATCAGAAAGAAAAATCCAAAAGCCCGGCCAAAGAAGTGCCGAGCGGCGGTGATTCAAAAAGCCAGGGCGACACCAAAAGTCAGAGCGATGCGAAATCTAGCGGCCAATCACAGGGCCAGAGCAAAGATAGCGGTAAAAGCAGCAGCGACAGTTCGAAAAGCCATAGCGTTATTGGTGCGGTGAAAGATGTGGTTGCGGGCGCCAAAGATGTGGCAGGCAAAGCGGTGGAAGATGCGCCGTCCATTATGCATACCGCCGTGGATGCGGTTAAAAATGCAGCGACCACCGTTAAAGATGTGGCGAGCAGCGCAGCCAGCACCGTGGCGGAAAAAGTAGTGGATGCGTATCACTCAGTTGTCGGCGATAAAACTGATGATAAAAAAGAAGGCGAACATTCGGGCGATAAAAAAGATGATAGCAAAGCGGGTAGCGGCAGCGGCCAGGGCGGCGACAACAAAAAATCAGAAGGTGAAACGTCCGGCCAGGCGGAAAGCAGCTCAGGTAACGAAGGTGCCGCCCCGGCGAAAGGCCGCGGCCGTGGTCGCCCGCCAGCGGCCGCAAAAGGCGTTGCCAAAGGTGCCGCCAAAGGCGCGGCGGCATCAAAAGGCGCGAAATCGGGTGCGGAATCAAGTAAAGGCGGCGAACAGAGCAGCGGCGATATCGAAATGGCGGATGCTAGCAGCAAAGGCGGCAGCGATCAACGTGACAGTGCCGCCACCGTGGGCGAAGGTGGCGCGTCGGGCAGCGAAGGTGGTGCGAAAAAAGGCCGTGGCCGCGGTGCCGGCAAAAAAGCGGATGCGGGCGATACGAGCGCGGAACCGCCGCGCCGCAGCAGTCGTTTAACCTCAAGCGGCACCGGCGCCGGCAGCGCGCCGGCAGCGGCCAAAGGCGGCGCCAAACGCGCGGCCAGCAGCTCGAGCACCCCGAGCAATGCCAAAAAACAGGCGACCGGCGGCGCGGGCAAAGCGGCCGCAACCAAAGCGACCGCCGCGAAAAGTGCGGCGTCTAAAGCTCCGCAAAATGGCGCGGGCGCGAAAAAAAAAGGCGGCAAAGCAGGTGGCCGCAAACGCAAA
3.4. You have a sequence! Now what? The next step is to express this sequence in a host (transcription) and get the protein (translation). Since I optimized the sequence for E. coli, I would use an E. coli expression system, and the cell’s transcription/translation machinery will produce the protein. I will achieve this in the following steps:
- Constructing a recombinant vector (Dsup gene + expression plasmid) via Gibson Assembly. It will comprise of the Dsup gene, a promoter, ribosome binding site, selectable marker (antibiotic resistance) genes, and a His-tag sequence.
- Transformation of E.coli cells by introducing the vector. Cells containing the plasmid are selected using antibiotic media.
- Transcription: inside the bacterial cell, RNA polymerase recognizes the promoter on the plasmid and transcribes the Dsup gene into mRNA.
- Translation: ribosomes bind the ribosome binding site on the mRNA and translate codons into amino acids using tRNAs. The growing polypeptide folds into the functional Dsup protein inside the cell.
- The protein is purified and obtained by lysing the cells, and passing the cell lysates through affinity chromatography with a His-tag. Since the Dsup gene was designed with a His-tag sequence in the vector, the protein will also have it, and this will help it to be separated (purified).
Part 4: Prepare a Twist DNA Synthesis Order
Step 1) Create a Twist account and a Benchling account ✅
Step 2) Build Your DNA Insert Sequence I have used my codon-optimized dsup sequence for an E.coli expression system. On Benchling, I sequentially inserted the following sequences (mentioned in the HW) into the “Create DNA / RNA sequence” tab:
- Promoter (e.g. BBa_J23106): TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC
- RBS (e.g. BBa_B0034 with spacers for optimal expression): CATTAAAGAGGAGAAAGGTACC
- Start Codon: ATG
- Coding Sequence (my codon optimized dsup for a protein of interest)
- 7x His Tag (to enable protein purification from E. coli): CATCACCATCACCATCATCAC
- Stop Codon: TAA
- Terminator (e.g. BBa_B0015): CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
 During the process of annotating:
During the process of annotating:
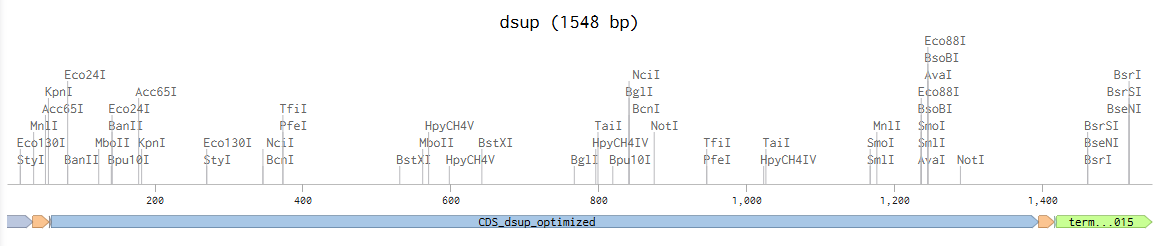
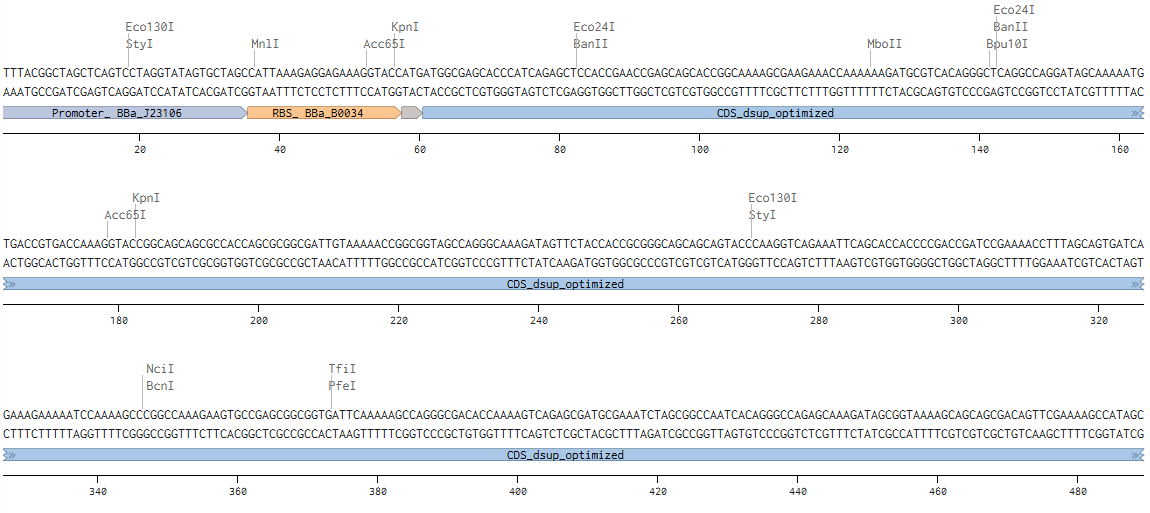 Linear Map (link: https://benchling.com/s/seq-FJPRVyjCrav3opFmzzZ2?m=slm-MQBEBBrWm3w4d8JMFsGv):
Linear Map (link: https://benchling.com/s/seq-FJPRVyjCrav3opFmzzZ2?m=slm-MQBEBBrWm3w4d8JMFsGv):
Step 3) On Twist Biosciences:
Selecting “Genes” and then “Clonal Genes” Option

Importing the Benchling Casette Sequence (FASTA)
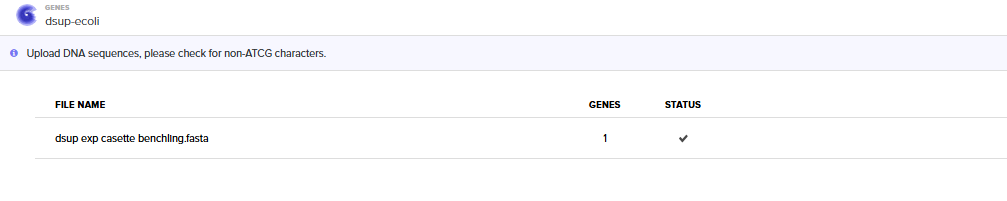
Choosing Twist Vector
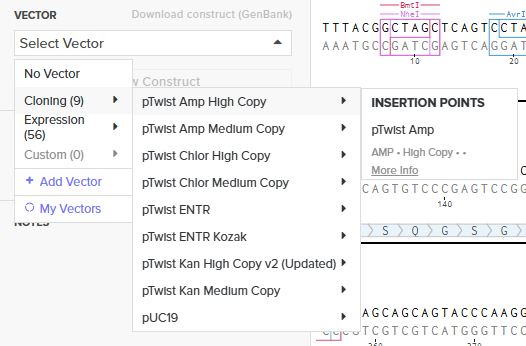
Downloading GenBank sequence of the construct
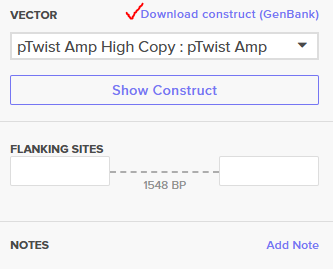
Here is what the recombinant vector looks like in Twist:
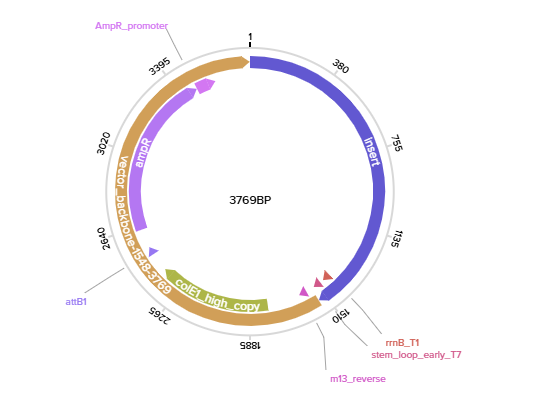
Step 4) Back to Benchling:
After importing the Twist construct (GenBank file), this is the final plasmid + expression casette:
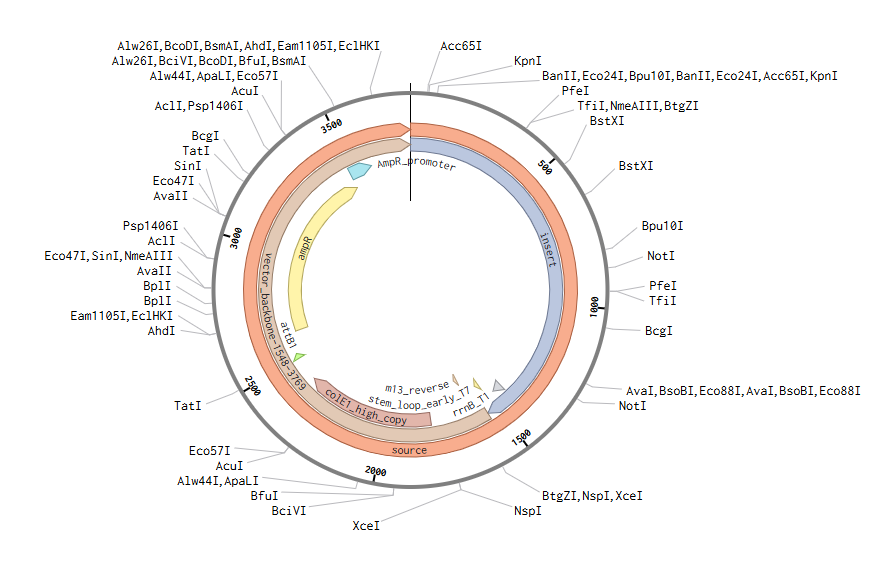
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would want to sequence the tardigrade dsup gene (damage suppressor). The Dsup protein binds and physically shields DNA from hydroxyl radicals generated by radiation and oxidative stress. This makes it super interesting for some of my envisioned projects on:
- Space biology (could it be used to protect astronauts from space radiation? maybe in engineered probiotics or human gene therapy for long-term missions?)
- Radioprotectant for cancer therapy (protecting healthy cells from radiotherapy?)
- Biomanufacturing (dsup-engineered human cells that are radiation-resistant?)
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would choose Oxford Nanopore sequencing (3rd generation) because it provides very long read lengths and direct sequencing of DNA withough any amplification, which is ideal for verifying complex constructs like the Dsup gene and related assemblies. Nanopore sequencing reduces bias and enables rapid real-time data generation. However, it has a higher raw error rate compared to short-read sequencing (1st + 2nd gen) and requires computational base-calling, so I would use it primarily for assembly confirmation and structure verification.
The essential steps (I used ChatGPT to simplify this) are given below:
- Library preparation: attaching specific sequence adapters to the DNA so it can interact with pores
- DNA passes through protein nanopores: each single-stranded DNA molecule travels through a tiny biological pore embedded in a membrane
- Electrical signal detection: as each nucleotide passes through the pore, it causes a distinctive change in ionic current (measured in real-time)
- Base-calling: signal data is processed by software to convert electrical signals into a DNA sequence
- Data analysis: aligning the reads to the intended design to confirm correct assembly
Output: set of DNA read files (FASTQ) containing the nucleotide sequences of DNA fragments + associated quality scores. These reads can be aligned to the designed Dsup sequence to verify that the synthesized gene matches the intended design. Other outputs like consensus sequences and variant files summarize the final DNA sequence and any differences.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would synthesize a fusion gene encoding a chimeric Dsup–HMGN protein for expression in human cells. Dsup (from tardigrades) protects DNA from radiation damage, but since it is not a human protein it may not localize efficiently in human chromatin and could trigger immune responses. To improve this, I would fuse Dsup to the nucleosome-binding domain of a human HMGN protein, which naturally binds chromatin in human cells. HMGN are present in vertebrates, including humans, and have some sequence similarity with dsup (Chavez et al, 2019).
The goal would be to create a protein that:
- targets human DNA effectively
- protects DNA during radiation exposure
In terms of potential applications, this could protect healthy tissue during radiotherapy and astronauts from space radiation.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use phosphoramidite chemical DNA synthesis followed by enzymatic assembly (workflow used by Twist Bioscience). Why?
- My construct (~1.5–2 kb fusion gene) is too long to synthesize in one piece
- Chemical synthesis reliably produces short oligos (≈150–200 bp)
- These fragments can then be assembled into the full gene using PCR assembly/Gibson assembly
- It allows codon optimization and seamless fusion of Dsup + linker + HMGN
So my worflow would look like this:
- Design codon-optimized sequence (human expression)
- Chemically synthesize short DNA oligos
- Assemble into full gene
- Clone into plasmid
- Verify sequence using Oxford Nanopore sequencing
Why Oxford Nanopore?
- Can read the entire plasmid in one read (long-read sequencing)
- Confirms correct assembly and orientation
- Detects large insertions, deletions, or rearrangements that Sanger may miss
Essential steps of the chosen sequencing method (Oxford Nanopore; repeated from above)
- Library preparation: attaching specific sequence adapters to the DNA so it can interact with pores
- DNA passes through protein nanopores: each single-stranded DNA molecule travels through a tiny biological pore embedded in a membrane
- Electrical signal detection: as each nucleotide passes through the pore, it causes a distinctive change in ionic current (measured in real-time)
- Base-calling: signal data is processed by software to convert electrical signals into a DNA sequence
- Data analysis: aligning the reads to the intended design to confirm correct assembly
Limitations of the sequencing method in terms of speed, accuracy, scalability: (I consulted the internet and ChatGPT for this)
Accuracy
- Raw read accuracy lower than short-read sequencing
- Homopolymers (AAAAA…) are error-prone
- Usually solved by high coverage consensus
Speed
- Fast setup and real-time results
- But computational basecalling adds analysis time
Scalability
- Excellent for plasmids and constructs
- Not ideal for very high-throughput small variant detection compared to Illumina
[References]
- Carolina Chavez, Grisel Cruz-Becerra, Jia Fei, George A Kassavetis, James T Kadonaga (2019) The tardigrade damage suppressor protein binds to nucleosomes and protects DNA from hydroxyl radicals eLife 8:e47682 https://doi.org/10.7554/eLife.47682
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would focus on editing human genomes to improve health, increase disease resistance, and extend healthy lifespan. With my background in cancer research and interest in space biology, I’m especially intrigued by the tardigrade Dsup protein, which protects DNA from radiation damage. Understanding or applying similar mechanisms could help humans better withstand cosmic radiation during space travel and even serve as radioprotective strategies in cancer therapy, all while enhancing our natural resilience to disease and aging.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-Cas9 to edit human genomes because it is precise, efficient, and versatile, allowing targeted insertion of protective genes like the tardigrade Dsup protein to enhance DNA resilience, disease resistance, and longevity.
- How does your technology of choice edit DNA? What are the essential steps? (Used ChatGPT to simplify and refine concepts)
CRISPR uses a guide RNA (gRNA) to direct the Cas9 enzyme to a specific DNA sequence, where Cas9 creates a double-strand break. The cell then repairs the break via homology-directed repair (HDR), allowing precise insertion of a donor DNA sequence, such as my codon-optimized Dsup gene.
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Design Steps:
- Identify the target DNA sequence in the human genome.
- Design a specific guide RNA complementary to the target.
- If introducing a new gene (e.g., Dsup), design a donor DNA template with homology arms for HDR.
Inputs Needed:
- gRNA sequence targeting the genomic site.
- Cas9 enzyme or Cas9-expressing plasmid.
- Donor DNA template (for dsup gene).
- Human cells for editing, cultured in vitro.
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
- HDR efficiency can be low, especially in non-dividing cells.
- Off-target effects may cause unintended mutations.
- Delivering CRISPR components into human tissues is challenging.
- Ethical and safety concerns limit clinical applications in humans.