Omama Syed — HTGAA Spring 2026
About me
Cancer researcher 🧫 🧬 & aspiring space synthetic biologist 🌑 🌠 🌌
Cancer researcher 🧫 🧬 & aspiring space synthetic biologist 🌑 🌠 🌌
Week 1 HW: Principles and Practices
Week 1 Assignment Q1) First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about. Engineered Cyanobacteria as Oxygen-Generating Systems for Extraterrestrial Environments
Week 2 HW: DNA Read/Write/Edit
Part 1: Benchling & In-silico Gel Art For this part, I created an account on Benchling –> imported the Lambda DNA –> played around with 7 restriction enzymes to get the gel patterns from the digests. Here’s the enzymes I used and the resulting gel pattern of digests:


Engineered Cyanobacteria as Oxygen-Generating Systems for Extraterrestrial Environments
With renewed interest in space exploration following missions such as NASA’s Artemis II program [1], I can’t help but wonder how far we have come in space research and what else we have yet to accomplish in leaving the Earth and settling on other planets. Beyond the immediate and well-studied challenges of spaceflight, such as radiation exposure, bone loss, and other risks to human health in space [2], I have often found myself thinking about a more distant future: what would it be like to stand on the surface of another planet? And could humans one day (hopefully soon!) be able to breathe there without the constant protection of a spacesuit? Through this course, and particularly through learning more about synthetic biology, that once-distant idea feels less like science fiction and more like a long-term scientific possibility. For this assignment, I therefore propose the idea of engineering cyanobacteria-based biological oxygen generation systems for use on the Moon, Mars, or other extraterrestrial bodies. These organisms would be genetically optimized to carry out photosynthesis under extreme conditions—such as high radiation, low pressure, limited water availability, large temperature fluctuations, and reduced light—converting locally available carbon dioxide and resources into oxygen-rich, breathable air.
Cyanobacteria are photosynthetic bacteria that played a foundational role in shaping life on Earth – 2.5 billion years ago they rapidly produced all the oxygen in an event called the Great Oxygenation Event and birthing complex life. Today, they are found across a range of environments, from marine bodies, rocks, and soil environments to extreme temperature regions such as hot springs and the Arctic [3]. For these qualities, they are viewed as promising candidates for space-related applications. Several studies have already shown that cyanobacteria can survive and maintain photosynthetic and nitrogen-fixing activity in simulated Martian environments, highlighting their potential role in future life-support or terraforming strategies [4].
A core bottleneck in long-duration space exploration has been sustainable life-support. Current systems on space stations rely on mechanical methods such as electrolysis, or on oxygen supplied from Earth—all of which are costly, resource-intensive, and difficult to scale [5]. While recent technologies like NASA’s MOXIE experiment have demonstrated that oxygen can be chemically extracted from the Martian atmosphere, these systems remain energy-intensive and limited in output [6]. By contrast, a biologically driven approachcould offer a more self-sustaining and scalable alternative, potentially reducing mission risk and supporting longer-term human presence beyond Earth.
At the same time, intentionally deploying engineered life beyond Earth raises some tough ethical, ecological, and governance challenges. Living systems can evolve, spread, and behave unpredictably, and once released, they may irreversibly alter extraterrestrial environments. For this reason, governance cannot be treated as an afterthought, but must be considered a central design constraint in the development of extraterrestrial bioengineering technologies.
To ensure that extraterrestrial bioengineering contributes to an ethical future, governance must balance innovation with planetary protection, safety, equity, and long-term responsibility. I propose three governance goals, most of which are directly aligned with COSPAR’s planetary protection policies:
Goal 1: Prevent Biological Harm and Irreversible Planetary Contamination
Sub-goals:
Goal 2: Ensure Responsible Innovation Through Transparency, Oversight, and Accountability
Sub-goals:
Goal 3: Promote Equity and Collective Stewardship of Extraterrestrial Environments
Sub-goals:
Action 1: Risk Review Before Using Engineered Organisms in Space
Purpose
At the moment, planetary protection rules are mainly designed to prevent accidental contamination from Earth microbes, but they do not fully account for situations where scientists intentionally design organisms to survive and function beyond Earth. Before engineered cyanobacteria are used on the Moon, Mars, or elsewhere, there should be a required risk review to ask straightforward but important questions: What does the organism do? Where will it be used? What could go wrong? And how would those risks be managed? The goal is to make sure decisions are made carefully and responsibly, rather than being rushed by competition or technological excitement.
Design
Assumptions
Risks of Failure & “Success”
Action 2: Containment-First Deployment with Reversible Design Requirements
Purpose
Because living systems can evolve and spread in unexpected ways, releasing engineered organisms directly into a planetary environment carries serious risks. Therefore, early uses of engineered cyanobacteria should remain contained and reversible, rather than open or permanent. Instead of aiming for large-scale atmospheric change right away, cyanobacteria would first be used in closed or semi-closed systems, such as bioreactors or controlled habitats. These systems could be monitored, adjusted, or shut down if problems arise. Designing for reversibility helps reduce the risk of causing permanent harm before we fully understand the consequences.
Design
Assumptions
Risks of Failure & “Success”
Action 3: International Open-Source Registry and Tiered Licensing System
Purpose
To ensure transparency, prevent monopolization, and enable oversight, all engineered cyanobacteria strains intended for space use should be traceable and licensed.
Design
Assumptions
Risks of Failure & “Success

I would prioritize responsible innovation with clear accountability, supported by shared oversight. When it comes to introducing engineered life beyond Earth, it doesn’t feel like something that should move as fast as possible, even if the technology allows it. A risk review creates space to think carefully about safety and long-term consequences before taking irreversible steps.
Shared oversight adds another layer of care by making these decisions collective rather than competitive. Instead of one actor deciding the future of an entire planetary environment, responsibility is spread across many voices. The trade-off is time: broader discussion and staged approval slow things down and can feel frustrating in a field driven by ambition and speed. This approach also assumes cooperation between countries and institutions, which is not guaranteed. Still, given how permanent the consequences could be, moving more thoughtfully seems worth the cost.
References
[1] National Aeronautics and Space Administration. (n.d.). Artemis II. NASA. https://www.nasa.gov/mission/artemis-ii/
[2] Tomsia, M., Cieśla, J., Śmieszek, J., Florek, S., Macionga, A., Michalczyk, K., & Stygar, D. (2024). Long-term space missions’ effects on the human organism: What we do know and what requires further research. Frontiers in Physiology, 15, 1284644. https://doi.org/10.3389/fphys.2024.1284644
[3] Bekker, A., Holland, H. D., Wang, P.-L., Rumble, D. R. III, Stein, H. J., Hannah, J. L., Coetzee, L. L., & Beukes, N. J. (2004). Dating the rise of atmospheric oxygen. Nature, 427(6970), 117–120. https://doi.org/10.1038/nature02260
[4] Coleine, C., Delgado-Baquerizo, M., Rosado, A. S., & Zerboni, A. (2025). The role of extremophile microbiomes in terraforming Mars. Communications Biology, 8(1), 1588. https://doi.org/10.1038/s42003-025-08973-1
[5] Jones, H., NASA Ames Research Center, Anderson, G., & Paragon Space Development Corporation. (2017). Need for cost optimization of space life support systems. In 47th International Conference on Environmental Systems. https://ntrs.nasa.gov/api/citations/20170010166/downloads/20170010166.pdf
[6] Hoffman, J. A., Hecht, M. H., Rapp, D., Hartvigsen, J. J., SooHoo, J. G., Aboobaker, A. M., McClean, J. B., Liu, A. M., Hinterman, E. D., Nasr, M., Hariharan, S., Horn, K. J., Meyen, F. E., Okkels, H., Steen, P., Elangovan, S., Graves, C. R., Khopkar, P., Madsen, M. B., . . . Eisenman, D. J. (2022). Mars Oxygen ISRU Experiment (MOXIE)—Preparing for human Mars exploration. Science Advances, 8(35), eabp8636. https://doi.org/10.1126/sciadv.abp8636
[7] COSPAR, 2026. COSPAR Policy on Planetary Protection, Space Research Today, Volume 224, 17-39. https://doi.org/10.60970/012026SRT224/PPP
[8] The Antarctic Treaty. (1961). United Nations Treaty Series, 72, No. 5778. https://treaties.un.org/doc/Publication/UNTS/Volume%20402/volume-402-I-5778-English.pdf
[9] Robert.Wickramatunga. (n.d.). Outer Space Treaty. https://www.unoosa.org/oosa/en/ourwork/spacelaw/treaties/outerspacetreaty.html
Professor Jacobson
Biological DNA synthesis uses DNA polymerase, which has an error rate of 1:10^5, that is it adds 1 incorrect base every 100,000 bases. Comparing this with the 3.2 Gb (3.2 x 109 bp) human genome, this enzyme would result in tens of thousands of errors every genome replication cycle, which would be unsustainable. This discrepancy is dealt with by the DNA polymerase itself, which has a dual polymerase and exonuclease function. It is capable of extending the DNA molecule in the 5’ 3’ direction, while also proof reading via removing incorrect and adding correct bases in the 3’ 5’ direction. That is to say, the polymerase has a natural self-correcting/proofreading mechanism for removing an erroneous base every time it adds it to the replicating DNA strand. The polymerase plus its proof-reading system results in an error rate of 1:106, which still gives a significant over 3000 errors per cell division. This is further reduced by the MutS repair system, wherein a group of proteins (MutS, MutL MutH) recognize mismatches in DNA replication, cleave the region with the incorrect bases, and allow the DNA polymerase to extend it with the right set of bases.
For an average human protein, the DNA code would be 1036 bp long, which corresponds to roughly 345 codons (and subsequently, amino acids). Since the genetic code is redundant i.e more than 1 codon can give the same amino acid, there are many Due to codon degeneracy, each amino acid can be encoded by multiple synonymous codons. Assuming an average of ~3 codons per amino acid, the number of possible DNA sequences that could encode the same protein is approximately 3345 , an astronomically large number. In practice, most of these sequences are not biologically viable due to physical and biochemical constraints. One of these is GC content, where strands with extremely high and low GC contents in their sequence limit effective DNA transcription. GC content also affects secondary structure formation, with certain GC-rich/poor sequences allowing for hairpins and loops to form, again not allowing for the genetic code to be translated into the protein of interest. Moreover, the ribozyme RNAse III preferentially cuts at certain RNA sequences or secondary structures (RNA Cleavage Rules), making many theoretical codes fail.
Dr. LeProust
The most commonly used method for oligonucleotide synthesis currently is the phosphoramidite method, developed by Caruthers in 1981. This method involves a cyclic process performed on solid-phase support (CPG) where nucleotides are added one at a time in a repeating four-step cycle: (1) coupling with phosphoramidite to add the next nucleotide, (2) capping any unreacted sites to prevent errors, (3) oxidation to stabilize the phosphate linkage, and (4) deblocking to prepare for the next cycle. This cycle is repeated N times to build an oligonucleotide of N bases in length. The phosphoramidite method became the foundation for automated DNA synthesis when Applied Biosystems (ABI) introduced the first automated DNA synthesizer in 1983, and it remains the standard chemistry used in modern synthesis platforms, including high-throughput approaches like microarrays and silicon-based synthesis technologies.
Making oligonucleotides longer than 200 nucleotides via direct synthesis is extremely difficult due to the cumulative effect of incomplete coupling reactions at each synthesis cycle. In the phosphoramidite method, each nucleotide addition cycle has a coupling efficiency of approximately 98-99%, meaning that 1-2% of growing chains fail to add the correct base at each step. While this seems like a high success rate, the effect compounds over many cycles—after 200 cycles, the proportion of full-length, error-free product becomes vanishingly small, with the majority of molecules being truncated or containing errors. This is evident from the significant achievement of synthesizing 700-mers for the first time using enhanced chemistry, which required substantial optimization to achieve 97% full-length material. The baseline synthesis process shows clear accumulation of truncation products for sequences approaching 500 nucleotides, and even with enhanced chemistry, a greater than 10-fold increase in PCR yield was necessary to obtain better uniformity and more full-length materials. Essentially, as oligo length increases beyond 200nt, the yield of correct, full-length product drops exponentially while error rates accumulate, making direct synthesis increasingly impractical.
A 2000bp gene cannot be made via direct oligonucleotide synthesis because this length far exceeds the practical limitations of the phosphoramidite method. With the most advanced chemistry enhancements, direct synthesis is limited to approximately 200-700 nucleotides maximum, making a 2000bp sequence 3-10 times longer than what's achievable in a single synthesis reaction. Instead, longer genes must be constructed through an assembly approach such as classical gene synthesis methods by first designing multiple shorter, overlapping oligonucleotides that are then pieced together. Modern gene synthesis workflows, such as Twist's platform, use a multi-step process: first synthesizing ultra-long oligonucleotides (which are still limited in length), then using enzymatic assembly methods to join these fragments together, and finally using long-read sequencing to verify the final assembled product. This assembly strategy allows for the production of gene fragments up to 5kb with low error rates (1:7,500 ), but these are explicitly described as assembled products rather than directly synthesized single molecules.
George Church
The 20 amino acids found in nature are further divided into two groups based on their role in growth and nitrogen balance: essential amino acids and non-essential amino acids. Essential amino acids (EAAs) are those whose carbon skeletons are not synthesized de novo by animal cells, or are insufficiently synthesized de novo relative to metabolic needs (Wu, 2009). In other words, they cannot be produced by the body and therefore must be obtained from external sources (food). The 10 essential amino acids in all animals include Phe, Val, Thr, Trp, Ile, Met, His, Arg, Leu, and Lys. The "Lysine Contingency" is a concept from the Jurassic Park films in which the dinosaurs are genetically engineered to be unable to naturally produce the amino acid lysine and must be fed it in their diets. The scientists added this feature as a kill-switch: if the dinosaurs escaped their confines, they would be unable to survive in nature without human-supplemented lysine in their food. However, this concept is scientifically flawed because lysine is already an essential amino acid that isn't naturally synthesized by the body and can only be obtained externally through diet. The film's inaccuracy (portraying lysine as though it were normally non-essential) demonstrates the filmmakers' negligence in aligning the biotech/synbio-focused plot with biological reality. To non-science audiences, this may seem like a clever plot device, but as someone with a biology background who enjoyed these films growing up (and can even say they inspired me to pursue biotech), this now seems like a cringe-worthy and amusing mistake in such a major franchise.
Citations:
Hou, Y., & Wu, G. (2018). Nutritionally essential amino acids. Advances in Nutrition, 9(6), 849–851. https://doi.org/10.1093/advances/nmy054
https://jurassicpark.fandom.com/wiki/Lysine_contingency
(I acknowledge that AI tools such as ChatGPT, Claude, and DeepSeek were used to assist with conceptual understanding and language refinement. The content, ideas, and intellectual contributions of this work remain entirely my own.)
For this part, I created an account on Benchling –> imported the Lambda DNA –> played around with 7 restriction enzymes to get the gel patterns from the digests. Here’s the enzymes I used and the resulting gel pattern of digests:
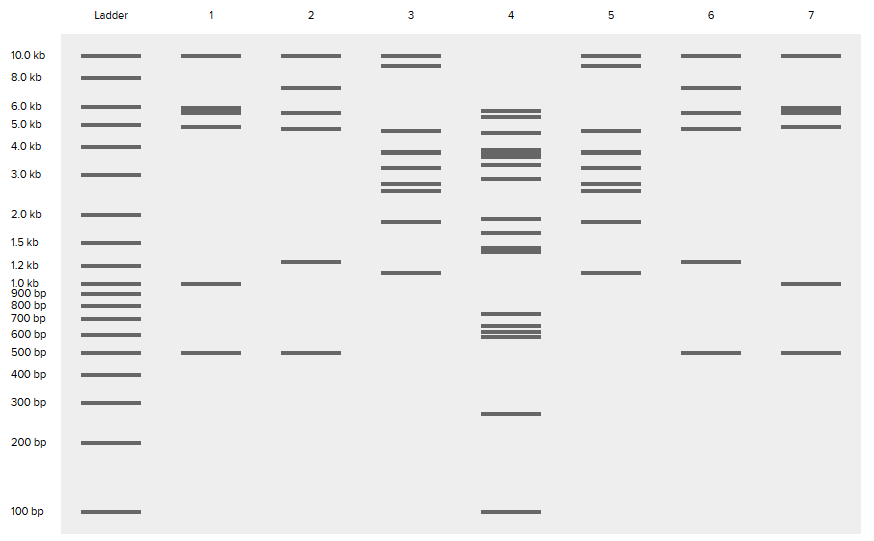
(I envisioned creating a dragonfly, so if you look closely I hope you can see it 😄)
3.1. Choose your protein
The protein I chose for this assignment is the Tardigrade Dsup (Damage Suppressor), which is a unique DNA-binding protein produced only by tardigrades (Phylum Tardgrada) that enables them to survive ROS and radiation-induced DNA damage (Hashimoto et.al 2016). It’s one of the key reasons these tiny organisms can tolerate the extreme conditions of space (Jonsson et.al 2008). What I found especially fascinating is that when Dsup was expressed in human cultured cells (HEK293), it also reduced X-ray- and hydrogen-peroxide-induced DNA damage by lowering DNA breaks and fragmentation. Furthermore, a lot of exciting studies on Dsup-based gene and mRNA delivery approaches as potential radioprotective therapies, which are especially relevant to cancer treatment (Cui et. al. 2025). The (UniProt sequence)[https://www.uniprot.org/uniprotkb/P0DOW4/entry] of Dsup is pasted below:
MASTHQSSTEPSSTGKSEETKKDASQGSGQDSKNVTVTKGTGSSATSAAIVKTGGSQGKDSSTTAGSSSTQGQKFSTTPTDPKTFSSDQKEKSKSPAKEVPSGGDSKSQGDTKSQSDAKSSGQSQGQSKDSGKSSSDSSKSHSVIGAVKDVVAGAKDVAGKAVEDAPSIMHTAVDAVKNAATTVKDVASSAASTVAEKVVDAYHSVVGDKTDDKKEGEHSGDKKDDSKAGSGSGQGGDNKKSEGETSGQAESSSGNEGAAPAKGRGRGRPPAAAKGVAKGAAKGAAASKGAKSGAESSKGGEQSSGDIEMADASSKGGSDQRDSAATVGEGGASGSEGGAKKGRGRGAGKKADAGDTSAEPPRRSSRLTSSGTGAGSAPAAAKGGAKRAASSSSTPSNAKKQATGGAGKAAATKATAAKSAASKAPQNGAGAKKKGGKAGGRKRK
References
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Using the “Reverse Translate” tool of the Sequence Manipulation Suite (https://www.bioinformatics.org/sms2/rev_trans.html)
atggcgagcacccatcagagcagcaccgaaccgagcagcaccggcaaaagcgaagaaacc aaaaaagatgcgagccagggcagcggccaggatagcaaaaacgtgaccgtgaccaaaggc accggcagcagcgcgaccagcgcggcgattgtgaaaaccggcggcagccagggcaaagat agcagcaccaccgcgggcagcagcagcacccagggccagaaatttagcaccaccccgacc gatccgaaaacctttagcagcgatcagaaagaaaaaagcaaaagcccggcgaaagaagtg ccgagcggcggcgatagcaaaagccagggcgataccaaaagccagagcgatgcgaaaagc agcggccagagccagggccagagcaaagatagcggcaaaagcagcagcgatagcagcaaa agccatagcgtgattggcgcggtgaaagatgtggtggcgggcgcgaaagatgtggcgggc aaagcggtggaagatgcgccgagcattatgcataccgcggtggatgcggtgaaaaacgcg gcgaccaccgtgaaagatgtggcgagcagcgcggcgagcaccgtggcggaaaaagtggtg gatgcgtatcatagcgtggtgggcgataaaaccgatgataaaaaagaaggcgaacatagc ggcgataaaaaagatgatagcaaagcgggcagcggcagcggccagggcggcgataacaaa aaaagcgaaggcgaaaccagcggccaggcggaaagcagcagcggcaacgaaggcgcggcg ccggcgaaaggccgcggccgcggccgcccgccggcggcggcgaaaggcgtggcgaaaggc gcggcgaaaggcgcggcggcgagcaaaggcgcgaaaagcggcgcggaaagcagcaaaggc ggcgaacagagcagcggcgatattgaaatggcggatgcgagcagcaaaggcggcagcgat cagcgcgatagcgcggcgaccgtgggcgaaggcggcgcgagcggcagcgaaggcggcgcg aaaaaaggccgcggccgcggcgcgggcaaaaaagcggatgcgggcgataccagcgcggaa ccgccgcgccgcagcagccgcctgaccagcagcggcaccggcgcgggcagcgcgccggcg gcggcgaaaggcggcgcgaaacgcgcggcgagcagcagcagcaccccgagcaacgcgaaa aaacaggcgaccggcggcgcgggcaaagcggcggcgaccaaagcgaccgcggcgaaaagc gcggcgagcaaagcgccgcagaacggcgcgggcgcgaaaaaaaaaggcggcaaagcgggc ggccgcaaacgcaaa
3.3. Codon optimization
When expressing a foreign gene in a host organism (chassis), it’s important to consider codon usage bias, which refers to the host’s preference of translating a synonymous codon for an amino acid over one that is in the genetic sequence, which is not frequently translated by the host. This is where codon optimization comes into play, by replacing some codons with those the host prefers to translate (but not changing the amino acid sequence), to ensure that the host organism optimally translates our protein of interest.
Supposing I want to acquire an engineered Dsup protein for therapeutic use, then I would choose [E.coli] as the chassis/delivery system. E.coli is one of the standard and most reliable chassis in synthetic biology - thoroughly studied, well characterized, and it grows rapidly in large volumes inexpensively.
Using the Codon Optimization Tool of [VectorBuilder]:
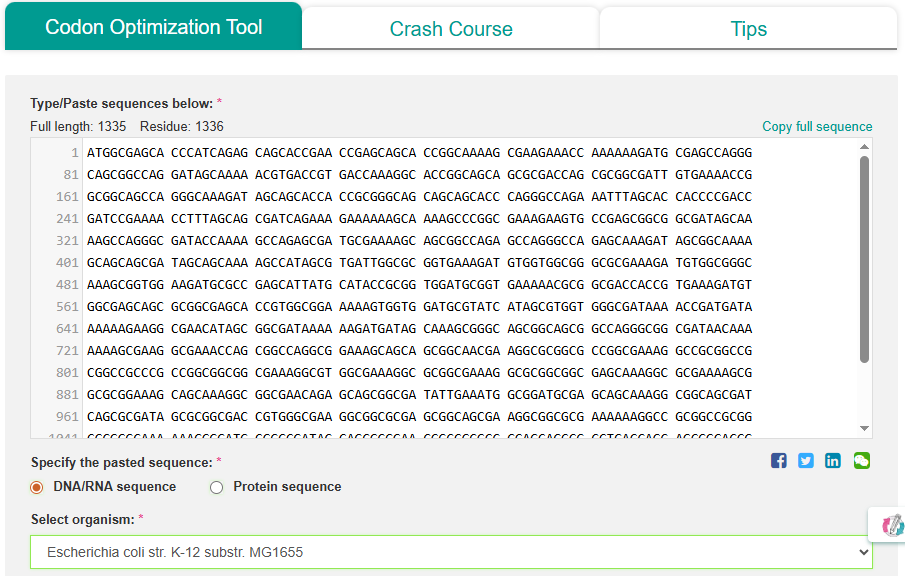
I get the following improved sequence: 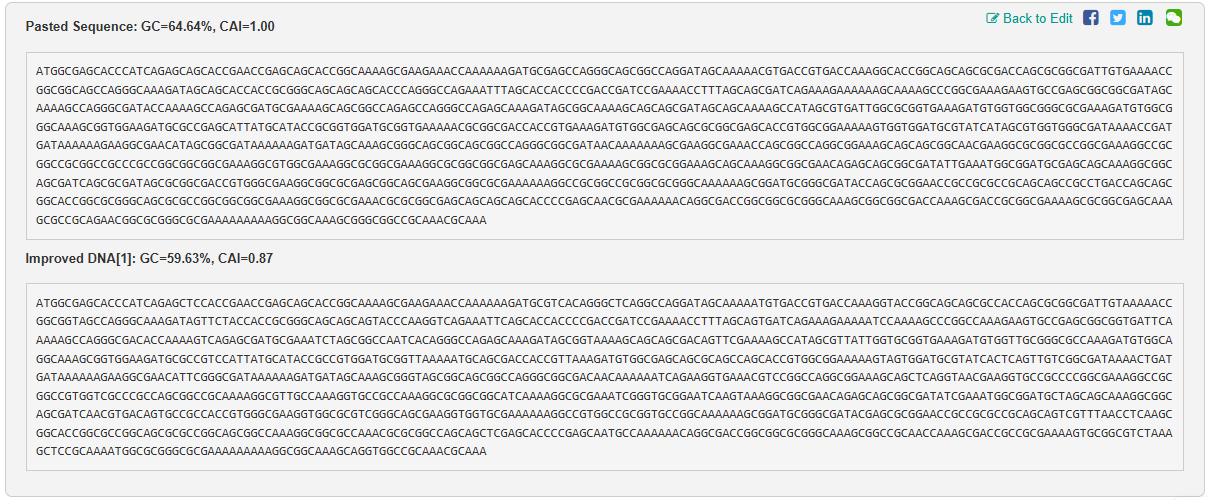
ATGGCGAGCACCCATCAGAGCTCCACCGAACCGAGCAGCACCGGCAAAAGCGAAGAAACCAAAAAAGATGCGTCACAGGGCTCAGGCCAGGATAGCAAAAATGTGACCGTGACCAAAGGTACCGGCAGCAGCGCCACCAGCGCGGCGATTGTAAAAACCGGCGGTAGCCAGGGCAAAGATAGTTCTACCACCGCGGGCAGCAGCAGTACCCAAGGTCAGAAATTCAGCACCACCCCGACCGATCCGAAAACCTTTAGCAGTGATCAGAAAGAAAAATCCAAAAGCCCGGCCAAAGAAGTGCCGAGCGGCGGTGATTCAAAAAGCCAGGGCGACACCAAAAGTCAGAGCGATGCGAAATCTAGCGGCCAATCACAGGGCCAGAGCAAAGATAGCGGTAAAAGCAGCAGCGACAGTTCGAAAAGCCATAGCGTTATTGGTGCGGTGAAAGATGTGGTTGCGGGCGCCAAAGATGTGGCAGGCAAAGCGGTGGAAGATGCGCCGTCCATTATGCATACCGCCGTGGATGCGGTTAAAAATGCAGCGACCACCGTTAAAGATGTGGCGAGCAGCGCAGCCAGCACCGTGGCGGAAAAAGTAGTGGATGCGTATCACTCAGTTGTCGGCGATAAAACTGATGATAAAAAAGAAGGCGAACATTCGGGCGATAAAAAAGATGATAGCAAAGCGGGTAGCGGCAGCGGCCAGGGCGGCGACAACAAAAAATCAGAAGGTGAAACGTCCGGCCAGGCGGAAAGCAGCTCAGGTAACGAAGGTGCCGCCCCGGCGAAAGGCCGCGGCCGTGGTCGCCCGCCAGCGGCCGCAAAAGGCGTTGCCAAAGGTGCCGCCAAAGGCGCGGCGGCATCAAAAGGCGCGAAATCGGGTGCGGAATCAAGTAAAGGCGGCGAACAGAGCAGCGGCGATATCGAAATGGCGGATGCTAGCAGCAAAGGCGGCAGCGATCAACGTGACAGTGCCGCCACCGTGGGCGAAGGTGGCGCGTCGGGCAGCGAAGGTGGTGCGAAAAAAGGCCGTGGCCGCGGTGCCGGCAAAAAAGCGGATGCGGGCGATACGAGCGCGGAACCGCCGCGCCGCAGCAGTCGTTTAACCTCAAGCGGCACCGGCGCCGGCAGCGCGCCGGCAGCGGCCAAAGGCGGCGCCAAACGCGCGGCCAGCAGCTCGAGCACCCCGAGCAATGCCAAAAAACAGGCGACCGGCGGCGCGGGCAAAGCGGCCGCAACCAAAGCGACCGCCGCGAAAAGTGCGGCGTCTAAAGCTCCGCAAAATGGCGCGGGCGCGAAAAAAAAAGGCGGCAAAGCAGGTGGCCGCAAACGCAAA
3.4. You have a sequence! Now what? The next step is to express this sequence in a host (transcription) and get the protein (translation). Since I optimized the sequence for E. coli, I would use an E. coli expression system, and the cell’s transcription/translation machinery will produce the protein. I will achieve this in the following steps:
Step 1) Create a Twist account and a Benchling account ✅
Step 2) Build Your DNA Insert Sequence I have used my codon-optimized dsup sequence for an E.coli expression system. On Benchling, I sequentially inserted the following sequences (mentioned in the HW) into the “Create DNA / RNA sequence” tab:
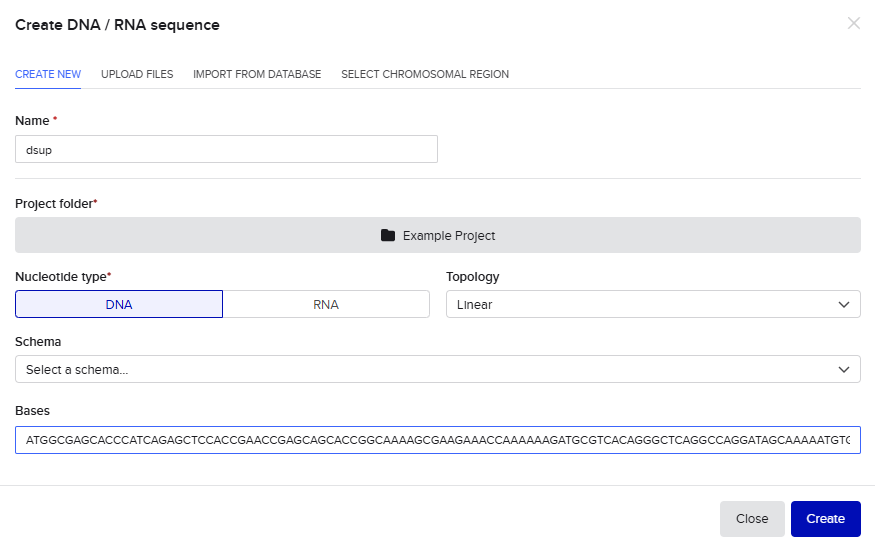 During the process of annotating:
During the process of annotating:
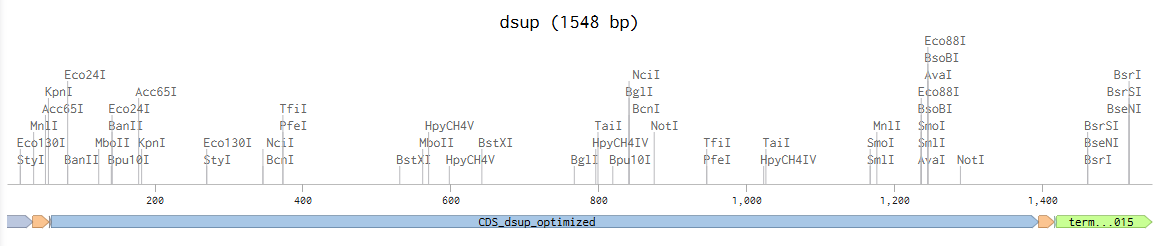
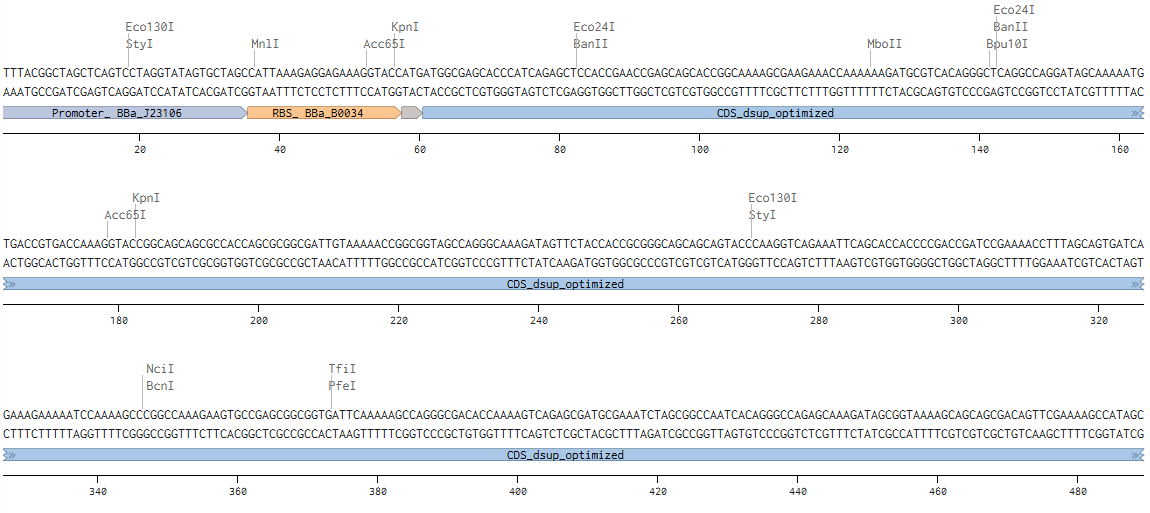 Linear Map (link: https://benchling.com/s/seq-FJPRVyjCrav3opFmzzZ2?m=slm-MQBEBBrWm3w4d8JMFsGv):
Linear Map (link: https://benchling.com/s/seq-FJPRVyjCrav3opFmzzZ2?m=slm-MQBEBBrWm3w4d8JMFsGv):
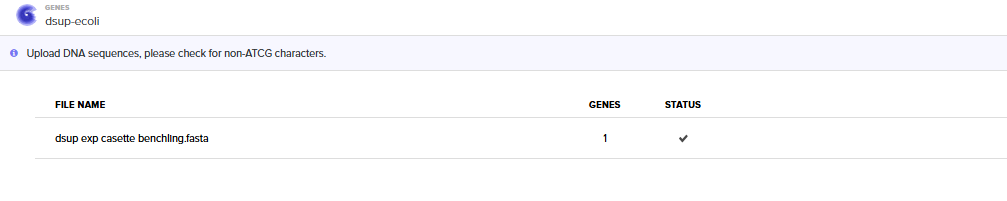
Step 3) On Twist Biosciences:
Selecting “Genes” and then “Clonal Genes” Option

Importing the Benchling Casette Sequence (FASTA)
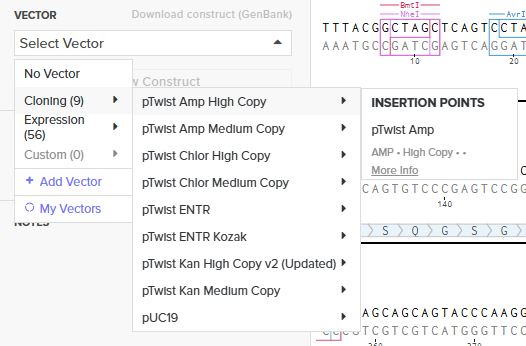
Choosing Twist Vector
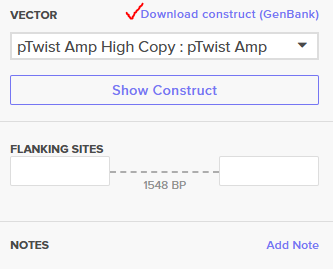
Downloading GenBank sequence of the construct
Here is what the recombinant vector looks like in Twist:
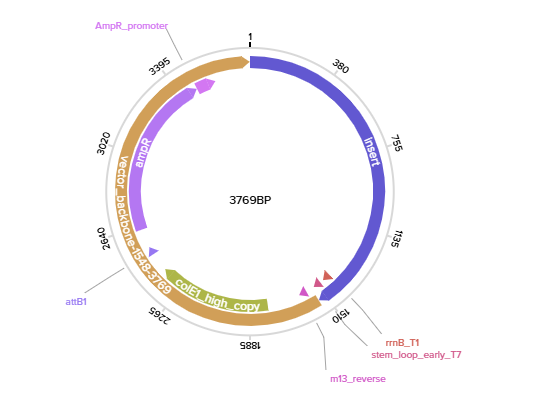
Step 4) Back to Benchling:
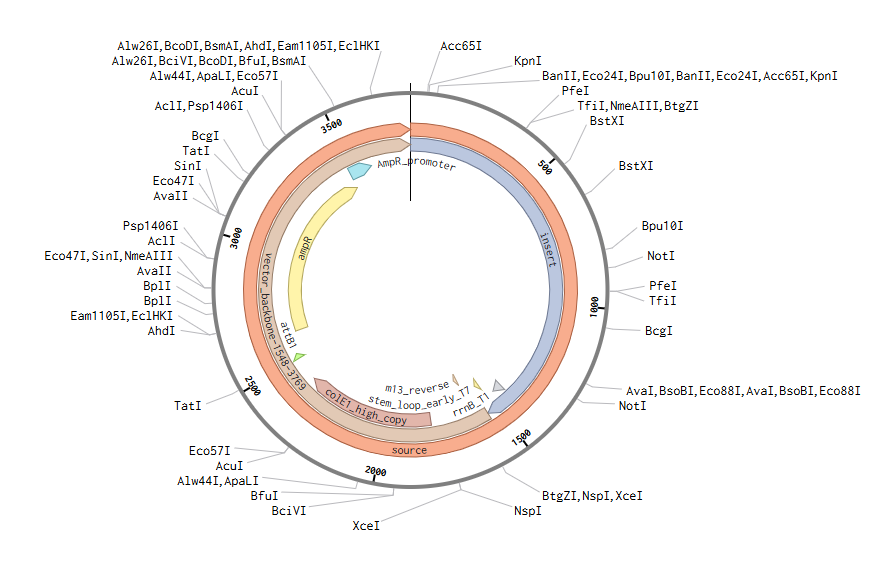
After importing the Twist construct (GenBank file), this is the final plasmid + expression casette:
(i) What DNA would you want to sequence (e.g., read) and why?
I would want to sequence the tardigrade dsup gene (damage suppressor). The Dsup protein binds and physically shields DNA from hydroxyl radicals generated by radiation and oxidative stress. This makes it super interesting for some of my envisioned projects on:
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would choose Oxford Nanopore sequencing (3rd generation) because it provides very long read lengths and direct sequencing of DNA withough any amplification, which is ideal for verifying complex constructs like the Dsup gene and related assemblies. Nanopore sequencing reduces bias and enables rapid real-time data generation. However, it has a higher raw error rate compared to short-read sequencing (1st + 2nd gen) and requires computational base-calling, so I would use it primarily for assembly confirmation and structure verification.
The essential steps (I used ChatGPT to simplify this) are given below:
Output: set of DNA read files (FASTQ) containing the nucleotide sequences of DNA fragments + associated quality scores. These reads can be aligned to the designed Dsup sequence to verify that the synthesized gene matches the intended design. Other outputs like consensus sequences and variant files summarize the final DNA sequence and any differences.
(i) What DNA would you want to synthesize (e.g., write) and why?
I would synthesize a fusion gene encoding a chimeric Dsup–HMGN protein for expression in human cells. Dsup (from tardigrades) protects DNA from radiation damage, but since it is not a human protein it may not localize efficiently in human chromatin and could trigger immune responses. To improve this, I would fuse Dsup to the nucleosome-binding domain of a human HMGN protein, which naturally binds chromatin in human cells. HMGN are present in vertebrates, including humans, and have some sequence similarity with dsup (Chavez et al, 2019).
The goal would be to create a protein that:
In terms of potential applications, this could protect healthy tissue during radiotherapy and astronauts from space radiation.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use phosphoramidite chemical DNA synthesis followed by enzymatic assembly (workflow used by Twist Bioscience). Why?
So my worflow would look like this:
Why Oxford Nanopore?
Essential steps of the chosen sequencing method (Oxford Nanopore; repeated from above)
Limitations of the sequencing method in terms of speed, accuracy, scalability: (I consulted the internet and ChatGPT for this)
Accuracy
Speed
Scalability
[References]
(i) What DNA would you want to edit and why?
I would focus on editing human genomes to improve health, increase disease resistance, and extend healthy lifespan. With my background in cancer research and interest in space biology, I’m especially intrigued by the tardigrade Dsup protein, which protects DNA from radiation damage. Understanding or applying similar mechanisms could help humans better withstand cosmic radiation during space travel and even serve as radioprotective strategies in cancer therapy, all while enhancing our natural resilience to disease and aging.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-Cas9 to edit human genomes because it is precise, efficient, and versatile, allowing targeted insertion of protective genes like the tardigrade Dsup protein to enhance DNA resilience, disease resistance, and longevity.
CRISPR uses a guide RNA (gRNA) to direct the Cas9 enzyme to a specific DNA sequence, where Cas9 creates a double-strand break. The cell then repairs the break via homology-directed repair (HDR), allowing precise insertion of a donor DNA sequence, such as my codon-optimized Dsup gene.
Design Steps:
Inputs Needed: