Week 2 Lecture HW: DNA Read Write and Edit
Pre-lecture 2 homework questions:
Question 1: What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy? Response: The error rate of DNA polymerase varies between 1 in 104 to 1 in 105. The length of the human genome is 3 * 109 base pairs, this would mean hundereds of mutations every cell division. However, biology dealt with that discrepancy through proofreading, which decreases the error rate to about 1 in 107, along with other correcting methods like mismatch repair, DNA damage checkpoints, and if there is still severe mutation detected, the cell will go into apoptosis.
Question 2: How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest? Response: The genetic code is degenerate, meaning multiple codons can encode the same amino acid. With 61 codons specifying 20 amino acids, each amino acid is encoded by about three codons on average. For a typical human protein of roughly 400 amino acids, this results in approximately 3400 (about 10190) possible DNA sequences that could produce the same protein. In practice, many of these sequences do not function effectively. Cells exhibit codon bias, preferring certain codons because the corresponding tRNAs are more abundant, which improves translation efficiency. Some sequences also create mRNA structures that hinder ribosome movement or reduce stability, leading to lower protein production. also, specific nucleotide patterns can unintentionally introduce regulatory signals such as premature stop cues or splice sites. For these reasons, although many DNA sequences are theoretically possible, only a subset will reliably generate the desired protein within a biological system.
Questions from Dr. LeProust:
- Most commonly used oligo synthesis method? response: The most commonly used oligonucleotide synthesis method is solid-phase phosphoramidite synthesis.
- Why it’s hard to make oligos >200 nt directly? Response: It is difficult to synthesize oligos longer than about 200 nucleotides because each nucleotide addition is not perfectly efficient. Even with ~99% coupling efficiency, small errors accumulate at every step, causing many strands to be truncated or contain mistakes by the end of the process. Longer sequences also become harder to chemically handle and purify, reducing overall yield and quality.
- Why you can’t make a 2000 bp gene by direct oligo synthesis? Response: A 2000 bp gene cannot be made directly through oligo synthesis because the stepwise chemical process introduces small errors during each nucleotide addition. As the sequence length increases, these errors accumulate, leading to a very low proportion of full-length, correct DNA strands. The resulting mixture would contain many truncated or incorrect sequences, making purification impractical. Instead, long genes are typically assembled from shorter, high-quality oligos using methods such as PCR or gene assembly.
Question from Dr. George Church: What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”? Response: The ten essential amino acids in animals are histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine, and arginine (arginine is especially essential during growth). They are considered essential because animals cannot synthesize them in sufficient amounts and must obtain them through diet. This weakens the idea behind a “lysine contingency,” which proposes engineering organisms to depend on lysine so they cannot survive outside controlled environments. Since lysine is already common in many natural environments and diets, relying on it alone would not provide strong containment. An organism could potentially obtain lysine from surrounding biological material, making the safeguard less reliable. Effective biocontainment typically requires dependencies on nutrients that are rare or entirely synthetic rather than widely available amino acids.
Week 2 Homework
Part 1:
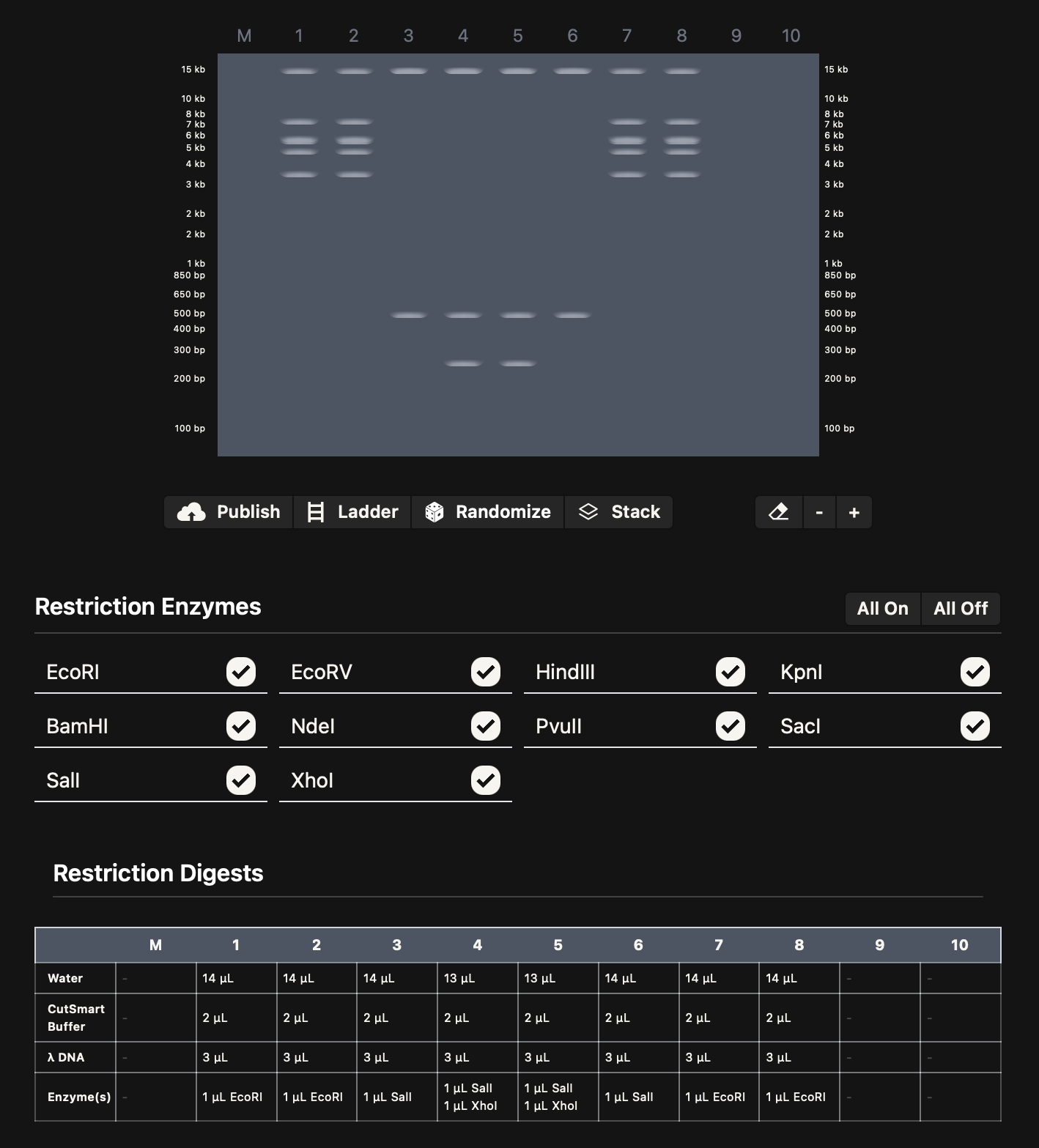
I imported the lambda sequence on Benchling, and started experimenting on Ronan’s website rcdonovan.com until I found a way to make a smiley face. I then used the restriction enzymes on Benchling to get the virtual digest from there. Below are images for my results

Part 3:
The protein of choice for me is green fluorescent protein (GFP), because it is widely used as a reporter in molecular and synthetic biology to visualize gene expression and protein localization. Its function is easy to assay, and it is well-characterized. I got the sequence from Uniprot
Protein sequence:
MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
Reverse translated DNA sequence: I got the sequence from Here
atgagcaaaggcgaagaactgtttaccggcgtggtgccgattctggtggaactggatggcgatgtgaacggccataaatttagcgtgagcggcgaaggcgaaggcgatgcgacctatggcaaactgaccctgaaatttatttgcaccaccggcaaactgccggtgccgtggccgaccctggtgaccacctttagctatggcgtgcagtgctttagccgctatccggatcatatgaaacagcatgatttttttaaaagcgcgatgccggaaggctatgtgcaggaacgcaccattttttttaaagatgatggcaactataaaacccgcgcggaagtgaaatttgaaggcgataccctggtgaaccgcattgaactgaaaggcattgattttaaagaagatggcaacattctgggccataaactggaatataactataacagccataacgtgtatattatggcggataaacagaaaaacggcattaaagtgaactttaaaattcgccataacattgaagatggcagcgtgcagctggcggatcattatcagcagaacaccccgattggcgatggcccggtgctgctgccggataaccattatctgagcacccagagcgcgctgagcaaagatccgaacgaaaaacgcgatcatatggtgctgctggaatttgtgaccgcggcgggcattacccatggcatggatgaactgtataaa
3.3 Codon Optimization
Codon optimization is needed because different organisms prefer different codons due to tRNA abundance. Using non-preferred codons can slow translation, reduce protein yield, or cause misfolding.
I optimized the GFP coding sequence for Escherichia coli, since it is commonly used for recombinant protein expression due to its fast growth, low cost, and well-developed genetic tools. Codon optimization increases translation efficiency and protein output in the chosen host.
This is the tool I used for optimization: Vectorbuilder
Optimized sequence:
atgagcaaaggcgaagaactgtttaccggcgtggtgccgattctggtggaactggatggcgatgtgaatggccataaatttagcgtgagcggcgaaggtgaaggcgatgcgacctatggcaaactgaccctgaaatttatctgcaccaccggtaaactgccggtgccgtggccgaccctggtgaccaccttcagctacggcgtgcagtgttttagccgctacccggatcatatgaaacagcatgatttttttaaaagcgcgatgccggaaggctatgtgcaggaacgcaccatttttttcaaagatgatggcaattacaaaacccgtgccgaagtgaaattcgaaggcgataccctggtgaatcgcattgaactgaaaggcattgattttaaagaagatggtaacattctgggccacaaactggaatacaactataacagccataacgtgtacattatggcggataaacagaaaaatggcattaaagtgaactttaaaattcgccataacattgaagatggctcagtgcagctggcggatcactatcagcagaacaccccgattggcgatggcccggttctgctgccggataaccactatctgagcacccagagcgcgctgtcgaaagatccgaacgaaaaacgcgatcacatggtgctgctggaatttgtgaccgccgcgggcatcacccatggtatggatgaactgtataaa
I noticed that the optimized sequence was almost identical to the unoptimized one. This is probably because the GFP is very well known and is already highly optimized for E. choli.
3.4 Producing the Protein
To produce GFP, the optimized DNA sequence can be cloned into an expression vector under a strong promoter. In a cell-dependent system, such as E. coli, the DNA is transcribed into mRNA by RNA polymerase and translated by ribosomes into protein. The GFP protein then folds into its functional fluorescent form.
3.5 Multiple Proteins from One Gene
In natural systems, a single gene can produce multiple proteins through mechanisms such as alternative splicing, alternative promoters, or alternative start codons. These processes allow different mRNA transcripts to be produced from the same DNA sequence, which are then translated into distinct protein isoforms with different functions or localizations.
Part 4:
At this point I saw that GFP is used as an example, however I will still continue my work on it because of how much I am interested in its uses and I have already invested time on it. I followed the steps, made TWIST & Benchling accounts, imported the genome to Benchling, added the sequence and annotations for the promoter, RBS, start codon, sequence, HIS tags, stop codon, and terminator. Exported it as FASTA, imported it on TWIST, added the pTwist Amp High Copy vector. Exported it as Genbank, and opened it on Benchling to see the plasmid.
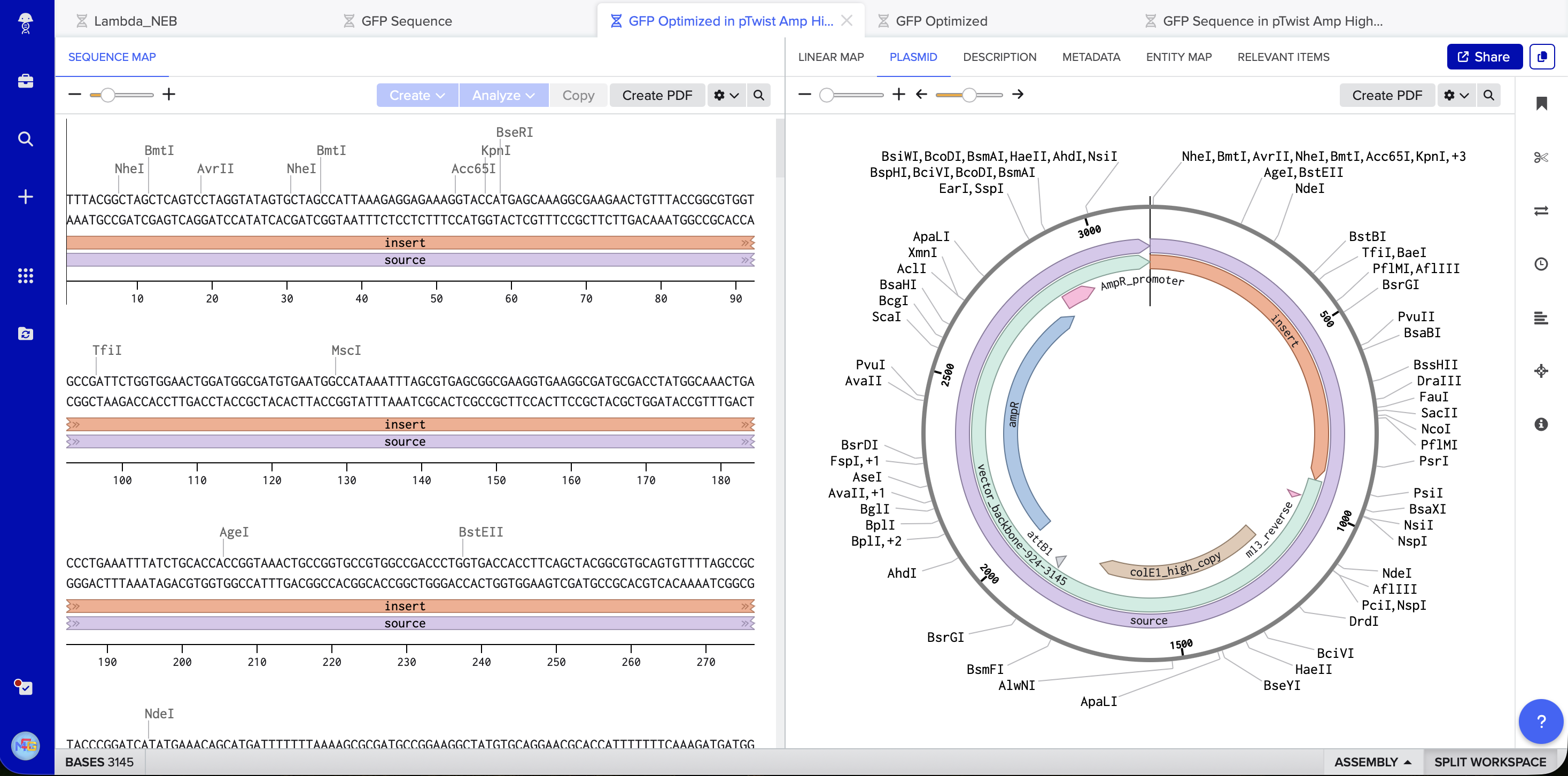
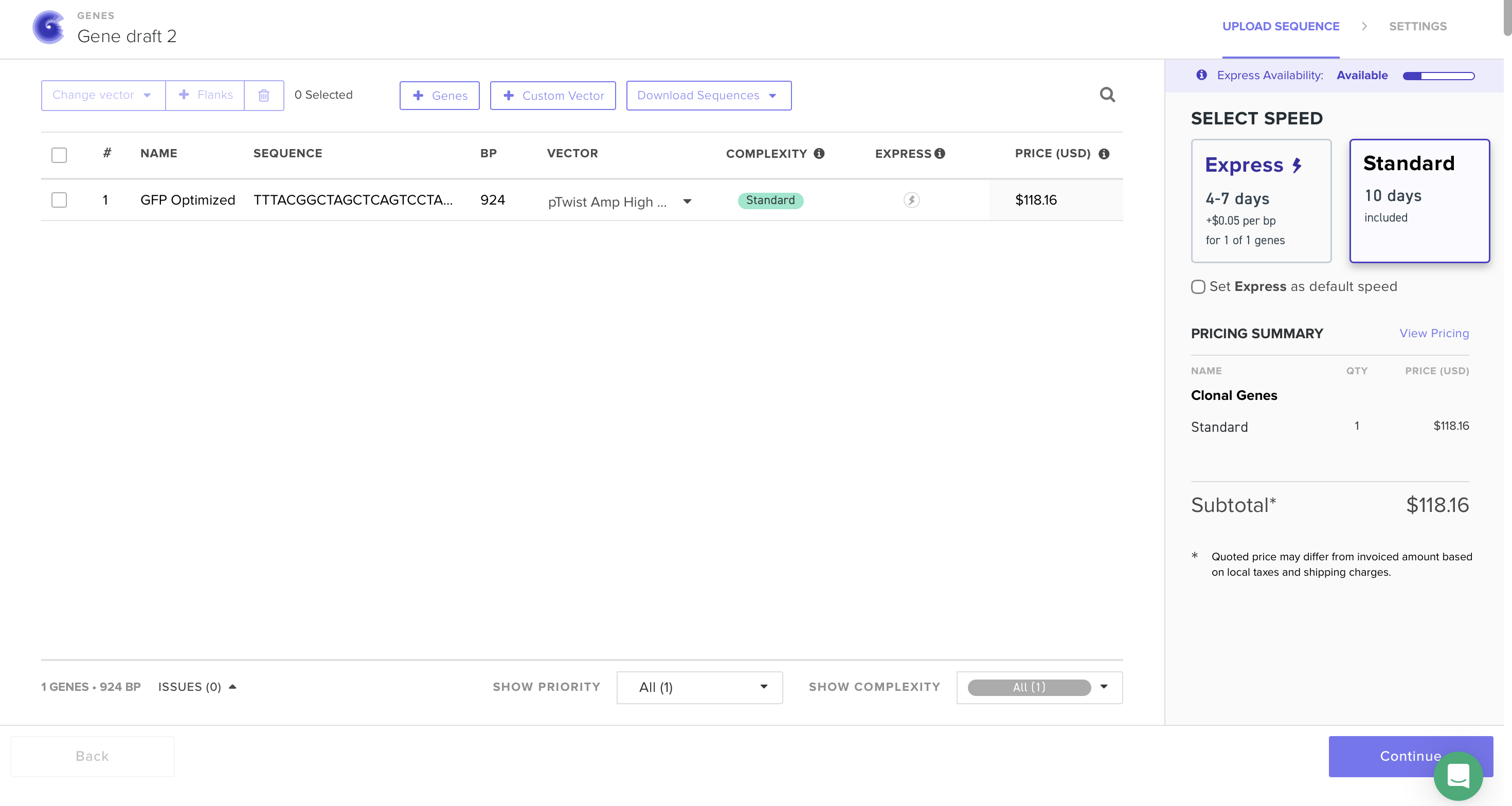
5.1 (i) I would sequence the human BRCA1 gene. It matters because BRCA1 variants strongly link to hereditary breast and ovarian cancer. Reading this DNA lets us detect pathogenic mutations, estimate cancer risk, and guide screening or preventive care. It also helps choose therapy since BRCA1 status affects response to PARP inhibitors.
(ii) I would use Illumina sequencing because it is high accuracy and high throughput which is important for detecting small mutations such as single nucleotide variants or small insertions and deletions.
Illumina sequencing is a second-generation sequencing technology because it sequences millions of DNA fragments simultaneously using amplified clusters on a sequencing flow cell.
The input is genomic DNA extracted from human cells, such as blood or saliva. Preparation steps:
- DNA extraction from the sample.
- Fragmentation of genomic DNA into shorter pieces (usually ~200–500 bp).
- Adapter ligation, where synthetic adapter sequences are attached to the ends of DNA fragments.
- PCR amplification to increase the number of DNA fragments containing the BRCA1 region.
- Loading onto a sequencing flow cell, where fragments bind and form clusters through bridge amplification.
Illumina sequencing uses sequencing-by-synthesis:
- DNA polymerase incorporates nucleotides into the growing DNA strand.
- Each nucleotide (A, T, C, G) has a different fluorescent label.
- After each incorporation, a camera detects the fluorescence signal.
- The signal identifies which base was added at that position.
Repeating this cycle allows the instrument to determine the DNA sequence.
- The output is millions of short DNA reads with associated quality scores, typically stored in FASTQ files. These reads are then aligned to the human reference genome to identify mutations or variants in the BRCA1 gene.
5.2 DNA Write
(i) I would synthesize a genetic circuit that allows bacteria to detect pollution or toxins and produce a fluorescent signal. This could help monitor contamination in environmental samples. Such biosensors could help detect pollution or toxins in water or soil quickly and cheaply.
(ii) I would use solid-phase phosphoramidite DNA synthesis combined with gene assembly (the method used by companies like Twist Bioscience).
Essential steps
- Chemical synthesis of short oligonucleotides (~150–200 nt) using phosphoramidite chemistry.
- Cleavage and purification of the oligos.
- Assembly of longer genes using PCR-based assembly or Gibson Assembly.
- Cloning into plasmid vectors for propagation in bacteria.
Limitations • Length limits: individual oligos cannot exceed ~200 nt. • Error accumulation during synthesis. • Cost and time increase with longer constructs. • Very large constructs (whole genomes) require complex assembly.
5.3 DNA Edit
(i) I would edit the PCSK9 gene in humans to reduce cholesterol levels and lower the risk of cardiovascular disease. PCSK9 regulates the number of LDL receptors on liver cells. Certain natural mutations that disable PCSK9 lead to lower LDL cholesterol levels and a reduced risk of heart disease. By editing the PCSK9 gene to mimic these naturally occurring protective mutations, it may be possible to create a long-term treatment for high cholesterol.
(ii) I would use CRISPR-Cas9 genome editing because it allows targeted modification of specific genes such as PCSK9.
- How the technology edits DNA
- A guide RNA (gRNA) is designed to match the DNA sequence within the PCSK9 gene.
- The Cas9 enzyme binds to the guide RNA to form a complex.
- The complex finds the matching sequence in the genome.
- Cas9 creates a double-strand break at that location.
- The cell repairs the break through non-homologous end joining (NHEJ), which can disrupt the gene and reduce PCSK9 activity.
Reducing PCSK9 function increases the number of LDL receptors on liver cells, helping remove LDL cholesterol from the bloodstream.
- Preparation and inputs
To perform the edit, several components are required: • A guide RNA targeting the PCSK9 gene • The Cas9 enzyme or a plasmid encoding Cas9 • A delivery system such as lipid nanoparticles or viral vectors • Target cells, typically liver cells where PCSK9 is expressed
The guide RNA must be carefully designed to ensure specificity for the PCSK9 gene.
- Limitations
There are several limitations to this editing approach: • Off-target edits may occur at similar DNA sequences elsewhere in the genome • Editing efficiency may vary depending on the cell type and delivery method • Delivery challenges, especially when targeting specific tissues in the body • Ethical and regulatory considerations related to editing human genes
Despite these limitations, editing PCSK9 is considered a promising approach for treating high cholesterol and preventing cardiovascular disease.
Sources used for homework: Google searches and slides for Lecture 2