Week 4 HW: Protein Design Part I

Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Eating a 500-gram piece of meat provides you with approximately 6.62 x 10²³ molecules of amino acids. Since raw meat is roughly 22% protein, you are consuming about 110 grams of actual protein; dividing this by the average mass of 100 Daltons per amino acid tells us you have 1.1 moles of them. By multiplying that by Avogadro’s constant, we find that you are swallowing about 662 sextillion molecules, a number so large it exceeds the number of stars in the observable universe
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans do not become the animals they eat because their digestive systems break down foreign DNA and proteins into basic amino acids and nutrients, which are then used to build human-specific cells. Essentially, the body digests the information (DNA) of the food, removing its ability to turn the consumer into the consumed.
Also answered in this thread
3. Why are there only 20 natural amino acids?
While over 500 amino acids exist in nature, only these 20 are universally encoded by the genetic code, representing a “frozen accident” of evolution that optimized functionality while minimizing metabolic costs.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein and why you selected it
The protein I selected is Green Fluorescent Protein (GFP) from the jellyfish Aequorea victoria. GFP naturally emits bright green fluorescence when exposed to ultraviolet or blue light. It is widely used in molecular biology as a reporter protein to visualize gene expression, protein localization, and cellular processes in living organisms. I chose GFP because it is one of the most important tools in modern biotechnology and synthetic biology, and I previously used it in earlier parts of this assignment.
2. Amino acid sequence
The amino acid sequence of GFP was obtained from UniProt (UniProt ID: P42212)). The sequence begins as follows:
MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTT FSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELK GTDEKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIG DGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK

- Protein length and most frequent amino acid: The GFP protein contains 238 amino acids. Using the amino acid frequency analysis tool in the provided Google Colab notebook, I counted the occurrence of each amino acid in the sequence. The most frequent amino acid in GFP is glycine (G), appearing 22 times.

- Number of protein sequence homologs: Using the UniProt BLAST search tool, I compared the GFP sequence against protein databases to identify homologous proteins. The search returned 205 hits of homologs across different organisms.

- Protein family: GFP belongs to the Green Fluorescent Protein family, which includes naturally occurring and engineered fluorescent proteins used as biological markers. Members of this family share a conserved beta-barrel structure that protects the internal chromophore responsible for fluorescence.
3. Identify the structure page of your protein in RCSB
The 3D structure of GFP can be found in the RCSB Protein Data Bank(https://www.rcsb.org/structure/1GFL)
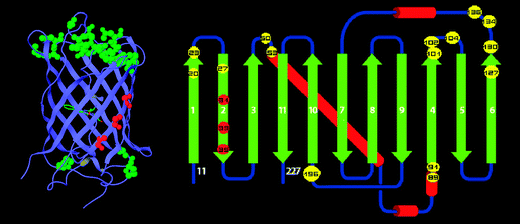
When the structure was solved and quality: The crystal structure of GFP was solved in 1996. The structure has a resolution of approximately 1.9 Å, which is considered high quality because lower resolution values indicate more precise structural data. This high resolution makes GFP a reliable model for structural and molecular visualization studies.
Other molecules present in the structure: Besides the protein itself, the solved structure contains the internal chromophore, which is formed from amino acids within the protein after folding. Some crystal structures may also include water molecules or ions that help stabilize the structure during crystallography experiments.
4. Open the structure of your protein in any 3D molecule visualization software
Using PyMOL, I visualized the GFP structure in three different representations:
- Cartoon representation to highlight overall folding

- Ribbon representation to show secondary structures

- Ball-and-stick representation

After coloring the protein by secondary structure, I observed that the structure contains significantly more beta sheets than alpha helices. These beta sheets form a characteristic barrel structure that surrounds the chromophore in the center of the protein.

When coloring the protein by residue type, I observed that hydrophobic residues are mostly located in the interior of the protein structure, where they help stabilize folding through hydrophobic interactions. Hydrophilic residues are primarily distributed on the outer surface, where they can interact with water and the surrounding environment.

When visualizing the surface of the protein, the structure forms a compact barrel-like shape with small surface cavities and indentations.

Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scan

Using the ESM2 protein language model, I generated a deep mutational scan of my protein, Green Fluorescent Protein. The model predicts how favorable different amino-acid substitutions are at each position in the sequence. I noticed that mutations in residues located near the core of the protein were predicted to be much less favorable than mutations on the surface. For example, substitutions near the chromophore region showed very low likelihood scores, suggesting that these residues are important for maintaining the protein’s structure and fluorescence.
Latent Space Analysis

The notebook also allowed me to embed protein sequences into a reduced dimensional space to compare their similarities. When visualizing these embeddings, proteins with similar sequences and functions tended to cluster together. When I located GFP in the map, it appeared close to other fluorescent or beta-barrel proteins. This suggests the model captures meaningful relationships between proteins based only on their sequence information.
C2. Protein Folding

I used ESMFold to predict the structure of GFP from its amino-acid sequence and compared the prediction with the experimentally solved structure in the RCSB Protein Data Bank. The predicted structure closely resembled the original structure, especially the characteristic beta-barrel fold. When I introduced small mutations into the sequence, the predicted structure stayed mostly similar. Larger sequence changes, however, caused the model to predict structures that deviated more from the original fold.
C3. Protein Generation
Inverse Folding (Protein Generation) Using ProteinMPNN, I generated possible sequences that could fold into the same backbone structure as GFP. The predicted sequences were similar to the original sequence in several important regions, particularly residues located in the protein core. Some positions varied more, which suggests those areas may tolerate mutations without disrupting the overall structure. When I tested one of the generated sequences with ESMFold, the predicted structure still looked very similar to the original GFP fold.

Original GFP Sequence: MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
New Sequence: SYPGEELFTGKVPIEVELDGDVNGKKFSVKGEGEGDATKGTITLKLICTTGKLPVPWPTLIDTFSGGLPCFTRYPPHMRQHDFFKSAMPEGYKQERTITFEGDGKYETRSEVKMEGDTLVNRIELKGSGFKEDGNILGHKLEFSFNSYKVNITADAAANGIKETYTLELKLKDGSVQKAKVDRKVTPIGDGPVLLPEPHYIEVEVKLSKDPNEKRDHVVIEQKSTAAGIE