Week 4 HW: Protein Design
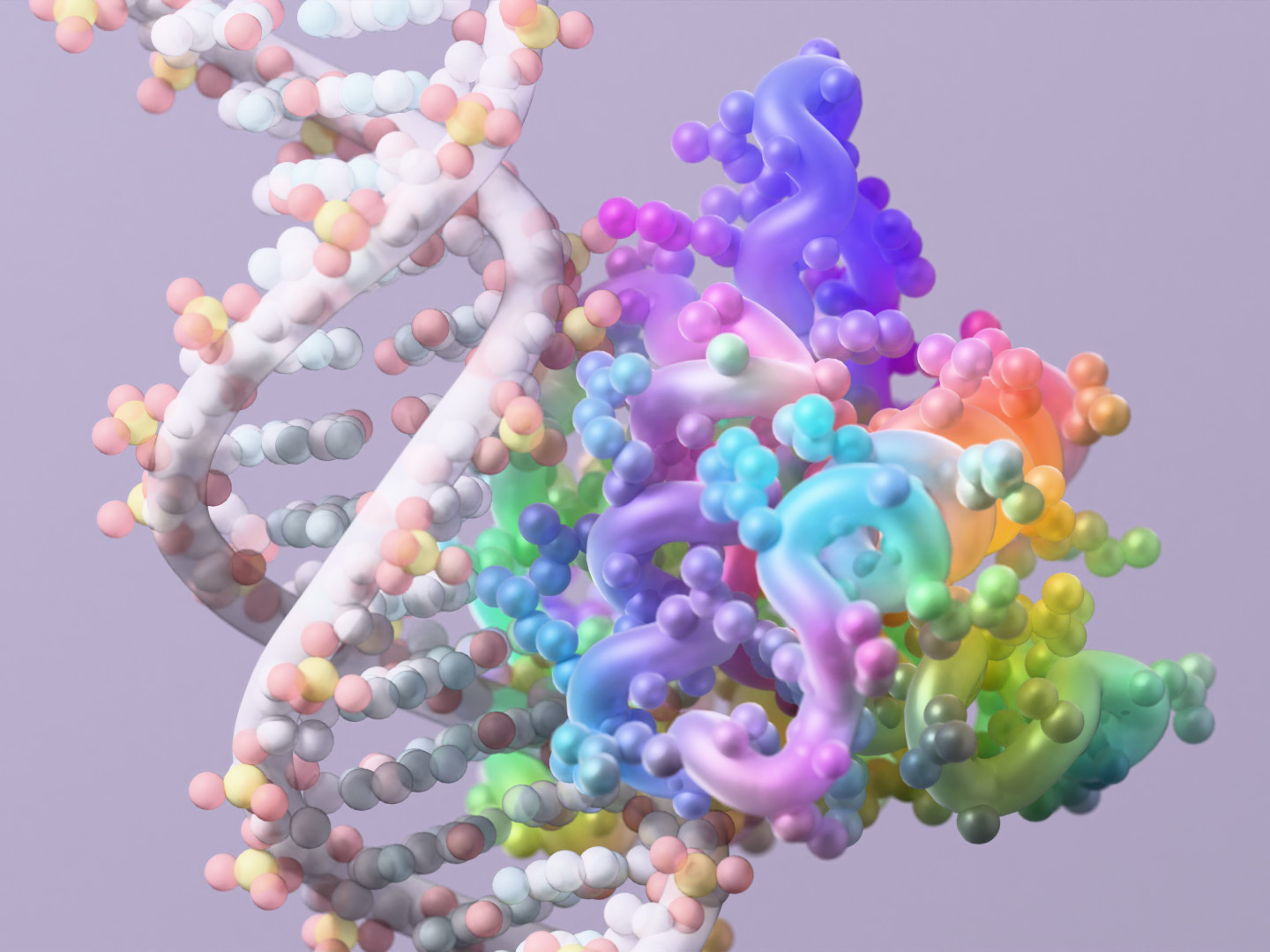
A protein created by RFdiffusion3, a newly released protein design tool from Nobel laureate David Baker’s lab, interacting with DNA. (UW Institute for Protein Design / Ian C. Haydon Image)
Protein Design I
Objective:
- Learn basic concepts:
- amino acid structure
- 3D protein visualization
- the variety of ML-based design tools
Part A. Conceptual Questions
1/ How many molecules of amino acids do you take with a piece of 500 grams of meat?

2/ Why do humans eat beef but do not become a cow, eat fish but do not become fish?

3/ Why are there only 20 natural amino acids?

4a/ Can you make other non-natural amino acids? Design some new amino acids.
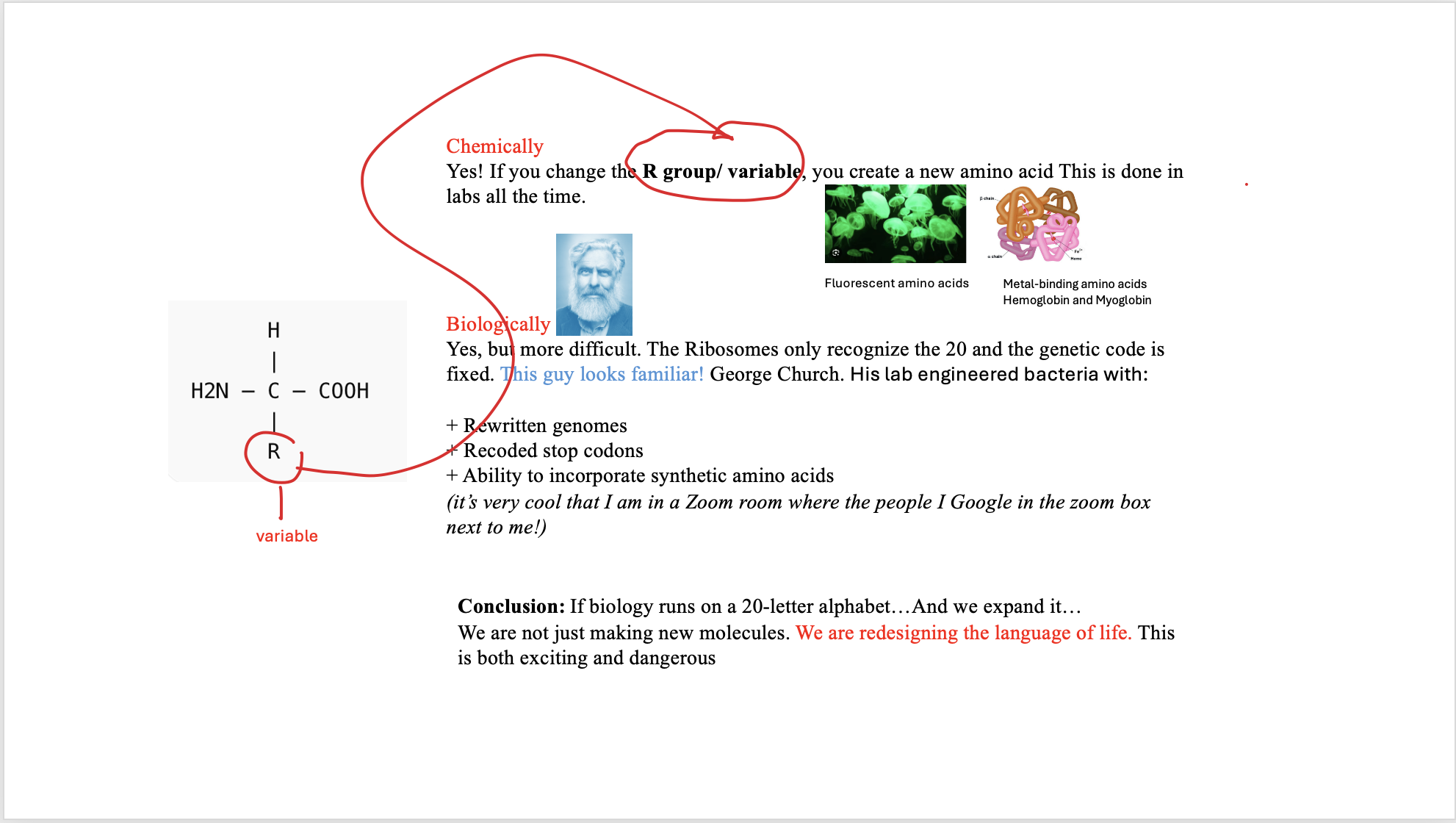
4b/ Design some new amino acids.

5/ Where did amino acids come from before enzymes that make them, and before life started?

Ran out of time. Will return!
6/ If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
7/ Can you discover additional helices in proteins?
8/ Why are most molecular helices right-handed?
9/ Why do β-sheets tend to aggregate?
10/ What is the driving force for β-sheet aggregation?
11/ Why do many amyloid diseases form β-sheets?
12/ Can you use amyloid β-sheets as materials?
13/ Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
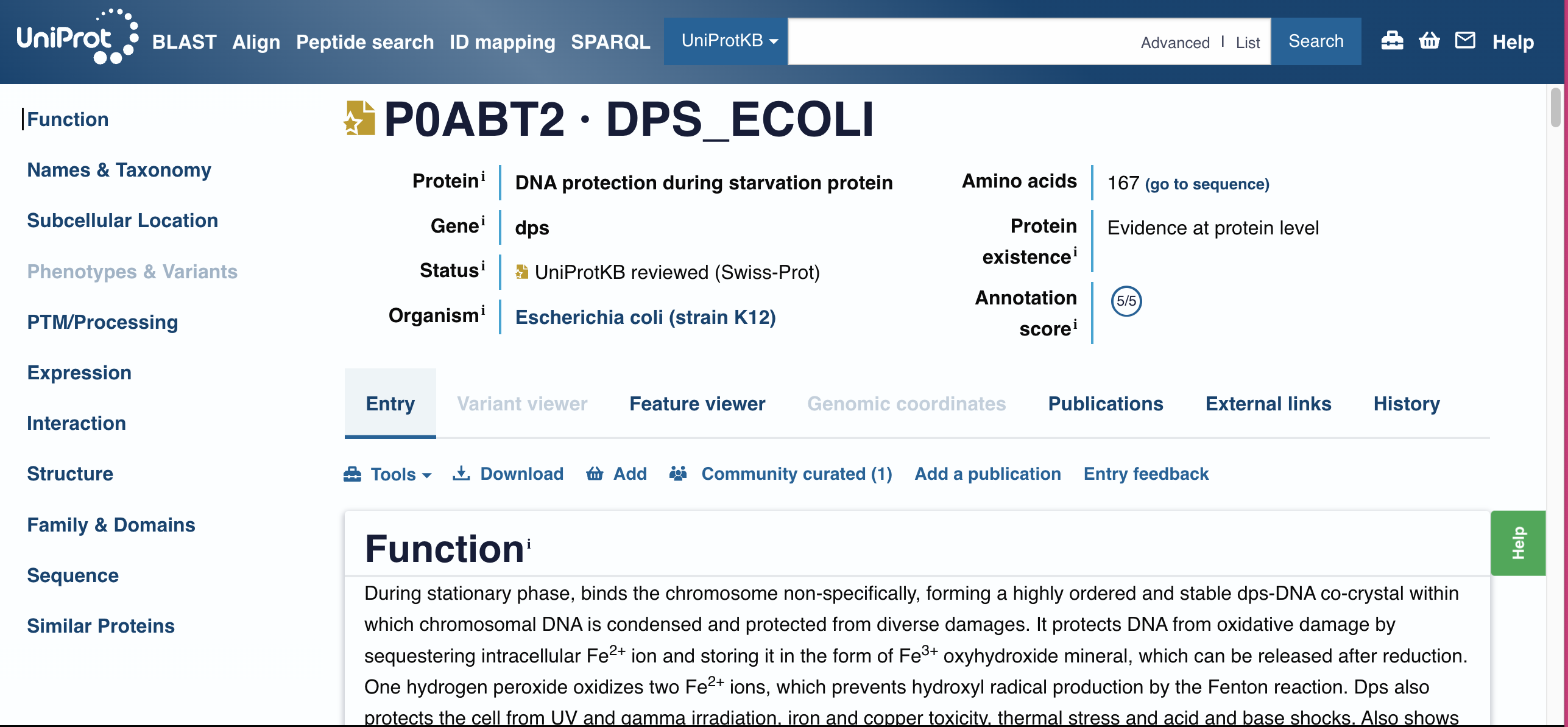
Dps (DNA-binding protein from starved cells)
I selected it because Dps is a bacterial stress protein that shows up strongly when cells are under starvation, oxidative stress, and other harsh conditions—exactly the kinds of conditions microbes face in disrupted environments (like post-fire soils). It binds and compacts DNA, helping preserve genetic information when conditions are bad. That’s very “molecular archive” which is something I proposed in mt final project. In addition, I became really interested in DNA storage after I went to the first BIOPUNK lab meeting. All 16 GB of English Wikipedia have been encoded into DNA? Dps is a natural DNA packaging and protection system. Dps is a gorgeous bridge between information and material protection before a fire.
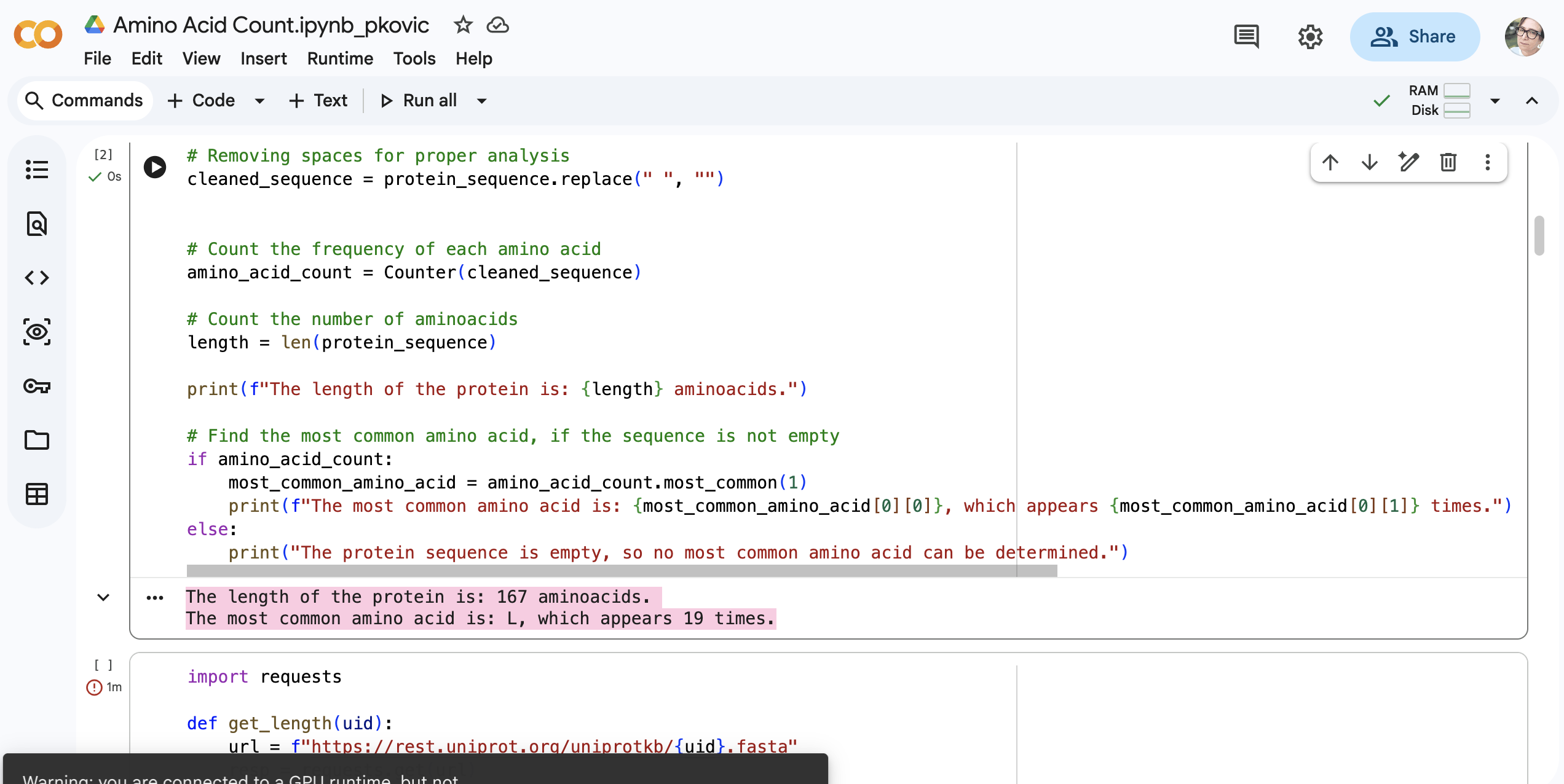
Identify the amino acid sequence of your protein.
MSTAKLVKSKATNLLYTRNDVSDSEKKATVELLNRQVIQFIDLSLITKQAHWNMRGANFIAVHEMLDGFRTALIDHLDTMAERAVQLGGVALGTTQVINSKTPLKSYPLDIHNVQDHLKELADRYAIVANDVRKAIGEAKDDDTADILTAASRDLDKFLWFIESNIE
How long is it?
715 amino acids The length of the protein is: 167 aminoacids?
What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The most common amino acid is: L, which appears 19 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
250
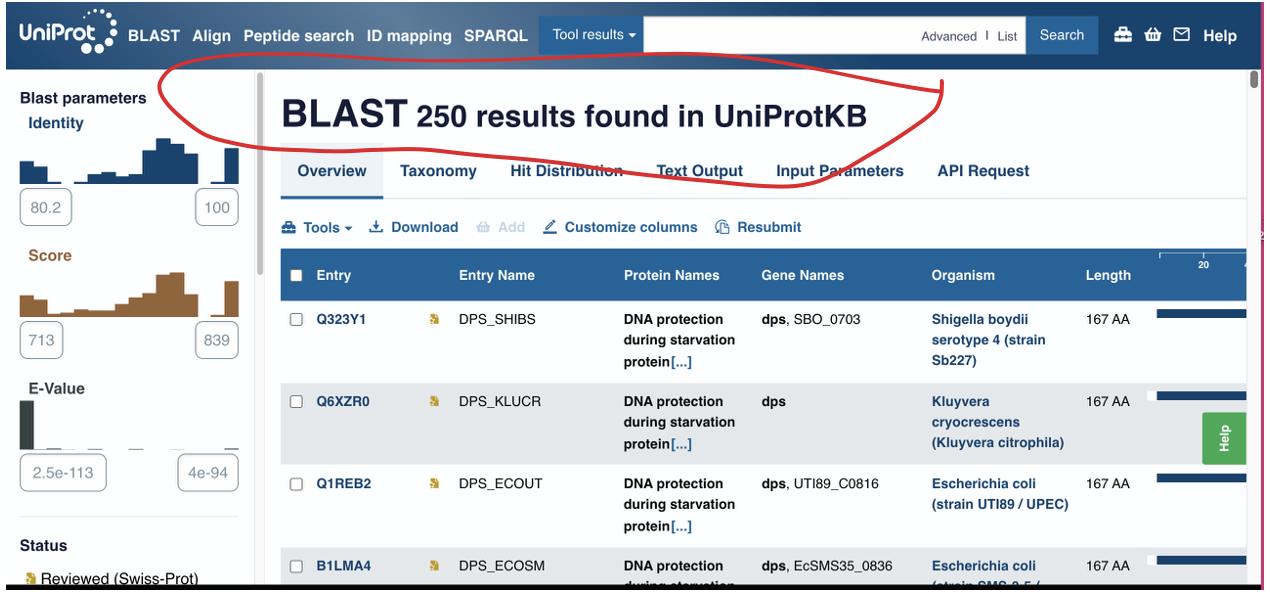
Does your protein belong to any protein family?
Dps / Ferritin-like superfamily Dps family (DNA-binding proteins from starved cells)
Identify the structure page of your protein in RCSB
The structure of Dps from Escherichia coli can be found in the RCSB Protein Data Bank under PDB ID 9ZC2. The structure was determined using single-particle cryo-electron microscopy at 1.3 Å resolution, indicating excellent structural quality.
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
It is 1.3 Å resolution. That’s good@
Are there any other molecules in the solved structure apart from protein?
Ligand Interaction (FE)
Does your protein belong to any structure classification family?
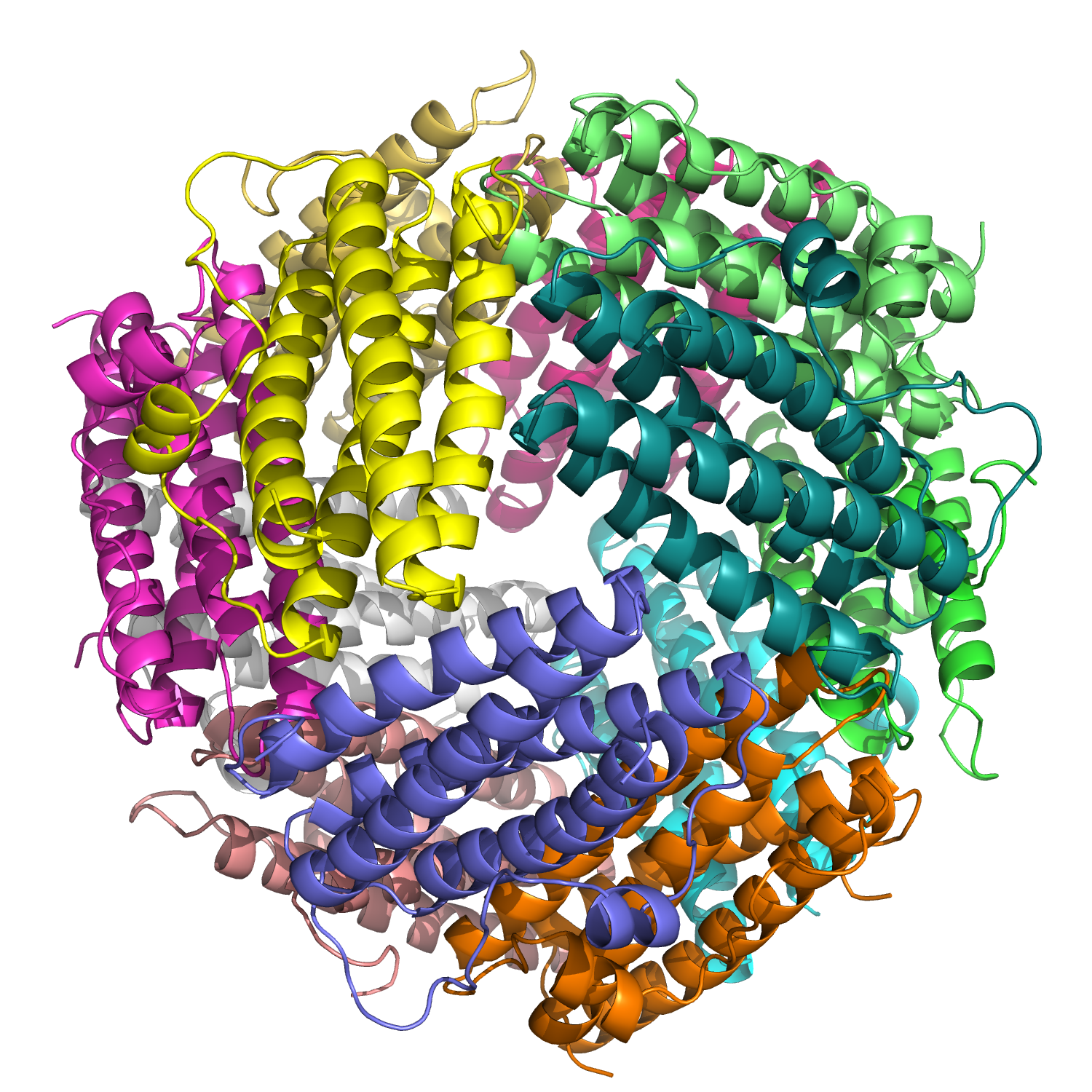
Dps belongs to the ferritin-like structural fold and assembles into a homo-dodecamer (A12) with tetrahedral symmetry. The biological assembly consists of 12 identical alpha-helical subunits forming a hollow spherical cage, characteristic of the ferritin-like superfamily.The structure contains iron (Fe) ions in addition to the protein subunits, consistent with the iron-binding function of Dps.
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
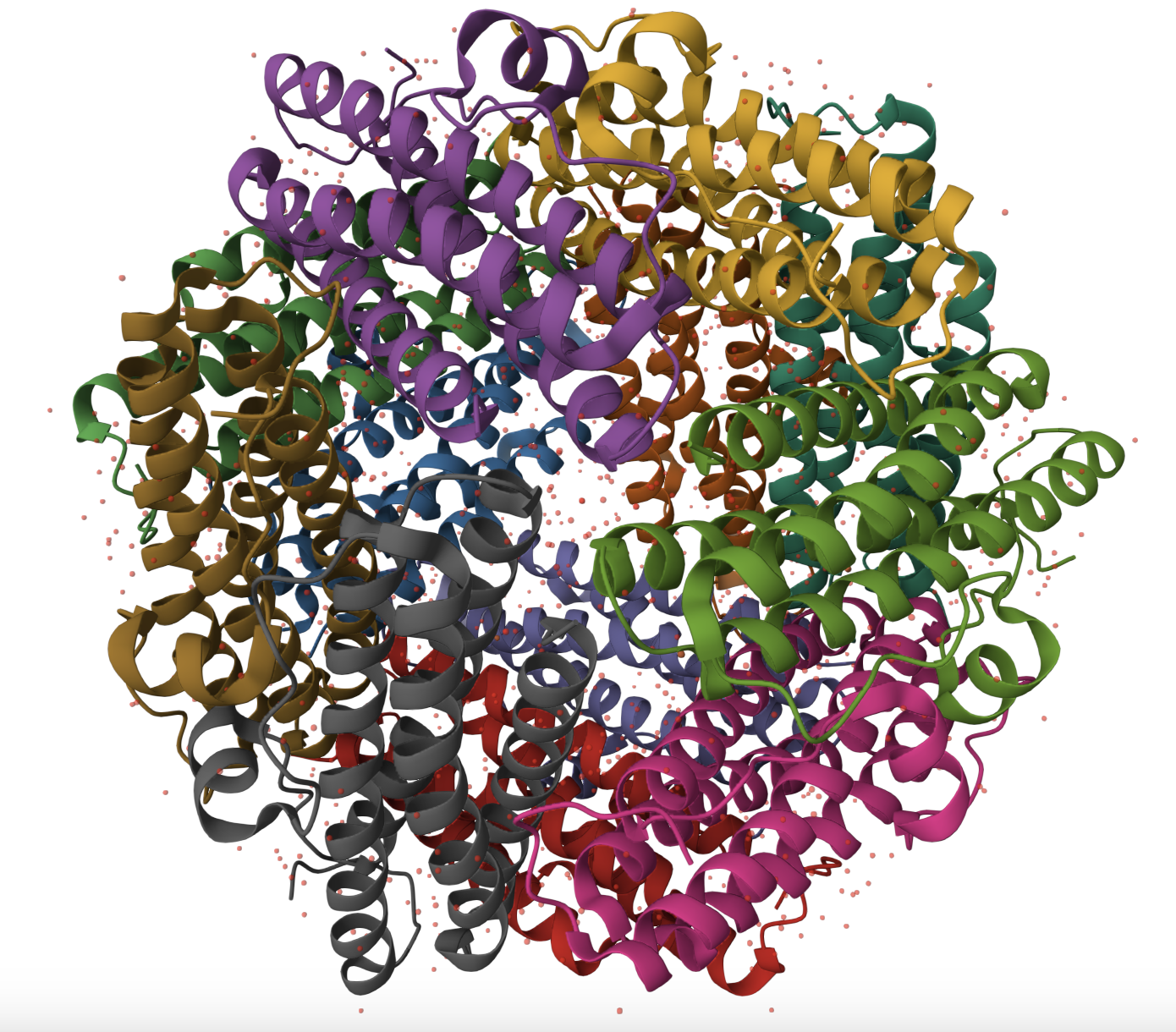
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Dps =
- Mostly alpha helices
- Connected by short loops
- Assembled into a 12-subunit spherical cage
- With metal ions inside
Color the protein by secondary structure. Does it have more helices or sheets?
Completely helices
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
A moderate amount of evenly distributed tiny dots. Chat gPT says: Coloring the protein by residue type reveals that hydrophobic residues are predominantly buried within the interior of each helical subunit, stabilizing the protein core. Hydrophilic and charged residues are enriched on the outer surface and within the internal cavity. This distribution is consistent with Dps being a soluble cytoplasmic protein that binds DNA and coordinates iron, both of which require surface-exposed charged residues
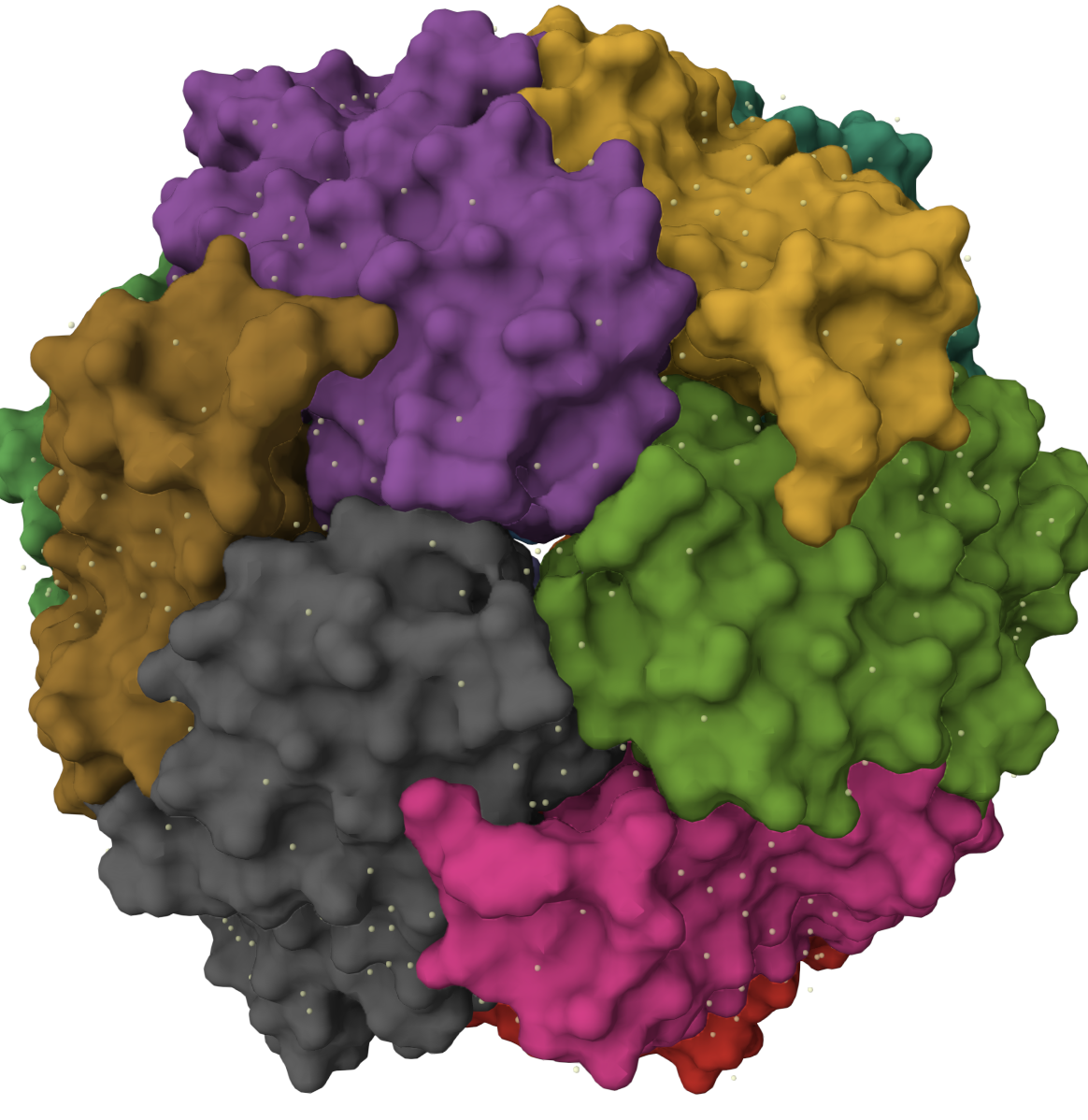
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
I see a center hole and small “insetion like shapes between the sub unites.Chat GPT says Surface visualization reveals that Dps forms a hollow spherical assembly with a large central cavity. Small pores and indentations are visible at the interfaces between subunits, particularly at symmetry axes. These openings likely facilitate iron transport into the internal cavity, consistent with the protein’s role in iron sequestration and stress protection.
Part C. Using ML-Based Protein Design Tools
Assignees for this section MIT/Harvard students Required Committed Listeners Required In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU. Choose your favorite protein from the PDB. We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website: C1. Protein Language Modeling Picture Source: Bordin, Nicola et al (2023). Novel machine learning approaches revolutionize protein knowledge. Trends in Biochemical Sciences, Volume 48, Issue 4, 345 - 359 Picture Source: Bordin, Nicola et al (2023). Novel machine learning approaches revolutionize protein knowledge. Trends in Biochemical Sciences, Volume 48, Issue 4, 345 - 359
Deep Mutational Scans Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. Can you explain any particular pattern? (choose a residue and a mutation that stands out) (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment. Latent Space Analysis Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods: do they approximate similar proteins? Place your protein in the resulting map and explain its position and similarity to its neighbors. C2. Protein Folding Picture Source: Lin et al (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Picture Source: Lin et al (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model.
Folding a protein Fold your protein with ESMFold. Do the predicted coordinates match your original structure? Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations? C3. Protein Generation Picture Source: 1. Post from Sergey Ovchinnikov 2. Roney, Ovchinnikov et al (2022). State-of-the-art estimation of protein model accuracy using AlphaFold. Phys. Rev. Lett. 129, 238101 Picture Source: 1. Post from Sergey Ovchinnikov 2. Roney, Ovchinnikov et al (2022). State-of-the-art estimation of protein model accuracy using AlphaFold. Phys. Rev. Lett. 129, 238101
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one. Input this sequence into ESMFold and compare the predicted structure to your original. Part D. Group Brainstorm on Bacteriophage Engineering Assignees for this section MIT/Harvard students Optional Committed Listeners Required Find a group of ~3–4 students Read through the Phage Reading material listed under “Reading & Resources” below. Review the Bacteriophage Final Project Goals for engineering the L Protein: Increased stability (easiest) Higher titers (medium) Higher toxicity of lysis protein (hard) Brainstorm Session Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”). Write a 1-page proposal (bullet points or short paragraphs) describing: Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”). Why do you think those tools might help solve your chosen sub-problem? Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”). Include a schematic of your pipeline. This resource may be useful: HTGAA Protein Engineering Tools Each individually put your plan on your HTGAA website Include your group’s short plan for engineering a bacteriophage