Week # 1 Homework Principles & Practices A look at the ethics, safety and security considerations for a biological engineering application with the proposed governance policy goals and actions.
Most countries like Kenya in the developing countries have a waste problem that causes a lot of health issues to the people who live near them while damaging the ecosytem around them that creates a burden for the country in dealing with the financial implications. Synthetic genomic has made it possible through the use of biological organisms that clean up environmental waste and simultaneously produce energy, making this one of the most active fields in biotechnology often referred to as the Circular Bioeconomy. In the latest research which is moving toward Genetically Modified Organisms (GMOs) that can perform multiple tasks at once. Using CRISPR-Cas9, scientists have been able to ceate “super-microbes” that can: • Detect a specific pollutant (like a biosensor). • Break down that pollutant (bioremediation). • Synthesize a fuel molecule (valorization) simultaneously. There is the need to produce biofuels more sustainably than the traditional way with the use of synthetic biology. The problem in Kenya right now we have a lot of second hand clothes that are piled up as waste in dump site, also plastics chocking waterways and scattered all over the streets with no central place to collect them or few collection centers. E-Waste where Kenya generates over 53,000 tonnes annually creating a waste problem. The new technology from synthetic biology would help to eradicate the problem and at the same time generate energy that will help counter the large import bill for gasoline, diesel and kerosine we purchase every year.
Week # 2 Homework DNA READ, WRITE & EDIT A look at the sequencing and synthesis workflows, restriction digests and gel electrophoresis, and early genome-editing frameworks.
Part 1: Benchling & In-silico Gel Art See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details. Overview: • Make a free account at benchling.com • Import the Lambda DNA. • Simulate Restriction Enzyme Digestion with the following Enzymes: ◦ EcoRI ◦ HindIII ◦ BamHI ◦ KpnI ◦ EcoRV ◦ SacI ◦ SalI • Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. • You might find Ronan’s website a helpful tool for quickly iterating on designs!
Week # 3 Lab Automation LAB AUTOMATION To get hands-on (or at least code-on) with pipetting robots.
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot. 0. Review this week’s recitation and this week’s lab for details on the Opentrons and programming it. 1. Generate an artistic design using the GUI at opentrons-art.rcdonovan.com. 2. Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons. ◦ You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept. ◦ If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead. Ask for help early! 3. If the Python component is proving too problematic even with AI and human assistance, download the full Python script from the GUI website and submit that: Use the download icon pointed to by the red arrow in this diagram. The Python component was problematic and I sent the the python script (1 OTDesign_02-26-26_22-49-52.py)
Week # 4 Protein Design Part I PROTEIN DESIGN PART I To look at how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions.
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip) How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
For a Tilapia Fish: Assuming : meat = 20% protein by weight; average amino acid ≈ 100 Da (g/mol). Calculation: • Protein mass = 500 g × 0.20 = 100 g • Moles of amino-acid residues = 100 g ÷ 100 g·mol⁻¹ = 1.00 mol • Number of amino-acid molecules using Avogadro’s number ≈ 1.00 × ≈ 6.02 × 1023 = 6.02 × 1023 amino-acid molecules. Why do humans eat beef but do not become a cow, eat fish but do not become fish? The beef meat is in the form of amino acids that our body needs which is broken down by the enzymes in our stomach to the amino acids required by our body. The amino acids are the building blocks of DNA. Beef also provides protein, zinc and several D vitamins used for muscle health, iron that boosts our immune system
Week # 5 Protein Design Part II PROTEIN DESIGN PART II To learn how cutting-edge AI and protein language models are used to design functional proteins and peptides “in silico”.
Part A: SOD1 Binder Peptide Design (From Pranam) Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation. Your challenge: 1. Design short peptides that bind mutant SOD1. 2. Then decide which ones are worth advancing toward therapy. You will use three models developed in our lab: • PepMLM: target sequence-conditioned peptide generation via masked language modeling • PeptiVerse: therapeutic property prediction • moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Week # 6 Genetic Circuits Part I GENETIC CIRCUITS PART I To learn core molecular biology tools and techniques for processing and assembling DNA, including PCR and Gibson Assembly.
Assignment: DNA Assembly Answer these questions about the protocol in this week’s lab: What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion High-Fidelity PCR Master Mix is a comprehensive formulation that supplies all the essential components required for precise and efficient DNA amplification through the polymerase chain reaction (PCR). The mixture contains Phusion polymerase, an enzyme renowned for its exceptional accuracy in synthesizing new DNA strands during the amplification process. It also includes deoxynucleotide triphosphates (dNTPs), which serve as the molecular building blocks that polymerase incorporates into the growing DNA chains. Additionally, magnesium chloride (MgCl₂) is present as a critical cofactor—an enabling molecule that the polymerase enzyme requires to function optimally and catalyze the formation of new DNA bonds. Finally, the formulation includes a reaction buffer solution that maintains the proper chemical environment throughout the PCR process. This buffer preserves stable pH levels and regulates salt concentration, ensuring that all enzymatic reactions proceed smoothly and that the overall amplification process achieves maximum efficiency. In essence, Phusion High-Fidelity PCR Master Mix eliminates the need to manually combine individual components—it is a ready-to-use formulation where all necessary ingredients are already optimized and proportioned for reliable, high-fidelity DNA amplification.
Week # 7 Genetic Circuits Part II GENETIC CIRCUITS PART II To learn neuromorphic genetic circuits, showing how engineered gene networks can implement neural-network “perceptron”-like computation and learning
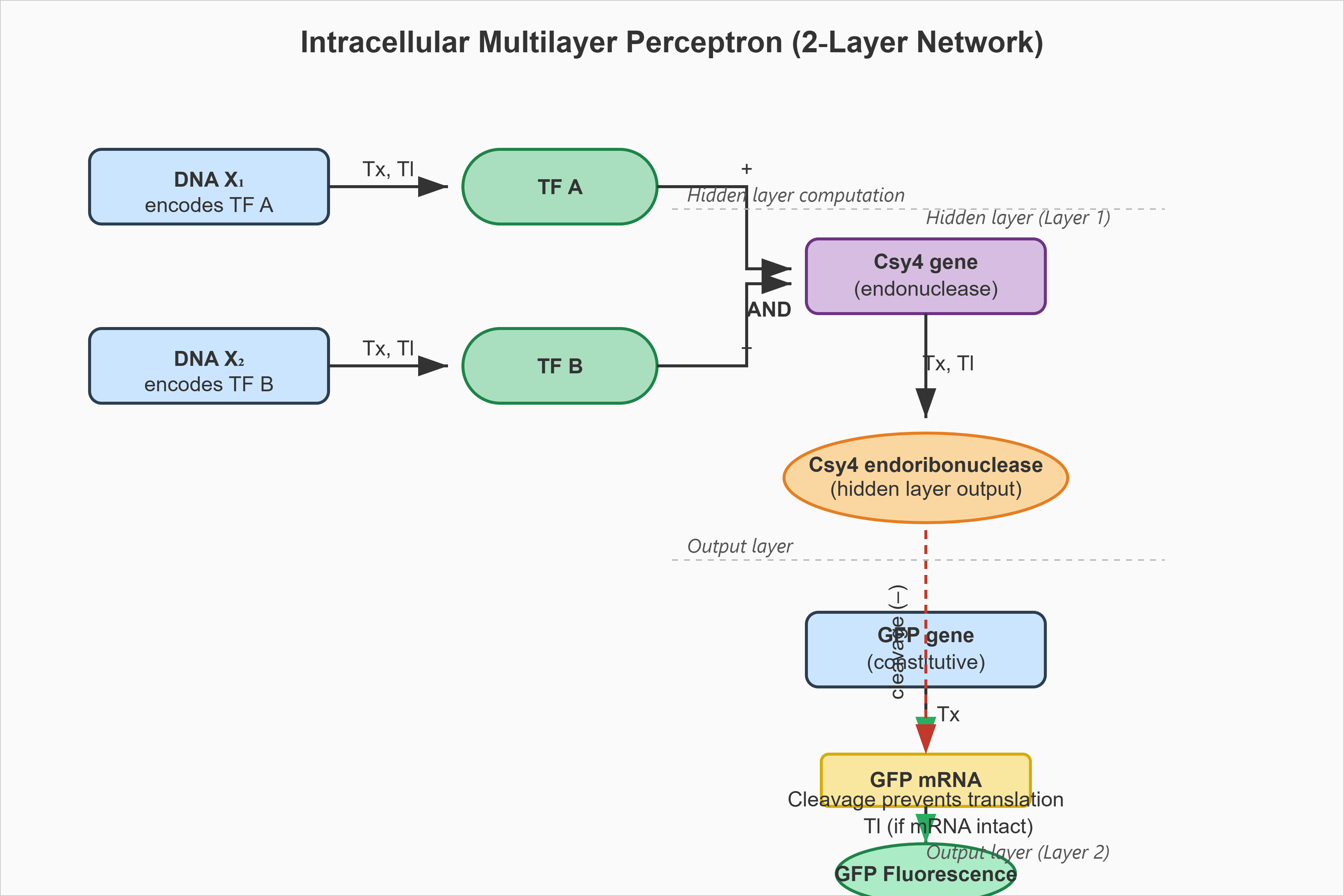
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? To understand the advantages of IANNs (In silico Artificial Neural Networks / Integrated Artificial Neural Networks in synthetic biology) over traditional Boolean genetic circuits, it helps to look at how biological computing is evolving. Traditional genetic circuits act like classic computer chips: they take inputs (like the presence of a specific molecule) and use logic gates (AND, OR, NOT) to produce a definitive, binary ON/OFF response. IANNs, however, mimic the brain’s neural networks using biological components. Here is why IANNs are a massive step up from traditional Boolean genetic circuits:
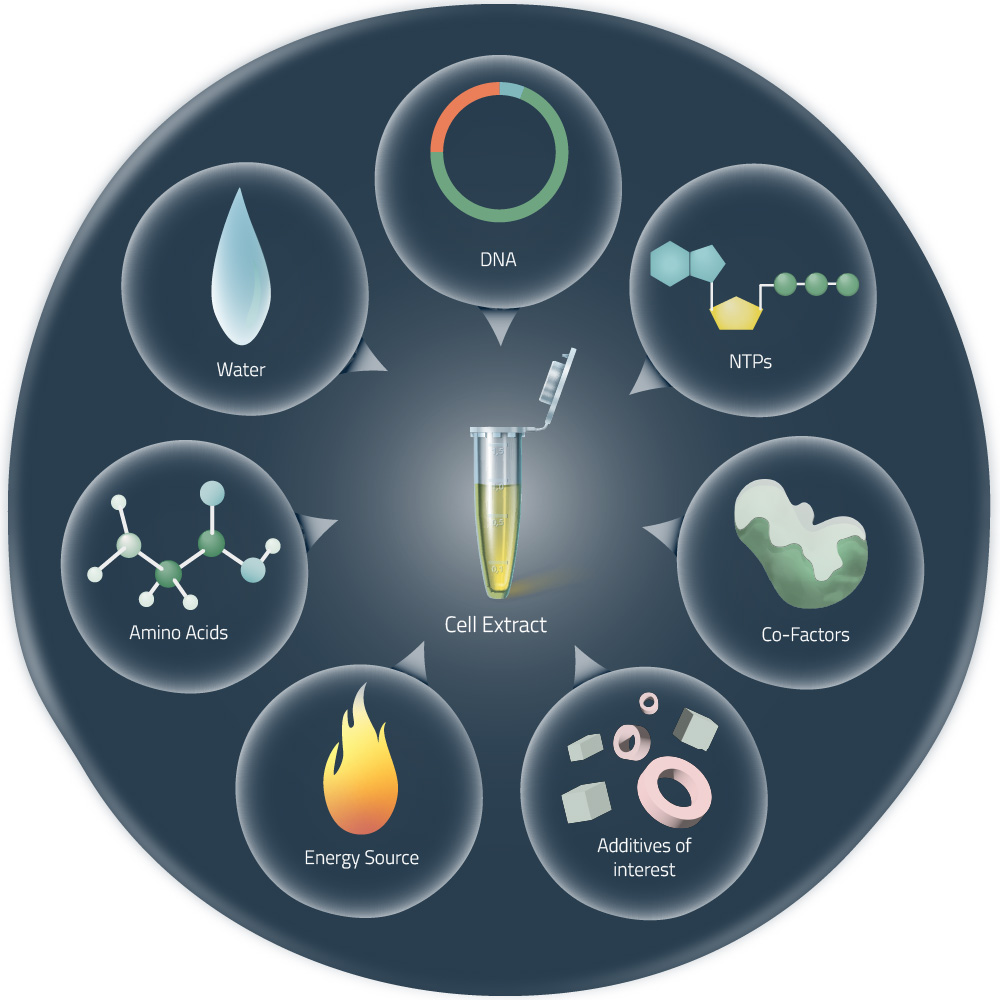
Week # 9 Cell Free Systems CELL FREE SYSTEMS To learn synthesis of proteins using cellular machinery outside of a cell.
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-Free Protein Synthesis Advantages Cell-free protein synthesis (CFPS) provides substantial benefits compared to conventional cell-based protein production methods, particularly in terms of experimental flexibility and precise control over reaction parameters. In contrast to traditional in vivo approaches that require cell transformation, growth in culture media, and cell disruption, CFPS enables rapid protein production without these intermediate steps, significantly accelerating the research timeline.
Week # 10 Imaging and Measurement IMAGING AND MEASUREMENT To learn a range of advanced technologies to do precision measurement of proteins at atomic scales, characterizing chemical composition, and detecting protein sequence and structure.
Homework: Waters Part I — Molecular Weight
Week # 11 Building Genomes BUILDING GENOMES To inspire collaboration and creativity while designing a scientifically rigorous cell-free fluorescent protein optimization experiment together.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork 1. Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST. ◦ A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse. https://rcdonovan.com/synbiobeta I contributed 3 on the in the middle of the artwork
Subsections of Homework
Week 1: PRINCIPLES & PRACTICES
Week # 1 Homework
Principles & Practices
A look at the ethics, safety and security considerations for a biological engineering application with the proposed governance policy goals and actions.
Most countries like Kenya in the developing countries have a waste problem that causes a lot of health issues to the people who live near them while damaging
the ecosytem around them that creates a burden for the country in dealing with the financial implications. Synthetic genomic has made it possible through
the use of biological organisms that clean up environmental waste and simultaneously produce energy, making this one of the most active fields in
biotechnology often referred to as the Circular Bioeconomy.
In the latest research which is moving toward Genetically Modified Organisms (GMOs) that can perform multiple tasks at once. Using CRISPR-Cas9,
scientists have been able to ceate “super-microbes” that can:
• Detect a specific pollutant (like a biosensor).
• Break down that pollutant (bioremediation).
• Synthesize a fuel molecule (valorization) simultaneously.
There is the need to produce biofuels more sustainably than the traditional way with the use of synthetic biology. The problem in Kenya right now we have
a lot of second hand clothes that are piled up as waste in dump site, also plastics chocking waterways and scattered all over the streets with no
central place to collect them or few collection centers. E-Waste where Kenya generates over 53,000 tonnes annually creating a waste problem. The new
technology from synthetic biology would help to eradicate the problem and at the same time generate energy that will help counter the large import bill
for gasoline, diesel and kerosine we purchase every year.
The research being done on biological “waste-to-fuel” systems has now led to a major shift from laboratory “proof of concept” to integrated
biorefineries where organisms don’t just clean the environment,they act as the living hardware for fuel production.
The discovery of a technology through research of Microbial Fuel Cell has made it possible to turn waste into electricity or hydrogen directly without
burning anything which is being piloted in waste water plants. Used clothes wastes from Gikomba and Dandora can be turned into Bioethanol, Sewage & Heavy
metals from Nairobi River can be turned into Biofuels, Hydrogen or electricity, Plastics in the creation of Bio-oil, Organic waste producing Biomethane.
Since the GMO organisms will be bioengineerd to scout for the waste in different damp sites there would be the need to ensure the environment around the site
is protected, with the technology being used to make sure the community around benefit from it and have the area restored and once done the organisms can be
engineered to sense the completion of there task and intergrate into the ecosystem without harming it.
Environment
• How would the damp site be free of the bioengineered organisms after conversion to biofuel?
• How will the biofuel be evacuated fron the damp site without harming the ecosystem?
Equitable use of technology
• Will the GMO be made available to the public?
• How will the technology be used in the area where it is needed and will the community benefit from the biofuel?
Biosecurity
• The technology needs to be safe to handle and use without leading to biological disasters.
• The GMO should not be able to mutate and create a situation where they alter other organisms in the ecosystem.
Looking at the three different potential governance “actions” with the four aspects below (Purpose, Design, Assumption, Risks of Failure & “Success”)
Researchers
• There is the need to show the standards and how the super microbes will be handled and produced either locally or imported.
• Publications from reputable institutions to show how they are able to use similar microbes in a safe way with a manual compiled for the laboratory
use of them.
• A database that has all the known super microbes that are able to produce and how to be handled, the risks and best practices.
Microbiome Companies
• There needs to be a way the regulators can look into auditing where the companies are following the law and standards set by researchers.
• Public participation is needed to educate the community in the areas where they plan to use their technology.
• The need to be informed each stage on what is going on with the project once it commences till the end.
Government Regulators
• The agencies tasked to monitor will assess using their standards and gauge on what needs to be done with the super microbes registered on the
quantities used.
• Always monitoring that safety is observed for the audits conducted abruptly without notice to ensure safety of the products they claim to use and
guidelines set.
• Ensure the people on the site working are of the recommended number and not overcrowded and following ecological standards and public participation.
Waste to Biofuels
Researchers
Microbiome Companies
Government Regulators
Enhance Biosecurity
• Monitoring
1
1
1
• Response
1
Equality of use
• By preventing incident
3
3
1
• By helping respond
4
4
1
Environment Protection
• Monitoring
1
1
1
• Response
4
4
4
Other considerations
• Minimizing costs and burdens to stakeholders
1
• Feasibility?
1
1
• Not impede research
1
• Promote constructive applications
1
1
The researchers would be the laboratories that test and develop the microbes either in an institution like a University or private entity. The Microbiome
companies design the microbiomes and have organisnm engineers who develop new organism using biology,they vary in size from small to large scale. The
government regulators look into getting approvals and can use third party firms to enforce the regulations.
To get approval in the use of synthetic genomics there are three primary regulators with the process streamlined under the 2022 Genome editing guidelines.
The first step is The National Biosafety Authority (NBA) where one gets the permit from for the lab research and the risk assessment. The second step is
National Environment Management Authority (NEMA) for the environmental impact assessment and the need for a permit to discharge the treated byproduct and
bioprospecting permit for microbes. The third step is The Energy & Petroleum Regulatory Authority(EPRA) where you get the Biofuel production license,
Construction Permit and KEBS standardization.
The economic risks would be the bioavailability of plastic as an engineered microbe cannot eat a plastic bottle unless it is shredded and pretreated
(Hydrothermal pretreatment).
With the introduction of Carbon Credits by the Kenyan governmet in the Climate Change Act,can lead to a saving of 30% towards the operational costs. Based on the scoring above the goverment would need to know how the super microbes function and have the community know the benefits of the use of them in
clearing the waste. The Researchers and Microbiome companies need to return at least 5-10 % of the biofuels as a way for giving back to the community so as
to minimize pushback. Since there is the incentive offered by the government on the use of local microbe,research can be done to see how they can be
engineered to reduce the initial set up cost.
Homework Answers for Professor Jacobson
Nature’s machinery for copying DNA is called polymerase.
What is the error rate of polymerase? The error rate of polymerase is 1 mistake per 10*6 base pairs.
How does this compare to the length of the human genome? DNA polymerase which is an enzyme is approximately 10 – 15 nanometers (nm) in length while the
human genome which is the template is approximately 2 meters when stretched out. A scale ratio of 1: 108 x longer (200m /10 nm = 20,000,000x)
How does biology deal with that discrepancy? It does this by not relying on a single enzyme. It uses a highly organized, factory-like system with four key
strategies:
The first is the multiple origins of replication instead of a single one, each origin has two replication forks creating a replication bubble where
thousands of DNA polymerase molecules can work simultaneously across all chromosomes where each has a small copying segment.
The second, is it doesn’t work alone as is part of a complex of proteins known as a replisome a key component is the sliding clamp (PCNA in humans)
where the donut- shaped protein encircles the DNA and tethers the polymerase to the template which leads to the increase in processivity making one
polymerase can be able to add thousands of nucleotides without falling off, turning it from a slow inefficient enzyme to a high speed, long distance
replicator.
Third, different polymerases have specialized roles with the leading strand being synthesized continuously by a highly processive polymerase and the
lagging strand synthesized in short Okazaki fragments that require different coordinated processes. The main replicative polymerases have proofreading
ability (3’→5’ exonuclease activity). Where they are able to mmediately back up and fix a mistake, ensuring speed doesn’t come at the cost of
catastrophic error rates (final error rate: ~1 in 10 billion bases)
Fourth, the compartmentalization and packaging of DNA has mabe the 2 meters of DNA not in a loose tangle. It istightly wound and packaged with proteins
into chromosomes inside the nucleus (~10 µm wide) where the replisome has to navigate this dense chromatin structure, with the Helicases unwinding it,
topoisomerases relieve twisting stress, and other proteins modify the packaging to allow access. This organization makes it possible to bring distant
genomic regions into physical proximity while making the logistics of finding origins and assembling machinery more efficient.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of
these different codes don’t work to code for the protein of interest? The different ways to code an average human protein is 10*214 , which is a very
large number and impractical. Most of the codes fail in producing a functional protein in the living cell. The reason why most of the codes don’t work
is not all of them are created equal where codon usage bias can affect translaton speed leading to misfolded or incomplete proteins. The second, is where
mRNA has a secondary structure where it folds back on itself and form shapes like hair pins and loops and if a randomly chosen DNA sequence creates an
mRNA strand that folds at the beginning, the ribosome can’t get on the track and start reading. The third is splicing (cutting and pasting) RNA before
it is translated where specific sequences that signal where an intron (junk DNA) finishes ans an exon(coding DNA) starts, with many containing the code
for a “splice-site” in the middle of the gene and throw away, leaving a fragment which is a useless protein. The fourth, DNA is chemically stable cause
of the G-C pairs, where it is held together by hydrogen bonds, G-C pairs have three bonds, while A-T pairs only have two. Where too high of a G-C content the DNA ‘zipper’ would be really hard to open whereas a low A-T rich would make it unstable. The sixth is the human immune system through evolution,
recognizes the Cytosine followed by a Guanine (CpG) as a pattern of a viral or bacterial infection and if the code has too many of these “CpG islands” triggers the cell that its under attack by a virus and could lead to gene silencing or an inflammatory response to “shield” itself.
Homework Answers for Dr. LeProust
What’s the most commonly used method for oligo synthesis currently? The oligonucleotide synthesis is the phosphoramidite method, using thr Solid Phase
Synthesis (SPS). Naturally DNA builds in the 5’ to 3’ direction, while laboratory method builds the chain backward—from the 3’ end to the 5’ end. The
method has a four step cycle, where it happens on a solid support (usually controlled-pore glass or polystyrene beads). The addition of a single
nucleotide, the machine must complete a full revolution of these four chemical steps:
The first step known as Deblocking (Deprotection) where the nucleotide is already attached to the solid support. Its 5’-hydroxyl group is “blocked” by
a protective chemical called DMT (dimethoxytrityl) to prevent it from reacting prematurely. An acid is added to wash away the DMT, leaving a “naked” 5’-
OH group ready for the next link.
The second step whrere the next nucleotide (a phosphoramidite monomer) is added to the chamber along with an activator. The 5’ end of the growing chain
binds to the 3’ end of the new monomer. Leading to an Efficiency usually >99%, but in chemistry, 100% is impossible.
The third step is capping where a small percentage of the chains that failed to couple in Step 2, they must be “capped” (usually with acetic anhydride).
This prevents them from reacting in future cycles, which would result in “deletion mutants”—strands that are missing a middle letter.
The fourth step is oxidation where a bond formed during coupling is a bit unstable (a phosphite triester). An iodine solution is added to oxidize this
bond, turning it into a stable phosphate triester which is the familiar backbone of DNA.
Why is it difficult to make oligos longer than 200nt via direct synthesis? The phosphoramidite synthesis has a specific limit where errors accumulate
with every step, making it hard to build a single strand longer than 200–300 bases with high purity. Since every time you add a base, there is the of
lose a tiny bit of your starting material because the reaction never goes to 100% completion.
Secondly, the Capping step of the cycle stops “failed” chains from growing further which leads to Purifying the “perfect” sequence away from the
“almost perfect” ones becoming a nightmare. Comparing it to trying to find a specific grain of sand in a pile of slightly smaller grains of sand
Thirdly, The first step of the cycle (Deblocking) uses an acid to remove the protective DMT group that DNA doesn’t actually like. Every time you
expose the growing chain to acid, there is the risk of depurination where you accidentally snip a Guanine or Adenine base off the sugar backbone. If for
comparison, a 200nt strand being subjected to the first base to 200 rounds of acid wash. By the time you reach the end, the beginning of your sequence is
often chemically “chewed up.”
Fourth, the time and mechanical failure as synthesizing 200 bases takes a long time (often 10–15 hours) and the longer the run, the higher the
possibility of a “mechanical failure” where a bubble in the line, a slight drop in temperature, or a reagent running dry. Bringing about a failure at base 190 leading to wasting the entire 14-hour run and all the expensive chemicals used up to that point.
Why can’t you make a 2000bp gene via direct oligo synthesis?
First, its statistically impossible even if your machine was the most efficient running 99.5% efficiency per base addition where only 0.004% of the
molecules in your final mixture would actually be the correct 2,000bp length. The other 99.996% would be “truncated” sequence with broken fragments that
are missing one or more bases.The massive mound of chemical errors makes it impossible to find the perfect bases.
Second, there is the physical crowding and stuttering while the DNA chain grows to 2,000 bases, it doesn’t just stay a neat and organized. It starts to
fold, tangle, and stick to the solid support (the glass or plastic bead it’s being built on). This leads to the “top” of the growing DNA chain becomes
physically hard for new chemicals to reach because it’s buried in a crowd of other DNA strands. At the addition of the 2,000th base, the 1st base has
been washed in acid 2,000 times making the chemical integrity of the beginning of the gene completely compromised.
Homework Answers for George Church
(Using Google & Prof . Church’s slide #4) What are the 10 essential amino acids in all animals and how does this affect my view of the “ Lysine
Contigency”? Animals require 10 essential amino acids from their diet since they cannot synthesize them. These are universally needed across all animals
like mammals, birds, and fish for protein synthesis and growth.
Essential Amino Acids List
The 10 essential ones, remembered by the acronym “PVT TIM HALL,” are:
Phenylalanine
Valine
Tryptophan
Threonine
Isoleucine
Methionine
Histidine
Arginine
Leucine
Lysine
Lysine Contingency as explained in “Jurassic Park”, was the “lysine contingency” genetically modified dinosaurs to be unable to produce lysine, an
essential amino acid, making them dependent on park-supplied supplements to prevent escape and survival in the wild. It failed scientifically as all
animals, including dinosaurs as modeled, already couldn’t synthesize lysine and obtain it from protein-rich foods like meat or plants, abundant in
ecosystems. Removing synthesis offers no control, as lysine is widespread, rendering the plan ineffective as dinosaurs would simply eat lysine-
containing prey or vegetation.
The use of LLM to help with finding information and reporting
Week 2: DNA READ, WRITE, AND EDIT
Week # 2 Homework
DNA READ, WRITE & EDIT
A look at the sequencing and synthesis workflows, restriction digests and gel electrophoresis, and early genome-editing frameworks.
Part 1: Benchling & In-silico Gel Art
See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details. Overview:
• Make a free account at benchling.com
• Import the Lambda DNA.
• Simulate Restriction Enzyme Digestion with the following Enzymes:
◦ EcoRI
◦ HindIII
◦ BamHI
◦ KpnI
◦ EcoRV
◦ SacI
◦ SalI
• Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
• You might find Ronan’s website a helpful tool for quickly iterating on designs!
I was able to make a free account on Benchling and imported the Lamda DNA sequence as seen below.
A pattern below showing the simulation for each of the enzymes producing different fragment patterns created from the restriction enzyme digest with the following enzymes:
◦ EcoRI
◦ HindIII
◦ BamHI
◦ KpnI
◦ EcoRV
◦ SacI
◦ SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
The instructions on the lab experiment designed in Part 1 and outlined in the Gel Art: Restriction Digests and Gel Electrophoresis protocol. Since I had
no access to the lab experiment, the simulation of the gel shows how Lamda DNA would have been digested by the seven different restriction enzymes as
seen from the gel electrophoresis plate. The individual lanes show how each enzyme cut the DNA, using the NEB2-log as the ladder on the left as a
reference for size. The patterns are used to verify the sequence and map the DNA.
Part 3: DNA Design Challenge
3.1 Choose your protein.
Protein: Amyloid beta precursor protein
Organism: Homo sapiens
GenBank: BDX53017.1
AA Sequence
I chose this protein since numerous studies have placed the protein leading to a molecular pathway mechanism that leads to neurodegeneration,
synaptic failure and the clinical onset of Alzheimer’s disease.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of
the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
[Example from our group homework, you may notice the particular format — The example below came from UniProt]
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Searching Online on how to Reverse Translate, I found the following website that has the tool to help me reverse translate: https://www.bioinformatics.org/sms2/rev_trans.html with the results shown in the image below.
Reverse Translate results
Results for 770 residue sequence “BDX53017.1 amyloid beta precursor protein [Homo sapiens]” starting “MLPGLALLLL”
reverse translation of BDX53017.1 amyloid beta precursor protein [Homo sapiens] to a 2310 base sequence of most likely codons.
atgctgccgggcctggcgctgctgctgctggcggcgtggaccgcgcgcgcgctggaagtg
ccgaccgatggcaacgcgggcctgctggcggaaccgcagattgcgatgttttgcggccgc
ctgaacatgcatatgaacgtgcagaacggcaaatgggatagcgatccgagcggcaccaaa
acctgcattgataccaaagaaggcattctgcagtattgccaggaagtgtatccggaactg
cagattaccaacgtggtggaagcgaaccagccggtgaccattcagaactggtgcaaacgc
ggccgcaaacagtgcaaaacccatccgcattttgtgattccgtatcgctgcctggtgggc
gaatttgtgagcgatgcgctgctggtgccggataaatgcaaatttctgcatcaggaacgc
atggatgtgtgcgaaacccatctgcattggcataccgtggcgaaagaaacctgcagcgaa
aaaagcaccaacctgcatgattatggcatgctgctgccgtgcggcattgataaatttcgc
ggcgtggaatttgtgtgctgcccgctggcggaagaaagcgataacgtggatagcgcggat
gcggaagaagatgatagcgatgtgtggtggggcggcgcggataccgattatgcggatggc
agcgaagataaagtggtggaagtggcggaagaagaagaagtggcggaagtggaagaagaa
gaagcggatgatgatgaagatgatgaagatggcgatgaagtggaagaagaagcggaagaa
ccgtatgaagaagcgaccgaacgcaccaccagcattgcgaccaccaccaccaccaccacc
gaaagcgtggaagaagtggtgcgcgaagtgtgcagcgaacaggcggaaaccggcccgtgc
cgcgcgatgattagccgctggtattttgatgtgaccgaaggcaaatgcgcgccgtttttt
tatggcggctgcggcggcaaccgcaacaactttgataccgaagaatattgcatggcggtg
tgcggcagcgcgatgagccagagcctgctgaaaaccacccaggaaccgctggcgcgcgat
ccggtgaaactgccgaccaccgcggcgagcaccccggatgcggtggataaatatctggaa
accccgggcgatgaaaacgaacatgcgcattttcagaaagcgaaagaacgcctggaagcg
aaacatcgcgaacgcatgagccaggtgatgcgcgaatgggaagaagcggaacgccaggcg
aaaaacctgccgaaagcggataaaaaagcggtgattcagcattttcaggaaaaagtggaa
agcctggaacaggaagcggcgaacgaacgccagcagctggtggaaacccatatggcgcgc
gtggaagcgatgctgaacgatcgccgccgcctggcgctggaaaactatattaccgcgctg
caggcggtgccgccgcgcccgcgccatgtgtttaacatgctgaaaaaatatgtgcgcgcg
gaacagaaagatcgccagcataccctgaaacattttgaacatgtgcgcatggtggatccg
aaaaaagcggcgcagattcgcagccaggtgatgacccatctgcgcgtgatttatgaacgc
atgaaccagagcctgagcctgctgtataacgtgccggcggtggcggaagaaattcaggat
gaagtggatgaactgctgcagaaagaacagaactatagcgatgatgtgctggcgaacatg
attagcgaaccgcgcattagctatggcaacgatgcgctgatgccgagcctgaccgaaacc
aaaaccaccgtggaactgctgccggtgaacggcgaatttagcctggatgatctgcagccg
tggcatagctttggcgcggatagcgtgccggcgaacaccgaaaacgaagtggaaccggtg
gatgcgcgcccggcggcggatcgcggcctgaccacccgcccgggcagcggcctgaccaac
attaaaaccgaagaaattagcgaagtgaaaatggatgcggaatttcgccatgatagcggc
tatgaagtgcatcatcagaaactggtgttttttgcggaagatgtgggcagcaacaaaggc
gcgattattggcctgatggtgggcggcgtggtgattgcgaccgtgattgtgattaccctg
gtgatgctgaaaaaaaaacagtataccagcattcatcatggcgtggtggaagtggatgcg
gcggtgaccccggaagaacgccatctgagcaaaatgcagcagaacggctatgaaaacccg
acctataaattttttgaacagatgcagaac
reverse translation of BDX53017.1 amyloid beta precursor protein [Homo sapiens] to a 2310 base sequence of consensus codons.
atgytnccnggnytngcnytnytnytnytngcngcntggacngcnmgngcnytngargtn
ccnacngayggnaaygcnggnytnytngcngarccncarathgcnatgttytgyggnmgn
ytnaayatgcayatgaaygtncaraayggnaartgggaywsngayccnwsnggnacnaar
acntgyathgayacnaargarggnathytncartaytgycargargtntayccngarytn
carathacnaaygtngtngargcnaaycarccngtnacnathcaraaytggtgyaarmgn
ggnmgnaarcartgyaaracncayccncayttygtnathccntaymgntgyytngtnggn
garttygtnwsngaygcnytnytngtnccngayaartgyaarttyytncaycargarmgn
atggaygtntgygaracncayytncaytggcayacngtngcnaargaracntgywsngar
aarwsnacnaayytncaygaytayggnatgytnytnccntgyggnathgayaarttymgn
ggngtngarttygtntgytgyccnytngcngargarwsngayaaygtngaywsngcngay
gcngargargaygaywsngaygtntggtggggnggngcngayacngaytaygcngayggn
wsngargayaargtngtngargtngcngargargargargtngcngargtngargargar
gargcngaygaygaygargaygaygargayggngaygargtngargargargcngargar
ccntaygargargcnacngarmgnacnacnwsnathgcnacnacnacnacnacnacnacn
garwsngtngargargtngtnmgngargtntgywsngarcargcngaracnggnccntgy
mgngcnatgathwsnmgntggtayttygaygtnacngarggnaartgygcnccnttytty
tayggnggntgyggnggnaaymgnaayaayttygayacngargartaytgyatggcngtn
tgyggnwsngcnatgwsncarwsnytnytnaaracnacncargarccnytngcnmgngay
ccngtnaarytnccnacnacngcngcnwsnacnccngaygcngtngayaartayytngar
acnccnggngaygaraaygarcaygcncayttycaraargcnaargarmgnytngargcn
aarcaymgngarmgnatgwsncargtnatgmgngartgggargargcngarmgncargcn
aaraayytnccnaargcngayaaraargcngtnathcarcayttycargaraargtngar
wsnytngarcargargcngcnaaygarmgncarcarytngtngaracncayatggcnmgn
gtngargcnatgytnaaygaymgnmgnmgnytngcnytngaraaytayathacngcnytn
cargcngtnccnccnmgnccnmgncaygtnttyaayatgytnaaraartaygtnmgngcn
garcaraargaymgncarcayacnytnaarcayttygarcaygtnmgnatggtngayccn
aaraargcngcncarathmgnwsncargtnatgacncayytnmgngtnathtaygarmgn
atgaaycarwsnytnwsnytnytntayaaygtnccngcngtngcngargarathcargay
gargtngaygarytnytncaraargarcaraaytaywsngaygaygtnytngcnaayatg
athwsngarccnmgnathwsntayggnaaygaygcnytnatgccnwsnytnacngaracn
aaracnacngtngarytnytnccngtnaayggngarttywsnytngaygayytncarccn
tggcaywsnttyggngcngaywsngtnccngcnaayacngaraaygargtngarccngtn
gaygcnmgnccngcngcngaymgnggnytnacnacnmgnccnggnwsnggnytnacnaay
athaaracngargarathwsngargtnaaratggaygcngarttymgncaygaywsnggn
taygargtncaycaycaraarytngtnttyttygcngargaygtnggnwsnaayaarggn
gcnathathggnytnatggtnggnggngtngtnathgcnacngtnathgtnathacnytn
gtnatgytnaaraaraarcartayacnwsnathcaycayggngtngtngargtngaygcn
gcngtnacnccngargarmgncayytnwsnaaratgcarcaraayggntaygaraayccn
acntayaarttyttygarcaratgcaraay
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The
Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using
one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to
the protein sequence you chose above.
Lysis protein DNA sequence
atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttac
caatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon
optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for
and why?
It is done to come up with codons that are more frequently used by the host organism so as it can efficiently translate the protein and produce the
desired protein in higher levels. As codon that are rare or less favoured would affect the production of the protein in the organism and the programs
helps in identifying the best fit. I chose E.coli cause it is a cheaper alternative in terms of cost and is widely used. I also the use of human to use
in the trial of the protein in a mammalian system.
Lysis protein DNA sequence with Codon-Optimization
ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCA
CCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into
your protein. You may describe either cell-dependent or cell-free methods, or both.
Cells rely on DNA as an instruction manual for protein synthesis. The DNA sequence is first transcribed into messenger RNA (mRNA), which acts as a
working blueprint. The cell then translates this mRNA template step-by-step to assemble the desired protein.
The production of amyloid beta precursor protein (APP) follows these standard genetic expression processes. Interestingly, a single APP gene can
generate several different protein variants through a mechanism called alternative splicing, which occurs during and after transcription. When
scientists create recombinant APP in laboratory settings, they must carefully manage multiple factors—including how much protein is produced, whether
it folds correctly, and how chemical modifications are added after the protein is initially made. One particularly important modification is
glycosylation (the addition of sugar molecules). These chemical alterations are essential because they affect how APP sits in the cell membrane and how
it gets broken down into amyloid-beta (Aβ), a protein fragment associated with Alzheimer’s disease.
Central Dogma Process
DNA encoding APP is transcribed into pre-mRNA by RNA polymerase II, which includes exons and introns. Alternative splicing (e.g., inclusion/exclusion
of exon 15) generates multiple mature mRNA isoforms, which ribosomes then translate into distinct APP protein variants (e.g., APP695, APP751,
APP770) differing in length and function.
Cell-Dependent Methods
HEK293 or CHO cells are optimal for mammalian expression due to proper folding and secretion. Transfect with pcDNA3.1-APP plasmids under CMV
promoter, induce with IPTG if hybrid, and purify via Ni-NTA (His-tagged) or immunoprecipitation; yields reach 10-50 mg/L with glycosylation intact
for secretase cleavage studies.
Cell-Free Methods
Rabbit reticulocyte lysate or wheat germ extracts excel for rapid prototyping. Mix PCR-amplified APP DNA (T7 promoter) with lysate, Mg2+/NTPs, and
translate in 1-2 hours; add microsomes for membrane insertion. Best for isotopic labeling (15N-APP) without cellular toxicity, yielding 1-5 μg/μL
but lacking full glycosylation.
Recommendation
For authentic APP with Aβ-processing fidelity, use HEK293 cell-dependent systems over cell-free, as they support splicing machinery and PTMs essential
for multiple isoforms. Cell-free suits quick screening or labeled protein.
Cell-Dependent Methods
HEK293 and CHO cells are the preferred choice for producing APP proteins in mammalian systems because they naturally promote proper protein folding
and enable the release of proteins from cells. Researchers introduce APP genes into these cells using pcDNA3.1 plasmids controlled by the CMV promoter.
If using a hybrid system, IPTG can trigger protein production. Once synthesized, the APP protein is isolated using purification techniques like
Ni-NTA chromatography (which targets His-tags) or immunoprecipitation. This approach produces substantial quantities—between 10-50 mg/L—and crucially,
the proteins retain their sugar modifications (glycosylation), which are necessary for studying how secretase enzymes break down APP into amyloid-beta.
Cell-Free Methods
Cell-free protein synthesis offers a faster, simpler alternative using extracts from rabbit reticulocytes or wheat germ instead of living cells.
Scientists amplify APP DNA (using T7 promoter sequences) and combine it with the cell extract along with magnesium and nucleotides, completing
protein synthesis in just 1-2 hours. Adding microsomes (membrane fragments) allows the newly made protein to insert into a membrane-like environment.
This method is ideal for rapidly testing concepts and for creating labeled proteins enriched with isotopes like 15N without harming cells. However,
yields are more modest at 1-5 μg/μL, and the proteins don’t receive the complete sugar modifications that cellular systems provide.
Recommendation
For studying APP that behaves authentically and processes into amyloid-beta correctly, cell-based systems using HEK293 cells are superior to
cell-free approaches. This is because living cells contain the machinery to perform alternative splicing and add critical chemical
modifications—processes essential for generating the multiple APP variants. Cell-free systems work best for quick preliminary experiments or when
producing specialized labeled proteins.
Differential Processing of Amyloid-β Precursor Protein Directs Human Embryonic Stem Cell Proliferation and Differentiation into Neuronal Precursor Cells-(https://pmc.ncbi.nlm.nih.gov/articles/PMC2749153/)
This is a practice exercise, not necessarily your real Twist order!
4.1. Create a Twist account, and Benchling account
4.2. Build Your DNA Insert Sequence
For example, let’s make a sequence that will make E. coli glow fluorescent green under UV light by constitutively (always) expressing sfGFP (a green fluorescent protein):
4.6. Choose Your Vector
For this demonstration, choose a Twist cloning vectors like pTwist Amp High Copy.
Go back to your Benchling account. Inside of a folder, click the import DNA/RNA sequence button and upload the GenBank file you just downloaded.
This is the plasmid you just built with your expression cassette included. Congratulations on building your first plasmid!
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease
research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I wanted to research on the effects of microplastics and nanoplastics to our DNA as a lot of diseases are in the population and there is a
possibility plastics in our environment could be contributing. I used Large Language Models to help me get the information. The detection of
plastics particles is done through spectroscopic ways with methods like Raman or FTIR(Fourier-Transform Infrared,while sequencing methods won’t be
able to physically detect plastics. I am curious to find out the research on whether it is true plastics are affecting DNA and other body functions.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA
and why?
Next-Generation Sequencing for Microplastic Research
Advanced sequencing technologies, particularly Illumina’s second-generation platforms, enable researchers to assess the genetic damage, health impacts,
and changes in gene activity triggered by exposure to microplastics and nanoplastics. Each approach requires distinct preparation procedures,
specific chemical processes during sequencing, and generates different types of data. The core technique relies on synthesis-based sequencing,
which simultaneously reads millions of short DNA segments with exceptional accuracy. These methods reveal molecular differences and distinct
mutation patterns in DNA, helping scientists identify whether cells have been damaged or exposed to harmful plastics. The main sequencing
approaches detailed below measure how micro- and nanoplastics affect living organisms:
Key Sequencing Technologies
Whole Genome Sequencing (WGS): This method scans the entire genetic code to identify point mutations and large-scale chromosomal rearrangements
(including single base changes, paired base alterations, and insertions/deletions) that result from exposure to toxic compounds released by plastics,
such as Bisphenol A (BPA).
Metagenomic Sequencing (16S/ITS): This technique examines changes in bacterial populations living in the human gut and identifies genes that
allow microorganisms to break down plastic particles after they are consumed.
RNA-Seq Technologies (Standard and Low-Input): These approaches measure the abundance of thousands of messenger RNA molecules across an entire
genome, revealing how microplastic exposure alters which genes are active in tissues and laboratory-grown cell structures.
Single-Cell RNA Sequencing (scRNA-seq): This specialized method examines individual cells within the kidney and liver, providing a detailed snapshot
of genetic activity in specific cell types, including immune cells and kidney filtration cells, after plastic exposure.
ChIP-Seq: This technique identifies where proteins bind to DNA, enabling researchers to determine whether observed changes in gene activity result
directly from signaling pathways or arise indirectly from the cell’s stress response.
Workflow for Next-Generation Sequencing Technologies
Implementing these sequencing technologies requires a structured, multi-step procedure that begins with preparing biological samples and concludes
with computational analysis of the results.
Sample Preparation and Cell Isolation
The first stage involves preparing biological tissue samples for analysis. Tissues—such as kidney samples from laboratory mice—are broken down
into individual cells using automated equipment. When researchers need to study single cells in isolation, they employ microfluidic devices, which
are miniaturized systems capable of sorting and capturing individual cells with precision.
RNA/DNA Extraction and Quality Control
High-quality genetic material must be isolated from the prepared samples before sequencing can begin. For projects involving RNA, the extracted
material must meet strict quality standards; specifically, the RNA Integrity Number (RIN) should be 8 or higher to ensure that the RNA molecules
remain intact and suitable for accurate analysis.
Library Construction
This critical step prepares the genetic material for sequencing. The DNA or RNA is fragmented into manageable pieces, and for RNA-based studies,
reverse transcription converts RNA back into DNA. Adapter sequences and unique identifying barcodes are then attached to each fragment, allowing
researchers to sort and track the samples during and after sequencing.
Sequencing Run
The prepared libraries are pooled together and loaded onto high-capacity sequencing instruments, such as the Illumina HiSeq X or NovaSeq 6000.
These machines generate sequences of both ends of each DNA fragment (paired-end reads), producing massive amounts of genetic information in a single run.
Bioinformatics Analysis
The raw sequencing data undergoes three distinct computational stages:
Primary Analysis: The sequencing instrument produces raw data files in FASTQ format, which contain the DNA sequences alongside quality scores indicating
how confident the machine is in each nucleotide reading.
Secondary Analysis: The raw sequences are compared and aligned to a reference genome (such as the pig genome, Sus scrofa, or mouse genome,
Mus musculus) using specialized software tools like HISAT2. Alternatively, researchers may assemble the sequences without a reference by comparing them to each other.
Tertiary Analysis: The aligned data is processed to measure how actively each gene is being expressed—using metrics such as FPKM or TPM—and to identify which genes show significantly different activity levels between samples (Differentially Expressed Genes, or DEGs).
Data Outputs Revealing Molecular Damage from Plastic Exposure
These advanced sequencing technologies generate comprehensive molecular information that maps the biological harm caused by plastic particles at the genetic and cellular levels.
Gene Expression Matrices
Large-scale datasets are produced that document which genes become more active (upregulated) or less active (downregulated) when organisms are exposed to microplastics and nanoplastics. These matrices provide a complete picture of how plastic exposure alters the cell’s genetic activity across thousands of genes simultaneously.
Visual Subpopulation Mapping
Computational algorithms such as UMAP and t-SNE transform complex genetic data into visual plots that reveal how plastic exposure changes the composition
of different cell types within tissues. For example, researchers can observe how exposure increases the proportion of specialized immune cells like
CD8⁺ effector T cells, which are involved in fighting infections or damaged cells.
Mutational Signatures
Plastic exposure creates distinctive patterns of DNA mutations—such as the conversion of cytosine bases to adenine bases (C>A substitutions)—that serve as
a “fingerprint” of exposure to specific plastic contaminants like Bisphenol A (BPA) or styrene oxide. These characteristic mutation patterns help scientists identify which type of plastic damage has occurred and what toxic compounds were responsible.
Pathway Enrichment Analysis
Specialized bioinformatics tools like KEGG (Kyoto Encyclopedia of Genes and Genomes) and GO (Gene Ontology) analysis identify which fundamental
biological processes are being disrupted by plastic exposure. Common disrupted pathways include oxidative phosphorylation (energy production in cells),
the MAPK signaling pathway (cellular communication), and chemical carcinogenesis (processes that can lead to cancer).
Microbial Diversity Indices
Statistical measures quantify the balance between harmful and beneficial bacteria in the human gut microbiome following plastic ingestion. These indices reveal whether plastic consumption shifts the microbial ecosystem toward pathobionts (disease-promoting microorganisms) or maintains a healthy population
of beneficial bacteria.
Base Calling in Next-Generation Sequencing
The identification of individual DNA or RNA bases—a process called base calling—represents the foundational analytical step in all
next-generation sequencing (NGS) workflows. This process takes place directly within the sequencing instrument (such as those from Illumina, PacBio,
or Oxford Nanopore) and converts the biological signals detected by the machine into a readable digital genetic code.
Primary Analysis and Signal Generation
On-Platform Processing
Base calling occurs as “Primary Analysis,” meaning it happens in real-time while the biological sample is being sequenced inside the machine. The
sequencing platform simultaneously reads and identifies nucleotides as the process unfolds.
Technology-Specific Sequencing Methods
Different sequencing platforms employ distinct approaches:
Short-read sequencing (Illumina): DNA molecules are cut into small fragments ranging from 200 to 500 base pairs long. The sequencing machine then
reads these fragments from both ends (paired-end reads), typically generating sequences of approximately 150 base pairs in length, and
systematically identifies each nucleotide in order.
Long-read sequencing (PacBio and Oxford Nanopore): These technologies sequence complete, unbroken DNA molecules. PacBio’s Single Molecule Real-Time
(SMRT) sequencing produces average read lengths of about 20 kilobases, while Oxford Nanopore generates ultra-long reads averaging around 100 kilobases, allowing researchers to capture much larger stretches of genetic information in a single read.
Detection of Mutation Patterns
The sequencing process identifies specific genetic variations caused by environmental exposures, such as Single Base Substitutions (SBS). These
are characteristic mutation patterns at particular locations (such as guanine residues) that indicate damage from toxic chemicals derived from plastics.
Output and File Formats
FASTQ Files
The direct result of base calling is raw “reads” that are stored in FASTQ format—the standard file format for sequencing data.
Data Content
Each FASTQ file contains two essential pieces of information for every base identified: the sequence of nucleotides (represented as A, C, G, or T) and
an associated quality score, known as a Phred quality score or Q-score, which indicates the confidence level of that base call.
Measuring Accuracy and Quality Control
Quality Scoring System
To verify that the base calling process is reliable, each identified base is assigned a probability score indicating the likelihood that the
identification is correct.
The Standard for High-Quality Data
A Q30 score is the widely accepted benchmark for excellent data quality, representing a 99.9% accuracy rate in base identification. This threshold
ensures that the sequencing data is trustworthy for downstream analysis.
Quality Filtering
During the next phase of analysis, bioinformatics software programs like fastp or FastQC examine the raw data and filter out low-quality reads where
base calling may have been uncertain or unreliable, ensuring only high-confidence data moves forward.
Downstream Assembly and Analysis of Decoded Bases
After the sequencing machine completes base calling and produces individual reads, the data enters Secondary Analysis, where bioinformatics tools
organize and interpret the complete genetic information.
Reference-Based Genome Alignment
The decoded genetic sequences are compared against and mapped to a reference genome—such as the human genome (GRCh38) or the pig genome (Sus scrofa)—
to determine the correct location of each read within the organism’s full genetic blueprint.
De Novo Assembly
When researchers lack a suitable reference genome for comparison, an alternative strategy is employed: the decoded reads are further subdivided
into overlapping short segments called k-mers. These k-mers are then reassembled using computational algorithms and graph-based methods to
reconstruct longer continuous sequences, known as contigs, that represent the original genetic material.
Also answer the following questions:
1. Is your method first-, second- or third-generation or other? How so?
2. What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
3. What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
4. What is the output of your chosen sequencing technology?
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes,
and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to
novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli,
etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity
to actually have Twist synthesize these DNA constructs! :)
See some famous examples of DNA design
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
1. What are the essential steps of your chosen sequencing methods?
2. What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such
DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is
also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or
in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
1. How does your technology of choice edit DNA? What are the essential steps?
2. What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
3. What are the limitations of your editing methods (if any) in terms of efficiency or precision?
CRISPR Base Editing for Precise DNA Modification
CRISPR base editing offers a highly precise approach to DNA modification that changes individual bases without breaking both strands of the DNA
molecule. This technique represents a significant advancement in genetic engineering because it enables targeted alterations with minimal disruption to the overall genetic structure.
How Base Editing Works
The method combines two key components: a specially engineered Cas enzyme and a deaminase protein. The deaminase acts as a molecular converter,
transforming one DNA base into another—such as changing cytosine to thymine (C→T) or adenine to guanine (A→G). To target a specific gene like
myostatin, researchers design a guide RNA that directs the editing machinery to the correct location. The entire system is then introduced into cells
using a plasmid vector and a physical delivery method such as electroporation, which creates temporary pores in the cell membrane to allow the
genetic material to enter.
PAM Sequence Requirements and Solutions
One constraint of base editing is that the Cas enzyme requires a specific DNA sequence called a PAM (Protospacer Adjacent Motif) located near the
target site to function properly. This requirement previously limited the number of editable locations throughout the genome. However, newer Cas9
variants—such as SpRY—can recognize and work with a broader range of PAM sequences, dramatically expanding the number of genomic locations available
for targeting and editing.
Improving Precision and Safety
Advanced bioinformatics tools like BE-HIVE and Honeycomb have enhanced the effectiveness of base editing by predicting the most promising edit sites and simultaneously reducing the risk of unintended mutations at off-target locations, making the entire process more reliable and safer.
References
DNA Sequencing at 40: Past, Present, and Future (2017) Shendure, J., Balasubramanian, S., Church, G. et al. https://doi.org/10.1038/nature24286
DNA Synthesis Technologies to Close the Gene Writing Gap (2023), Hoose, A., Vellacott, R., Storch, M. et al. https://doi.org/10.1038/s41570-022-00456-9
Recombineering and MAGE (2021), Wannier T, et al. Nat Rev Methods Primers, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9083505/
CRISPR Technology: A Decade of Genome Editing is Only the Beginning, Wang, Doudna, et al., https://www.science.org/doi/10.1126/science.add8643
DNA Sequencing Technologies, How They Differ, and Why It Matters https://www.fjc.gov/content/361255/dna-sequencing-technologies-how-they-differ-and-why-it-matters
sangeranalyseR: Simple and Interactive Processing of Sanger … https://pmc.ncbi.nlm.nih.gov/articles/PMC7939931/
Next-Generation Sequencing Technology: Current Trends … - PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/
Overview of PacBio SMRT sequencing: principles, workflow, and … https://www.cd-genomics.com/pacbio-smrt-system-single-molecule-real-time-sequencing.html
PacBio sequencing output increased through uniform and … https://www.nature.com/articles/s41598-021-96829-z
Library preparation for nanopore sequencing https://oxfordnanoporedx.com/products/prepare
What are the different types of DNA sequencing technologies? https://www.thermofisher.com/us/en/home/life-science/sequencing/sequencing-learning-center/sequencing-basics/dna-sequencing-technologies.html
DNA Sequencing Fact Sheet https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Fact-Sheet
DNA Sequencing: How to Choose the Right Technology https://frontlinegenomics.com/dna-sequencing-how-to-choose-the-right-technology/
Sample Preparation - GENEWIZ from Azenta Life Sciences https://www.genewiz.com/public/resources/sample-submission-guidelines/sanger-sequencing-sample-submission-guidelines/sample-preparation
Sanger Sequencing: Introduction, Principle, and Protocol | CD Genomics Blog https://www.cd-genomics.com/blog/sanger-sequencing-introduction-principle-and-protocol/
[PDF] Sanger Sequencing Best and Worst Practices - rtsf@msu.edu https://rtsf.natsci.msu.edu/_assets/files/genomics/Sanger_Sequencing_Best_and_Worst_Practices_Guide_25April2024.pdf
[PDF] Sanger Sequencing Handbook - FULL SERVICE https://www.biotech.cornell.edu/sites/default/files/2020-08/Full_service_Sanger_Handbook.pdf
Sanger Sequencing - Sample Prep & Data Analysis with BLAST https://www.youtube.com/watch?v=ez-_YtHm9pk
The use of LLM to help with finding information and reporting
Week 3 LAB AUTOMATION
Week # 3 Lab Automation
LAB AUTOMATION
To get hands-on (or at least code-on) with pipetting robots.
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
0. Review this week’s recitation and this week’s lab for details on the Opentrons and programming it.
1. Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
2. Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your
design using the Opentrons.
◦ You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good
job writing functional Python, while you probably need to take charge of the art concept.
◦ If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may
do that instead.
Ask for help early!
3. If the Python component is proving too problematic even with AI and human assistance, download the full Python script from the GUI website and
submit that:
Use the download icon pointed to by the red arrow in this diagram. The Python component was problematic and I sent the the python script
(1 OTDesign_02-26-26_22-49-52.py)
One of the great parts about having an automated robot is being able to precisely mix, deposit, and run reactions without much intervention, and design
and deploy experiments remotely.
For this week, we’d like for you to do the following:
1. Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
The research papers are referenced below and using cancer research using opentron as described in sections below:
Automating Cancer Research Through Robotic Laboratory Systems
Laboratory automation transforms manual pipetting and sample handling into standardized, repeatable robotic processes that enhance throughput
and consistency in cancer research. Common platforms such as the Opentrons OT-2 and OT-3 are increasingly deployed to automate large-scale drug
screening experiments, three-dimensional organoid cultivation, and protein analysis from clinical samples.
Hardware and Software Architecture
Effective automation requires integrating a robotic arm with specialized modules designed to replicate the conditions and functions of a
traditional laboratory workbench.
The Robotic Platform
The Opentrons OT-2 serves as the central automation unit, featuring a motorized arm that moves along three axes (X, Y, and Z coordinates) and
can accommodate up to two electronic pipetting heads—either single-channel or eight-channel configurations—to transfer liquids between containers.
Supporting Hardware Modules
Cancer research protocols typically require specialized add-on modules to perform specific tasks:
Temperature Modules: These maintain biological reagents and cell culture plates at precise temperatures, such as 4°C for refrigerated storage or 37°C
for maintaining cells at body temperature.
Magnetic Modules: These devices use magnetic fields to capture and manipulate magnetic beads, which are essential for isolating DNA and RNA or
enriching specific proteins from samples.
Thermocyclers: Integrated PCR machines mounted directly on the robotic platform allow for on-deck amplification of genetic material during library preparation without removing samples.
Software Control and Customization
Researchers can program the Opentrons system using the Python programming language through the Opentrons Python API, which permits conditional
instructions and calculations that adjust volumes dynamically. For simpler applications, user-friendly no-code platforms like OT2-CherryPick
provide accessible interfaces that require no programming expertise, making the system suitable for straightforward tasks such as transferring
samples between plates or mapping sample locations.
Converting Manual Protocols into Automated Workflows
Translating a published cancer research method into an automated robotic protocol requires careful deconstruction into standardized components.
Liquid Class Calibration
Different biological liquids behave uniquely during pipetting. Viscous solutions like basement membrane extract or volatile substances like ethanol
require customized pipetting speeds and discharge volumes to guarantee accuracy and prevent errors.
Deck Mapping and Coordinate Assignment
Every location on a 96-well or 384-well plate must be precisely mapped to exact spatial coordinates (x, y, z positions) so the robotic arm can access
each well with precision.
Converting Manual Steps into Computational Logic
Manual instructions such as “perform three washes with phosphate-buffered saline (PBS)” are transformed into Python programming loops that
automatically repeat the washing sequence across all plate positions.
Real-World Cancer Research Applications
3D Organoid and Microtissue Development
Expanding three-dimensional cell models is essential for capturing the complexity and variation seen in actual tumors. The Scaffold-supported Platform
for Organoid-based Tissues (SPOT) uses the Opentrons OT-2 to automate the creation and maintenance of organoids grown from patient tumor samples.
This automated method produces results comparable to manual methods while streamlining multiple steps—including tissue generation, adding test drugs, and breaking down the gel matrix for downstream analysis of individual cells. This integration reduces labor and improves consistency.
High-Throughput Protein Analysis in Cancer Immunotherapy
Identifying disease-associated proteins in blood plasma from cancer patients requires processing large numbers of samples rapidly. Automated workflows
on the OT-2 have successfully streamlined the entire analysis pipeline—from preparing samples through to loading them onto specialized mass spectrometry instruments—enabling analysis of up to 192 patient samples within a 6-hour window. This capability was applied to examine how immune checkpoint
inhibitors alter the protein composition of blood plasma in patients with advanced melanoma.
Automated Management of Cancer Cell Lines
Maintaining living cancer cells in culture—whether they grow attached to surfaces or suspended in liquid—presents a significant operational challenge.
The Automated Cell Culture Splitter (ACCS), developed using the Opentrons OT-2, incorporates an integrated imaging system that counts living cells in
real-time, allowing the robot to automatically seed new plates at precisely controlled cell densities. This approach reduces hands-on labor by more than
61% while achieving remarkably consistent seeding across wells, with variation remaining below 11%.
Testing and Quality Assurance Before Running Experiments
Before executing a protocol with valuable or limited patient samples, researchers must validate the automated workflow through multiple verification steps:
Virtual Simulation: Software tools like opentrons_simulate perform a computer-based “dry run” of the protocol to identify potential coordinate errors
or physical collisions between the robotic arm and laboratory equipment before the robot actually moves.
Water Runs: The complete protocol is executed using water colored with dyes, allowing researchers to visually confirm that the correct volumes are
being transferred and that solutions are mixing properly throughout the process.
Real-Time Observation: Imaging modules integrated into the system monitor the status of cells or organoids during automated runs, ensuring that cultures
are progressing normally and providing immediate feedback if adjustments are needed.
References
Avci, M. B. (2026). An integrated platform for liquid handling and cell imaging in life science applications. PMC.
Cao, R., Li, N. T., Latour, S., Cadavid, J. L., Tan, C. M., Forman, A., Jackson, H. W., & McGuigan, A. P. (2023). An automation workflow for high‐throughput manufacturing and analysis of scaffold‐supported 3D tissue arrays. Advanced Healthcare Materials, 12. https://doi.org/10.1002/adhm.202202422
Courville, G., Vaid, S., Toruño, A., Lebel, P., Cabrera, J. P., Raghavan, P., Jacobsen, A., Bell, G., Leonetti, M. D., & Gómez-Sjöberg, R. (2024). Open-source cell culture automation system with integrated cell counting for passaging microplate cultures. PNAS Nexus. https://doi.org/10.1101/2024.12.27.629034
Fusco, R. (2026). OT2-CherryPick: A zero-install web platform for orchestrating complex liquid handling on the Opentrons OT-2. ChemRxiv. https://doi.org/10.26434/chemrxiv.15000637
Kverneland, A. H., Harking, F., Vej-Nielsen, J. M., Huusfeldt, M., Bekker-Jensen, D. B., Svane, I. M., Bache, N., & Olsen, J. V. (2023). Fully automated workflow for integrated sample digestion and Evotip loading enabling high-throughput clinical proteomics. PubMed. https://doi.org/10.1101/2023.12.22.573056
2. Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts,
3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at
the start of lecture and does not need to be tested on the Opentrons yet.
Automating Pancreatic Cancer Research with Opentrons
Converting a manual cancer assay into an automated liquid-handling system involves standardizing sample preparation, distributing reagents, mixing
samples, managing incubation periods, and preparing materials for final analysis. The Opentrons platform facilitates this transformation by offering
a collection of pre-built protocols, user-friendly workflow design tools without requiring programming, and a Python-based application programming
interface for custom development. The system is specifically designed to support the types of experiments common in cancer research, including
genomic analysis, cell biology studies, and assays using cultured cells.
Essential Components for Automation
Successfully automating a pancreatic cancer research workflow depends on establishing a clear scientific objective, breaking the experimental procedure
into discrete robotic steps, and assigning appropriate equipment to each stage.
Starting with a Clear Research Goal
The first step is identifying the specific biological question driving the research. Pancreatic cancer investigations typically focus on one of three
main approaches: analyzing individual cells to understand their molecular characteristics, testing patient-derived tumor models to see how they respond
to drugs, or preparing genetic material for next-generation sequencing analysis. The Opentrons system excels in situations where you need to perform
the same pipetting task reliably across numerous wells, multiple patient samples, or many different experimental conditions.
Converting the Assay into Discrete Robotic Operations
Breaking down the experiment into individual automation steps is essential. A typical automated pancreatic cancer assay includes setting up
plates, equalizing reagent concentrations, moving cells or tissue samples between containers, creating series of decreasing concentrations for
dose-response studies, breaking open cells or preserving cell structures, isolating target molecules using magnetic bead separation, amplifying
DNA segments, and constructing libraries for sequencing. The Opentrons protocol library contains established workflows for nucleic acid
isolation, sequencing library preparation, protein detection assays (ELISA), and cell-based experiments—all fundamental building blocks for
pancreatic cancer research.
Connecting Assay Steps to Hardware Resources
Each step of the assay must be matched to the appropriate robotic tools and accessories. Specify which liquid-dispensing devices (pipettes),
storage containers for pipette tips, temperature-control modules, or magnetic separation tools the protocol requires, then input the exact locations
and volumes within the robotic workspace. Both the Flex platform and Opentrons’ Python API support automation ranging from straightforward liquid
transfers to highly customized workflows, including connection to external instruments or software systems.
Research Applications for Pancreatic Cancer
Single-Cell Analysis and Sequencing Library Preparation
The most promising automation opportunity for pancreatic cancer research centers on single-cell multiomics—techniques that reveal multiple
molecular characteristics (genomics, proteomics, etc.) from individual cells—and automating the capture and library-preparation steps required
for sequencing. This focus is particularly valuable because understanding tumor heterogeneity (the differences between cancer cells within a single
tumor) and moving discoveries from research to clinical practice both depend on these methods. A notable example is the partnership between BD and
Opentrons to automate cell isolation and sequencing library construction on the Opentrons Flex platform, targeting both fundamental disease research
and pharmaceutical development.
Three-Dimensional Tumor Models and Drug Testing
Another significant application is automating the creation and screening of 3D tumor models—including spheroids grown under conditions mimicking the
oxygen-poor, fibrotic tumor microenvironment. Published research on pancreatic cancer has demonstrated that high-throughput automation of spheroid
platforms improves the reliability and scalability of tumor biology studies. Although these studies may not exclusively use Opentrons, the same
core automation principles apply: standardized dispensing of liquids, precisely timed incubations, and controlled sample movement all reduce
experimental inconsistency and strengthen the robustness of drug screening results.
Building a Practical Automated Workflow
A realistic Opentrons-based workflow for pancreatic cancer research follows this sequence:
Sample intake and standardization: Receive and prepare patient samples so they are comparable across the experiment.
Automated reagent distribution: Dispense solutions into 96- or 384-well plates with precision.
Cell or tissue model setup: Seed cells or organize 3D spheroids if modeling tumor structure.
Controlled waiting periods: Allow reactions to proceed off the robot or using integrated heating modules.
Magnetic bead-based purification: Isolate target molecules using magnetic separation.
Genetic amplification or sequencing preparation: Set up DNA amplification or library construction for sequencing.
Sample finalization and export: Seal plates and prepare them for downstream analysis.
This structured approach is particularly valuable for pancreatic cancer research because patient samples are frequently scarce and genetically diverse,
so automation conserves sample material, eliminates human pipetting errors, and ensures that replicate samples are processed identically.
Implementing Automation
Choosing a Starting Point
Begin by checking whether the Opentrons protocol library contains a workflow similar to the assay, because using an existing protocol is the fastest way
to access a validated and tested starting point. For custom or modified assays, a Python protocol that specifies the containers, pipettes,
liquid properties, transfer volumes, mixing instructions, and module operations, then test it thoroughly with small-scale trials before expanding to
larger experiments.
Development Strategy
A proven approach to establishing a reliable automated assay follows these stages:
Start with a single plate and one type of sample to confirm basic functionality.
Test how much liquid remains unusable, verify transfer precision, and confirm timing is appropriate.
Incorporate positive and negative control samples and replicate wells.
Confirm that the automated method produces biological results identical to hand-performed pipetting.
Only after verifying the assay’s stability should you increase the scale to more samples or plates.
Why Opentrons Works for Pancreatic Cancer Research
The Opentrons system is particularly well-suited for pancreatic cancer workflows that require many repetitive liquid-handling steps—such as
single-cell genetic analysis, sample preparation, protein measurement assays, or testing how patient tumor organoids respond to drug treatments.
The advantage extends beyond simply completing work faster; consistency across wells and between experimental runs is equally important, especially
in pancreatic cancer research where tumor microenvironment variations and limited patient material mean that experimental errors are costly and difficult
to replicate.
A practical example illustrates this value: an automated pipeline could receive patient-derived pancreatic cancer cells, apply chemicals to dissociate
them into single cells, adjust cell numbers so they are consistent across samples, distribute cells into test wells, set up a series of drug
concentrations to evaluate treatment responses, and prepare sequencing libraries from surviving cells. This type of integrated workflow aligns
with Opentrons’ flexible automation framework and reflects the emerging focus on fully automated, multiomics analysis in cancer research.
The use of LLM to help with finding information and reporting
Week 4: PROTEIN DESIGN PART I
Week # 4 Protein Design Part I
PROTEIN DESIGN PART I
To look at how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions.
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) For a Tilapia Fish:
Assuming : meat = 20% protein by weight; average amino acid ≈ 100 Da (g/mol).
Calculation:
• Protein mass = 500 g × 0.20 = 100 g
• Moles of amino-acid residues = 100 g ÷ 100 g·mol⁻¹ = 1.00 mol
• Number of amino-acid molecules using Avogadro’s number ≈ 1.00 × ≈ 6.02 × 1023
= 6.02 × 1023 amino-acid molecules.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The beef meat is in the form of amino acids that our body needs which is broken down by the enzymes in our stomach to the amino acids required by our body. The amino acids are the building blocks of DNA. Beef also provides protein, zinc and several D vitamins used for muscle health, iron that boosts our immune system
Why are there only 20 natural amino acids?
Through evolutionary selection there are 20 natural amino acids that are encoded by the genetic code used by all life forms for protein synthesis,
although there are more than 500 amino acids that exist in nature.
Can you make other non-natural amino acids? Design some new amino acids.
There are scientists who have designed new amino acids like the Cyclopropyl‑alanine (cPrAla) whose properties are small, rigid , steric probe that
increases backbone constraint. Its uses are conformational stabilization and protease resistance.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids could have been synthesized through multiple non-biological chemical pathways on the early Earth. Electric discharges and ultraviolet
radiation acting on simple atmospheric gases generated diverse mixtures of organic compounds, including compounds like alanine and glycine.
For these organic building blocks to combine into short peptide chains, the process likely required special activation mechanisms—such as
energized compounds like thioesters or cyanamide—along with repeated cycles of drying and rewetting, or assistance from mineral surfaces acting
as catalysts. Importantly, enzymes like those found in modern life were not yet available to facilitate these reactions. Evidence suggests that the
raw materials for life may have arrived from space itself: meteorites containing carbon-based compounds (such as the Murchison meteorite) and material
from comets both carry amino acids and organic molecules, demonstrating that similar chemical reactions can occur naturally beyond Earth and could
have supplied the ingredients for the early planet. Additional experimental pathways support this scenario. Ultraviolet light shining on frozen water or heating formamide (a simple organic compound) produces amino acids and related molecules under conditions that are colder or more oxidizing than those
used in earlier laboratory experiments.
Finally, geothermal hot vents and rocky mineral surfaces—particularly those containing metal sulfides and clay minerals—would have naturally
concentrated these chemical precursors and promoted their assembly into peptide chains.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
It would be the Left‑handed — D‑amino acids form the mirror‑image (left‑handed) α‑helix corresponding to the right‑handed helix made from L‑amino
acids (same geometry mirrored). It is the one preferred for drug design as its more resistant to breakdown.
Can you discover additional helices in proteins?
It is possible by the use of a deep learning system such as Alpha Fold to predict a protein’s 3D atomic structure from its amino acid sequence.
Why are most molecular helices right-handed?
Proteins naturally favor a right-handed helical structure due to the molecular properties of the L-amino acids from which they are built. The
three-dimensional arrangement of atoms in L-amino acids—their stereochemical configuration and the steric (spatial) and electronic (charge-based)
forces acting on them—determines which backbone torsion angles (φ, ψ) are energetically favorable. These preferred angles naturally align with a
right-handed helical geometry, which represents the lowest-energy, most stable configuration.
This structural preference is further reinforced by the D-sugars present in the DNA and RNA backbone. The presence of D-sugars (rather than their
mirror-image L-form) in nucleic acids creates a similar geometric environment that also favors and supports the right-handed helical arrangement, making
it consistent with the overall architecture of proteins built from L-amino acids.
In essence, the molecular “handedness” of biological building blocks—L-amino acids and D-sugars—creates a coordinated system where all the natural
forces involved push the same direction, promoting right-handed helical structures as the thermodynamically favored state.
Why do β-sheets tend to aggregate?
Aromatic interactions: The π–π stacking of aromatic amino acids (phenylalanine, tyrosine, and tryptophan) creates additional stabilization through
their interactions, further reinforcing stacked sheet structures.
Nucleation and templating: Once an initial molecular nucleus forms, subsequent strands can attach to this template, which lowers the energy barrier
for further assembly and accelerates the growth of the fibril structure—a process driven by kinetic facilitation.
Side-chain packing and van der Waals forces: The complementary, regularly repeating interfaces between side chains (sometimes called “steric zippers”)
allow for maximal close-contact packing between molecules, which maximizes the weak dispersion forces (van der Waals interactions) that contribute
to overall stability.
Backbone hydrogen bonding: Extended networks of hydrogen bonds form between the backbone atoms of adjacent strands (specifically between N–H groups and
C=O groups across different molecules), which significantly stabilize the β-sheet structure between strands.
Dehydration and entropic effects: Thermodynamic stability increases when water molecules are expelled from the interfaces between molecules and when
the total surface area exposed to solvent decreases.
Hydrophobic effect: When nonpolar (hydrophobic) side chains are buried inside the assembled structure as sheets stack together, ordered water molecules
are released into solution. This release of water increases the system’s entropy, providing a large favorable thermodynamic driving force.
Polar side-chain networks: Amino acids with polar side chains (such as glutamine and asparagine) form interconnected hydrogen-bonding patterns that
act cooperatively to stabilize and strengthen the fibril structure.
Why do many amyloid diseases form β-sheets?
β-sheets are the thermodynamically favored conformation for proteins involved in amyloid diseases due to the multiple stabilizing factors that
work together. The backbone hydrogen bonding between extended strands creates a very stable intermolecular network. Additionally, the hydrophobic effect
is particularly powerful—when nonpolar amino acid side chains are buried within stacked β-sheets, water molecules are released, which provides a
large entropic gain that drives assembly.
Kinetic factors further promote β-sheet formation. Once a small nucleus of β-sheet structure forms, it acts as a template. New protein molecules can
rapidly attach to this growing template with a lower activation energy than forming the initial nucleus, creating a positive feedback loop that
accelerates fibril growth.
The β-sheet structure is also highly resistant to degradation. The extensive hydrogen-bonding networks and tightly packed interfaces make these
aggregates very difficult for cellular machinery to break down or clear, allowing amyloid fibrils to accumulate over time.
Additionally, certain proteins are inherently prone to adopting β-sheet conformations due to their amino acid sequences and three-dimensional properties.
In amyloid diseases, proteins that normally fold into other structures (like α-helices) become misfolded under stress conditions and instead
spontaneously assemble into the more stable β-sheet configuration.
In essence, the combination of thermodynamic stability, kinetic acceleration through templating, and cellular difficulty in clearing these structures
makes β-sheets the inevitable outcome for proteins that misfold in amyloid diseases.
◦ Can you use amyloid β-sheets as materials?
Amyloid β-sheets are gaining recognition as valuable materials for practical applications across multiple industries, leveraging their
distinctive structural and functional characteristics.
Healthcare Applications
Amyloid β-sheets can be modified and designed to serve as delivery vehicles for pharmaceuticals, enabling them to direct treatments to particular cell
types or anatomical locations. Their exceptional mechanical robustness and structural durability make them ideal candidates for use as supportive
frameworks in regenerative medicine, where they can facilitate the growth and organization of new tissue.
Sensing and Detection
The self-assembling capability of amyloid β-sheets offers promise for creating highly sensitive detection systems. These structures can be incorporated
into biosensing devices that identify and measure biological molecules or contaminants present in the environment with enhanced precision.
Environmental and Sustainability Applications
As there is growing interest in environmentally responsible material alternatives, amyloid β-sheets represent a promising option for developing
compostable or biodegradable materials that could replace conventional petroleum-based plastics. This application addresses the need for
sustainable solutions to reduce plastic waste and environmental contamination.
In summary, the stability, mechanical properties, and programmable assembly of amyloid β-sheets position them as versatile engineering materials that
can address challenges in medicine, environmental monitoring, and sustainable manufacturing.
Design a β-sheet motif that forms a well-ordered structure.
The foundation of creating a well-ordered β-sheet involves establishing an alternating pattern of water-repelling and water-attracting amino acids along
the protein backbone. By arranging hydrophobic (nonpolar) residues to point inward and hydrophilic (polar) residues to point outward, you create a
striped arrangement where the nonpolar side chains cluster together in the interior of the sheet while polar groups remain accessible to the
aqueous surroundings. This pattern naturally promotes the formation of stable, regular β-sheet structures.
To further enhance stability, aromatic amino acids should be distributed at regular intervals throughout the sequence. These aromatic residues (such
as phenylalanine and tyrosine) interact through π–π stacking, which reinforces the stacking of sheets on top of each other and creates additional
geometric constraints that lock the structure into place.
A Practical Example
A straightforward and effective sequence would be: [I-V-F-L-Y-L-F-V-I-V-F-L-Y-L-F-V], composed primarily of isoleucine, valine, leucine, phenylalanine,
and tyrosine. These residues are small and hydrophobic, allowing them to pack tightly together while aromatic residues facilitate lateral interactions.
For applications requiring additional functionality, incorporating charged amino acids like glutamate at regular intervals adds surface solubility a
nd potential binding sites without disrupting the core sheet geometry. A modified version might be: [I-E-F-L-Y-L-F-E-I-E-F-L-Y-L-F-E].
Why This Design Works
Hydrophobic side-chain burial drives rapid self-assembly because water molecules trapped within the nonpolar interior are released as the structure
forms, providing a large entropy gain. The repeating, predictable pattern ensures that once initial nucleation occurs, subsequent protein molecules
attach to the growing structure in an orderly fashion, promoting uniform, large-scale assembly. The complementary packing of side chains maximizes
weak intermolecular forces, while the extensive hydrogen-bonding networks between backbones of adjacent strands create remarkable stability.
Customization Options
To strengthen the material further, you can increase the proportion of aromatic and β-branched residues to achieve denser packing. For
tailored applications, replace specific residues with histidine or aspartate to create binding sites for metal ions, or introduce serine or threonine for
potential chemical modifications. To control the final size of assembled structures, strategically insert proline residues to act as “breaks” that
limit sheet expansion and create defined structural units.
This rational design approach—carefully balancing hydrophobic and hydrophilic properties while incorporating multiple stabilizing
interactions—represents the practical framework for engineering functional amyloid-based materials.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it.
PD-1 (programmed cell death protein 1) was selected as the subject of study. This protein is an inhibitory receptor located on the cell membrane of
T cells and other immune cells. When PD-1 binds to its ligands—PD-L1 or PD-L2—the binding signal suppresses the activation and proliferation of
T cells, essentially acting as a “brake” on immune responses.
This checkpoint mechanism normally serves an important regulatory function, but tumors and certain pathogens have learned to exploit it. By producing
and displaying PD-L1 or PD-L2 on their surfaces, cancer cells and infectious agents can hijack this natural control system. They force T cells to express
PD-1 and engage it with the pathogen’s or tumor’s PD-ligands, effectively disabling the immune response that would otherwise attack them.
However, blocking the interaction between PD-1 and its ligands can reverse this immune suppression. When the PD-1/PD-L1 (or PD-L2) connection is
prevented, T cells regain their ability to recognize and attack cancer cells or infected cells. This principle forms the basis of checkpoint
inhibitor immunotherapies, which have proven highly effective in treating certain cancers and enhancing immune responses against persistent infections.
◦ When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better
(Resolution: 2.70 Å)
It was resolved and Deposited : 2019-10-10 and Released: 2019-11-27 to the public.
The Resolution: 1.18 Å of high quality and precise as it is below 2.70 Å
◦ Are there any other molecules in the solved structure apart from protein?
Yes, Chloride Ion and Water.
◦ Does your protein belong to any structure classification family?
Yes,the extracellular domain of PD‑1 has an immunoglobulin V‑set (IgV) fold and is classified with Ig‑like domains in structural databases
(SCOP/CATH/Pfam). It is placed in the Ig‑superfamily structural class (consistent with its membership in the CD28/CTLA‑4 receptor family).
In the structure classification family database it is SCOP ID: 8059476 and SCOP ID: 8059477
Open the structure of your protein in any 3D molecule visualization software:
◦ PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
◦ Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
◦ Color the protein by secondary structure. Does it have more helices or sheets?
It has no alpha helices and 2 beta sheets. Adopts an immunoglobulin domain fold with a two-layer beta sandwich architecture.
◦ Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
It has a hydrophobic core to support the beta sandwich architecture and the hydrophilic residues on the surface ◦ Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes, as seen above.
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
I went through the steps in the jupiter notebook as shown in the recitation and created the mutation scan heat map below for protein PD-1. There were errors encountered as I run the code for each section,but Gemini was able to rectify the problem and run without problems producing the image below.
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Log-likelihood ratio (LLR) used in mutation analysis to help quantify the impact of mutations on protein function by comparing the likelihood of mutant versus wild-type residues. Using Gemini, the Most Favorable Mutation will be the one with the highest LLR where a change from Valine (V) to Leucine (L) at position 187, with an LLR of 5.4932. This suggests that substituting Valine with Leucine at this position could potentially be beneficial.. The Least Favorable Mutation with lowest LLR is a change from Methionine (M) to Isoleucine (I) at position 1, with an LLR of -16.6267. This indicates that this particular substitution is highly unfavorable.
c. (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
2. Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
Folding a protein
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
2. Input this sequence into ESMFold and compare the predicted structure to your original.
References
The use of LLM to help with finding information and reporting
Week 5: PROTEIN DESIGN PART II
Week # 5 Protein Design Part II
PROTEIN DESIGN PART II
To learn how cutting-edge AI and protein language models are used to design functional proteins and peptides “in silico”.
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
1. Design short peptides that bind mutant SOD1.
2. Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
• PepMLM: target sequence-conditioned peptide generation via masked language modeling
• PeptiVerse: therapeutic property prediction
• moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
References
The use of LLM to help with finding information and reporting
Week 6: GENETIC CIRCUITS PART I
Week # 6 Genetic Circuits Part I
GENETIC CIRCUITS PART I
To learn core molecular biology tools and techniques for processing and assembling DNA, including PCR and Gibson Assembly.
Assignment: DNA Assembly
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix is a comprehensive formulation that supplies all the essential components required for precise and efficient
DNA amplification through the polymerase chain reaction (PCR).
The mixture contains Phusion polymerase, an enzyme renowned for its exceptional accuracy in synthesizing new DNA strands during the amplification
process. It also includes deoxynucleotide triphosphates (dNTPs), which serve as the molecular building blocks that polymerase incorporates into the
growing DNA chains. Additionally, magnesium chloride (MgCl₂) is present as a critical cofactor—an enabling molecule that the polymerase enzyme requires to
function optimally and catalyze the formation of new DNA bonds.
Finally, the formulation includes a reaction buffer solution that maintains the proper chemical environment throughout the PCR process. This
buffer preserves stable pH levels and regulates salt concentration, ensuring that all enzymatic reactions proceed smoothly and that the
overall amplification process achieves maximum efficiency.
In essence, Phusion High-Fidelity PCR Master Mix eliminates the need to manually combine individual components—it is a ready-to-use formulation where
all necessary ingredients are already optimized and proportioned for reliable, high-fidelity DNA amplification.
What are some factors that determine primer annealing temperature during PCR?
The temperature at which primers attach to target DNA during PCR is determined by several interconnected factors that collectively influence how
effectively and specifically the primers bind to their complementary DNA sequences.
The melting temperature (Tm) of the primers is a central parameter that determines this annealing temperature. Melting temperature represents the
specific thermal point at which approximately half of all DNA-primer complexes denature and separate from each other. This value is not fixed—it
varies depending on the chemical composition of the primer sequence itself.
A major factor affecting Tm is the GC content of the primer. Since guanine-cytosine base pairs form stronger hydrogen bonds compared to adenine-thymine
base pairs, primers with a higher percentage of G and C nucleotides exhibit higher melting temperatures. Conversely, primers rich in adenine and
thymine have lower Tm values. This difference in bond strength directly correlates with thermal stability.
Beyond the primer sequence itself, the salt concentration within the PCR reaction buffer also significantly influences binding stability. Salts present
in the solution help reinforce and stabilize the interaction between primers and their target DNA by shielding the negative charges on the DNA
backbone, reducing electrostatic repulsion and promoting tighter primer-template binding.
In essence, optimizing the primer annealing temperature requires balancing the intrinsic properties of the primer sequence (particularly its GC content
and resulting Tm) with the chemical conditions of the reaction environment (salt concentration).
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Two Complementary Approaches to DNA Fragmentation
Two distinct strategies can be employed to generate linear DNA fragments: polymerase chain reaction (PCR) amplification and restriction enzyme
digestion. Each method operates on different principles and offers distinct advantages depending on the experimental objective.
PCR Method
PCR is a DNA amplification technique that simultaneously increases the quantity of DNA while allowing for deliberate sequence modifications.
During amplification, this method can introduce desired changes into the DNA—such as specific mutations or sequence overlaps that facilitate
subsequent cloning steps. A key characteristic of PCR is that it relies on primer binding to guide amplification rather than targeting specific
nucleotide sequences for cutting. This means the method is not constrained by the presence or absence of predefined restriction sites.
Restriction Enzyme Digestion
In contrast, restriction enzyme digestion employs specialized proteins that function as molecular scissors. These enzymes recognize and cut DNA
exclusively at specific sequence motifs that serve as their recognition sites. However, this method has a critical limitation: it can only be
used successfully if the target DNA contains those specific recognition sequences at the desired locations.
Choosing the Appropriate Method
PCR is the preferred approach when your goal is to alter or engineer the DNA sequence or when the precise locations where you want to separate the DNA
are unknown or unavailable as restriction sites. Restriction digestion becomes the better choice when you have already identified the exact locations where cuts should be made and the DNA contains appropriate restriction sites at those positions.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Preparing DNA Fragments for Gibson Assembly
To successfully prepare DNA fragments for Gibson cloning, several critical requirements must be met to ensure seamless assembly of the final construct.
Overlapping Sequence Regions
Each DNA fragment must contain complementary overlapping sequences at its ends, typically ranging from 20 to 40 base pairs in length. These
overlapping regions are essential for the assembly process—they allow the individual DNA pieces to recognize and bind to each other, providing
the molecular “glue” that holds the fragments together during Gibson cloning.
Absence of Interfering Restriction Sites
It is crucial to verify that the DNA fragments do not contain internal restriction enzyme recognition sites that could be problematic. If such sites
are present within the fragments themselves, they could lead to unwanted cutting or interference with the joining process, compromising the success of
the cloning procedure.
Proper Fragment Orientation
The fragments must be arranged and oriented in the correct order and direction relative to one another. This proper orientation ensures that when the
pieces assemble, they form a functional gene or plasmid with the correct genetic sequence and regulatory elements. Incorrect orientation would result in
non-functional or defective constructs.
In summary, successful Gibson cloning depends on careful design of overlapping sequences, verification of fragment integrity, and precise positioning of all component fragments.
How does the plasmid DNA enter the E. coli cells during transformation?
Getting Plasmid DNA into Bacterial Cells
During the transformation process, plasmid DNA successfully enters E. coli cells by temporarily disrupting the normally impermeable bacterial cell membrane, allowing DNA passage through the otherwise sealed barrier.
Two Primary Transformation Techniques
Two main strategies accomplish this membrane permeabilization:
Heat shock: This method applies a sudden, dramatic shift in temperature, destabilizing the membrane structure and opening transient channels through which DNA can enter.
Electroporation: This approach uses a brief, high-voltage electrical pulse that creates temporary microscopic pores across the cell membrane, providing pathways for the plasmid DNA to cross into the cytoplasm.
In both cases, the temporary openings allow DNA molecules to pass through the membrane barrier before the cell membrane reseals itself.
Post-Transformation Recovery
Following successful DNA uptake, the transformed bacterial cells require placement in a nutrient-rich growth medium where they can recover and
restore normal membrane integrity. During this recovery period, the cells repair the membrane damage and activate gene expression, including the
production of antibiotic resistance proteins encoded by genes on the plasmid.
This recovery step is essential for ensuring that successfully transformed cells can survive when exposed to selective conditions. Only cells that
have successfully integrated the plasmid will express the antibiotic resistance gene, allowing them to survive and grow on culture plates
containing antibiotics, while untransformed cells die.
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
When Golden Gate Assembly Is Advantageous
Golden Gate assembly is the preferred cloning method when your DNA sequences already contain Type IIS restriction sites and you need to assemble
multiple fragments simultaneously at high efficiency. Unlike Gibson assembly, which offers greater flexibility in fragment design, Golden Gate
is specifically optimized for situations where you have many DNA pieces—up to approximately 35 fragments—that need to be joined into a single construct
all at once. This method operates as a “one-pot” process, meaning all components react together in a single tube, streamlining the workflow compared
to multi-step procedures.
Essential Components and Mechanism
Golden Gate assembly requires several key ingredients: (1) DNA fragments obtained from PCR or previously cloned sources, each flanked by Type
IIS restriction enzyme recognition sites (such as BsaI or BsmBl); (2) a destination vector—a linearized plasmid backbone containing inward-facing Type
IIS recognition sites; (3) Type IIS restriction enzymes (for example, BsaI-HFv2 or BsmBl-v2) that cut DNA at specific recognition sequences;
(4) T4 DNA ligase, an enzyme that catalyzes strand joining; (5) reaction buffer to maintain proper chemical conditions; and (6) nuclease-free water as
the reaction medium.
The Assembly Process
All components are combined in a single reaction tube and then placed in a thermocycler, where the temperature alternates between 37°C (for
restriction enzyme cutting) and 16°C (for DNA ligation). This thermal cycling allows cutting and joining to occur sequentially within the same
reaction, maximizing efficiency.
The key to Golden Gate’s elegance is the Type IIS restriction enzymes, which cut DNA slightly offset from their recognition sequence, generating sticky
end overhangs that are perfectly complementary to adjacent DNA fragments. Once cutting is complete, the ligase enzyme seamlessly joins the sticky
ends together, assembling all fragments in the correct order and orientation. This approach is particularly powerful for combining multiple genetic
elements such as promoters, coding sequences, and regulatory regions. After the thermocycler reaction finishes, purification and bacterial
transformation procedures are essentially identical to Gibson assembly.
References
The use of LLM to help with finding information and reporting
Week 7: GENETIC CIRCUITS PART II
Week # 7 Genetic Circuits Part II
GENETIC CIRCUITS PART II
To learn neuromorphic genetic circuits, showing how engineered gene networks can implement neural-network “perceptron”-like computation and learning
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
To understand the advantages of IANNs (In silico Artificial Neural Networks / Integrated Artificial Neural Networks in synthetic biology) over traditional Boolean genetic circuits, it helps to look at how biological computing is evolving.
Traditional genetic circuits act like classic computer chips: they take inputs (like the presence of a specific molecule) and use logic gates (AND, OR, NOT) to produce a definitive, binary ON/OFF response. IANNs, however, mimic the brain’s neural networks using biological components.
Here is why IANNs are a massive step up from traditional Boolean genetic circuits:
Continuous (Analog) vs. Binary Processing
• Traditional Circuits: They are strictly binary. They require sharp thresholds to determine if a signal is a 1 or a 0. This is highly inefficient in biological systems because nature rarely operates in pure black and white.
• IANNs: They process analog signals. They can take continuous, graded inputs (e.g., varying concentrations of a toxin or biomarker) and produce a scaled, proportional output. This allows for much more nuanced decision-making, akin to how our own cells actually sense the environment.
Scalability and Resource Economy
• Traditional Circuits: Scaling up a Boolean circuit requires stacking more and more physical logic gates. In synthetic biology, every new gate requires distinct promoters, repressors, and plasmids. Cells quickly run out of metabolic energy (the “retroactivity” and “resource burden” problem), causing the circuit to crash.
• IANNs: They achieve complex computational depth using far fewer biological parts. By adjusting the “weights” of connections (e.g., tuning the binding affinities of a few regulatory proteins), a small network can perform tasks that would require dozens of traditional logic gates.
High-Dimensional Pattern Recognition
• Traditional Circuits: They struggle with complex pattern recognition. If you want a cell to detect a disease based on a combination of 5 different biomarkers, a Boolean circuit requires a massive, fragile web of nested AND gates.
• IANNs: Neural networks excel at fuzzy logic and multi-variate pattern recognition. They can integrate multiple noisy, weak inputs simultaneously, weigh their relative importance, and accurately classify a state (e.g., “Healthy” vs. “Cancerous”) even if one of the biomarkers is slightly off.
Robustness to Noise
• Traditional Circuits: Biological environments are incredibly noisy. Molecular fluctuations can easily cause a Boolean gate to misfire, flipping a 0 to a 1 and ruining the entire computational chain.
• IANNs: Because they are distributed networks, they possess inherent noise-filtering capabilities. The weights and non-linear activation functions average out random biological noise, making the overall system far more robust and less prone to catastrophic failure.
Trainability and Reconfigurability
• Traditional Circuits: If you want to change the function of a Boolean circuit (e.g., changing it from an AND gate to an OR gate), you usually have to physically re-engineer the DNA sequence, swap out promoters, and rebuild the cell line from scratch.
• IANNs: They can theoretically be “trained” or tuned. By subtly adjusting chemical inducers, light exposure (in optogenetic networks), or minor mutation rates, the same basic network structure can be repurposed to map entirely different input/output relationships without a complete structural overhaul.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
One of the most promising and impactful applications for an Integrated Artificial Neural Network (IANN) in synthetic biology is autonomous, multi-biomarker cancer diagnostics and targeted drug delivery.
In cancer therapy, a major hurdle is that tumor cells are highly adaptive and rarely defined by a single unique marker. Traditional Boolean logic gates require an exact combination of absolute ON/OFF signals (e.g., “Antigen A AND Antigen B must be present”). If a tumor cell downregulates just one antigen, a Boolean circuit fails to detect it.
An IANN solves this by acting as an intelligent, cell-based classifier that can process a “fuzzy” spectrum of environmental cues to accurately target tumor microenvironments while sparing healthy tissue.
The Application: Smart Living Therapeutics
Imagine engineering a patient’s T-cells or a non-pathogenic probiotic bacteria with a genetic IANN. This living therapeutic circulates through the body, constantly sampling the local environment to decide whether it is sitting next to a healthy cell or a malignant tumor.
Detailed Input/Output Behavior
An IANN mimics a computational neuron mathematically, where the output y is determined by the weighted sum of inputs passed through a non-linear activation function (like a sigmoidal Hill function):
The Inputs (Continuous Analog Signals)
Instead of binary 1s and 0s, the cell senses a continuous range of concentrations (x_i) via synthetic surface receptors and promoters:
• Input 1 (x_1): Hypoxia (Oxygen levels). Tumors are notoriously oxygen-deprived. The input is high when oxygen is low.
• Input 2 (x_2): Extracellular Lactate/Acidity. Tumor metabolism generates high levels of lactic acid, lowering local pH.
• Input 3 (x_3): Tumor-Associated Antigen (TAA). A surface protein commonly found on the specific cancer, but occasionally present on healthy cells in low amounts.
The Internal Processing (Biological Weights)
Inside the cell, these inputs trigger the production of specific regulatory proteins. The “weights” (w_i) are physically engineered into the DNA by adjusting the binding affinities of promoters or changing plasmid copy numbers. For instance, if Antigen presence (x_3) is the most reliable indicator, its promoter is engineered to have a high weight, meaning it drives transcription much more aggressively than the hypoxia signal.
The Output (Proportional Therapeutic Payload)
The final output (y) is the transcription and secretion of a localized therapeutic agent, such as an anti-tumor cytokine (e.g., IL-12) or a targeted toxin.
• Traditional Boolean Output: Either 100% drug release or 0% drug release.
• IANN Output: Graded and contextual. If the cell detects mild hypoxia and medium acidity, but zero tumor antigen, the network computes a low probability of cancer and releases no drug. If it detects high antigen, high acidity, and high hypoxia, it releases a maximum payload. If it encounters a complex intermediate profile, it secretes a proportional, moderate dose to safely address the threat without causing systemic toxicity.
Limitations Faced by Biological IANNs
While computationally elegant, implementing an IANN inside a living cell faces severe physical and biological constraints:
• The “Static Weight” Problem (No Real-Time Training): In computer software, a neural network learns by adjusting its weights via backpropagation over millions of iterations. In a living cell, you cannot easily “train” the network on the fly. Weights must be meticulously pre-calculated and hardcoded into the DNA architecture via genetic engineering before the cell enters the body.
• Metabolic Burden and Resource Competition: Every “node” and “weight” in a genetic neural network requires the host cell to transcribe RNA and translate proteins. If the IANN is too large, it will monopolize the cell’s ribosomes and ATP. The cell will either grow sluggishly, die, or evolutionarily mutate to eject the synthetic circuit entirely.
• Biological Crosstalk and Environmental Interference: Unlike clean code, a cell’s interior is packed with native signaling pathways. The synthetic transcription factors used to calculate the IANN’s weights might inadvertently bind to the cell’s native DNA, or native proteins might interfere with the circuit, wildly distorting the pre-calibrated math.
• Genetic Instability over Time: Living cells replicate and mutate. Because the IANN provides no survival advantage to the cell itself (only to the patient), natural selection incentivizes the cell to accumulate mutations that break the circuit to save energy.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Several fungal‑based materials are already being produced or prototyped, mainly using mycelium (the “root” network) or fungal biomass. They are used as biodegradable substitutes for plastics, foams, leather, paper, insulation, and even some construction elements.
Below are concrete examples, their uses, and how they compare to traditional counterparts.
Mycelium foam / packaging
Ecovative Design’s MycoFoam and MycoComposite packaging grown on agricultural waste (e.g., corn husks, hemp).
Mycelium “bricks” and cushioning blocks for protective packaging, replacing polystyrene (Styrofoam).
Uses:
Protective packaging for electronics, fragile goods, and shipping inserts.
Lightweight insulation or filler in construction panels.
Advantages vs. polystyrene / plastics:
Biodegradable and compostable; decomposes in weeks–months instead of centuries.
Grown at ambient temperature on low‑value waste (sawdust, straw), with low energy and carbon footprint.
Naturally fire‑resistant and termite‑resistant in some formulations, with good thermal and acoustic insulation.
Disadvantages:
More sensitive to moisture and humidity; can lose strength or compress if wet.
Lower mechanical strength and density than dense plastics; better for cushioning than load‑bearing structures.
Batch‑to‑batch variability in growth and density can complicate quality control.
MycoWorks’ Reishi mycelium leather for bags, shoes, and upholstery.
Other “myco‑leather” textiles grown as fungal mats on trays (often from species such as Fomes fomentarius or Phellinus spp.).
Uses:
Fashion (bags, shoes, jackets), furniture upholstery, and interior design.
Advantages vs. animal leather or synthetic leather:
No livestock rearing; lower land use, methane emissions, and water pollution than bovine leather.
Often biodegradable or compostable, unlike most synthetic (PU/PVC) leather.
Can be grown to precise shapes and textures, reducing mechanical cutting waste.
Disadvantages:
Limited durability and abrasion resistance compared with high‑grade bovine leather at present.
Cost and scale still higher than bulk synthetic leather; production is not yet at mass‑market polyester‑PU volumes.
May require coatings or treatments (e.g., for water resistance) that can reduce biodegradability.
Fungal‑derived paper‑like materials
Examples:
Fungal “paper” made from liquid fermentation of filamentous fungi (often Trametes or related polypores) into chitin–β‑glucan sheets.
Early stage materials for printing, filters, and coatings instead of wood‑pulp paper.
Uses:
Specialty printing surfaces, filtration membranes, and coatings.
Advantages vs. wood‑pulp paper:
Can be grown from waste streams (e.g., agricultural byproducts) instead of virgin trees.
Some fungal papers have higher toughness or porosity tailored for specific filtering or biomedical uses.
Disadvantages:
Not yet cost‑competitive for bulk printing or packaging paper.
Limited industrial supply chains and standardized processing compared with paper mills.
Mycelium “bricks” and construction panels
Examples:
Mycelium‑bound insulation panels and bricks grown on agricultural residues (hemp, straw, sawdust).
Acoustic panels from companies such as MOGU using mycelium‑based composites for interior sound‑absorbing surfaces.
Uses:
Thermal and acoustic insulation in walls, ceilings, and partitioning.
Interior cladding, acoustic panels, and non‑structural architectural elements.
Advantages vs. mineral wool / expanded polystyrene / concrete:
Very low embodied energy and carbon‑negative potential when grown on waste biomass.
Good thermal and acoustic performance per unit weight; lightweight and easy to handle.
Naturally biodegradable at end‑of‑life, unlike foam or mineral‑wool insulation.
Disadvantages:
Compressive strength far below concrete (mycelium bricks ~30 psi vs. concrete ~4000 psi).
Susceptible to moisture and long‑term fungal degradation if not properly sealed.
Limited load‑bearing capacity restricts use to non‑structural or low‑stress applications.
Other fungal “soft” materials
Examples:
Historical “felt‑like” textiles from polypore fruit bodies (e.g., Fomes fomentarius “amadou” or German Amou), now being revisited for niche textiles and fashion.
Fungal biomass for food and feed (e.g., Fusarium venenatum “Quorn” mycoprotein), which is a protein material but not usually classed as “structural.”
Uses:
Specialized textiles, cultural crafts, and decorative materials.
High‑protein foods and feedstocks.
Advantages vs. cotton / synthetic fibers:
Can be grown on waste streams with small land and water footprints.
Natural biodegradability and relatively low chemical input.
Disadvantages:
Limited mechanical strength and durability compared with conventional textiles.
Niche supply chains and limited industrial‑scale processing.
References
Flexible Fungal Materials: Shaping the Future https://www.sciencedirect.com/science/article/abs/pii/S0167779921000603
How Fungi Can Transform Waste Into Useful Materials https://joyfulmicrobe.com/how-fungi-can-transform-waste-into-useful-materials/
Will Buildings in the Future Be Built From Mushrooms? - RESET.ORG https://en.reset.org/mycelium-construction-material-benefit/
Current Insights in Fungal Importance—A Comprehensive … https://pmc.ncbi.nlm.nih.gov/articles/PMC10304223/
Benefits of Fungi for the Environment and Humans https://www.decadeonrestoration.org/stories/benefits-fungi-environment-and-humans
Growing sustainable materials from filamentous fungi | The Biochemist https://portlandpress.com/biochemist/article/45/3/8/233178/Growing-sustainable-materials-from-filamentous
Mycelium-Based Composites - Using Fungi as Building Materials https://www.youtube.com/watch?v=vWkGpbOXZj8
Fungus - Wikipedia https://en.wikipedia.org/wiki/Fungus
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
One would genetically engineer fungi to enhance or redirect their natural biology for industrial, medical, agricultural, or environmental applications. At the same time, fungi offer several unique advantages over bacteria for synthetic biology, especially for complex molecules and eukaryotic‑style processes.
Below I is an outline on using fungi then contrast fungal synthetic biology with bacterial systems.
What to engineer fungi to do
Hyper‑produce natural products and drugs
Fungi are already rich sources of antibiotics (e.g., penicillin), statins (lovastatin), immunosuppressants (cyclosporine), and anticancer scaffolds.
Engineer them to:
Overexpress or “wake up” silent biosynthetic gene clusters that do not normally make detectable compounds.
Combine pathways from different fungi or other organisms to create hybrid natural products with new bioactivities.
Why: 50% of approved clinical drugs are natural products or derivatives; engineered fungi can accelerate drug discovery and lower production costs.
Produce high‑value enzymes and acids
Filamentous fungi such as Aspergillus niger naturally secrete large amounts of industrial enzymes (amylases, cellulases, proteases) and organic acids (citric, aconitic, itaconic).
Engineering goals include:
Overexpressing enzymes for biomass deconstruction (e.g., in biorefineries).
Redirecting metabolism to increase titers of organic acids used as food additives or chemical‑building blocks.
Why: This enables cheaper, more sustainable routes to bulk chemicals and biocatalysts compared with chemical synthesis.
Build advanced materials and mycelium composites
Mycelium can be engineered to:
Modify chitin, β‑glucans, or hydrophobic surface proteins to tune water resistance, mechanical strength, and fire retardancy of mycelium bricks, foams, or textiles.
Express functional proteins (e.g., adhesion peptides, enzymes) to improve integration with other biomaterials or substrates.
Why:Fungal materials are low‑carbon, biodegradable alternatives to plastics and synthetics; genetic control can make them more robust and standardized.
Improve biocontrol and plant‑symbiosis traits
Entomopathogenic fungi (e.g., Beauveria, Metarhizium) can be engineered to:
Carry insecticidal toxins or plant‑defence elicitors to target pests more selectively.
Increase UV tolerance or persistence under field conditions.
Mycorrhizal or endophytic fungi can be tuned to:
Enhance phosphate/nitrogen uptake, stress tolerance, or pathogen resistance in crops.
Why:This reduces reliance on synthetic pesticides and fertilizers while keeping the system biologically specific.
Add “smart” metabolic or sensing functions
Fungi can be engineered with synthetic circuits for:
Biosensors that change color or emit light when they detect pollutants, plant pathogens, or soil nutrients.
Metabolic switches that turn on biodegradation pathways only in the presence of specific contaminants.
Why:Eukaryotic regulation (e.g., chromatin, promoters, secretion) allows more complex, context‑dependent behaviors than simple bacterial toggle switches.
The use of LLM to help with finding information and reporting
Week 9: CELL FREE SYSTEMS
Week # 9 Cell Free Systems
CELL FREE SYSTEMS
To learn synthesis of proteins using cellular machinery outside of a cell.
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control
over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-Free Protein Synthesis Advantages
Cell-free protein synthesis (CFPS) provides substantial benefits compared to conventional cell-based protein production methods, particularly in terms
of experimental flexibility and precise control over reaction parameters. In contrast to traditional in vivo approaches that require cell
transformation, growth in culture media, and cell disruption, CFPS enables rapid protein production without these intermediate steps,
significantly accelerating the research timeline.
Greater Experimental Control and Customization
CFPS operates in an open, accessible reaction environment where researchers enjoy extensive freedom to manipulate conditions on the fly. This
accessibility allows for straightforward modification of reaction parameters, supplementation with molecular chaperones and folding assistants, incorporation of non-standard amino acids not found in natural proteins, and inclusion of tagged or labeled molecular markers. These capabilities
make CFPS exceptionally well-suited for investigating translational control mechanisms, conducting ribosome display experiments for directed evolution,
or analyzing intricate protein-protein interaction networks.
Two Primary Applications Where CFPS Excels
Producing toxic or cytotoxic proteins: Cell-free systems enable the synthesis of proteins that would be harmful or lethal to living cells if produced
inside them. Since there is no intact cellular machinery to be damaged, toxic proteins can be safely manufactured in vitro.
Engineering protein sequences with labeled components: CFPS greatly simplifies the incorporation of specialized building blocks, such as amino acids
labeled with stable isotopes. This type of customization is vastly more straightforward to accomplish in a cell-free environment compared to achieving
the same modifications within living organisms, where metabolic pathways create complications and inefficiencies.
In essence, CFPS provides researchers with an unparalleled degree of control and experimental versatility that traditional cell-based systems simply cannot match.
Describe the main components of a cell-free expression system and explain the role of each component.
Cell-Free Extract
The cell-free extract represents the core biochemical engine of the CFPS system. This liquid extract is derived from disrupted cells and contains
the fundamental molecular machinery required for synthesizing proteins—specifically ribosomes (which build proteins), transfer RNAs (which deliver
amino acids), enzymes (which catalyze chemical reactions), and numerous regulatory factors. The source organism selected to prepare this extract depends
on the characteristics of the target protein: E. coli extracts are commonly used for straightforward proteins, rabbit reticulocyte extracts are chosen
when higher eukaryotic protein qualities are needed, and wheat germ extracts are employed for particularly complex or post-translationally modified proteins.
DNA Template
The DNA template serves as the genetic blueprint that contains all instructions for creating the desired protein. This template comprises two
essential regions: a promoter sequence that signals where transcription should begin and initiate RNA synthesis, and a coding sequence that encodes
the precise linear arrangement of amino acids that will form the target protein’s primary structure.
Energy Sources and Essential Cofactors
Protein synthesis is an energy-intensive process that demands substantial quantities of adenosine triphosphate (ATP) and guanosine triphosphate (GTP).
The CFPS system must continuously regenerate these energy molecules to sustain the reaction and prevent depletion. Beyond these energy sources, metal
ion cofactors—particularly magnesium and potassium—are critical for maintaining the chemical environment necessary for both transcription and translation
to proceed efficiently and accurately.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
The Critical Role of Energy Regeneration
Maintaining adequate ATP levels is essential for successful cell-free protein synthesis because adenosine triphosphate serves as the primary energy
currency driving both transcription (the conversion of DNA to RNA) and translation (the conversion of RNA to protein). During the CFPS reaction, ATP
is consumed rapidly and continuously, as nearly every step of protein synthesis requires energy input. If ATP levels are not actively replenished
and maintained, the biochemical machinery will exhaust its energy supply, the reaction will halt, and protein production will plummet significantly.
ATP Regeneration Strategy in E. coli Systems
To overcome this energy limitation, E. coli-based cell-free systems employ a coupling strategy using two key components: phosphoenolpyruvate (PEP) and
the enzyme pyruvate kinase. This enzymatic pair works together to continuously regenerate ATP from adenosine diphosphate (ADP), ensuring a steady supply
of fresh energy molecules throughout the reaction. By maintaining consistent ATP availability through this regeneration mechanism, the system can
sustain extended and robust protein synthesis, preventing the energy depletion that would otherwise terminate the reaction prematurely and severely
reduce overall protein yield. In essence, energy regeneration transforms CFPS from a short-lived, low-yield process into a sustainable,
high-productivity system capable of extended protein manufacturing.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free systems (e.g., E. coli extracts) are typically faster to produce protein because transcription and translation are closely
coupled, while eukaryotic systems separate these processes and are generally slower.
Prokaryotic extracts often yield higher total protein amounts and are less expensive to run than eukaryotic extracts.
Eukaryotic cell-free systems (e.g., rabbit reticulocyte, wheat germ, insect, or mammalian extracts) better support complex post-translational
modifications such as glycosylation and disulfide bond formation.
Prokaryotic systems lack most native eukaryotic chaperones and modification enzymes, which can limit correct folding and activity of many
eukaryotic proteins.
Eukaryotic systems can more accurately translate mRNAs with complex regulatory elements (e.g., internal ribosome entry sites, Kozak sequences) and
handle eukaryotic codon usage more naturally.
Prokaryotic systems are more amenable to genetic and biochemical optimization (e.g., energy regeneration, component supplementation) for
high-throughput screening and rapid prototyping.
Eukaryotic extracts are preferred for membrane proteins and multi-subunit complexes when proper folding, assembly, or membrane insertion requires
eukaryotic-specific factors or microsomes.
Prokaryotic systems often require codon optimization and may produce inclusion bodies or inactive protein for eukaryotic targets without additional
folding aids.
Eukaryotic systems are generally costlier, have lower batch-to-batch yields, and can be more variable, but they increase the likelihood of
obtaining functionally active eukaryotic proteins.
Choice depends on the goal: use prokaryotic systems for speed, yield, and cost-effective screening; use eukaryotic systems when native folding, modifications, or activity are required.
Recommended Proteins and Rationales
Prokaryotic System: Green Fluorescent Protein (GFP)
Why GFP is ideal for E. coli CFPS:
GFP is a small, robust protein (~27 kDa) that folds autonomously without requiring post-translational modifications or extensive chaperone assistance.
The chromophore (fluorescent group) forms spontaneously through auto-oxidation of its own amino acid sequence, making it self-contained and independent
of cellular machinery. The rapid synthesis capability of prokaryotic systems would allow researchers to quickly produce labeled protein variants for
high-throughput screening, fluorescence assays, or incorporation of non-canonical amino acids at specific positions to tune optical properties. The low
cost and ease of E. coli extract preparation make this system economically practical for producing large quantities needed for research or
diagnostic applications. Additionally, GFP doesn’t require disulfide bonds or glycosylation, which are unnecessary complications in a
prokaryotic environment.
Eukaryotic System: Antibody (Immunoglobulin)
Why antibodies are ideal for eukaryotic CFPS:
Antibodies are large, multimeric proteins (~150 kDa per molecule, often functioning as dimers or larger complexes) that require sophisticated
post-translational processing. They contain multiple disulfide bonds that stabilize the structure and are critical for antigen binding and immune
function—a feature that eukaryotic systems handle naturally through their oxidizing endoplasmic reticulum environment. Antibodies also undergo
glycosylation at specific sites, which is essential for their effector functions (complement activation, antibody-dependent cellular cytotoxicity, and
Fc receptor binding). The rich folding environment provided by eukaryotic extracts, with resident molecular chaperones like BiP and protein
disulfide isomerases, ensures proper tertiary and quaternary structure formation. While eukaryotic CFPS is more expensive and time-consuming, the
production of functional, correctly modified antibodies justifies this investment for therapeutic development, diagnostic reagents, or research
applications where authentic biological activity is non-negotiable.
Summary
Choose prokaryotic systems for rapid, simple, cost-effective production of robust proteins that self-fold and don’t require
post-translational modifications. Choose eukaryotic systems when investing in complex proteins that demand sophisticated folding assistance, disulfide
bond formation, or chemical modifications essential for biological function. The choice ultimately depends on balancing speed and cost against
biological complexity and functional requirements of your target protein.
Prokaryotic cell-free expression systems, such as those based on E. coli, are fast, cost-effective, and capable of producing high protein yields.
However, they lack the ability to perform post-translational modifications and often struggle with proper folding of complex or membrane-bound proteins.
In contrast, eukaryotic systems like wheat germ extract are slower and more expensive but offer better support for folding and modifications, making
them suitable for expressing complex eukaryotic proteins. For a prokaryotic system, I would choose to express
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
c. Could this function be realized by genetically modified natural cell?
d. Describe the desired outcome of your synthetic cell operation.
Design all components that would need to be part of your synthetic cell.
a. What would be the membrane made of?
b. What would you encapsulate inside? Enzymes, small molecules.
c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
d. How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Experimental details
a. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
b. How will you measure the function of your system?
AHL-Quenching Synthetic Cell for Biofilm Disruption
What the Cell Does
This engineered synthetic minimal cell detects the quorum-sensing molecule 3-oxo-C12-HSL, which is released by the pathogenic bacterium
Pseudomonas aeruginosa. When exposed to this signal, the cell produces the enzyme AiiA (a lactonase), which breaks down the AHL molecules within
the enclosed vesicle. As new AHL continuously enters from the surrounding environment, the trapped enzyme works to destroy it, establishing a depletion
zone that gradually reduces the available AHL in the nearby area. This reduction in quorum-sensing signaling dampens the pathogen’s ability to
express disease-causing proteins and build biofilm structures.
Input and Output
Input: The AHL signal (3-oxo-C12-HSL) from the surrounding liquid.
Output: Reduced AHL concentration in the environment; the lactonase enzyme stays trapped inside the vesicle and does not escape. The primary
measurable indicator is a decline in AHL levels over time.
Feasibility of Alternative Realizations
Using only cell-free transcription-translation in solution: This approach is technically possible—mixing DNA and AHL in a test tube would produce AiiA
and allow degradation. However, without encapsulation, the system lacks physical boundaries, the enzyme becomes diluted and exposed, and it cannot
be precisely delivered to an infection site. A vesicle creates a controlled, protected microreactor that can be positioned exactly where treatment is needed.
Using a genetically engineered living bacterium: An engineered E. coli strain programmed to express the LasR receptor and AiiA enzyme would
theoretically accomplish the same sensing and degradation. However, living microorganisms present significant concerns: they may multiply
uncontrollably, share modified genes with other bacteria, and face regulatory approval obstacles. A synthetic cell avoids these risks because it is
non-living, incapable of reproduction, and inherently safer for medical use.
Desired Outcome
Introducing a collection of these synthetic vesicles into a P. aeruginosa biofilm would lower AHL concentrations in that region to below the level
needed for quorum-sensing activation. This would suppress the production of harmful proteins (such as elastase and pyocyanin) and destabilize the
biofilm structure, making the bacteria vulnerable to destruction by the immune system or antimicrobial drugs.
Design of the Synthetic Cell
Membrane Composition
The synthetic cell consists of a single-layer lipid sphere containing:
• POPC (70% of lipid composition) – promotes fluid membrane behavior and is compatible with living tissue
• Cholesterol (30% of lipid composition) – strengthens the membrane structure and controls how easily molecules pass through
The AHL molecule is sufficiently lipid-soluble (logP ≈ 3.5) to naturally seep through the lipid barrier without requiring specialized transport proteins.
Encapsulated Components
Biological machinery: An E. coli cell-free system that includes the natural RNA polymerase (with σ⁷⁰ factor), ribosomes, and all components needed
for protein synthesis. This bacterial system is ideal because it recognizes standard bacterial promoters without needing more complex mammalian machinery.
Fuel and building blocks: Energy sources (PEP, ATP, GTP, UTP, CTP), amino acids, transfer RNA, nucleotides, creatine phosphate, and necessary
salts—all standard components for keeping bacterial lysate functional.
Genetic instructions: Two circular DNA plasmids:
pLuxR – continuously produces the LasR receptor protein using a powerful σ⁷⁰ promoter (J23119), with the lasR gene from
P. aeruginosa PAO1 (GenBank reference: NP_250121.1)
pAiiA produces the AiiA lactonase enzyme in response to AHL binding, controlled by the lasI promoter region (which contains the DNA binding
site for the LasR-AHL complex) and containing the aiiA gene from Bacillus sp. 240B1 (GenBank reference: AAF62398.1)
(These can be placed on separate plasmids for flexible tuning or combined on a single plasmid.)
Interaction with Surroundings
AHL freely passes through the membrane via passive diffusion; no special channels are required. By contrast, the lactonase protein cannot escape because
it carries no export signal and the membrane pores are too small (~0.5 nanometers) for proteins to exit. This design confines the synthetic cell to
act purely as an AHL sink, eliminating any release of the enzyme into the environment.
Experimental Details
Key Genetic and Chemical Components
Component Specification
POPC 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
Cholesterol Cholest-5-en-3β-ol
Promoter (constitutive) J23119 (Anderson collection)
lasR gene Pseudomonas aeruginosa PAO1, locus PA1430, protein NP_250121.1
PlasI promoter ~200 base pairs upstream of lasI (PA1432), includes las box binding site
aiiA gene Bacillus sp. 240B1 lactonase, GenBank reference AAF62398.1
Measuring Performance
Testing AHL removal from the environment: Expose a known quantity of synthetic vesicles to 1–5 µM of 3-oxo-C12-HSL in a buffered solution. At regular
time points, collect samples of the surrounding liquid and measure remaining AHL using either liquid chromatography–mass spectrometry (LC-MS/MS) or
a bacterial biosensor (E. coli carrying lasR and a fluorescent reporter gene under the lasI promoter). A decline in fluorescence demonstrates the
quenching effect.
Confirming enzyme activity inside the vesicle: Break open vesicles using freeze-thaw cycles and assess how efficiently AiiA degrades AHL by employing
a color-changing dye that reacts to the acid released when lactones hydrolyze, or by using a synthetic lactone compound linked to a fluorescent tag
(e.g., carboxyfluorescein) and monitoring the fluorescence change.
Evaluating impact on the pathogen: Combine the synthetic cells with live P. aeruginosa PAO1 and assess whether virulence markers
(elastase activity, pyocyanin pigment levels) decrease or whether the biofilm becomes thinner or less dense (using crystal violet staining).
This approach creates a clearly defined, non-replicating, safe therapeutic system that uses a minimal lipid compartment and standard bacterial
protein-making machinery to neutralize a pathogenic communication pathway.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material
itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using
cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
• Write a one-sentence summary pitch sentence describing your concept.
• How will the idea work, in more detail? Write 3-4 sentences or more.
• What societal challenge or market need will this address?
• How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
BioChroma—Biosensor in Enhanced Textiles for Athlete and Worker Health Monitoring
Product Concept
BioChroma is an innovative textile technology that incorporates freeze-dried, cell-free biosensing components directly into fabric fibers. The
system undergoes a lasting color shift when it encounters specific sweat-based health indicators, enabling athletes and industrial workers to
monitor hydration status and electrolyte balance in real time without needing electronic devices or specialized equipment.
Operational Mechanism
The system uses freeze-dried protein-synthesis machinery equipped with regulatory proteins that respond to specific molecular triggers and genes that
encode color-producing proteins. These components are packaged into tiny droplets suspended within a gel and then applied as a coating to sections of
fabric designed to absorb moisture. When sweat contacts the fabric, it rehydrates the dormant system, which then activates a series of reactions that
identify target molecules (such as lactate or sodium) and rapidly manufacture a colored pigment—all without needing living microorganisms or
electrical input. The chemical cascade is engineered to be one-directional and irreversible, ensuring that the color change becomes a permanent marker
of the wearer’s peak stress levels rather than a temporary indicator. By placing several different sensor zones at various locations on a single
garment, the design allows simultaneous tracking of multiple different biomarkers from different areas of the body.
The Problem and Market Opportunity
This technology tackles an important unmet need: many outdoor workers, military personnel, and endurance athletes are vulnerable to heat-related
medical emergencies but lack practical access to real-time body monitoring systems. Traditional electronic smart textiles are expensive, fragile, depend
on batteries, and may violate workplace safety rules. Additionally, these devices deteriorate when laundered and generate electronic waste. The
biological approach presented here offers a compelling alternative—significantly cheaper, single-use, and completely compostable—making
physiological monitoring accessible to broad consumer populations seeking practical health tracking options.
Overcoming Technical Challenges with Cell-Free Systems
To prevent unwanted activation and maintain long-term viability, the freeze-dried biosensing material is enclosed in a water-repellent silica coating
that dissolves only when exposed to sweat conditions (particular pH and salt concentration), guarding against false activation from rain or
everyday moisture and allowing the product to remain stable for more than two years at normal temperatures. The one-time-use nature of the system
is reframed as an intentional advantage, functioning similarly to radiation exposure badges that accumulate and record total stress over time. A
modular patch architecture means users can replace only the sensors that have been activated rather than discarding the entire garment. Additionally,
the dried biological material is treated with protective compounds (trehalose sugar and stabilizing proteins) that preserve enzyme function even after
the fabric experiences physical stress and repeated washing before the sensors are triggered.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space
competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in
space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to
practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the
other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration,
check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is s
ignificant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Cell-Free Biosensing for Radiation Damage Assessment in Space
Background
Astronauts traveling beyond Earth’s protective magnetic field are exposed to high-energy cosmic radiation that causes harmful changes to DNA,
elevating their lifetime risk of malignancy and cellular deterioration. Directly evaluating the biological impact of this damage in living cells
is challenging, particularly during space missions where resources are limited. Cell-free protein-making systems such as BioBits® offer a
practical solution: they enable rapid measurement of how radiation-damaged DNA impairs the production of mRNA and proteins, creating a simple, lightweight
detection method for monitoring genetic damage during extended journeys through deep space.
Molecular or Genetic Target
The experimental system uses a superfolder GFP (sfGFP) gene carried on a plasmid, which functions as a fluorescent signal indicating how efficiently
genes are transcribed and translated after the DNA has been damaged by radiation.
Connection to Space Biology Challenge
Radiation damage creates breaks and chemical alterations in the DNA backbone that obstruct the movement of RNA polymerase enzymes, thereby suppressing
mRNA production. When researchers introduce radiation-damaged sfGFP DNA into a BioBits reaction mixture, the reduced fluorescence signal directly
correlates with the proportion of DNA templates that have been rendered non-functional. This approach translates the amount of DNA injury into a
measurable decrease in gene expression, resembling what occurs when critical genes are damaged in the cells of actual space travelers.
Research Hypothesis and Objectives
The research team predicts that plasmids exposed to radiation mimicking galactic cosmic ray exposure will generate diminished amounts of sfGFP protein in
a cell-free reaction system, with the reduction increasing proportionally to radiation dose. Since BioBits reactions contain all necessary enzymes
for transcription and translation but lack the DNA repair machinery found in live cells, any reduction in fluorescence must come directly from
physical damage to the DNA template itself. By plotting the relationship between radiation dose and fluorescence loss, this assay will establish a
reliable, space-suitable method for quantifying how radiation affects gene expression capability, potentially enabling continuous health assessment
during long-term space missions.
Experimental Approach
Identical samples of isolated pSFGFP plasmid will be exposed to iron ion (⁵⁶Fe) radiation at four levels: no radiation (as a control), 5 gray, 10 gray,
and 20 gray. Each radiation-treated DNA sample (100 nanograms per reaction) will then be mixed into reconstituted BioBits components and allowed to
incubate for two hours at 30 degrees Celsius. Fluorescence intensity will be measured at the end of the reaction using a P51 Molecular Fluorescence
Viewer. The experiment will include three identical reactions per radiation dose plus a blank reaction containing no DNA to serve as a background
reference. The resulting fluorescence measurements will be plotted against radiation dose to establish a curve showing the progressive decline in
gene expression as DNA damage increases.
References
The use of LLM to help with finding information and reporting
Week 10: IMAGING AND MEASUREMENT
Week # 10 Imaging and Measurement
IMAGING AND MEASUREMENT
To learn a range of advanced technologies to do precision measurement of proteins at atomic scales, characterizing chemical composition, and detecting protein sequence and structure.
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
The molecular weight is 27875.41 Da
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
1. Determine for each adjacent pair of peaks using:
Let’s choose two adjacent peaks:
• m/z₁ = 1000.4947
• m/z₂ = 1037.4927
The molecular weight is 27875.41 Da
These represent two charge states: z and z+1
Week 11: BUILDING GENOMES
Week # 11 Building Genomes
BUILDING GENOMES
To inspire collaboration and creativity while designing a scientifically rigorous cell-free fluorescent protein optimization experiment together.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
1. Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
◦ A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse.
◦ If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
2. Make a note on your HTGAA webpages including:
◦ what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
◦ what you liked about the project, and
◦ what about this collaborative art experiment could be made better for next year.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): - Provides the cellular machinery needed for transcription and translation, including ribosomes, elongation factors, enzymes, and T7 RNA polymerase to drive expression from T7 promoters.
Salts and Buffer
Potassium Glutamate: - Maintains ionic strength and osmotic balance, helping transcription and translation machinery function properly.
-HEPES-KOH pH 7.5: - Buffers the reaction to keep pH stable, which is important for enzyme activity and protein synthesis.
Magnesium Glutamate: Supplies magnesium, a key cofactor that stabilizes ribosomes, nucleic acids, and many reaction enzymes.
Potassium phosphate monobasic: Contributes phosphate buffering and can help support energy-related chemistry in the reaction.
Potassium phosphate dibasic: Works with the monobasic form to set and maintain the desired pH and buffering capacity.
Energy and Nucleotides
Ribose: Provides a sugar backbone component for nucleotide-related chemistry and can support metabolic regeneration pathways.
Glucose: Serves as an energy source to help regenerate ATP and sustain the reaction.
AMP: Participates in energy metabolism and regeneration pathways that help maintain usable nucleotide pools.
CMP: Supports the nucleotide pool needed for RNA synthesis.
GMP: Supports the nucleotide pool needed for RNA synthesis.
UMP: Supports the nucleotide pool needed for RNA synthesis.
Guanine: Contributes to nucleotide salvage and replenishment of GTP-related pools.
Translation Mix
17 Amino Acid Mix: Supplies the protein-building substrates needed for translation into the target protein.
Tyrosine: Provides an additional amino acid component for protein synthesis.
Cysteine: Provides an additional amino acid component for protein synthesis and can be important for protein structure via disulfide bonds.
Additives
Nicotinamide: Supports NAD-related metabolic recycling, helping sustain energy generation in the reaction.
Backfill
Nuclease Free Water: Brings the reaction to final volume while avoiding nucleases that could degrade DNA or RNA templates.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The main difference is that the PEP-NTP master mix is designed for a fast, short reaction and uses phosphoenolpyruvate as the primary energy source, while the NMP-Ribose-Glucose master mix is built for a longer reaction and uses simpler metabolic substrates to sustain activity over many hours. In practical terms, the 1-hour mix favors rapid output, whereas the 20-hour mix favors longer-lasting protein synthesis and template usage.
Energy strategy
PEP-based systems usually generate energy more directly and quickly, which helps drive early, high-rate transcription and translation. NMP-Ribose-Glucose systems rely more on gradual metabolic recycling and downstream regeneration, so they tend to support a longer reaction window.
Reaction duration
The 1-hour optimized mix is tuned for speed, so it is useful when you want a quick readout. The 20-hour mix is tuned for persistence, so it is better when you want prolonged expression or higher total yield over time.
Practical implication
If you need a fast assay or screening result, the PEP-NTP mix is usually the better fit. If you need the reaction to keep running much longer, the NMP-Ribose-Glucose mix is the more suitable choice.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Transcription can still occur because guanine is a precursor, not the RNA building block itself. In the lysate, enzymes can convert guanine into GMP, then to GDP and GTP, and GTP is the actual nucleotide that RNA polymerase uses to build RNA.
Why guanine is enough
Free guanine can be salvaged into the nucleotide pool through the cell-free extract’s metabolic enzymes, so the reaction does not need GMP to be added separately. Once GMP is made, it can be phosphorylated to the triphosphate form needed for transcription.
What transcription needs
RNA synthesis requires the four ribonucleoside triphosphates: ATP, CTP, GTP, and UTP. So the important point is not whether GMP is added directly, but whether the system can maintain enough GTP availability for RNA polymerase to work.
In your mix
The presence of guanine suggests the master mix is designed to replenish guanine nucleotides metabolically rather than supplying GMP outright. That is consistent with a longer-running cell-free system that relies on internal recycling and salvage pathways.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP
mRFP1
mKO2
mTurquoise2
mScarlet_I
Electra2
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
Cell-free protein synthesis (CFPS) offers a unique environment for expressing fluorescent proteins (FPs). Because these systems lack the complex regulatory machinery of a living cell, the intrinsic biophysical properties of the protein itself—such as how fast it folds or how much oxygen it requires—become the primary drivers of the visible signal.
sfGFP (Superfolder GFP)
Property: Folding Robustness
sfGFP was specifically engineered to resist misfolding, allowing it to reach its functional state even when fused to poorly folding proteins or expressed at high speeds in cell-free systems. This makes it the “gold standard” for CFPS because it ensures that nearly all translated protein becomes fluorescently active rather than forming insoluble aggregates.
mRFP1 (Monomeric Red Fluorescent Protein 1)
Property: Maturation Time
As a first-generation monomeric red FP, mRFP1 suffers from a relatively slow maturation time (often exceeding one hour). In cell-free reactions, this creates a significant “time lag” between protein synthesis and signal detection, which can obscure the real-time dynamics of genetic circuits.
mKO2 (Monomeric Kusabira Orange 2)
Property: Acid Sensitivity (pK_a)
mKO2 is sensitive to pH changes, with a pK_a of approximately 5.0. While cell-free buffers are usually stable, the accumulation of metabolic byproducts (like organic acids) during long reactions can drop the pH, potentially quenching the mKO2 signal and leading to an underestimation of protein yield.
mTurquoise2
Property: Quantum Yield (Brightness)
mTurquoise2 possesses an exceptionally high quantum yield (0.93), making it one of the brightest cyan FPs available. This high intrinsic brightness allows for a very high signal-to-noise ratio in cell-free readouts, which is critical when working with low-yield reactions or microfluidic droplets where the total amount of protein is minimal.
mScarlet-I
Property: Maturation Kinetics
mScarlet-I is designed for high brightness and fast maturation (t_{1/2} \approx 36 minutes), which is significantly faster than many other red FPs. This rapid maturation makes it an ideal reporter for cell-free systems where users need to observe red fluorescence almost immediately after the start of translation.
Electra2
Property: Oxygen Dependence / Fast Maturation
Electra2 was specifically developed for ultra-fast readouts in time-resolved applications. Its primary functional advantage in cell-free systems is its optimized maturation speed, which minimizes the delay in signal acquisition for high-throughput screening and the characterization of rapid transcriptional-translational (TX-TL) bursts.
sfGFP folds rapidly and efficiently even under suboptimal conditions due to its superfolder mutations, making it ideal for cell-free systems where chaperone activity is limited. Its low pKa (around 3.1) ensures stable fluorescence across a wide pH range typical in these reactions.
mRFP1 has a relatively slow maturation time (around 60 minutes), which delays fluorescence readout in time-sensitive cell-free assays. It also shows moderate folding efficiency, potentially leading to lower yields in crowded lysate environments.
mKO2 exhibits fast maturation and high quantum yield (0.62), allowing quick orange fluorescence detection in cell-free setups. However, its slightly higher pKa (5.5) makes it more pH-sensitive than GFP variants, risking signal loss if the reaction acidifies.
mTurquoise2 matures very rapidly (half-time ~33 minutes) with minimal acid sensitivity (pKa 3.1), providing bright cyan signal early and reliably in cell-free transcription-translation. Its monomeric state prevents aggregation issues that plague some FPs in cell-free crowding.
mScarlet_I offers extremely fast maturation (<15 minutes reported in literature) and high photostability, enabling high-throughput cell-free screening with red-shifted emission. Its optimized folding minimizes misfolding in oxygen-variable cell-free conditions.
Electra2 (a newer far-red FP) shows oxygen-independent chromophore formation like other iLOV-derived proteins, crucial for anaerobic cell-free reactions. Its rapid folding supports expression monitoring without oxygenation dependence that hampers traditional FPs.


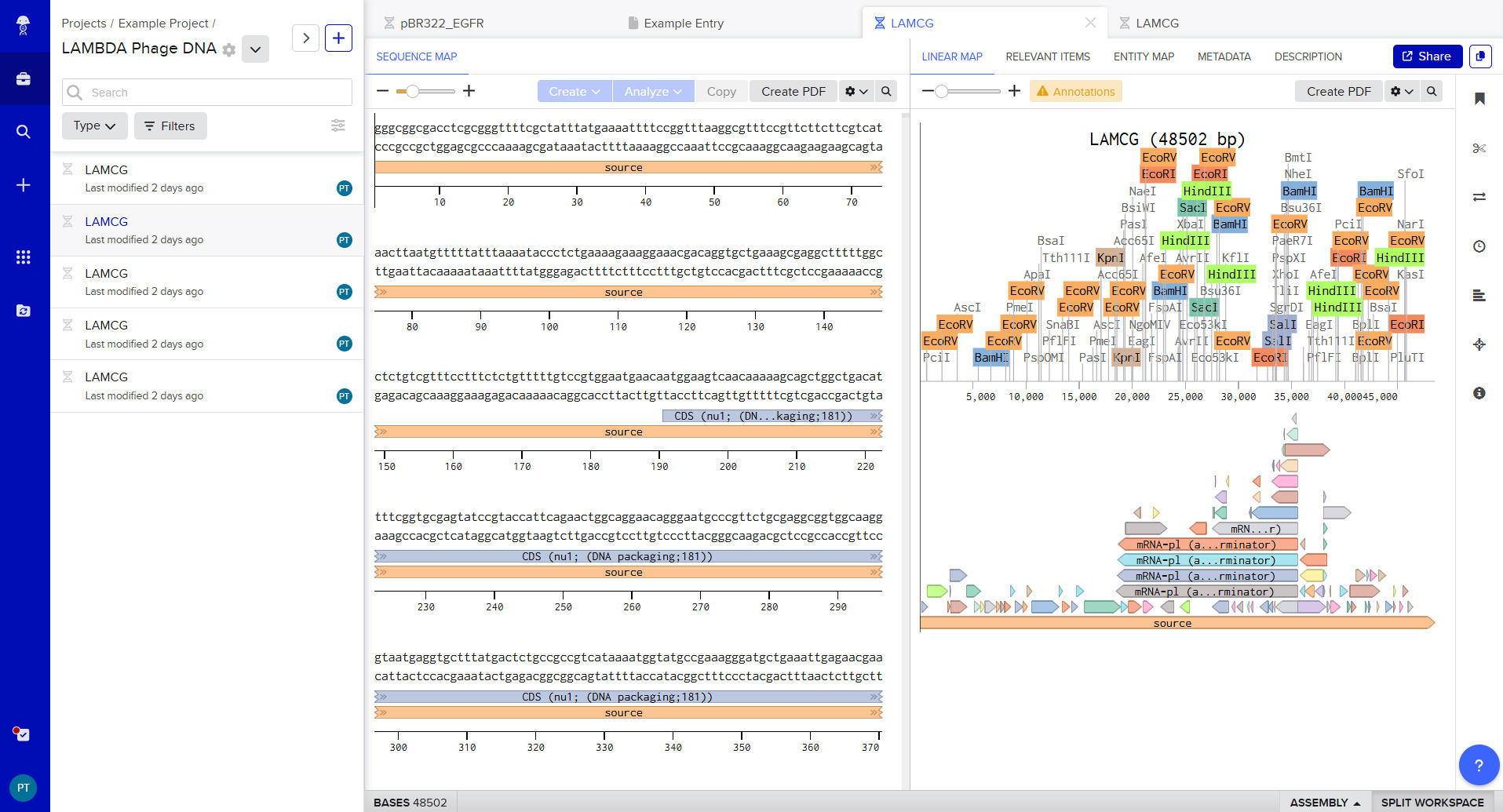
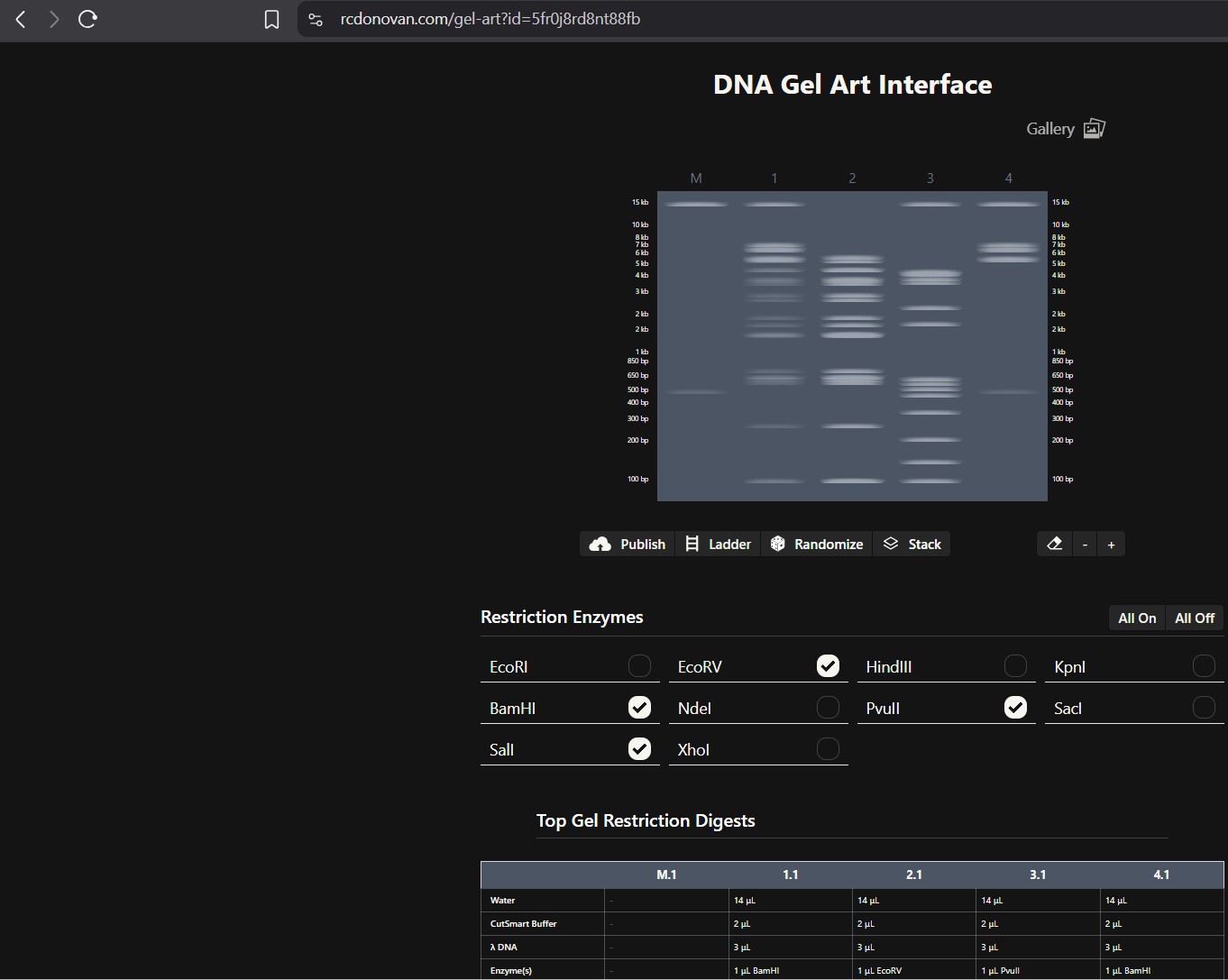

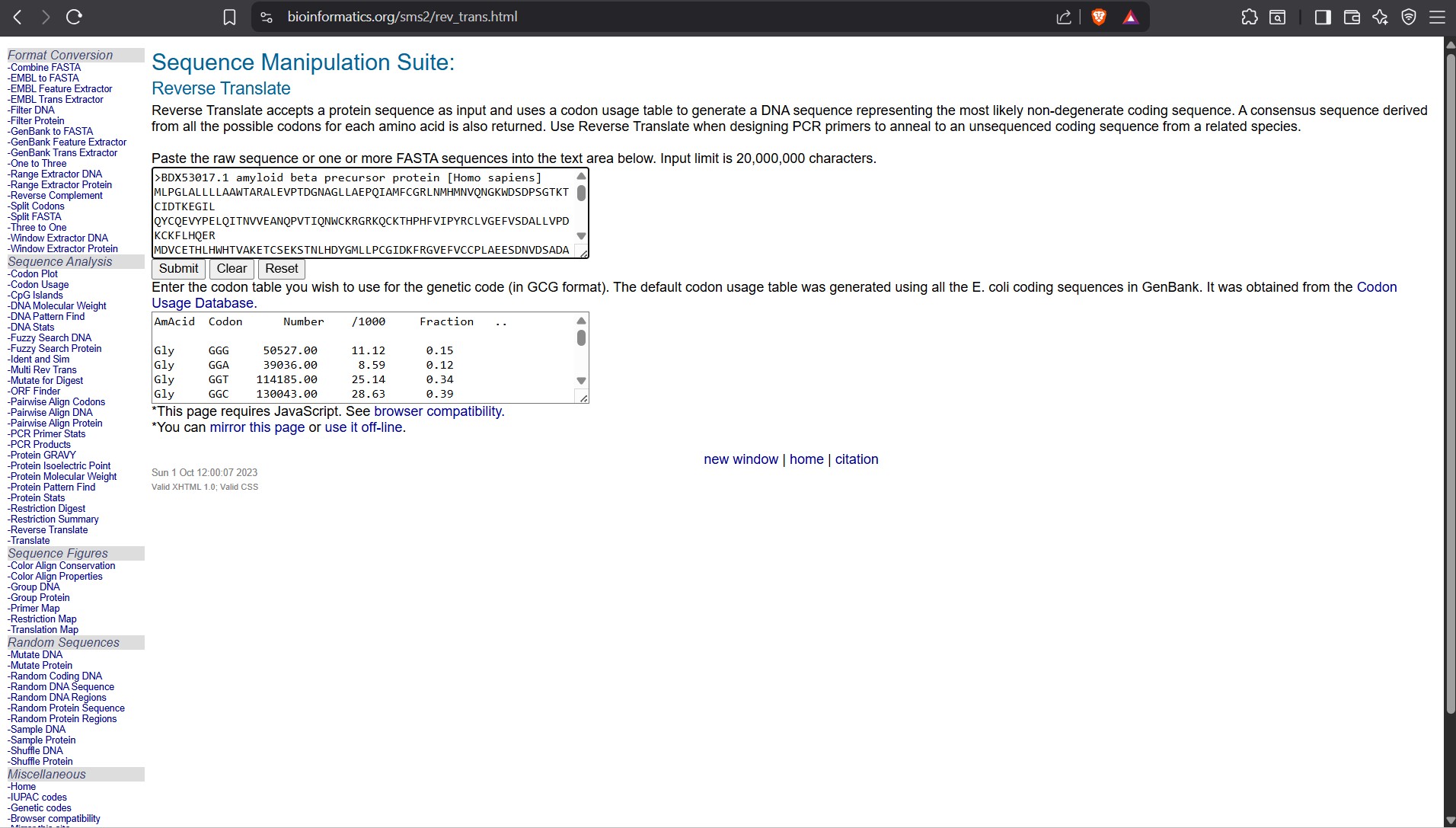
.jpg)
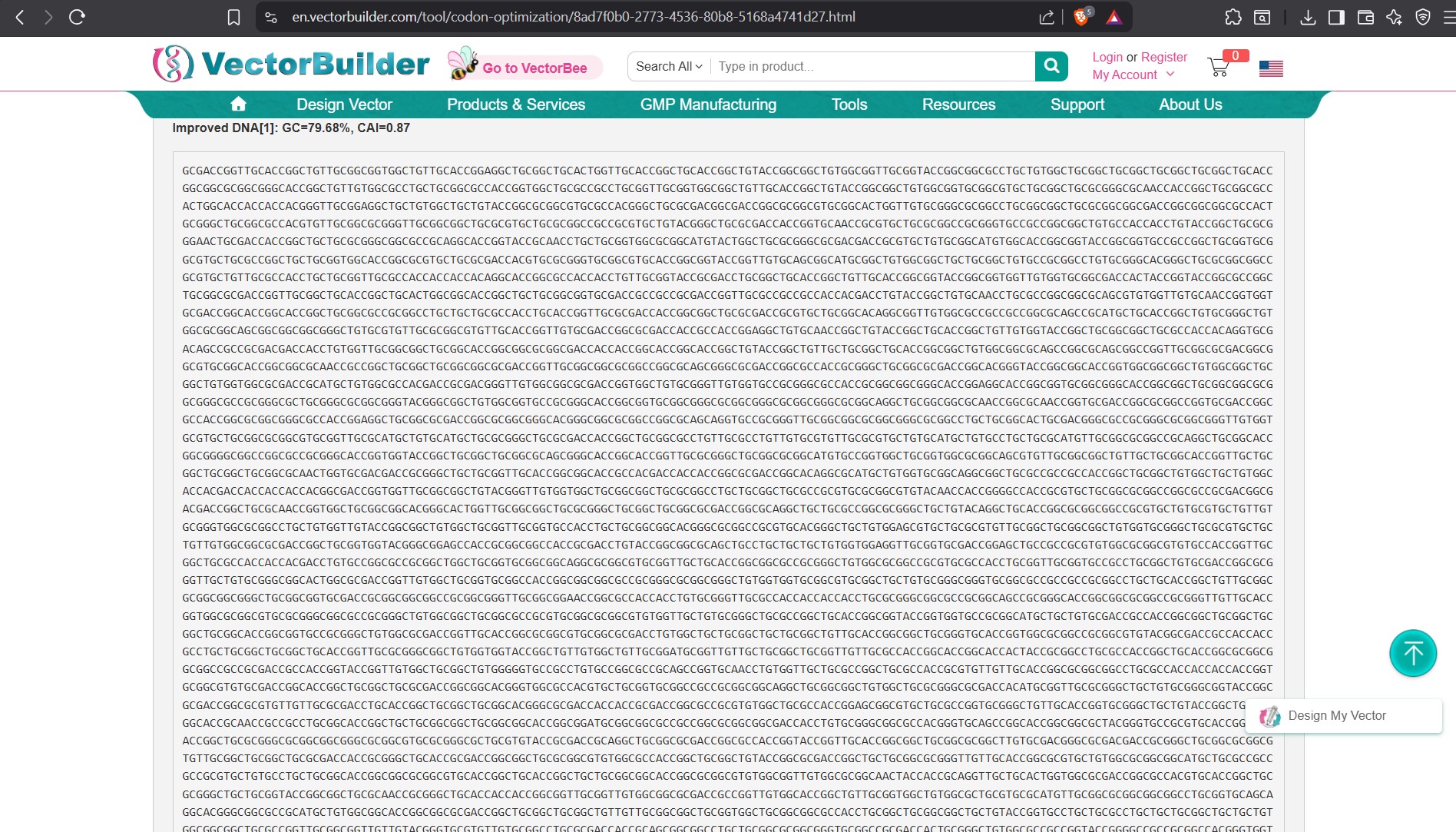
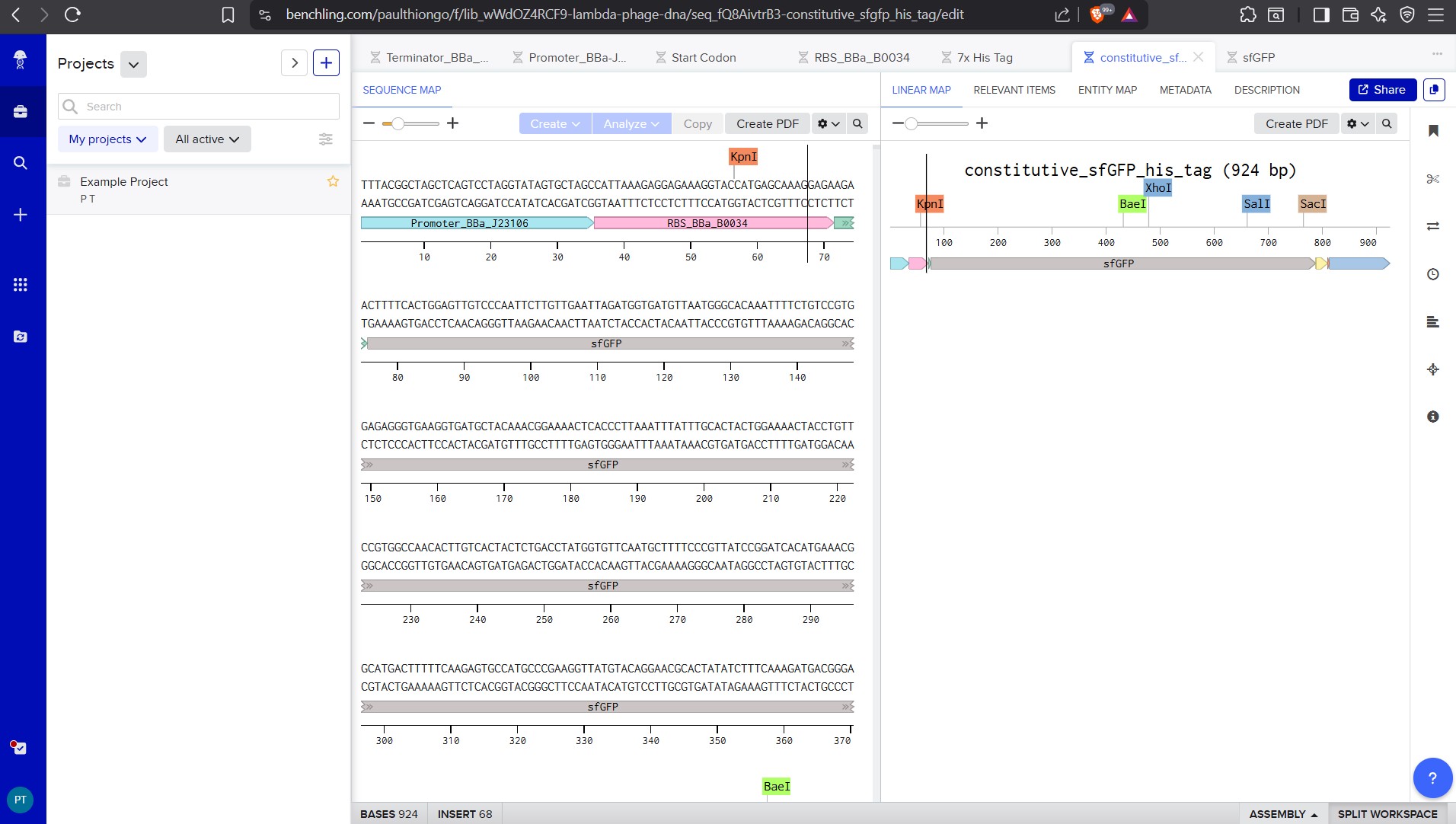
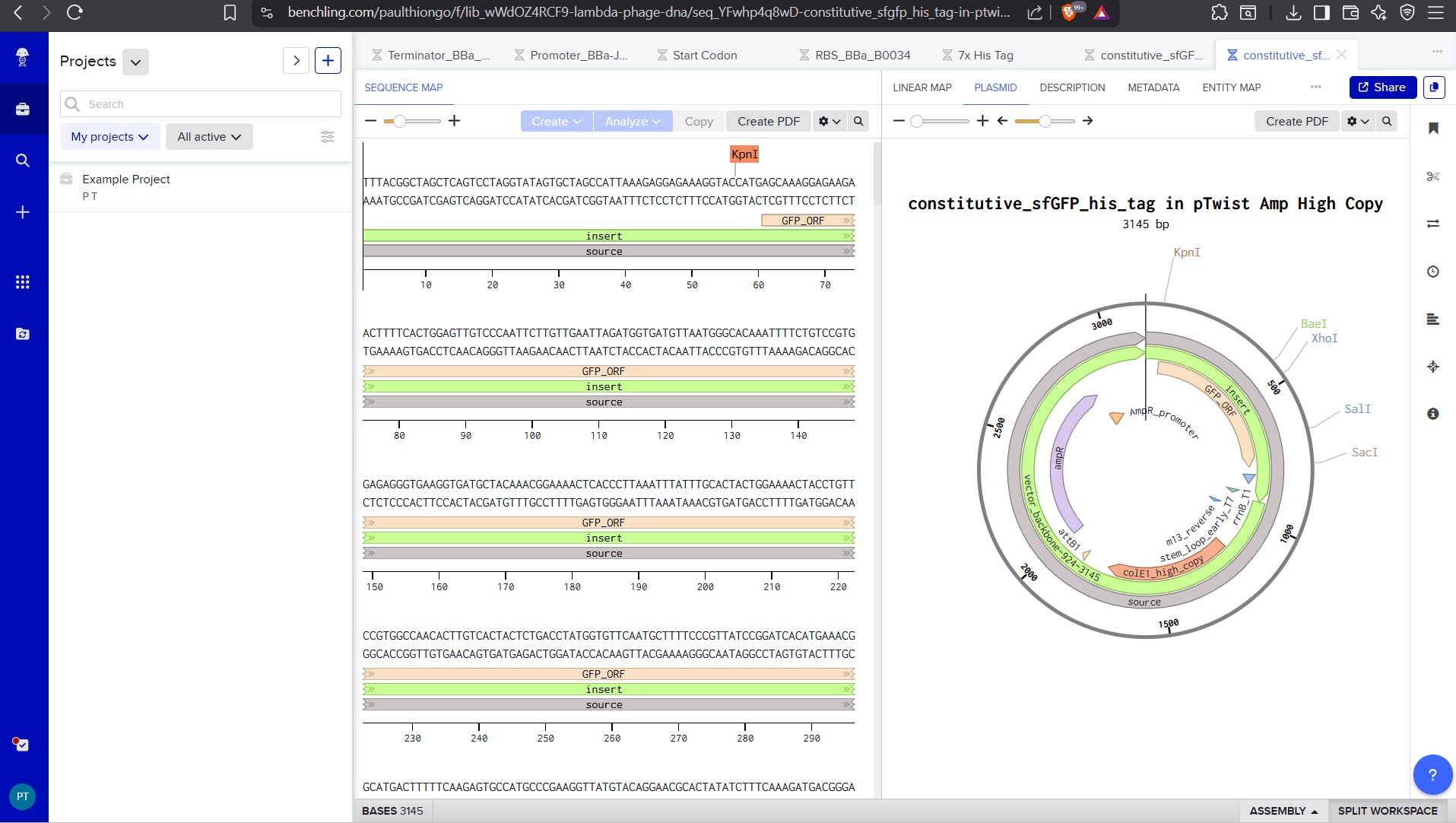

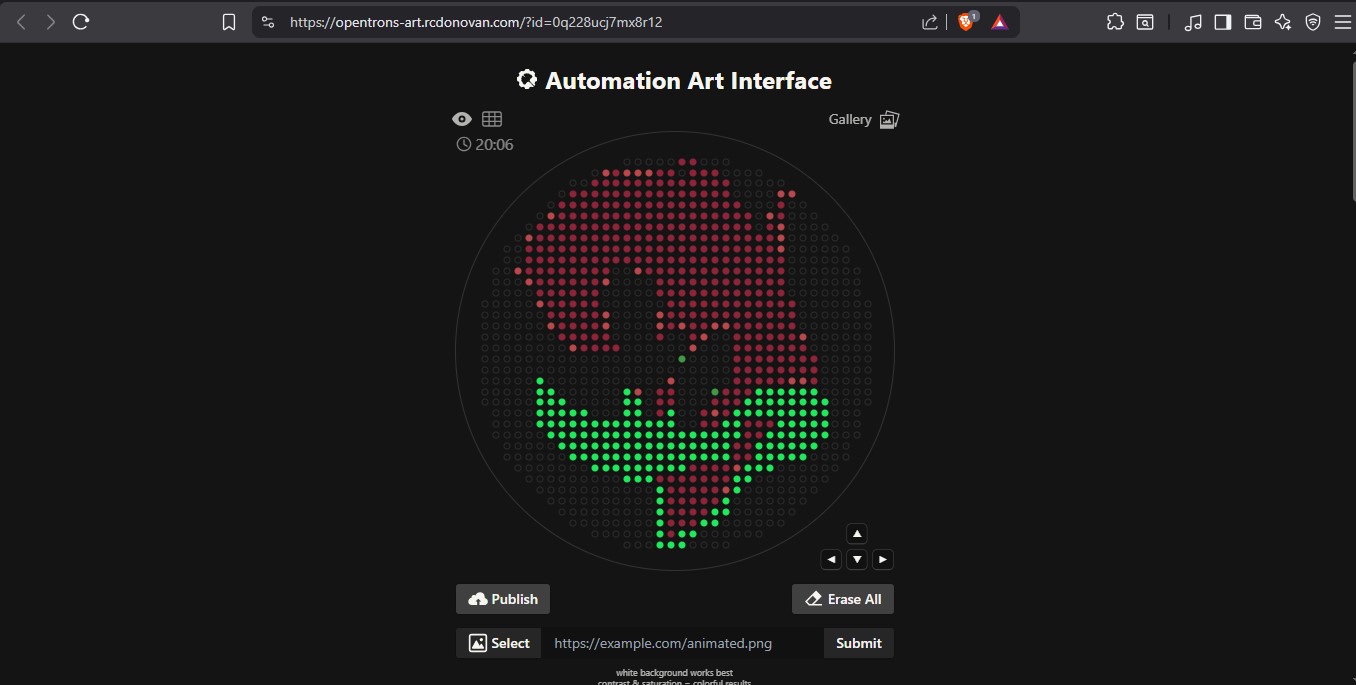
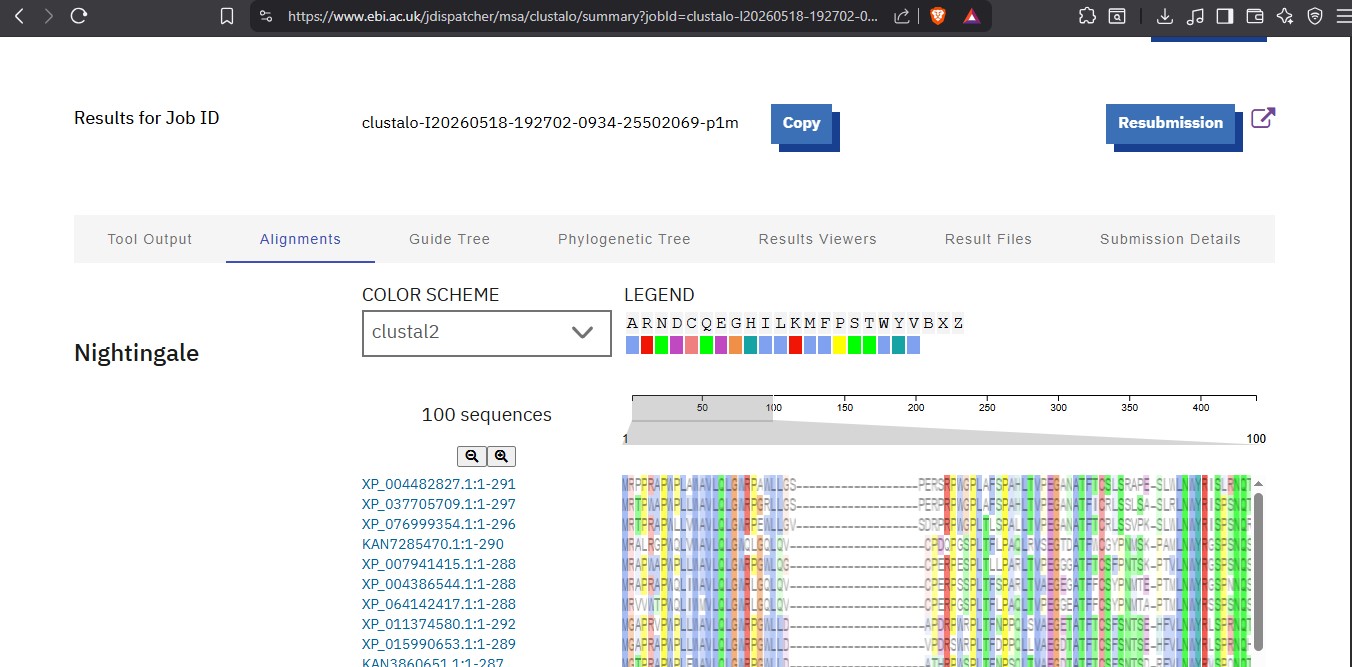
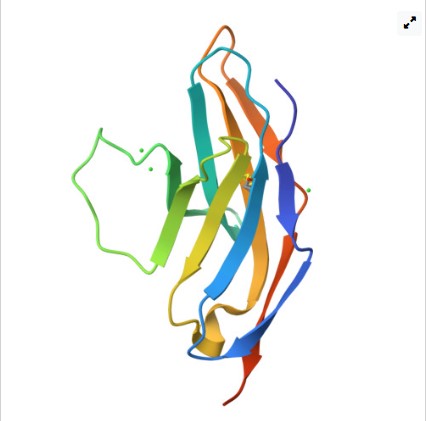
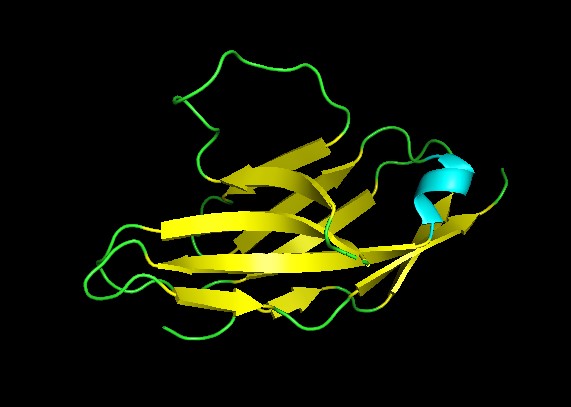
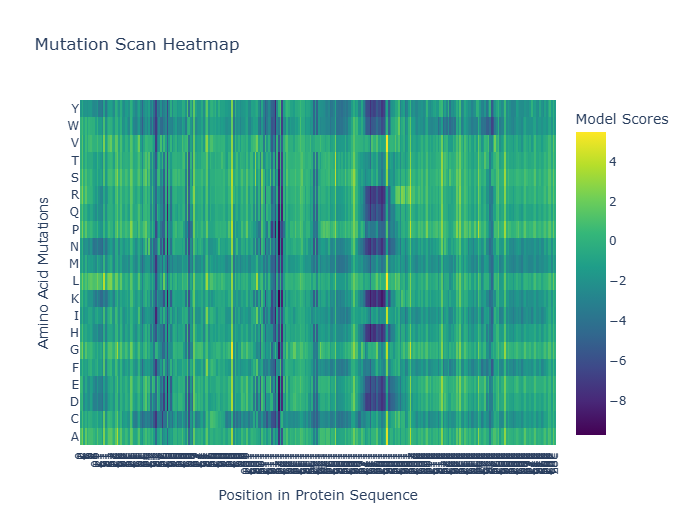
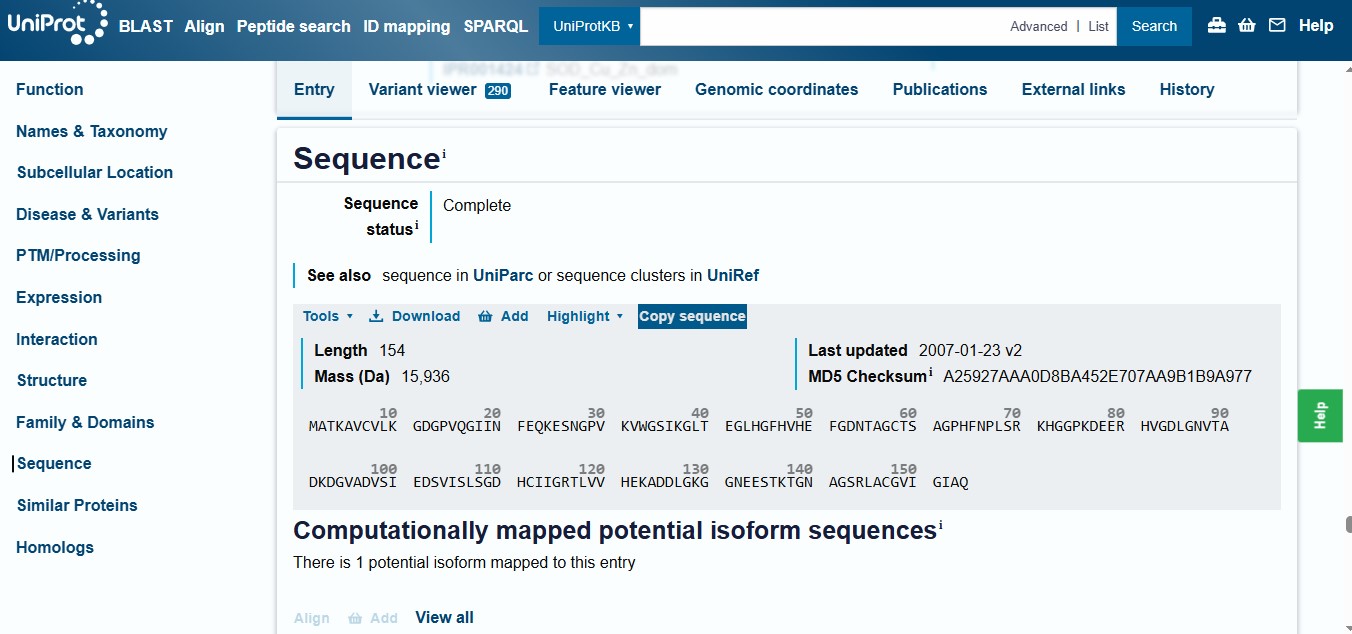
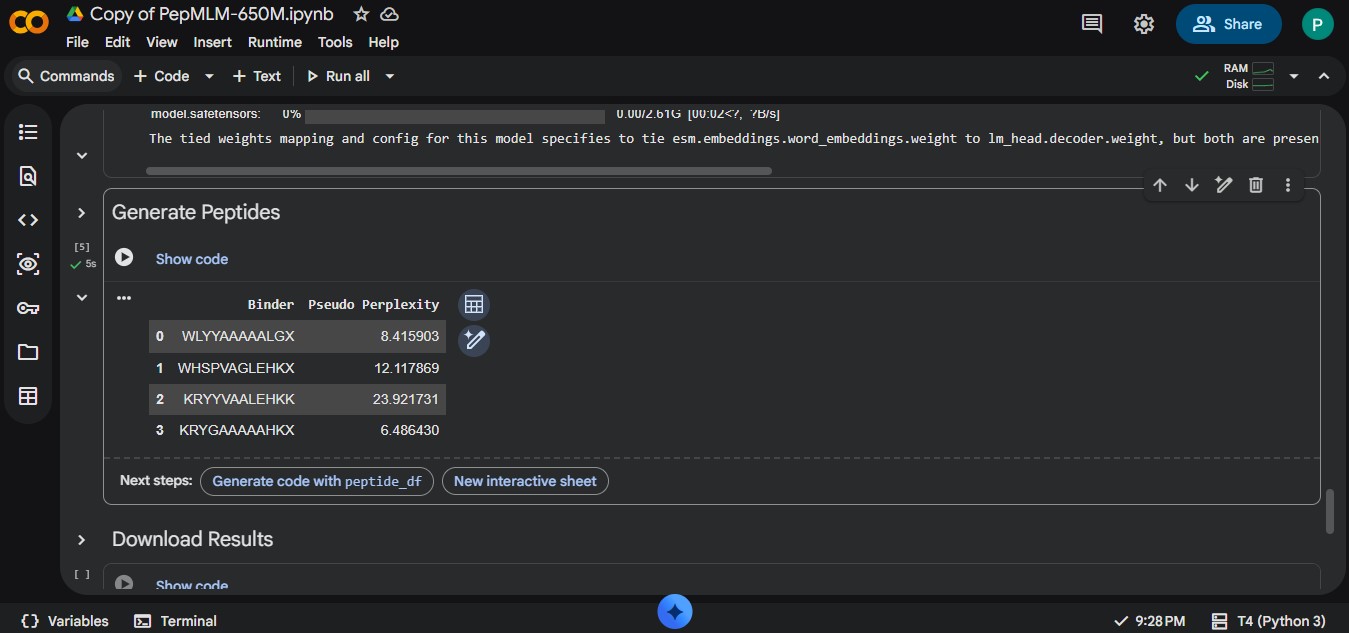
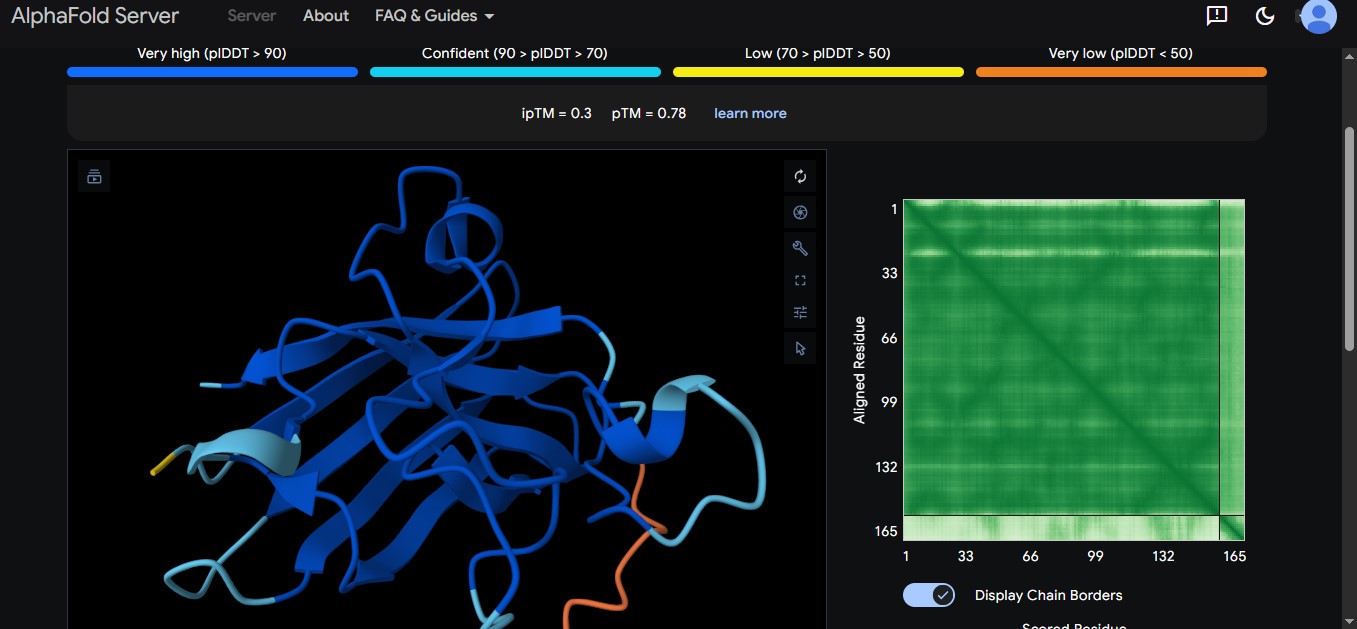
.jpg)
.jpg)