Week 2: DNA READ, WRITE, AND EDIT

Week # 2 Homework
DNA READ, WRITE & EDIT
A look at the sequencing and synthesis workflows, restriction digests and gel electrophoresis, and early genome-editing frameworks.
Part 1: Benchling & In-silico Gel Art
See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details. Overview: • Make a free account at benchling.com • Import the Lambda DNA. • Simulate Restriction Enzyme Digestion with the following Enzymes: ◦ EcoRI ◦ HindIII ◦ BamHI ◦ KpnI ◦ EcoRV ◦ SacI ◦ SalI • Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. • You might find Ronan’s website a helpful tool for quickly iterating on designs!
I was able to make a free account on Benchling and imported the Lamda DNA sequence as seen below.
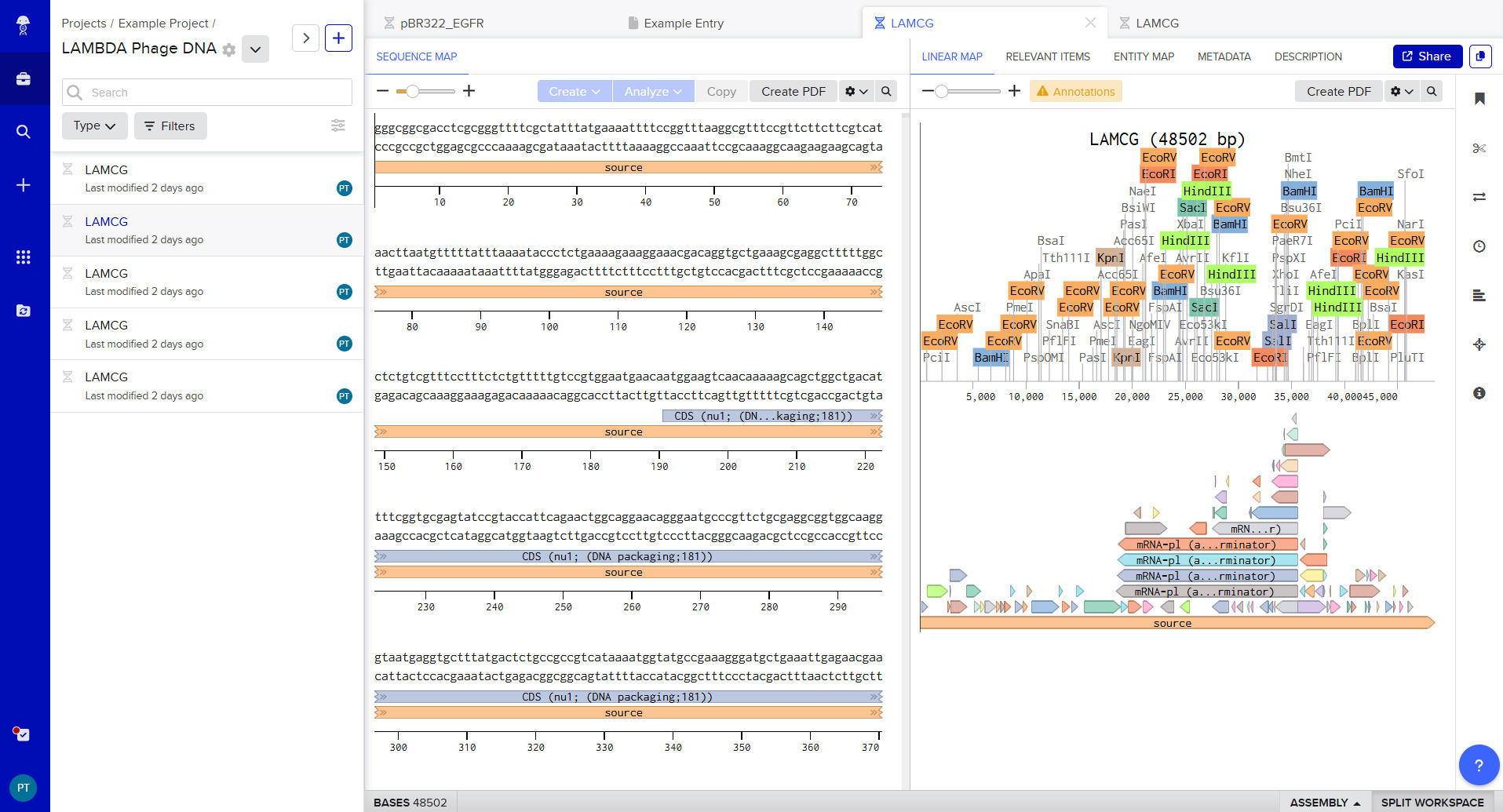
A pattern below showing the simulation for each of the enzymes producing different fragment patterns created from the restriction enzyme digest with the following enzymes: ◦ EcoRI ◦ HindIII ◦ BamHI ◦ KpnI ◦ EcoRV ◦ SacI ◦ SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
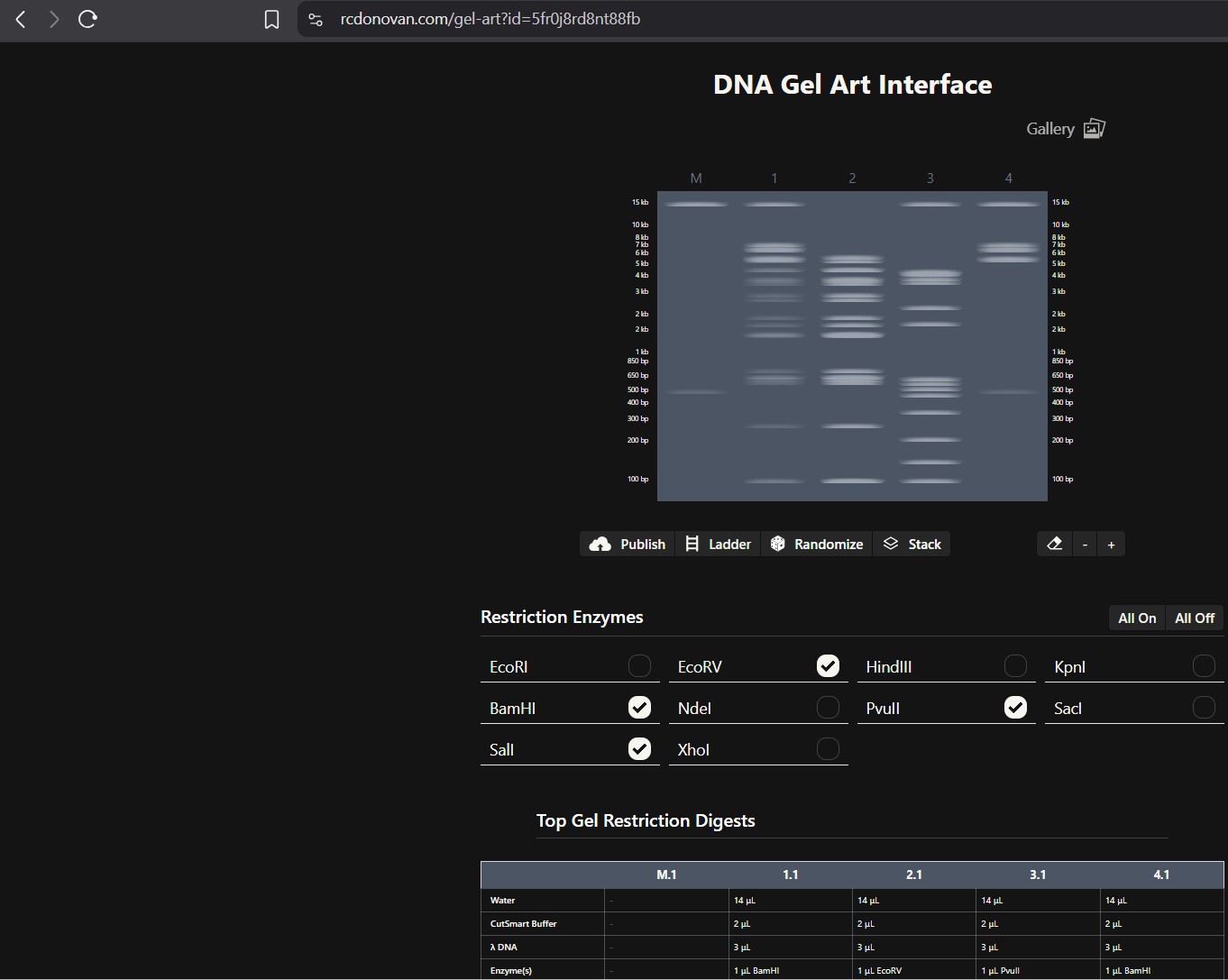
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis

The instructions on the lab experiment designed in Part 1 and outlined in the Gel Art: Restriction Digests and Gel Electrophoresis protocol. Since I had no access to the lab experiment, the simulation of the gel shows how Lamda DNA would have been digested by the seven different restriction enzymes as seen from the gel electrophoresis plate. The individual lanes show how each enzyme cut the DNA, using the NEB2-log as the ladder on the left as a reference for size. The patterns are used to verify the sequence and map the DNA.
Part 3: DNA Design Challenge
3.1 Choose your protein.
Protein: Amyloid beta precursor protein Organism: Homo sapiens GenBank: BDX53017.1 AA Sequence
BDX53017.1 amyloid beta precursor protein [Homo sapiens] MLPGLALLLLAAWTARALEVPTDGNAGLLAEPQIAMFCGRLNMHMNVQNGKWDSDPSGTKTCIDTKEGIL QYCQEVYPELQITNVVEANQPVTIQNWCKRGRKQCKTHPHFVIPYRCLVGEFVSDALLVPDKCKFLHQER MDVCETHLHWHTVAKETCSEKSTNLHDYGMLLPCGIDKFRGVEFVCCPLAEESDNVDSADAEEDDSDVWW GGADTDYADGSEDKVVEVAEEEEVAEVEEEEADDDEDDEDGDEVEEEAEEPYEEATERTTSIATTTTTTT ESVEEVVREVCSEQAETGPCRAMISRWYFDVTEGKCAPFFYGGCGGNRNNFDTEEYCMAVCGSAMSQSLL KTTQEPLARDPVKLPTTAASTPDAVDKYLETPGDENEHAHFQKAKERLEAKHRERMSQVMREWEEAERQA KNLPKADKKAVIQHFQEKVESLEQEAANERQQLVETHMARVEAMLNDRRRLALENYITALQAVPPRPRHV FNMLKKYVRAEQKDRQHTLKHFEHVRMVDPKKAAQIRSQVMTHLRVIYERMNQSLSLLYNVPAVAEEIQD EVDELLQKEQNYSDDVLANMISEPRISYGNDALMPSLTETKTTVELLPVNGEFSLDDLQPWHSFGADSVP ANTENEVEPVDARPAADRGLTTRPGSGLTNIKTEEISEVKMDAEFRHDSGYEVHHQKLVFFAEDVGSNKG AIIGLMVGGVVIATVIVITLVMLKKKQYTSIHHGVVEVDAAVTPEERHLSKMQQNGYENPTYKFFEQMQN
I chose this protein since numerous studies have placed the protein leading to a molecular pathway mechanism that leads to neurodegeneration, synaptic failure and the clinical onset of Alzheimer’s disease.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose. [Example from our group homework, you may notice the particular format — The example below came from UniProt]
sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1 METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLL EAVIRTVTTLQQLLT
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Searching Online on how to Reverse Translate, I found the following website that has the tool to help me reverse translate: https://www.bioinformatics.org/sms2/rev_trans.html with the results shown in the image below.
I used this tool: https://www.bioinformatics.org/sms2/rev_trans.html
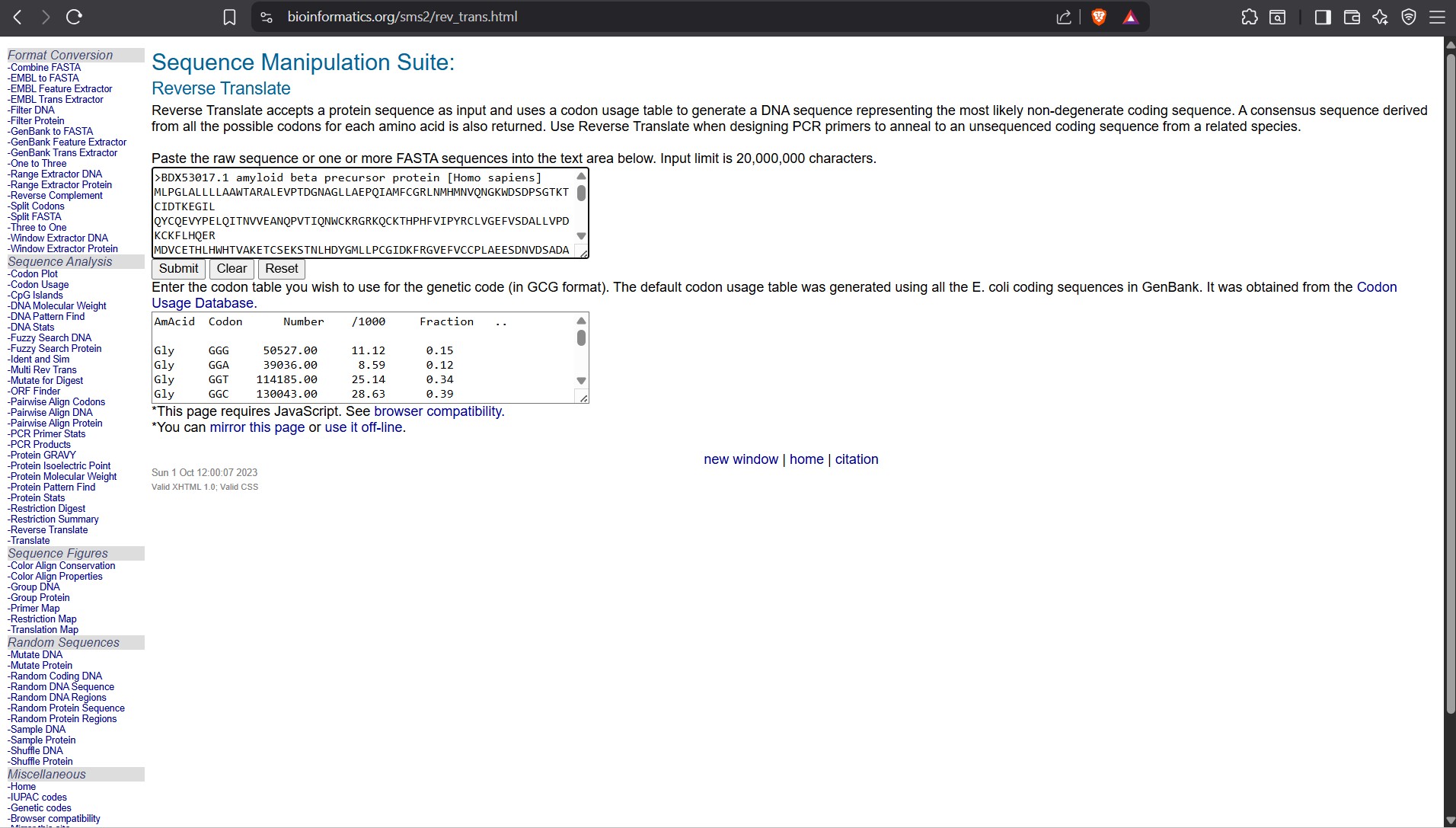
Reverse Translate results Results for 770 residue sequence “BDX53017.1 amyloid beta precursor protein [Homo sapiens]” starting “MLPGLALLLL”
reverse translation of BDX53017.1 amyloid beta precursor protein [Homo sapiens] to a 2310 base sequence of most likely codons. atgctgccgggcctggcgctgctgctgctggcggcgtggaccgcgcgcgcgctggaagtg ccgaccgatggcaacgcgggcctgctggcggaaccgcagattgcgatgttttgcggccgc ctgaacatgcatatgaacgtgcagaacggcaaatgggatagcgatccgagcggcaccaaa acctgcattgataccaaagaaggcattctgcagtattgccaggaagtgtatccggaactg cagattaccaacgtggtggaagcgaaccagccggtgaccattcagaactggtgcaaacgc ggccgcaaacagtgcaaaacccatccgcattttgtgattccgtatcgctgcctggtgggc gaatttgtgagcgatgcgctgctggtgccggataaatgcaaatttctgcatcaggaacgc atggatgtgtgcgaaacccatctgcattggcataccgtggcgaaagaaacctgcagcgaa aaaagcaccaacctgcatgattatggcatgctgctgccgtgcggcattgataaatttcgc ggcgtggaatttgtgtgctgcccgctggcggaagaaagcgataacgtggatagcgcggat gcggaagaagatgatagcgatgtgtggtggggcggcgcggataccgattatgcggatggc agcgaagataaagtggtggaagtggcggaagaagaagaagtggcggaagtggaagaagaa gaagcggatgatgatgaagatgatgaagatggcgatgaagtggaagaagaagcggaagaa ccgtatgaagaagcgaccgaacgcaccaccagcattgcgaccaccaccaccaccaccacc gaaagcgtggaagaagtggtgcgcgaagtgtgcagcgaacaggcggaaaccggcccgtgc cgcgcgatgattagccgctggtattttgatgtgaccgaaggcaaatgcgcgccgtttttt tatggcggctgcggcggcaaccgcaacaactttgataccgaagaatattgcatggcggtg tgcggcagcgcgatgagccagagcctgctgaaaaccacccaggaaccgctggcgcgcgat ccggtgaaactgccgaccaccgcggcgagcaccccggatgcggtggataaatatctggaa accccgggcgatgaaaacgaacatgcgcattttcagaaagcgaaagaacgcctggaagcg aaacatcgcgaacgcatgagccaggtgatgcgcgaatgggaagaagcggaacgccaggcg aaaaacctgccgaaagcggataaaaaagcggtgattcagcattttcaggaaaaagtggaa agcctggaacaggaagcggcgaacgaacgccagcagctggtggaaacccatatggcgcgc gtggaagcgatgctgaacgatcgccgccgcctggcgctggaaaactatattaccgcgctg caggcggtgccgccgcgcccgcgccatgtgtttaacatgctgaaaaaatatgtgcgcgcg gaacagaaagatcgccagcataccctgaaacattttgaacatgtgcgcatggtggatccg aaaaaagcggcgcagattcgcagccaggtgatgacccatctgcgcgtgatttatgaacgc atgaaccagagcctgagcctgctgtataacgtgccggcggtggcggaagaaattcaggat gaagtggatgaactgctgcagaaagaacagaactatagcgatgatgtgctggcgaacatg attagcgaaccgcgcattagctatggcaacgatgcgctgatgccgagcctgaccgaaacc aaaaccaccgtggaactgctgccggtgaacggcgaatttagcctggatgatctgcagccg tggcatagctttggcgcggatagcgtgccggcgaacaccgaaaacgaagtggaaccggtg gatgcgcgcccggcggcggatcgcggcctgaccacccgcccgggcagcggcctgaccaac attaaaaccgaagaaattagcgaagtgaaaatggatgcggaatttcgccatgatagcggc tatgaagtgcatcatcagaaactggtgttttttgcggaagatgtgggcagcaacaaaggc gcgattattggcctgatggtgggcggcgtggtgattgcgaccgtgattgtgattaccctg gtgatgctgaaaaaaaaacagtataccagcattcatcatggcgtggtggaagtggatgcg gcggtgaccccggaagaacgccatctgagcaaaatgcagcagaacggctatgaaaacccg acctataaattttttgaacagatgcagaac
reverse translation of BDX53017.1 amyloid beta precursor protein [Homo sapiens] to a 2310 base sequence of consensus codons. atgytnccnggnytngcnytnytnytnytngcngcntggacngcnmgngcnytngargtn ccnacngayggnaaygcnggnytnytngcngarccncarathgcnatgttytgyggnmgn ytnaayatgcayatgaaygtncaraayggnaartgggaywsngayccnwsnggnacnaar acntgyathgayacnaargarggnathytncartaytgycargargtntayccngarytn carathacnaaygtngtngargcnaaycarccngtnacnathcaraaytggtgyaarmgn ggnmgnaarcartgyaaracncayccncayttygtnathccntaymgntgyytngtnggn garttygtnwsngaygcnytnytngtnccngayaartgyaarttyytncaycargarmgn atggaygtntgygaracncayytncaytggcayacngtngcnaargaracntgywsngar aarwsnacnaayytncaygaytayggnatgytnytnccntgyggnathgayaarttymgn ggngtngarttygtntgytgyccnytngcngargarwsngayaaygtngaywsngcngay gcngargargaygaywsngaygtntggtggggnggngcngayacngaytaygcngayggn wsngargayaargtngtngargtngcngargargargargtngcngargtngargargar gargcngaygaygaygargaygaygargayggngaygargtngargargargcngargar ccntaygargargcnacngarmgnacnacnwsnathgcnacnacnacnacnacnacnacn garwsngtngargargtngtnmgngargtntgywsngarcargcngaracnggnccntgy mgngcnatgathwsnmgntggtayttygaygtnacngarggnaartgygcnccnttytty tayggnggntgyggnggnaaymgnaayaayttygayacngargartaytgyatggcngtn tgyggnwsngcnatgwsncarwsnytnytnaaracnacncargarccnytngcnmgngay ccngtnaarytnccnacnacngcngcnwsnacnccngaygcngtngayaartayytngar acnccnggngaygaraaygarcaygcncayttycaraargcnaargarmgnytngargcn aarcaymgngarmgnatgwsncargtnatgmgngartgggargargcngarmgncargcn aaraayytnccnaargcngayaaraargcngtnathcarcayttycargaraargtngar wsnytngarcargargcngcnaaygarmgncarcarytngtngaracncayatggcnmgn gtngargcnatgytnaaygaymgnmgnmgnytngcnytngaraaytayathacngcnytn cargcngtnccnccnmgnccnmgncaygtnttyaayatgytnaaraartaygtnmgngcn garcaraargaymgncarcayacnytnaarcayttygarcaygtnmgnatggtngayccn aaraargcngcncarathmgnwsncargtnatgacncayytnmgngtnathtaygarmgn atgaaycarwsnytnwsnytnytntayaaygtnccngcngtngcngargarathcargay gargtngaygarytnytncaraargarcaraaytaywsngaygaygtnytngcnaayatg athwsngarccnmgnathwsntayggnaaygaygcnytnatgccnwsnytnacngaracn aaracnacngtngarytnytnccngtnaayggngarttywsnytngaygayytncarccn tggcaywsnttyggngcngaywsngtnccngcnaayacngaraaygargtngarccngtn gaygcnmgnccngcngcngaymgnggnytnacnacnmgnccnggnwsnggnytnacnaay athaaracngargarathwsngargtnaaratggaygcngarttymgncaygaywsnggn taygargtncaycaycaraarytngtnttyttygcngargaygtnggnwsnaayaarggn gcnathathggnytnatggtnggnggngtngtnathgcnacngtnathgtnathacnytn gtnatgytnaaraaraarcartayacnwsnathcaycayggngtngtngargtngaygcn gcngtnacnccngargarmgncayytnwsnaaratgcarcaraayggntaygaraayccn acntayaarttyttygarcaratgcaraay
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Lysis protein DNA sequence atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttac caatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why? It is done to come up with codons that are more frequently used by the host organism so as it can efficiently translate the protein and produce the desired protein in higher levels. As codon that are rare or less favoured would affect the production of the protein in the organism and the programs helps in identifying the best fit. I chose E.coli cause it is a cheaper alternative in terms of cost and is widely used. I also the use of human to use in the trial of the protein in a mammalian system.
Improved DNA homo sapiens
I searched online for ways to Codon Optimize
and used the tool: https://en.vectorbuilder.com/tool/codon-optimization.html
.jpg)
Improved E.coli
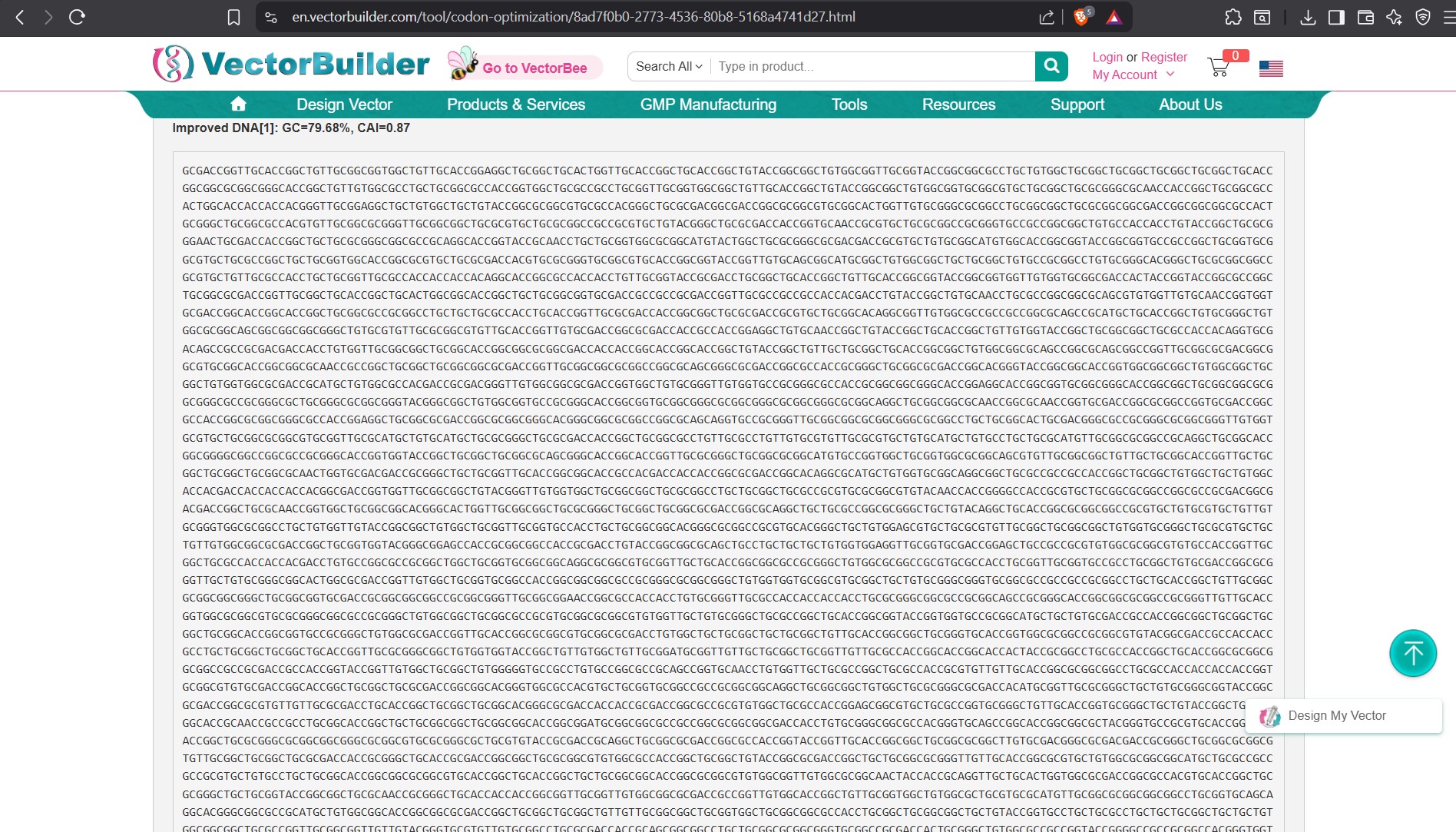
Improved DNA[1]: GC=58.01%, CAI=0.92 ATGCTGCCTGGACTGGCCCTCCTGCTGCTGGCCGCCTGGACCGCCAGGGCCCTGGAGGTGCCCACCGACGGTAACGCCGGCCTGCTGGCCGAGCCCCAGATCGCTATGTTCTGCGGGAGGCTGAACATGCACATGAACGTGCAGAACGGGA AGTGGGACTCCGACCCCTCCGGAACTAAGACTTGCATCGACACAAAAGAGGGAATTCTGCAGTACTGTCAGGAAGTGTACCCCGAGCTGCAGATCACTAATGTGGTGGAAGCTAATCAGCCAGTCACAATCCAGAACTGGTGCAAGAGAGGC AGAAAGCAGTGCAAAACCCACCCCCACTTCGTGATCCCATACAGGTGCCTGGTGGGCGAGTTCGTCTCTGACGCTCTGCTGGTGCCTGACAAATGCAAGTTCCTCCATCAGGAGAGGATGGACGTTTGTGAGACACACCTCCACTGGCACACC GTCGCCAAAGAGACATGCTCTGAGAAGAGTACTAACCTGCACGACTATGGGATGCTGCTGCCTTGTGGGATTGACAAGTTCCGGGGCGTGGAGTTCGTGTGCTGTCCCCTGGCCGAGGAGAGCGACAATGTGGACAGTGCCGACGCCGAGGAAG ACGACAGCGACGTGTGGTGGGGCGGCGCCGACACCGACTACGCCGATGGGAGCGAGGATAAAGTCGTGGAAGTCGCCGAGGAGGAGGAGGTGGCCGAGGTGGAGGAAGAAGAGGCCGACGACGATGAGGACGACGAGGATGGCGACGAGGTGGA GGAGGAAGCTGAGGAGCCATATGAAGAGGCAACAGAGCGGACCACCTCTATTGCGACCACAACCACCACCACCACTGAGAGCGTGGAGGAGGTGGTGAGGGAAGTGTGCTCTGAACAGGCCGAAACCGGGCCATGTAGAGCTATGATCTCCAGA TGGTATTTCGACGTCACAGAGGGCAAGTGCGCCCCCTTCTTTTACGGCGGCTGTGGCGGGAACCGGAACAATTTTGATACTGAGGAGTACTGCATGGCCGTCTGCGGCTCTGCAATGAGCCAGTCCCTGCTTAAAACTACCCAGGAGCCCCTGG CCAGGGACCCTGTGAAACTGCCCACCACCGCAGCCTCTACTCCCGATGCCGTGGACAAGTACCTGGAAACCCCCGGAGATGAGAACGAGCATGCCCACTTTCAGAAGGCAAAGGAAAGACTGGAGGCCAAGCACCGCGAAAGAATGTCCCAGGT GATGAGGGAATGGGAAGAGGCCGAGCGCCAGGCCAAGAACCTGCCCAAAGCCGACAAGAAGGCCGTGATCCAGCACTTTCAGGAAAAGGTGGAGTCTCTGGAGCAGGAGGCCGCCAATGAGAGACAGCAGCTGGTGGAGACCCACATGGCCCGC GTCGAGGCCATGCTGAATGACAGAAGGCGGCTGGCCCTGGAGAACTACATCACAGCCCTGCAGGCTGTGCCACCAAGGCCCAGGCATGTGTTTAACATGCTGAAAAAGTACGTGAGAGCCGAACAGAAGGATAGGCAGCACACACTGAAACATT TTGAGCACGTGCGGATGGTGGACCCCAAGAAGGCTGCACAGATCAGGTCTCAGGTGATGACCCATCTTAGAGTCATATACGAGAGAATGAACCAGTCCCTGAGCCTGCTGTATAACGTGCCCGCCGTGGCCGAGGAGATCCAGGACGAGGTGGA TGAGCTGCTGCAGAAGGAGCAGAATTATAGTGACGATGTGCTGGCCAACATGATCTCCGAGCCAAGAATCTCCTACGGAAACGACGCACTGATGCCCAGCCTGACCGAGACAAAGACAACAGTGGAGCTGCTGCCAGTGAATGGCGAATTTTCC CTGGACGATCTGCAGCCTTGGCACTCATTCGGCGCCGATAGCGTCCCTGCCAACACAGAGAACGAAGTGGAGCCTGTGGACGCCCGGCCTGCCGCAGACAGGGGCCTGACCACTAGGCCAGGATCCGGCCTGACCAACATCAAAACCGAGGAGA TCTCCGAGGTGAAGATGGATGCCGAGTTCAGACACGACAGCGGATACGAGGTGCACCACCAGAAGCTGGTGTTCTTTGCCGAGGATGTGGGAAGCAACAAGGGCGCAATCATCGGTCTGATGGTGGGCGGCGTGGTGATCGCCACCGTGATCGT GATCACCCTGGTGATGCTGAAAAAGAAGCAGTATACATCTATCCACCACGGCGTGGTGGAGGTGGATGCCGCCGTGACCCCCGAAGAGAGGCACCTGAGCAAGATGCAGCAGAACGGGTATGAGAATCCCACTTACAAATTCTTTGAGCAGATG CAGAAC
Improved DNA[1]: GC=53.81%, CAI=0.93 (Escherichia Coli) ATGCTGCCGGGCCTGGCGCTGCTGCTGCTGGCGGCGTGGACCGCGCGCGCGCTGGAAGTGCCGACCGACGGCAATGCGGGCCTGCTGGCCGAACCGCAGATTGCCATGTTTTGCGGCCGCCTGAATATGCATATGAACGTGCAGAATGGCAAAT GGGATAGCGATCCGAGCGGCACCAAAACGTGCATTGATACCAAAGAAGGCATTCTGCAGTACTGTCAGGAAGTGTATCCGGAACTGCAGATCACCAATGTGGTGGAAGCGAACCAGCCGGTGACCATTCAGAACTGGTGCAAACGCGGCCGCAA ACAGTGTAAAACCCATCCGCATTTTGTGATTCCGTATCGTTGCCTGGTGGGCGAGTTCGTTAGCGATGCCCTGCTGGTGCCGGATAAATGCAAATTTCTGCATCAGGAACGCATGGATGTGTGCGAAACCCATCTGCATTGGCATACTGTTGCA AAAGAAACCTGCTCAGAAAAAAGCACCAACCTGCATGATTATGGCATGCTGCTGCCGTGCGGCATTGATAAATTTCGCGGTGTTGAATTTGTGTGCTGCCCGCTGGCGGAAGAAAGCGATAACGTGGATAGTGCAGATGCGGAAGAAGATGACA GCGATGTGTGGTGGGGCGGCGCGGATACCGATTATGCGGACGGCAGCGAAGATAAAGTTGTGGAAGTGGCGGAGGAAGAAGAAGTGGCAGAAGTGGAAGAAGAAGAAGCCGATGATGATGAAGATGATGAAGATGGCGATGAAGTTGAAGAAGA AGCGGAAGAACCGTATGAAGAAGCGACGGAACGCACCACCAGCATTGCCACCACCACGACCACGACCACCGAAAGCGTGGAAGAAGTGGTGCGTGAAGTGTGCAGCGAACAGGCGGAAACCGGGCCGTGTCGTGCCATGATTAGCCGCTGGTAT TTTGATGTTACCGAAGGTAAATGCGCGCCGTTTTTTTATGGCGGCTGCGGTGGCAATCGTAACAACTTTGATACCGAAGAATACTGCATGGCCGTTTGCGGCAGCGCAATGTCGCAGAGCCTGCTGAAAACCACCCAGGAACCGCTGGCGCGCG ACCCGGTGAAACTGCCGACCACCGCAGCCAGCACCCCGGATGCCGTTGATAAATACCTGGAAACCCCGGGTGATGAAAATGAACATGCGCATTTTCAGAAAGCCAAAGAACGCCTGGAAGCGAAACATCGTGAACGCATGAGCCAGGTGATGCG CGAATGGGAAGAAGCGGAACGTCAGGCGAAAAACCTGCCGAAAGCGGACAAAAAGGCCGTGATTCAGCACTTTCAGGAGAAAGTGGAAAGCCTGGAGCAGGAAGCGGCCAATGAACGTCAGCAGCTGGTAGAAACCCACATGGCGCGCGTGGAA GCCATGCTGAACGATCGCCGTCGCTTGGCGCTGGAAAACTACATTACCGCGCTGCAGGCGGTGCCGCCGCGCCCGCGCCATGTGTTTAATATGCTGAAAAAATATGTGCGCGCCGAACAGAAAGATCGTCAGCACACCCTGAAACATTTTGAAC ACGTGCGCATGGTAGATCCGAAAAAAGCGGCACAGATTCGTAGCCAAGTGATGACCCACCTTCGCGTGATTTACGAACGCATGAACCAGAGCCTGAGCCTGCTGTATAACGTGCCGGCAGTGGCGGAAGAAATTCAGGATGAAGTGGACGAATT ACTGCAGAAAGAACAAAATTACAGCGATGATGTGCTGGCCAACATGATTTCGGAACCGCGCATTAGCTACGGCAATGATGCCCTGATGCCGAGCCTGACCGAAACCAAAACCACCGTTGAACTGCTGCCGGTAAATGGCGAATTCAGCCTGGAT GATCTGCAGCCGTGGCATAGCTTTGGCGCGGATAGCGTGCCGGCAAACACCGAAAATGAAGTTGAACCGGTGGATGCCCGTCCGGCGGCCGATCGTGGCCTGACGACCCGTCCGGGCAGCGGTCTGACCAACATTAAAACCGAAGAAATTAGCG AAGTGAAAATGGATGCGGAATTTCGCCACGATAGCGGCTATGAAGTGCACCATCAGAAACTGGTGTTCTTTGCGGAAGATGTGGGCAGTAACAAAGGCGCGATTATTGGCCTGATGGTGGGCGGCGTGGTAATCGCGACCGTCATTGTGATTAC CCTGGTGATGCTGAAAAAAAAACAGTATACCAGCATTCACCATGGCGTGGTGGAAGTGGATGCCGCAGTTACCCCGGAAGAACGCCATCTGAGCAAAATGCAGCAGAACGGCTACGAAAATCCGACCTATAAATTCTTTGAACAGATGCAGAAT
Lysis protein DNA sequence with Codon-Optimization ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCA CCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both. Cells rely on DNA as an instruction manual for protein synthesis. The DNA sequence is first transcribed into messenger RNA (mRNA), which acts as a working blueprint. The cell then translates this mRNA template step-by-step to assemble the desired protein.
The production of amyloid beta precursor protein (APP) follows these standard genetic expression processes. Interestingly, a single APP gene can generate several different protein variants through a mechanism called alternative splicing, which occurs during and after transcription. When scientists create recombinant APP in laboratory settings, they must carefully manage multiple factors—including how much protein is produced, whether it folds correctly, and how chemical modifications are added after the protein is initially made. One particularly important modification is glycosylation (the addition of sugar molecules). These chemical alterations are essential because they affect how APP sits in the cell membrane and how it gets broken down into amyloid-beta (Aβ), a protein fragment associated with Alzheimer’s disease.
Central Dogma Process DNA encoding APP is transcribed into pre-mRNA by RNA polymerase II, which includes exons and introns. Alternative splicing (e.g., inclusion/exclusion of exon 15) generates multiple mature mRNA isoforms, which ribosomes then translate into distinct APP protein variants (e.g., APP695, APP751, APP770) differing in length and function.
Cell-Dependent Methods HEK293 or CHO cells are optimal for mammalian expression due to proper folding and secretion. Transfect with pcDNA3.1-APP plasmids under CMV promoter, induce with IPTG if hybrid, and purify via Ni-NTA (His-tagged) or immunoprecipitation; yields reach 10-50 mg/L with glycosylation intact for secretase cleavage studies.
Cell-Free Methods Rabbit reticulocyte lysate or wheat germ extracts excel for rapid prototyping. Mix PCR-amplified APP DNA (T7 promoter) with lysate, Mg2+/NTPs, and translate in 1-2 hours; add microsomes for membrane insertion. Best for isotopic labeling (15N-APP) without cellular toxicity, yielding 1-5 μg/μL but lacking full glycosylation.
Recommendation For authentic APP with Aβ-processing fidelity, use HEK293 cell-dependent systems over cell-free, as they support splicing machinery and PTMs essential for multiple isoforms. Cell-free suits quick screening or labeled protein.
Cell-Dependent Methods HEK293 and CHO cells are the preferred choice for producing APP proteins in mammalian systems because they naturally promote proper protein folding and enable the release of proteins from cells. Researchers introduce APP genes into these cells using pcDNA3.1 plasmids controlled by the CMV promoter. If using a hybrid system, IPTG can trigger protein production. Once synthesized, the APP protein is isolated using purification techniques like Ni-NTA chromatography (which targets His-tags) or immunoprecipitation. This approach produces substantial quantities—between 10-50 mg/L—and crucially, the proteins retain their sugar modifications (glycosylation), which are necessary for studying how secretase enzymes break down APP into amyloid-beta.
Cell-Free Methods Cell-free protein synthesis offers a faster, simpler alternative using extracts from rabbit reticulocytes or wheat germ instead of living cells. Scientists amplify APP DNA (using T7 promoter sequences) and combine it with the cell extract along with magnesium and nucleotides, completing protein synthesis in just 1-2 hours. Adding microsomes (membrane fragments) allows the newly made protein to insert into a membrane-like environment. This method is ideal for rapidly testing concepts and for creating labeled proteins enriched with isotopes like 15N without harming cells. However, yields are more modest at 1-5 μg/μL, and the proteins don’t receive the complete sugar modifications that cellular systems provide.
Recommendation For studying APP that behaves authentically and processes into amyloid-beta correctly, cell-based systems using HEK293 cells are superior to cell-free approaches. This is because living cells contain the machinery to perform alternative splicing and add critical chemical modifications—processes essential for generating the multiple APP variants. Cell-free systems work best for quick preliminary experiments or when producing specialized labeled proteins.
References
- Amyloid Precursor Protein Processing and Bioenergetics - PMC (https://pmc.ncbi.nlm.nih.gov/articles/PMC5316384/)
- Amyloid beta: structure, biology and structure-based therapeutic …- (https://www.nature.com/articles/aps201728)
- Knockdown of Amyloid Precursor Protein: Biological … - (https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2022.835645/full)
- Amyloid-beta precursor protein - (https://en.wikipedia.org/wiki/Amyloid-beta_precursor_protein)
- Recombinant amyloid beta-peptide production by coexpression with an affibody ligand - (https://pmc.ncbi.nlm.nih.gov/articles/PMC2606684/)
- Can a single gene produce multiple proteins? - (https://scienceofbiogenetics.com/articles/investigating-the-phenomenon-can-a-single-gene-produce-multiple-protein-variations)
- Targeting Amyloid-β Precursor Protein, APP, Splicing with Antisense Oligonucleotides Reduces Toxic Amyloid-β Production- (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5986716/)
- A Cellular Model of Amyloid Precursor Protein Processing and Amyloid-β Peptide Production - (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3931259/)
- Differential Processing of Amyloid-β Precursor Protein Directs Human Embryonic Stem Cell Proliferation and Differentiation into Neuronal Precursor Cells-(https://pmc.ncbi.nlm.nih.gov/articles/PMC2749153/)
- Translation: DNA to mRNA to Protein - (https://www.nature.com/scitable/topicpage/translation-dna-to-mrna-to-protein-393/)
- Targeting Amyloid-β Precursor Protein, APP, Splicing…- PMC (https://pmc.ncbi.nlm.nih.gov/articles/PMC5986716/)
- Genetic and chemical disruption of amyloid precursor protein… - PMC (https://pmc.ncbi.nlm.nih.gov/articles/PMC10839650/)
- A novel method for expression and purification of authentic amyloid-β with and without 15 N labels- (https://www.sciencedirect.com/science/article/abs/pii/S1046592815001072)
- Transcription: an overview of DNA transcription (article) - (https://www.khanacademy.org/science/ap-biology/gene-expression-and-regulation/transcription-and-rna-processing/a/overview-of-transcription)
- Targeting Amyloid-β Precursor Protein, APP, Splicing with… (https://www.merckmillipore.com/SI/en/tech-docs/paper/1325529)
Part 4: Prepare a Twist DNA Synthesis Order
This is a practice exercise, not necessarily your real Twist order!
4.1. Create a Twist account, and Benchling account
4.2. Build Your DNA Insert Sequence
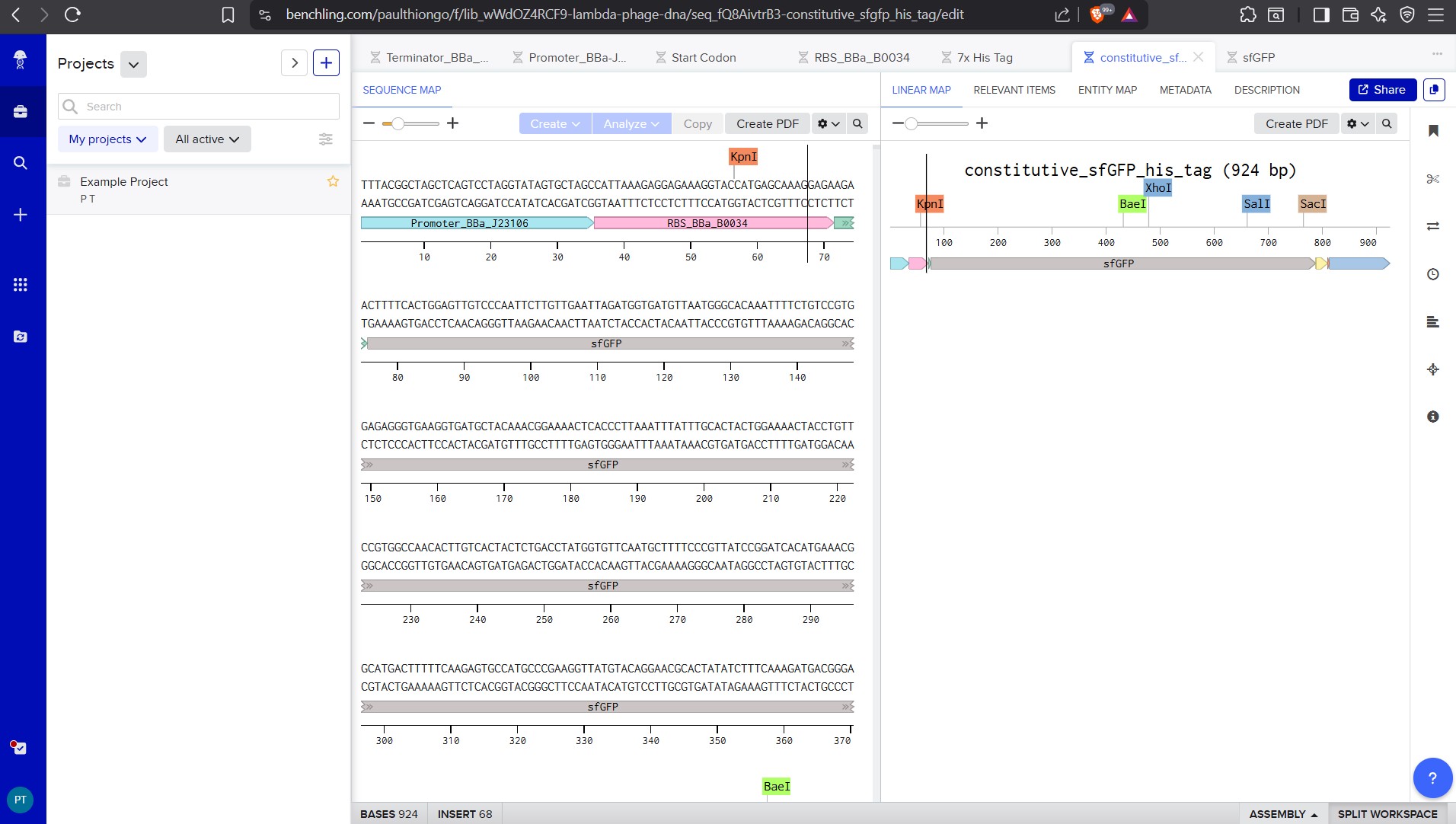
For example, let’s make a sequence that will make E. coli glow fluorescent green under UV light by constitutively (always) expressing sfGFP (a green fluorescent protein):
4.6. Choose Your Vector
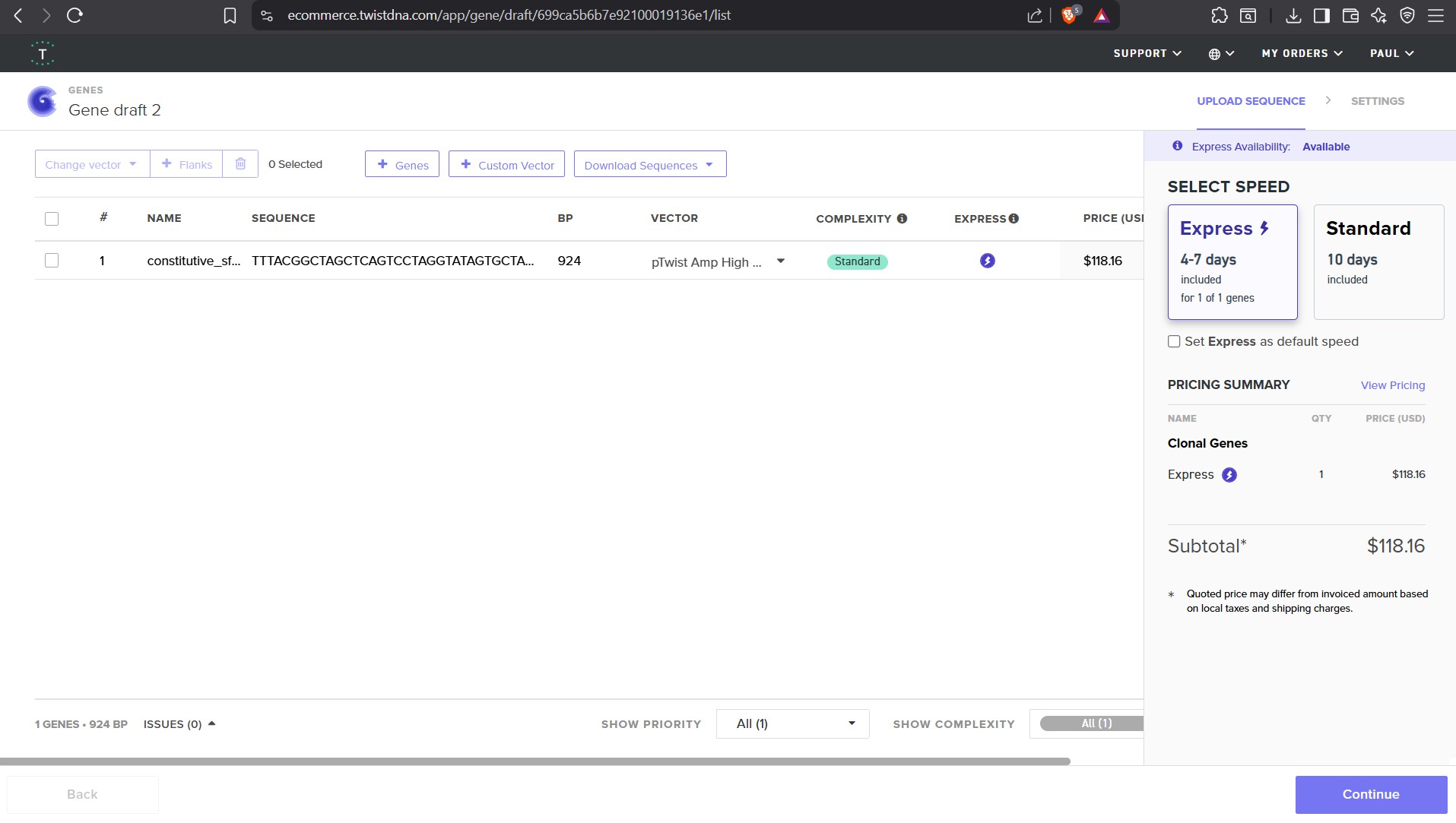
For this demonstration, choose a Twist cloning vectors like pTwist Amp High Copy.
Go back to your Benchling account. Inside of a folder, click the import DNA/RNA sequence button and upload the GenBank file you just downloaded.
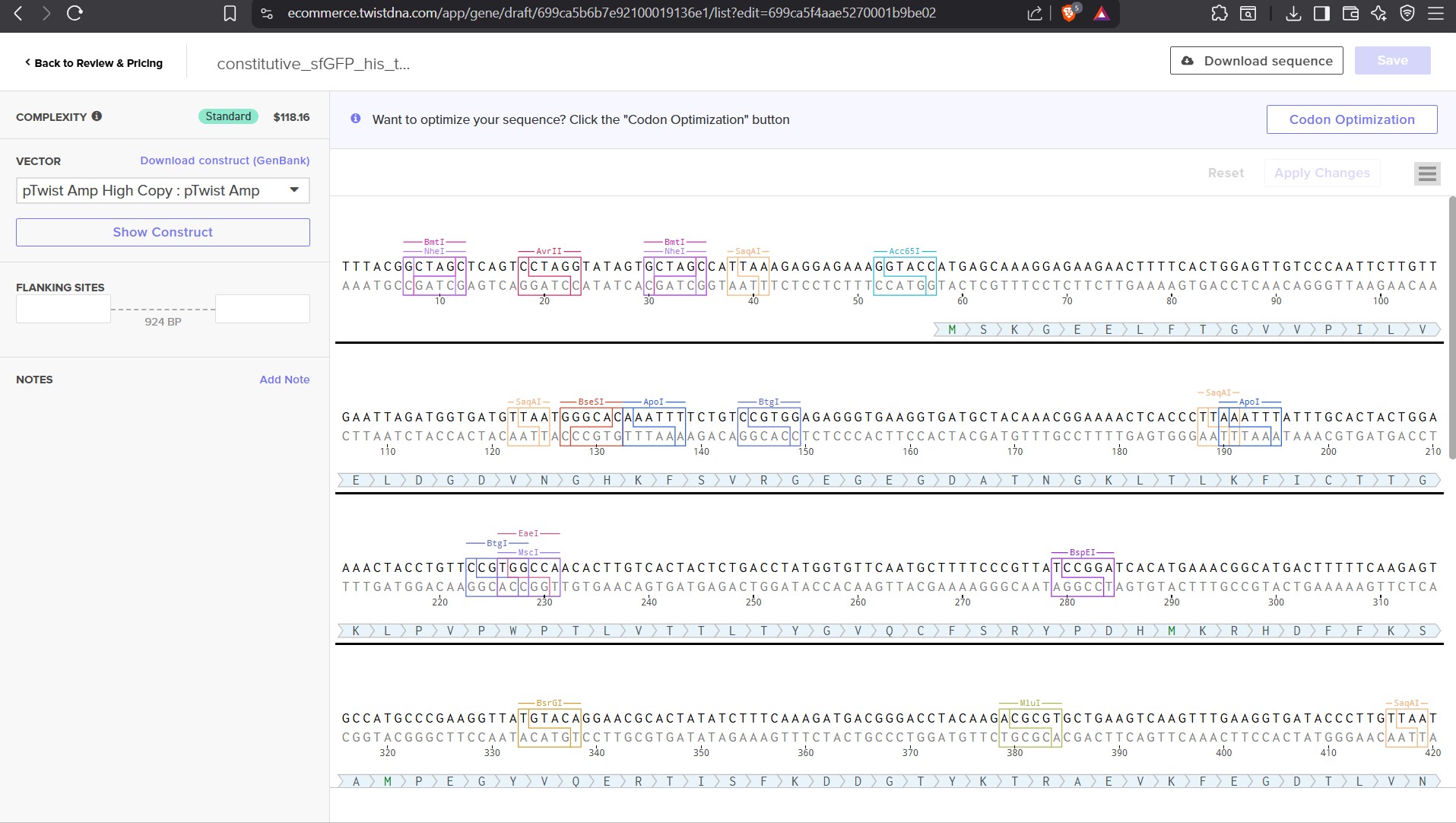
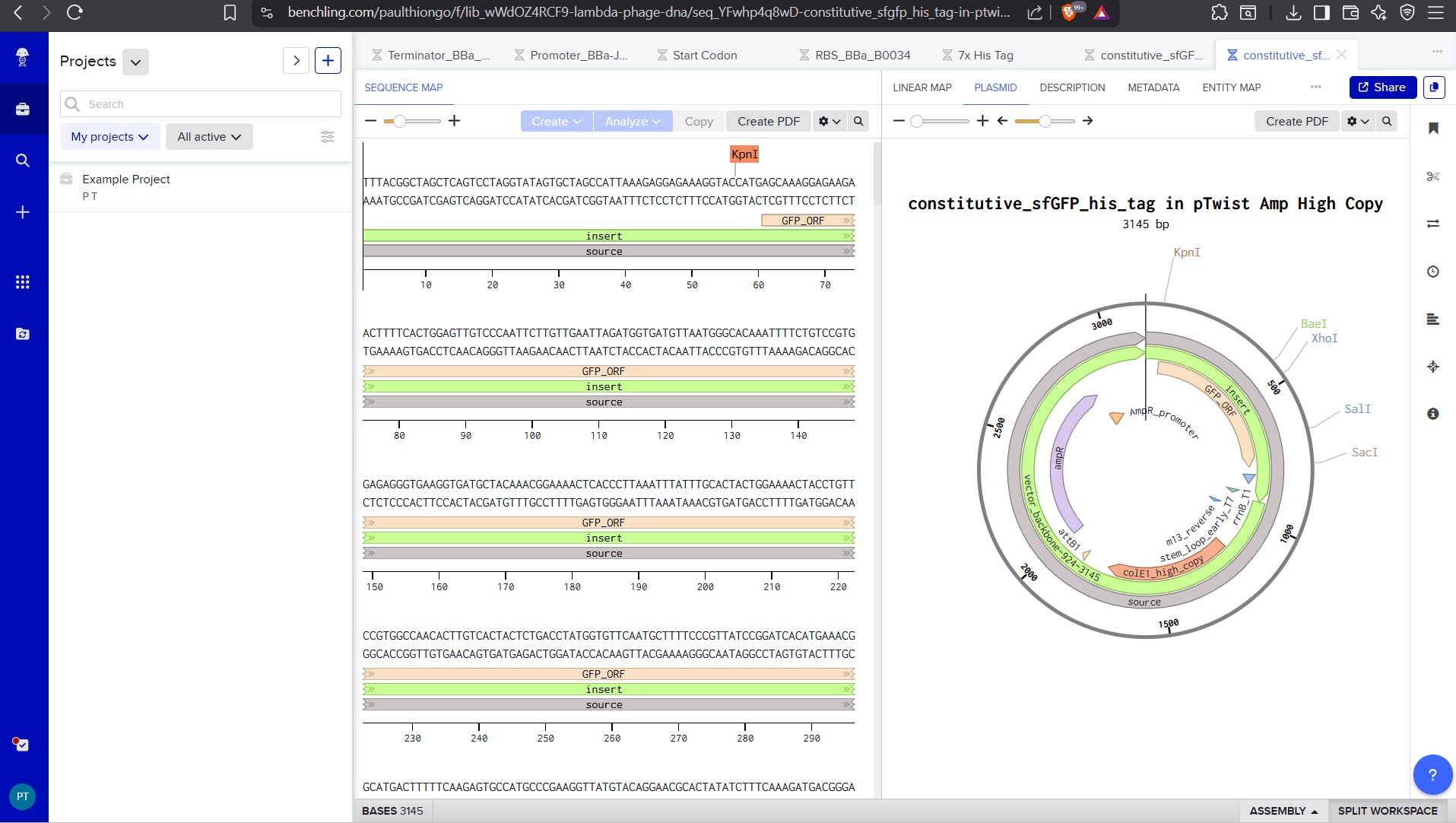
This is the plasmid you just built with your expression cassette included. Congratulations on building your first plasmid!
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank). I wanted to research on the effects of microplastics and nanoplastics to our DNA as a lot of diseases are in the population and there is a possibility plastics in our environment could be contributing. I used Large Language Models to help me get the information. The detection of plastics particles is done through spectroscopic ways with methods like Raman or FTIR(Fourier-Transform Infrared,while sequencing methods won’t be able to physically detect plastics. I am curious to find out the research on whether it is true plastics are affecting DNA and other body functions.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Next-Generation Sequencing for Microplastic Research Advanced sequencing technologies, particularly Illumina’s second-generation platforms, enable researchers to assess the genetic damage, health impacts, and changes in gene activity triggered by exposure to microplastics and nanoplastics. Each approach requires distinct preparation procedures, specific chemical processes during sequencing, and generates different types of data. The core technique relies on synthesis-based sequencing, which simultaneously reads millions of short DNA segments with exceptional accuracy. These methods reveal molecular differences and distinct mutation patterns in DNA, helping scientists identify whether cells have been damaged or exposed to harmful plastics. The main sequencing approaches detailed below measure how micro- and nanoplastics affect living organisms:
Key Sequencing Technologies Whole Genome Sequencing (WGS): This method scans the entire genetic code to identify point mutations and large-scale chromosomal rearrangements (including single base changes, paired base alterations, and insertions/deletions) that result from exposure to toxic compounds released by plastics, such as Bisphenol A (BPA). Metagenomic Sequencing (16S/ITS): This technique examines changes in bacterial populations living in the human gut and identifies genes that allow microorganisms to break down plastic particles after they are consumed. RNA-Seq Technologies (Standard and Low-Input): These approaches measure the abundance of thousands of messenger RNA molecules across an entire genome, revealing how microplastic exposure alters which genes are active in tissues and laboratory-grown cell structures. Single-Cell RNA Sequencing (scRNA-seq): This specialized method examines individual cells within the kidney and liver, providing a detailed snapshot of genetic activity in specific cell types, including immune cells and kidney filtration cells, after plastic exposure. ChIP-Seq: This technique identifies where proteins bind to DNA, enabling researchers to determine whether observed changes in gene activity result directly from signaling pathways or arise indirectly from the cell’s stress response.
Workflow for Next-Generation Sequencing Technologies Implementing these sequencing technologies requires a structured, multi-step procedure that begins with preparing biological samples and concludes with computational analysis of the results.
Sample Preparation and Cell Isolation The first stage involves preparing biological tissue samples for analysis. Tissues—such as kidney samples from laboratory mice—are broken down into individual cells using automated equipment. When researchers need to study single cells in isolation, they employ microfluidic devices, which are miniaturized systems capable of sorting and capturing individual cells with precision.
RNA/DNA Extraction and Quality Control High-quality genetic material must be isolated from the prepared samples before sequencing can begin. For projects involving RNA, the extracted material must meet strict quality standards; specifically, the RNA Integrity Number (RIN) should be 8 or higher to ensure that the RNA molecules remain intact and suitable for accurate analysis.
Library Construction This critical step prepares the genetic material for sequencing. The DNA or RNA is fragmented into manageable pieces, and for RNA-based studies, reverse transcription converts RNA back into DNA. Adapter sequences and unique identifying barcodes are then attached to each fragment, allowing researchers to sort and track the samples during and after sequencing.
Sequencing Run The prepared libraries are pooled together and loaded onto high-capacity sequencing instruments, such as the Illumina HiSeq X or NovaSeq 6000. These machines generate sequences of both ends of each DNA fragment (paired-end reads), producing massive amounts of genetic information in a single run.
Bioinformatics Analysis The raw sequencing data undergoes three distinct computational stages: Primary Analysis: The sequencing instrument produces raw data files in FASTQ format, which contain the DNA sequences alongside quality scores indicating how confident the machine is in each nucleotide reading. Secondary Analysis: The raw sequences are compared and aligned to a reference genome (such as the pig genome, Sus scrofa, or mouse genome, Mus musculus) using specialized software tools like HISAT2. Alternatively, researchers may assemble the sequences without a reference by comparing them to each other. Tertiary Analysis: The aligned data is processed to measure how actively each gene is being expressed—using metrics such as FPKM or TPM—and to identify which genes show significantly different activity levels between samples (Differentially Expressed Genes, or DEGs).
Data Outputs Revealing Molecular Damage from Plastic Exposure These advanced sequencing technologies generate comprehensive molecular information that maps the biological harm caused by plastic particles at the genetic and cellular levels.
Gene Expression Matrices Large-scale datasets are produced that document which genes become more active (upregulated) or less active (downregulated) when organisms are exposed to microplastics and nanoplastics. These matrices provide a complete picture of how plastic exposure alters the cell’s genetic activity across thousands of genes simultaneously.
Visual Subpopulation Mapping Computational algorithms such as UMAP and t-SNE transform complex genetic data into visual plots that reveal how plastic exposure changes the composition of different cell types within tissues. For example, researchers can observe how exposure increases the proportion of specialized immune cells like CD8⁺ effector T cells, which are involved in fighting infections or damaged cells.
Mutational Signatures Plastic exposure creates distinctive patterns of DNA mutations—such as the conversion of cytosine bases to adenine bases (C>A substitutions)—that serve as a “fingerprint” of exposure to specific plastic contaminants like Bisphenol A (BPA) or styrene oxide. These characteristic mutation patterns help scientists identify which type of plastic damage has occurred and what toxic compounds were responsible.
Pathway Enrichment Analysis Specialized bioinformatics tools like KEGG (Kyoto Encyclopedia of Genes and Genomes) and GO (Gene Ontology) analysis identify which fundamental biological processes are being disrupted by plastic exposure. Common disrupted pathways include oxidative phosphorylation (energy production in cells), the MAPK signaling pathway (cellular communication), and chemical carcinogenesis (processes that can lead to cancer).
Microbial Diversity Indices Statistical measures quantify the balance between harmful and beneficial bacteria in the human gut microbiome following plastic ingestion. These indices reveal whether plastic consumption shifts the microbial ecosystem toward pathobionts (disease-promoting microorganisms) or maintains a healthy population of beneficial bacteria.
Base Calling in Next-Generation Sequencing The identification of individual DNA or RNA bases—a process called base calling—represents the foundational analytical step in all next-generation sequencing (NGS) workflows. This process takes place directly within the sequencing instrument (such as those from Illumina, PacBio, or Oxford Nanopore) and converts the biological signals detected by the machine into a readable digital genetic code.
Primary Analysis and Signal Generation On-Platform Processing Base calling occurs as “Primary Analysis,” meaning it happens in real-time while the biological sample is being sequenced inside the machine. The sequencing platform simultaneously reads and identifies nucleotides as the process unfolds.
Technology-Specific Sequencing Methods Different sequencing platforms employ distinct approaches: Short-read sequencing (Illumina): DNA molecules are cut into small fragments ranging from 200 to 500 base pairs long. The sequencing machine then reads these fragments from both ends (paired-end reads), typically generating sequences of approximately 150 base pairs in length, and systematically identifies each nucleotide in order. Long-read sequencing (PacBio and Oxford Nanopore): These technologies sequence complete, unbroken DNA molecules. PacBio’s Single Molecule Real-Time (SMRT) sequencing produces average read lengths of about 20 kilobases, while Oxford Nanopore generates ultra-long reads averaging around 100 kilobases, allowing researchers to capture much larger stretches of genetic information in a single read. Detection of Mutation Patterns The sequencing process identifies specific genetic variations caused by environmental exposures, such as Single Base Substitutions (SBS). These are characteristic mutation patterns at particular locations (such as guanine residues) that indicate damage from toxic chemicals derived from plastics.
Output and File Formats FASTQ Files The direct result of base calling is raw “reads” that are stored in FASTQ format—the standard file format for sequencing data. Data Content Each FASTQ file contains two essential pieces of information for every base identified: the sequence of nucleotides (represented as A, C, G, or T) and an associated quality score, known as a Phred quality score or Q-score, which indicates the confidence level of that base call.
Measuring Accuracy and Quality Control Quality Scoring System To verify that the base calling process is reliable, each identified base is assigned a probability score indicating the likelihood that the identification is correct.
The Standard for High-Quality Data A Q30 score is the widely accepted benchmark for excellent data quality, representing a 99.9% accuracy rate in base identification. This threshold ensures that the sequencing data is trustworthy for downstream analysis.
Quality Filtering During the next phase of analysis, bioinformatics software programs like fastp or FastQC examine the raw data and filter out low-quality reads where base calling may have been uncertain or unreliable, ensuring only high-confidence data moves forward.
Downstream Assembly and Analysis of Decoded Bases After the sequencing machine completes base calling and produces individual reads, the data enters Secondary Analysis, where bioinformatics tools organize and interpret the complete genetic information.
Reference-Based Genome Alignment The decoded genetic sequences are compared against and mapped to a reference genome—such as the human genome (GRCh38) or the pig genome (Sus scrofa)— to determine the correct location of each read within the organism’s full genetic blueprint.
De Novo Assembly When researchers lack a suitable reference genome for comparison, an alternative strategy is employed: the decoded reads are further subdivided into overlapping short segments called k-mers. These k-mers are then reassembled using computational algorithms and graph-based methods to reconstruct longer continuous sequences, known as contigs, that represent the original genetic material.
Also answer the following questions: 1. Is your method first-, second- or third-generation or other? How so? 2. What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps. 3. What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)? 4. What is the output of your chosen sequencing technology?
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :) See some famous examples of DNA design
(ii) What technology or technologies would you use to perform this DNA synthesis and why? Also answer the following questions: 1. What are the essential steps of your chosen sequencing methods? 2. What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
(ii) What technology or technologies would you use to perform these DNA edits and why? Also answer the following questions: 1. How does your technology of choice edit DNA? What are the essential steps? 2. What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing? 3. What are the limitations of your editing methods (if any) in terms of efficiency or precision?
CRISPR Base Editing for Precise DNA Modification CRISPR base editing offers a highly precise approach to DNA modification that changes individual bases without breaking both strands of the DNA molecule. This technique represents a significant advancement in genetic engineering because it enables targeted alterations with minimal disruption to the overall genetic structure.
How Base Editing Works The method combines two key components: a specially engineered Cas enzyme and a deaminase protein. The deaminase acts as a molecular converter, transforming one DNA base into another—such as changing cytosine to thymine (C→T) or adenine to guanine (A→G). To target a specific gene like myostatin, researchers design a guide RNA that directs the editing machinery to the correct location. The entire system is then introduced into cells using a plasmid vector and a physical delivery method such as electroporation, which creates temporary pores in the cell membrane to allow the genetic material to enter.
PAM Sequence Requirements and Solutions One constraint of base editing is that the Cas enzyme requires a specific DNA sequence called a PAM (Protospacer Adjacent Motif) located near the target site to function properly. This requirement previously limited the number of editable locations throughout the genome. However, newer Cas9 variants—such as SpRY—can recognize and work with a broader range of PAM sequences, dramatically expanding the number of genomic locations available for targeting and editing.
Improving Precision and Safety Advanced bioinformatics tools like BE-HIVE and Honeycomb have enhanced the effectiveness of base editing by predicting the most promising edit sites and simultaneously reducing the risk of unintended mutations at off-target locations, making the entire process more reliable and safer.
References
- DNA Sequencing at 40: Past, Present, and Future (2017) Shendure, J., Balasubramanian, S., Church, G. et al. https://doi.org/10.1038/nature24286
- DNA Synthesis Technologies to Close the Gene Writing Gap (2023), Hoose, A., Vellacott, R., Storch, M. et al. https://doi.org/10.1038/s41570-022-00456-9
- Recombineering and MAGE (2021), Wannier T, et al. Nat Rev Methods Primers, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9083505/
- CRISPR Technology: A Decade of Genome Editing is Only the Beginning, Wang, Doudna, et al., https://www.science.org/doi/10.1126/science.add8643
- GenBank overview: https://www.ncbi.nlm.nih.gov/genbank/
- NCBI: https://www.ncbi.nlm.nih.gov/genome/
- Ensembl: https://useast.ensembl.org/index.html
- UCSC Genome Browser: https://genome.ucsc.edu/
- Protective and Enhancing Alleles: https://arep.med.harvard.edu/gmc/protect.html
- Overview of Next Generation Sequencing Technologies - PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC6020069/
- Sanger Sequencing Steps & Method - MilliporeSigma https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/sequencing/sanger-sequencing
- DNA Sequencing Technologies, How They Differ, and Why It Matters https://www.fjc.gov/content/361255/dna-sequencing-technologies-how-they-differ-and-why-it-matters
- sangeranalyseR: Simple and Interactive Processing of Sanger … https://pmc.ncbi.nlm.nih.gov/articles/PMC7939931/
- NGS Library Preparation - 3 Key Technologies https://www.illumina.com/techniques/sequencing/ngs-library-prep.html
- Next-Generation Sequencing Technology: Current Trends … - PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/
- Overview of PacBio SMRT sequencing: principles, workflow, and … https://www.cd-genomics.com/pacbio-smrt-system-single-molecule-real-time-sequencing.html
- PacBio sequencing output increased through uniform and … https://www.nature.com/articles/s41598-021-96829-z
- Library preparation for nanopore sequencing https://oxfordnanoporedx.com/products/prepare
- Output Structure - Oxford Nanopore Output Specifications https://nanoporetech.github.io/ont-output-specifications/latest/minknow/output_structure/
- What are the different types of DNA sequencing technologies? https://www.thermofisher.com/us/en/home/life-science/sequencing/sequencing-learning-center/sequencing-basics/dna-sequencing-technologies.html
- DNA Sequencing Fact Sheet https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Fact-Sheet
- DNA Sequencing: How to Choose the Right Technology https://frontlinegenomics.com/dna-sequencing-how-to-choose-the-right-technology/
- Sample Preparation - GENEWIZ from Azenta Life Sciences https://www.genewiz.com/public/resources/sample-submission-guidelines/sanger-sequencing-sample-submission-guidelines/sample-preparation
- Sanger Sequencing: Introduction, Principle, and Protocol | CD Genomics Blog https://www.cd-genomics.com/blog/sanger-sequencing-introduction-principle-and-protocol/
- [PDF] Sanger Sequencing Best and Worst Practices - rtsf@msu.edu https://rtsf.natsci.msu.edu/_assets/files/genomics/Sanger_Sequencing_Best_and_Worst_Practices_Guide_25April2024.pdf
- [PDF] Sanger Sequencing Handbook - FULL SERVICE https://www.biotech.cornell.edu/sites/default/files/2020-08/Full_service_Sanger_Handbook.pdf
- Sanger Sequencing - Sample Prep & Data Analysis with BLAST https://www.youtube.com/watch?v=ez-_YtHm9pk
- Videos https://www.neb.com/en/products/next-generation-sequencing-library-preparation/library-preparation-for-illumina
- The use of LLM to help with finding information and reporting