Week 4: PROTEIN DESIGN PART I
Week # 4 Protein Design Part I
PROTEIN DESIGN PART I
To look at how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions.
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
For a Tilapia Fish: Assuming : meat = 20% protein by weight; average amino acid ≈ 100 Da (g/mol). Calculation: • Protein mass = 500 g × 0.20 = 100 g • Moles of amino-acid residues = 100 g ÷ 100 g·mol⁻¹ = 1.00 mol • Number of amino-acid molecules using Avogadro’s number ≈ 1.00 × ≈ 6.02 × 1023 = 6.02 × 1023 amino-acid molecules.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
The beef meat is in the form of amino acids that our body needs which is broken down by the enzymes in our stomach to the amino acids required by our body. The amino acids are the building blocks of DNA. Beef also provides protein, zinc and several D vitamins used for muscle health, iron that boosts our immune system
- Why are there only 20 natural amino acids? Through evolutionary selection there are 20 natural amino acids that are encoded by the genetic code used by all life forms for protein synthesis, although there are more than 500 amino acids that exist in nature.
Can you make other non-natural amino acids? Design some new amino acids. There are scientists who have designed new amino acids like the Cyclopropyl‑alanine (cPrAla) whose properties are small, rigid , steric probe that increases backbone constraint. Its uses are conformational stabilization and protease resistance.
Where did amino acids come from before enzymes that make them, and before life started? Amino acids could have been synthesized through multiple non-biological chemical pathways on the early Earth. Electric discharges and ultraviolet radiation acting on simple atmospheric gases generated diverse mixtures of organic compounds, including compounds like alanine and glycine. For these organic building blocks to combine into short peptide chains, the process likely required special activation mechanisms—such as energized compounds like thioesters or cyanamide—along with repeated cycles of drying and rewetting, or assistance from mineral surfaces acting as catalysts. Importantly, enzymes like those found in modern life were not yet available to facilitate these reactions. Evidence suggests that the raw materials for life may have arrived from space itself: meteorites containing carbon-based compounds (such as the Murchison meteorite) and material from comets both carry amino acids and organic molecules, demonstrating that similar chemical reactions can occur naturally beyond Earth and could have supplied the ingredients for the early planet. Additional experimental pathways support this scenario. Ultraviolet light shining on frozen water or heating formamide (a simple organic compound) produces amino acids and related molecules under conditions that are colder or more oxidizing than those used in earlier laboratory experiments. Finally, geothermal hot vents and rocky mineral surfaces—particularly those containing metal sulfides and clay minerals—would have naturally concentrated these chemical precursors and promoted their assembly into peptide chains.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? It would be the Left‑handed — D‑amino acids form the mirror‑image (left‑handed) α‑helix corresponding to the right‑handed helix made from L‑amino acids (same geometry mirrored). It is the one preferred for drug design as its more resistant to breakdown.
Can you discover additional helices in proteins? It is possible by the use of a deep learning system such as Alpha Fold to predict a protein’s 3D atomic structure from its amino acid sequence.
Why are most molecular helices right-handed? Proteins naturally favor a right-handed helical structure due to the molecular properties of the L-amino acids from which they are built. The three-dimensional arrangement of atoms in L-amino acids—their stereochemical configuration and the steric (spatial) and electronic (charge-based) forces acting on them—determines which backbone torsion angles (φ, ψ) are energetically favorable. These preferred angles naturally align with a right-handed helical geometry, which represents the lowest-energy, most stable configuration. This structural preference is further reinforced by the D-sugars present in the DNA and RNA backbone. The presence of D-sugars (rather than their mirror-image L-form) in nucleic acids creates a similar geometric environment that also favors and supports the right-handed helical arrangement, making it consistent with the overall architecture of proteins built from L-amino acids. In essence, the molecular “handedness” of biological building blocks—L-amino acids and D-sugars—creates a coordinated system where all the natural forces involved push the same direction, promoting right-handed helical structures as the thermodynamically favored state.
Why do β-sheets tend to aggregate?
Aromatic interactions: The π–π stacking of aromatic amino acids (phenylalanine, tyrosine, and tryptophan) creates additional stabilization through their interactions, further reinforcing stacked sheet structures. Nucleation and templating: Once an initial molecular nucleus forms, subsequent strands can attach to this template, which lowers the energy barrier for further assembly and accelerates the growth of the fibril structure—a process driven by kinetic facilitation. Side-chain packing and van der Waals forces: The complementary, regularly repeating interfaces between side chains (sometimes called “steric zippers”) allow for maximal close-contact packing between molecules, which maximizes the weak dispersion forces (van der Waals interactions) that contribute to overall stability. Backbone hydrogen bonding: Extended networks of hydrogen bonds form between the backbone atoms of adjacent strands (specifically between N–H groups and C=O groups across different molecules), which significantly stabilize the β-sheet structure between strands. Dehydration and entropic effects: Thermodynamic stability increases when water molecules are expelled from the interfaces between molecules and when the total surface area exposed to solvent decreases. Hydrophobic effect: When nonpolar (hydrophobic) side chains are buried inside the assembled structure as sheets stack together, ordered water molecules are released into solution. This release of water increases the system’s entropy, providing a large favorable thermodynamic driving force. Polar side-chain networks: Amino acids with polar side chains (such as glutamine and asparagine) form interconnected hydrogen-bonding patterns that act cooperatively to stabilize and strengthen the fibril structure.
- Why do many amyloid diseases form β-sheets?
β-sheets are the thermodynamically favored conformation for proteins involved in amyloid diseases due to the multiple stabilizing factors that work together. The backbone hydrogen bonding between extended strands creates a very stable intermolecular network. Additionally, the hydrophobic effect is particularly powerful—when nonpolar amino acid side chains are buried within stacked β-sheets, water molecules are released, which provides a large entropic gain that drives assembly. Kinetic factors further promote β-sheet formation. Once a small nucleus of β-sheet structure forms, it acts as a template. New protein molecules can rapidly attach to this growing template with a lower activation energy than forming the initial nucleus, creating a positive feedback loop that accelerates fibril growth. The β-sheet structure is also highly resistant to degradation. The extensive hydrogen-bonding networks and tightly packed interfaces make these aggregates very difficult for cellular machinery to break down or clear, allowing amyloid fibrils to accumulate over time. Additionally, certain proteins are inherently prone to adopting β-sheet conformations due to their amino acid sequences and three-dimensional properties. In amyloid diseases, proteins that normally fold into other structures (like α-helices) become misfolded under stress conditions and instead spontaneously assemble into the more stable β-sheet configuration. In essence, the combination of thermodynamic stability, kinetic acceleration through templating, and cellular difficulty in clearing these structures makes β-sheets the inevitable outcome for proteins that misfold in amyloid diseases.
- ◦ Can you use amyloid β-sheets as materials? Amyloid β-sheets are gaining recognition as valuable materials for practical applications across multiple industries, leveraging their distinctive structural and functional characteristics.
Healthcare Applications Amyloid β-sheets can be modified and designed to serve as delivery vehicles for pharmaceuticals, enabling them to direct treatments to particular cell types or anatomical locations. Their exceptional mechanical robustness and structural durability make them ideal candidates for use as supportive frameworks in regenerative medicine, where they can facilitate the growth and organization of new tissue.
Sensing and Detection The self-assembling capability of amyloid β-sheets offers promise for creating highly sensitive detection systems. These structures can be incorporated into biosensing devices that identify and measure biological molecules or contaminants present in the environment with enhanced precision.
Environmental and Sustainability Applications As there is growing interest in environmentally responsible material alternatives, amyloid β-sheets represent a promising option for developing compostable or biodegradable materials that could replace conventional petroleum-based plastics. This application addresses the need for sustainable solutions to reduce plastic waste and environmental contamination. In summary, the stability, mechanical properties, and programmable assembly of amyloid β-sheets position them as versatile engineering materials that can address challenges in medicine, environmental monitoring, and sustainable manufacturing.
- Design a β-sheet motif that forms a well-ordered structure. The foundation of creating a well-ordered β-sheet involves establishing an alternating pattern of water-repelling and water-attracting amino acids along the protein backbone. By arranging hydrophobic (nonpolar) residues to point inward and hydrophilic (polar) residues to point outward, you create a striped arrangement where the nonpolar side chains cluster together in the interior of the sheet while polar groups remain accessible to the aqueous surroundings. This pattern naturally promotes the formation of stable, regular β-sheet structures. To further enhance stability, aromatic amino acids should be distributed at regular intervals throughout the sequence. These aromatic residues (such as phenylalanine and tyrosine) interact through π–π stacking, which reinforces the stacking of sheets on top of each other and creates additional geometric constraints that lock the structure into place.
A Practical Example A straightforward and effective sequence would be: [I-V-F-L-Y-L-F-V-I-V-F-L-Y-L-F-V], composed primarily of isoleucine, valine, leucine, phenylalanine, and tyrosine. These residues are small and hydrophobic, allowing them to pack tightly together while aromatic residues facilitate lateral interactions. For applications requiring additional functionality, incorporating charged amino acids like glutamate at regular intervals adds surface solubility a nd potential binding sites without disrupting the core sheet geometry. A modified version might be: [I-E-F-L-Y-L-F-E-I-E-F-L-Y-L-F-E].
Why This Design Works Hydrophobic side-chain burial drives rapid self-assembly because water molecules trapped within the nonpolar interior are released as the structure forms, providing a large entropy gain. The repeating, predictable pattern ensures that once initial nucleation occurs, subsequent protein molecules attach to the growing structure in an orderly fashion, promoting uniform, large-scale assembly. The complementary packing of side chains maximizes weak intermolecular forces, while the extensive hydrogen-bonding networks between backbones of adjacent strands create remarkable stability.
Customization Options To strengthen the material further, you can increase the proportion of aromatic and β-branched residues to achieve denser packing. For tailored applications, replace specific residues with histidine or aspartate to create binding sites for metal ions, or introduce serine or threonine for potential chemical modifications. To control the final size of assembled structures, strategically insert proline residues to act as “breaks” that limit sheet expansion and create defined structural units. This rational design approach—carefully balancing hydrophobic and hydrophilic properties while incorporating multiple stabilizing interactions—represents the practical framework for engineering functional amyloid-based materials.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
- Briefly describe the protein you selected and why you selected it. PD-1 (programmed cell death protein 1) was selected as the subject of study. This protein is an inhibitory receptor located on the cell membrane of T cells and other immune cells. When PD-1 binds to its ligands—PD-L1 or PD-L2—the binding signal suppresses the activation and proliferation of T cells, essentially acting as a “brake” on immune responses. This checkpoint mechanism normally serves an important regulatory function, but tumors and certain pathogens have learned to exploit it. By producing and displaying PD-L1 or PD-L2 on their surfaces, cancer cells and infectious agents can hijack this natural control system. They force T cells to express PD-1 and engage it with the pathogen’s or tumor’s PD-ligands, effectively disabling the immune response that would otherwise attack them. However, blocking the interaction between PD-1 and its ligands can reverse this immune suppression. When the PD-1/PD-L1 (or PD-L2) connection is prevented, T cells regain their ability to recognize and attack cancer cells or infected cells. This principle forms the basis of checkpoint inhibitor immunotherapies, which have proven highly effective in treating certain cancers and enhancing immune responses against persistent infections.
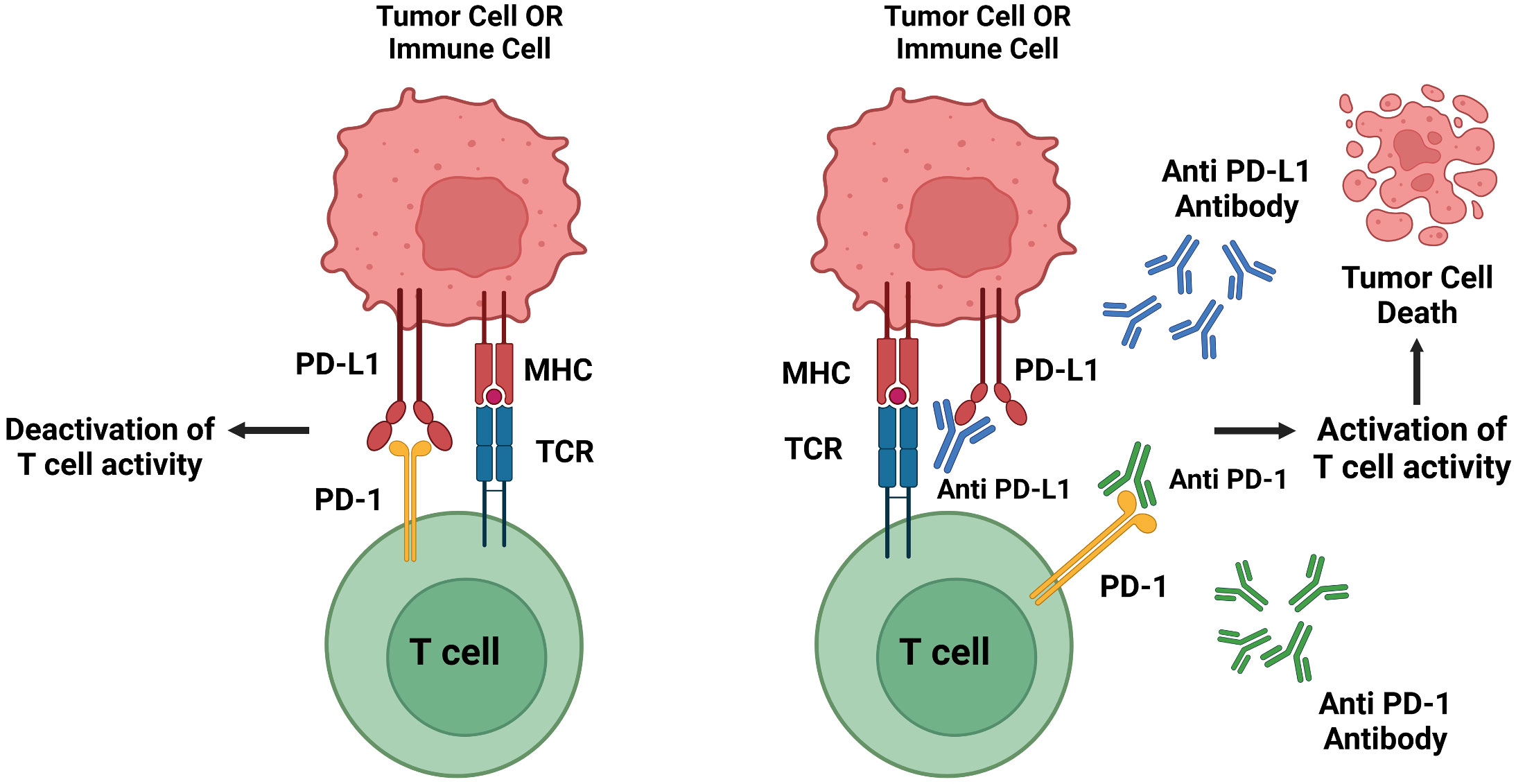
- Identify the amino acid sequence of your protein.
MQIPQAPWPVVWAVLQLGWRPGWFLDSPDRPWNPPTFSPALLVVTEGDNATFTCSFSNTSESFVLNWYRMSPSNQTDKLAAFPEDRSQPGQDCRFRVTQLPNGRDFHMSVVRARRNDSGTYLCGAISLAPKAQIKESLRAELRVTERRAEVPT AHPSPSPRPAGQFQTLVVGVVGGLLGSLVLLVWVLAVICSRAARGTIGARRTGQPLKEDPSAVPVFSVDYGELDFQWREKTPEPPVPCVPEQTEYATIVFPSGMGTSSPARRGSADGPRSAQPLRPEDGHCSWPL
◦ How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
PD-1 has a length of 288, analyzing using the notebook P appears to be the most frequent at 33.
◦ How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
I used the pBLAST tool to search for homologs and ClustalOmega to align and visualize them.
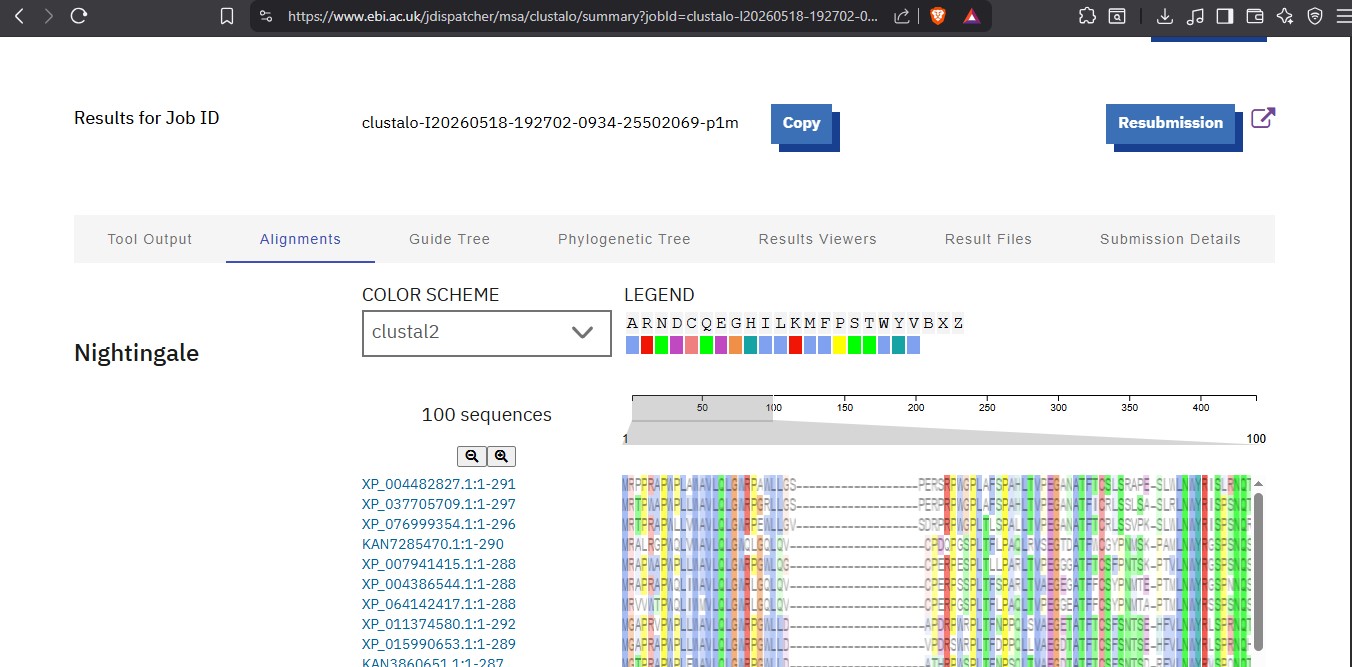 After visualizing there are 100 homologs.
After visualizing there are 100 homologs.
◦ Does your protein belong to any protein family?
Yes. It is a member of the of the immunoglobulin superfamily and of the CD28/CTLA‑4 family of T‑cell co‑stimulatory/inhibitory receptors.
- Identify the structure page of your protein in RCSB https://www.rcsb.org/structure/6UMU
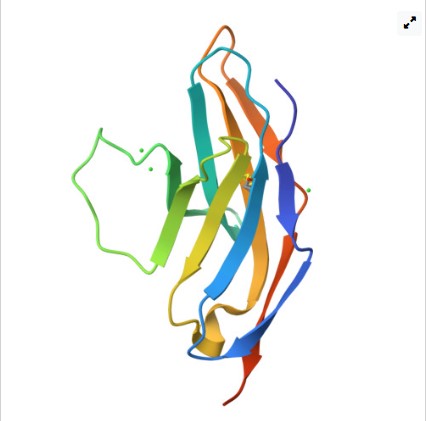
◦ When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better
(Resolution: 2.70 Å)
It was resolved and Deposited : 2019-10-10 and Released: 2019-11-27 to the public. The Resolution: 1.18 Å of high quality and precise as it is below 2.70 Å
◦ Are there any other molecules in the solved structure apart from protein?
Yes, Chloride Ion and Water. ◦ Does your protein belong to any structure classification family? Yes,the extracellular domain of PD‑1 has an immunoglobulin V‑set (IgV) fold and is classified with Ig‑like domains in structural databases (SCOP/CATH/Pfam). It is placed in the Ig‑superfamily structural class (consistent with its membership in the CD28/CTLA‑4 receptor family). In the structure classification family database it is SCOP ID: 8059476 and SCOP ID: 8059477
- Open the structure of your protein in any 3D molecule visualization software:
◦ PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
◦ Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
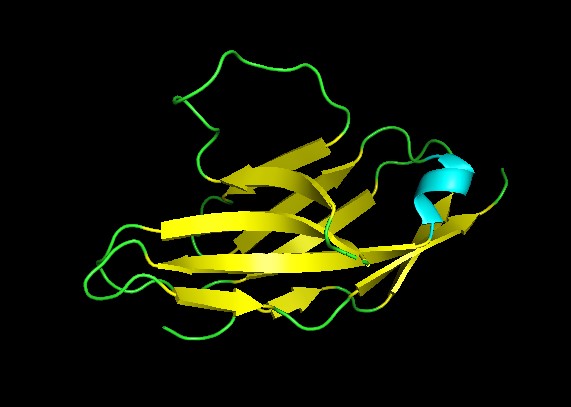
 ◦ Color the protein by secondary structure. Does it have more helices or sheets?
It has no alpha helices and 2 beta sheets. Adopts an immunoglobulin domain fold with a two-layer beta sandwich architecture.
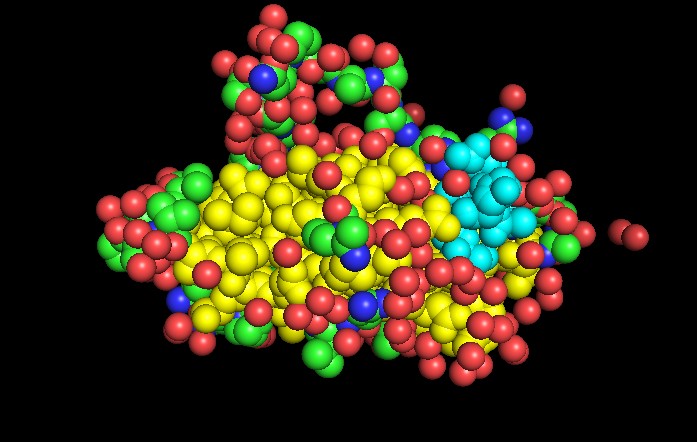
◦ Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
◦ Color the protein by secondary structure. Does it have more helices or sheets?
It has no alpha helices and 2 beta sheets. Adopts an immunoglobulin domain fold with a two-layer beta sandwich architecture.
◦ Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
It has a hydrophobic core to support the beta sandwich architecture and the hydrophilic residues on the surface
◦ Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
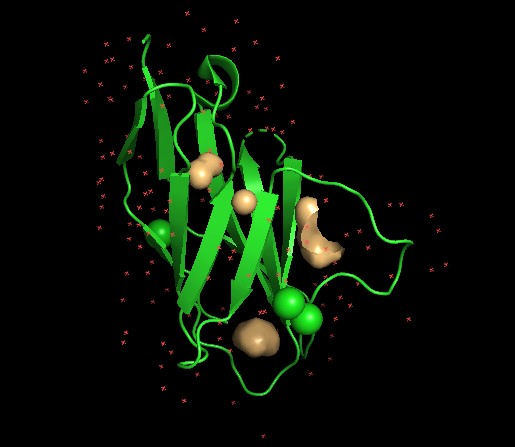 Yes, as seen above.
Yes, as seen above.
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
- Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
- Choose your favorite protein from the PDB.
- We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
- Deep Mutational Scans a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
I went through the steps in the jupiter notebook as shown in the recitation and created the mutation scan heat map below for protein PD-1. There were errors encountered as I run the code for each section,but Gemini was able to rectify the problem and run without problems producing the image below.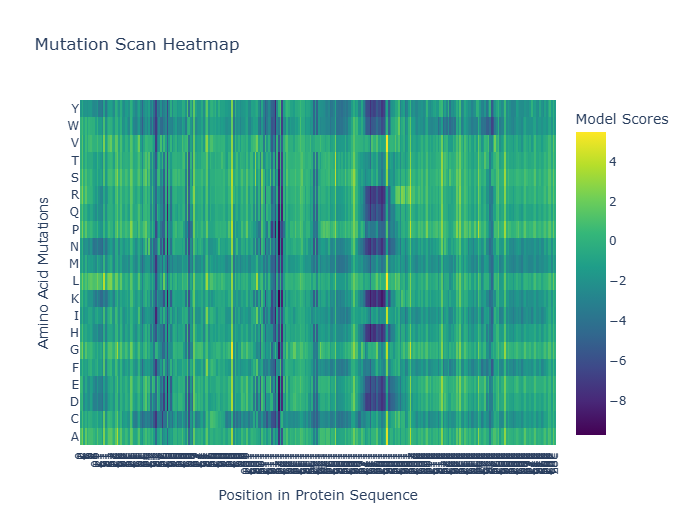
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Log-likelihood ratio (LLR) used in mutation analysis to help quantify the impact of mutations on protein function by comparing the likelihood of mutant versus wild-type residues. Using Gemini, the Most Favorable Mutation will be the one with the highest LLR where a change from Valine (V) to Leucine (L) at position 187, with an LLR of 5.4932. This suggests that substituting Valine with Leucine at this position could potentially be beneficial.. The Least Favorable Mutation with lowest LLR is a change from Methionine (M) to Isoleucine (I) at position 1, with an LLR of -16.6267. This indicates that this particular substitution is highly unfavorable.
c. (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
2. Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
Folding a protein 1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure? 2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations? C3. Protein Generation Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN 1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one. 2. Input this sequence into ESMFold and compare the predicted structure to your original.
References
- The use of LLM to help with finding information and reporting