Homework
Weekly homework submissions:
Week 01 HW: Principles and Practices
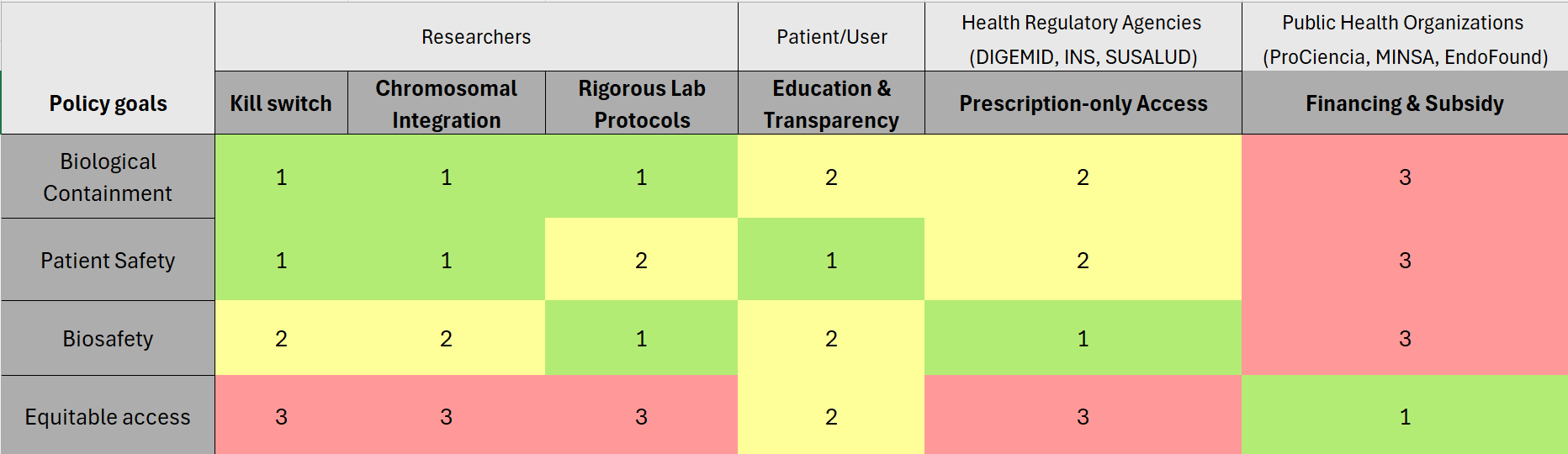
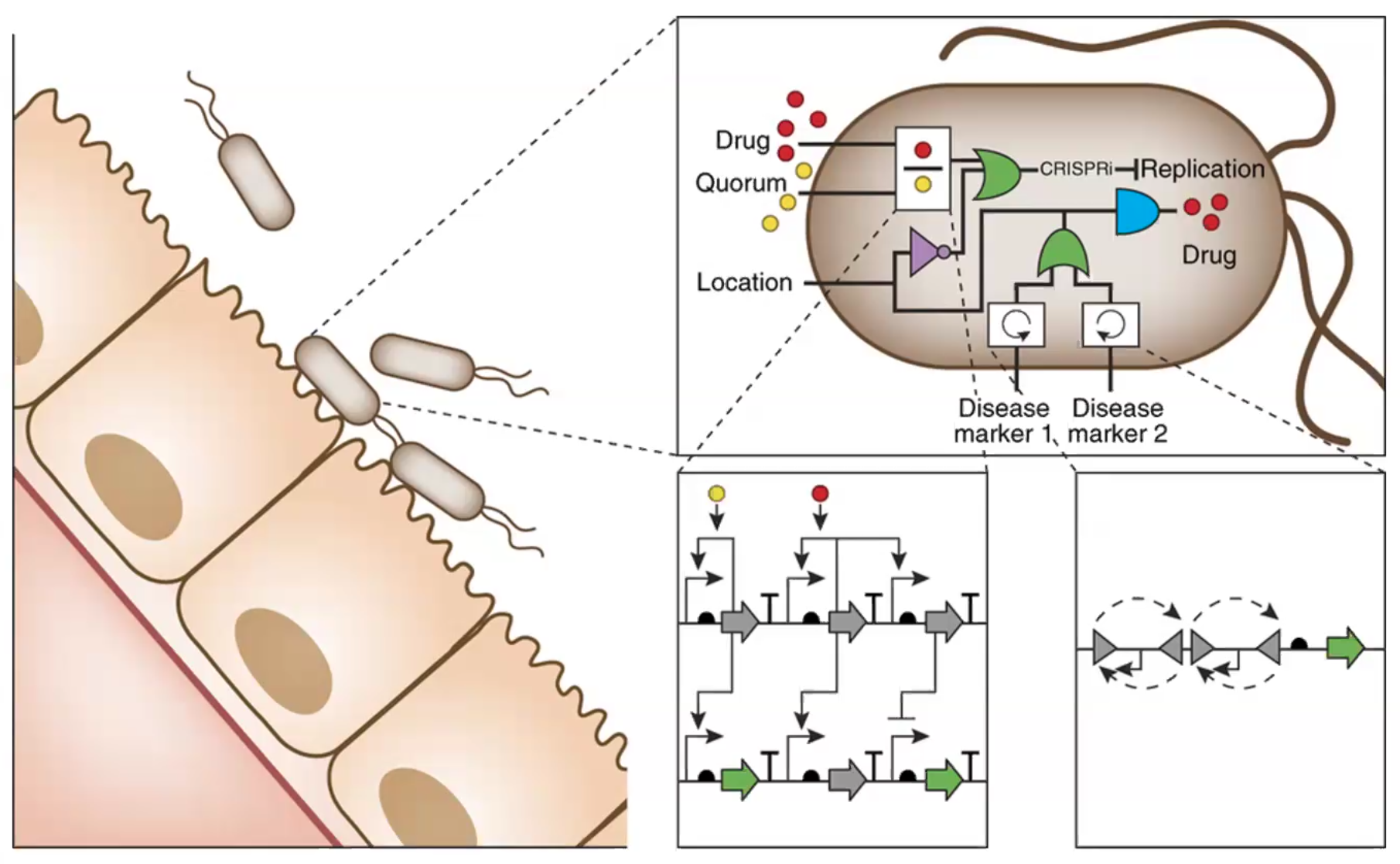
Class Assignment 1. First, describe a biological engineering application or tool you want to develop and why. Endometriosis is an inflammatory disease characterized by the endometrial-like tissue growth outside of the uterine cavity. This ectopic growth leads to hormonal imbalances, systemic inflammation, and debilitating pain during menstruation, sexual intercourse, and bodily functions . Although it affects 10–15% of reproductive age women, there is currently no cure and the diagnosis of this diseases remains a clinical challenge [1]. Current clinical management is limited to hormonal suppression, pain control and surgical excision [2]. Consequently, there is a critical need for non-invasive, targeted therapies that can modulate the immune response and minimize recurrence rates without compromising the patient’s reproductive health.
Week 02 HW: DNA READ, WRITE AND EDIT
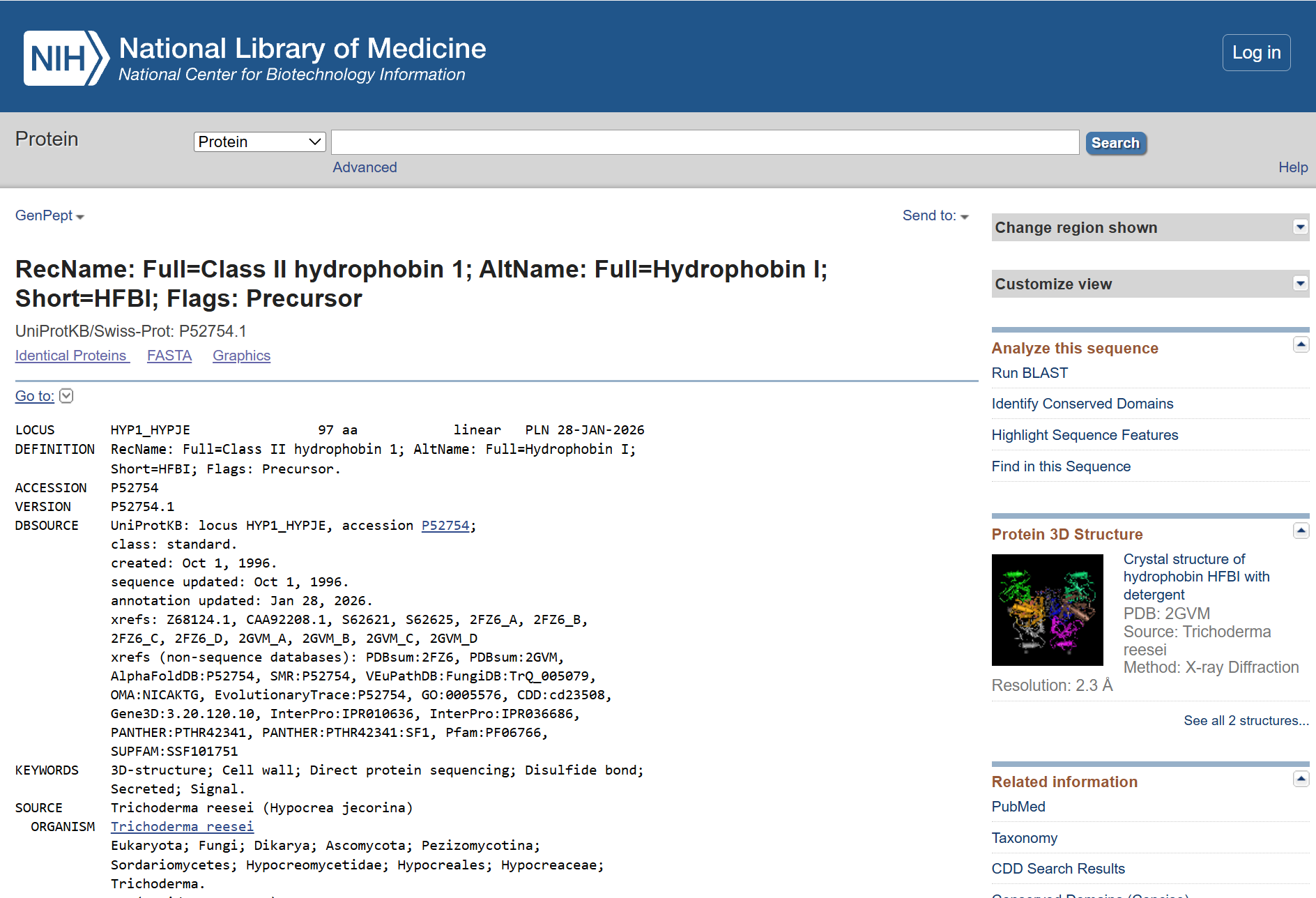
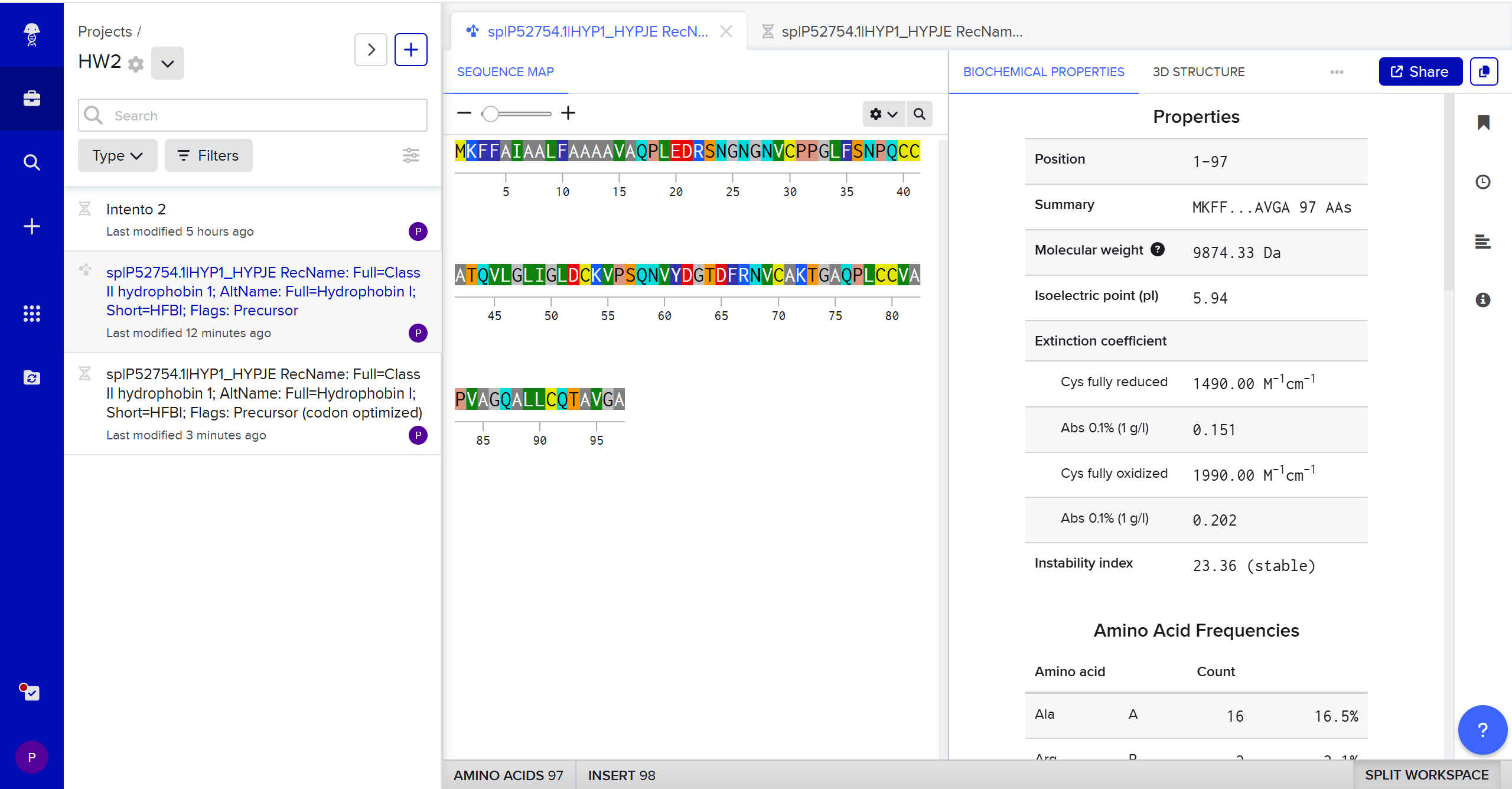
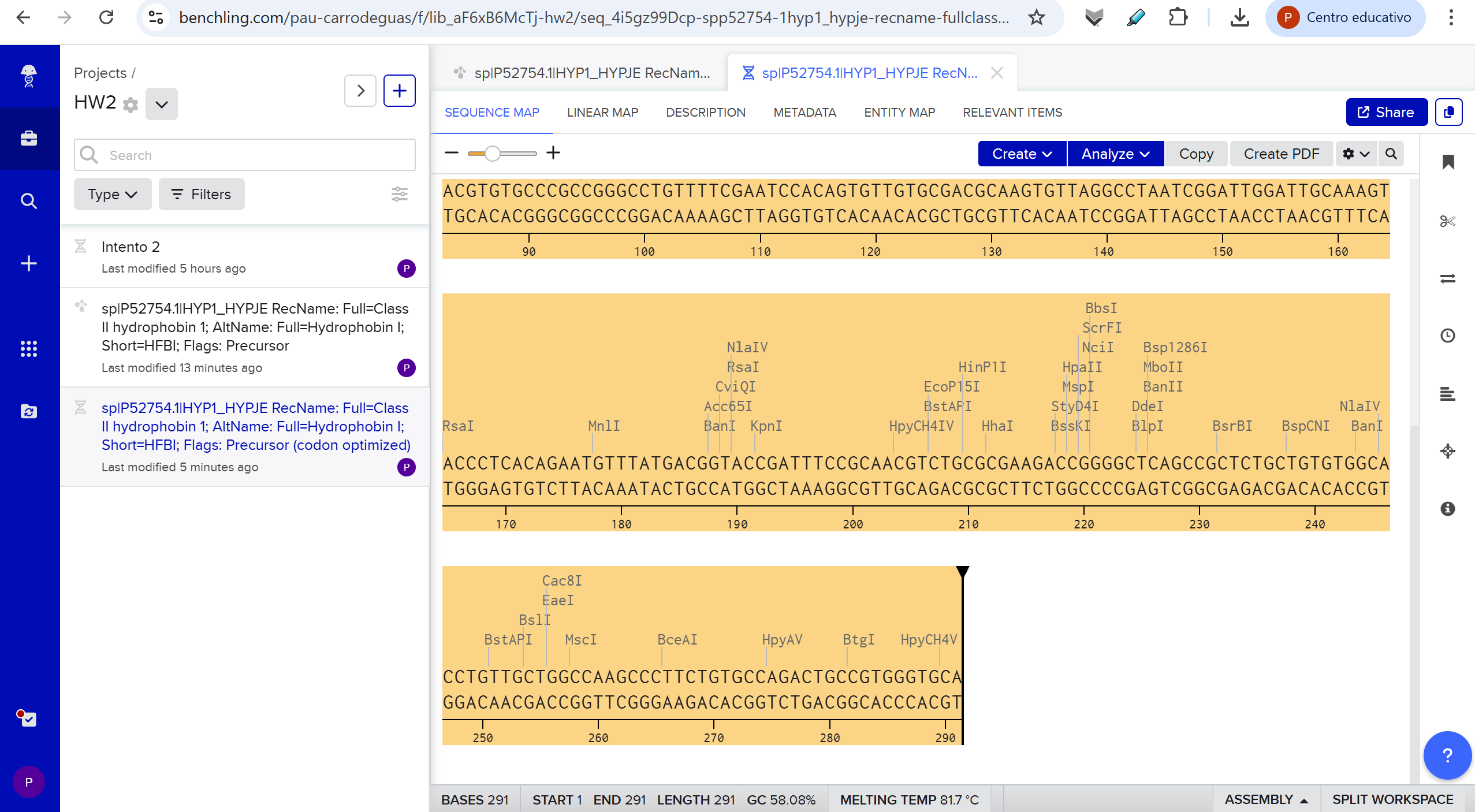
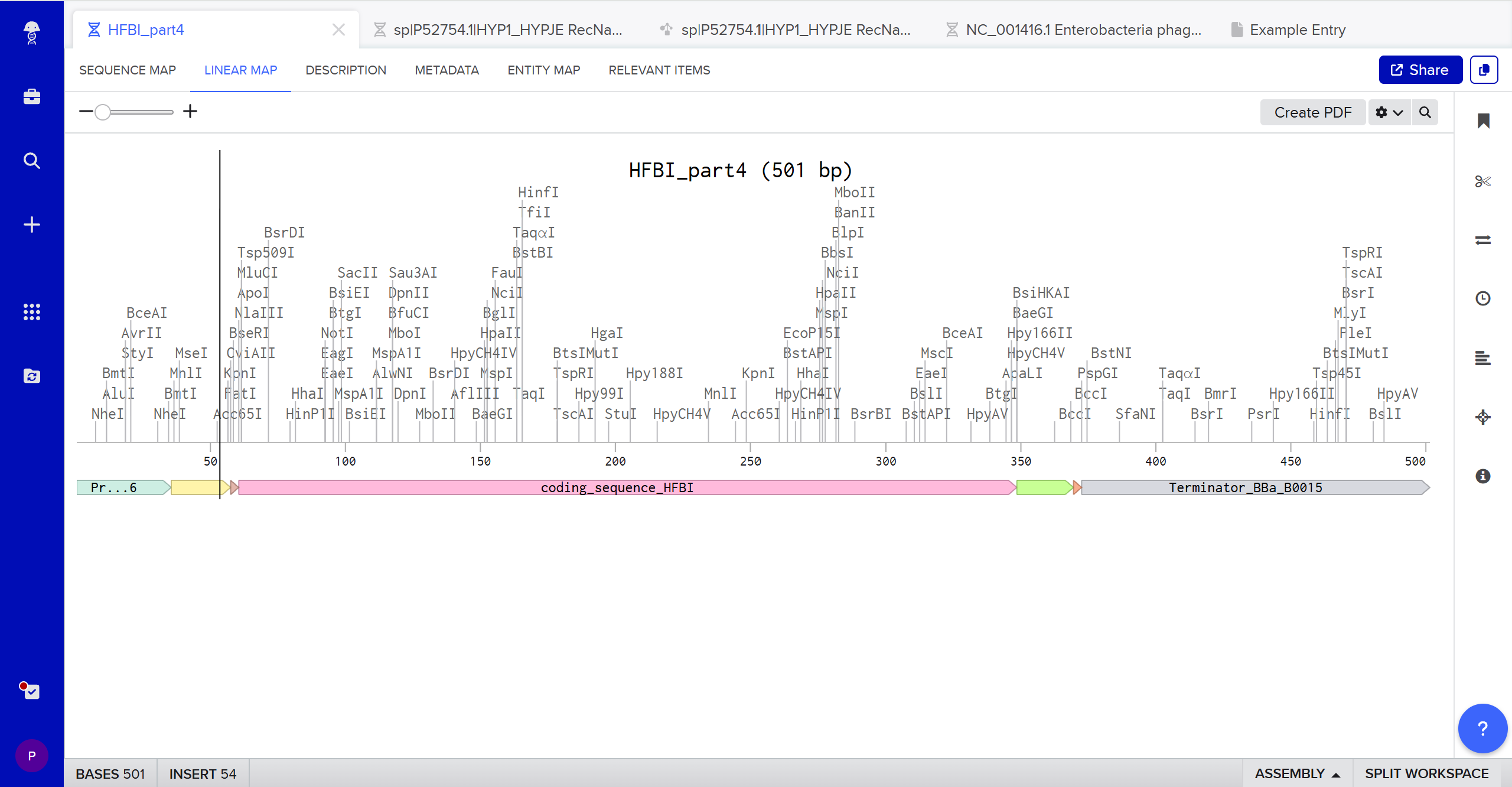
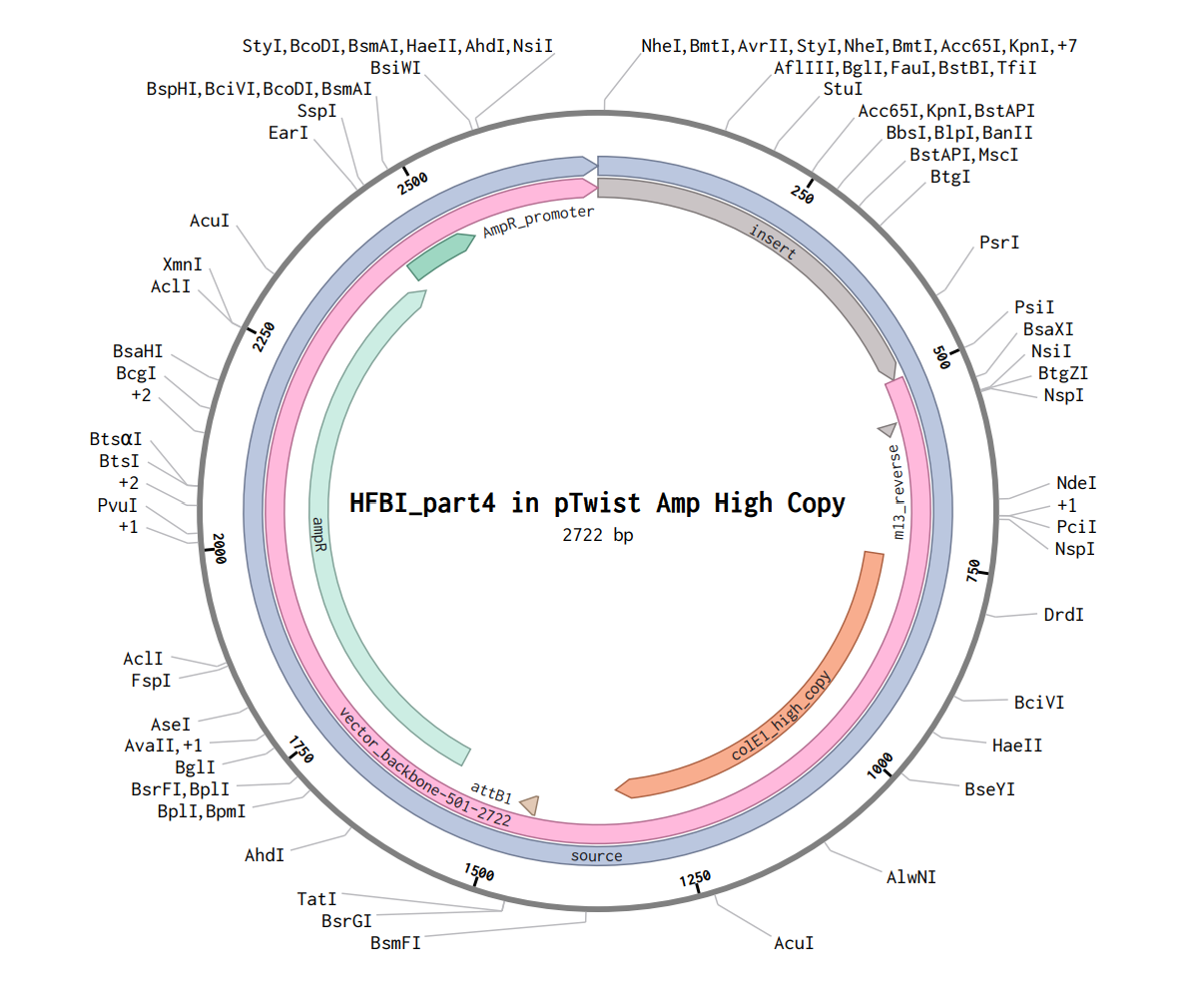
Part 1: Benchling & In-silico Gel Art This DNA gel art was designed in the style of Paul Vanouse’s Latent Figure Protocol. I chose to create the letter “P” as it is the initial of my name, Paula. To achieve this, I used Ronan’s website, which was a helpful tool for quickly iterating on the designs and determining the best enzyme combinations to form the silhouette of the letter.
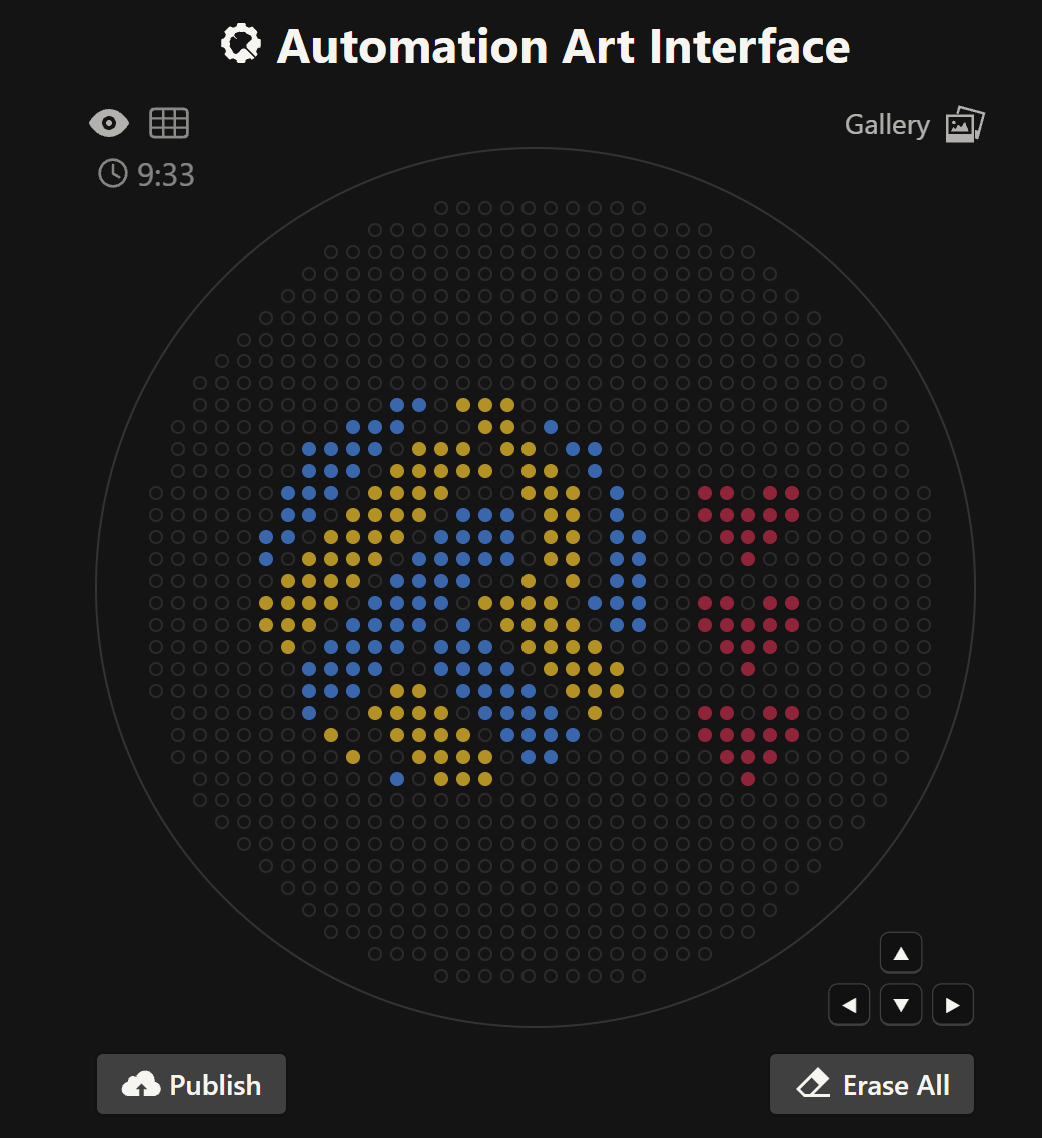
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot. 1. Generate an artistic design using the GUI at opentrons-art.rcdonovan.com. 2. Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons. Link to cell: https://colab.research.google.com/drive/1-f-vpwBCOx1gmlD5qXbW5z-7sLun1xwP?authuser=2#scrollTo=pczDLwsq64mk&line=1&uniqifier=1
Week 04 HW: Protein Design part I
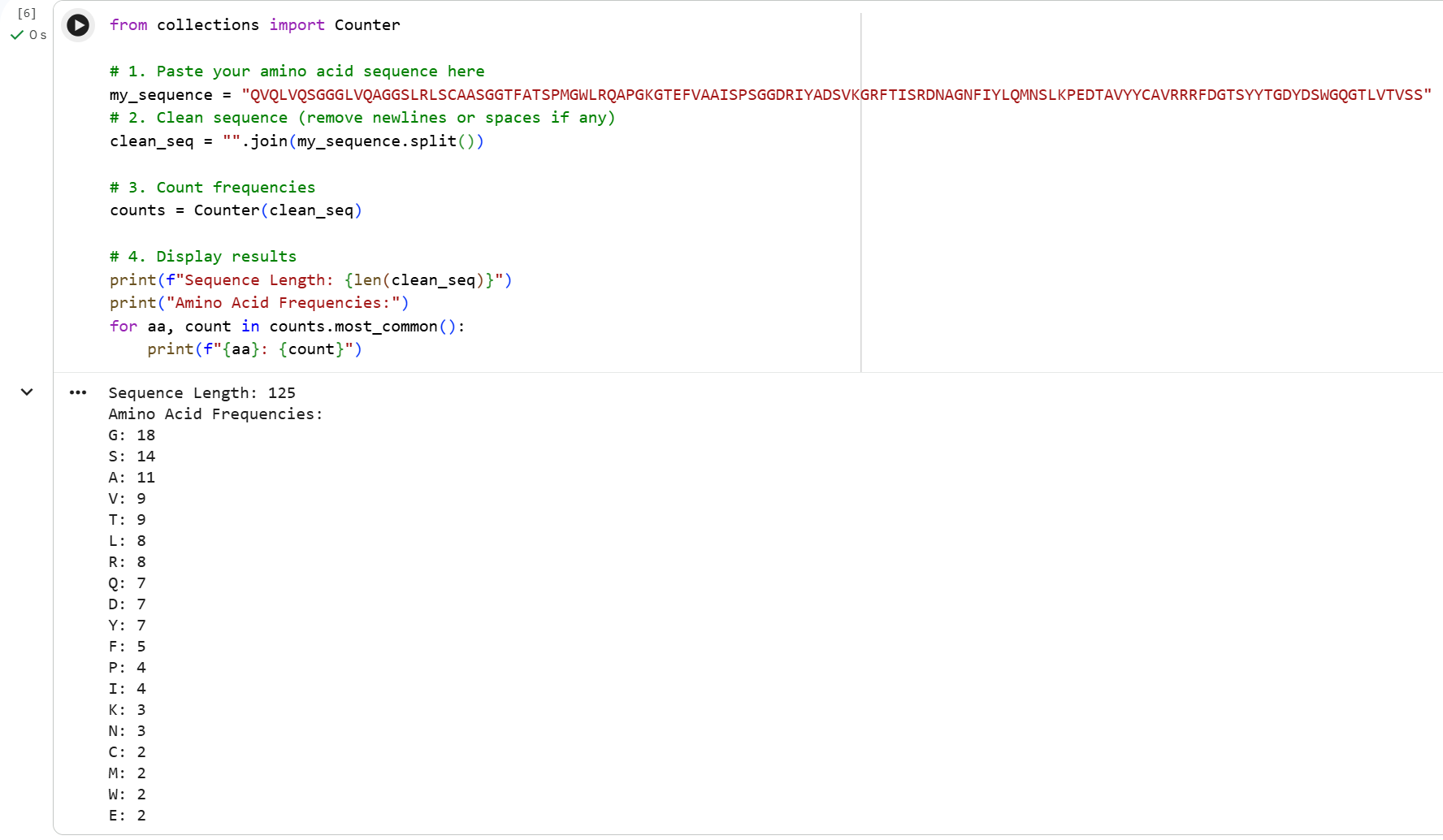
Part A. Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) The average composition of muscle without external fat cover is composed of approximately 70% water, 20% protein, and 9% fat (The exact values vary depending on the animal source) [1]. Therefore 500 g of meat provides 100 g of protein. Since proteins are chains of amino acids, once digested they break down into individual amino acid molecules. We are told the average molecular weight of an amino acid is ~100 Daltons, which means its molar mass is 100 g/mol.
Week 05 HW: Protein Design part II
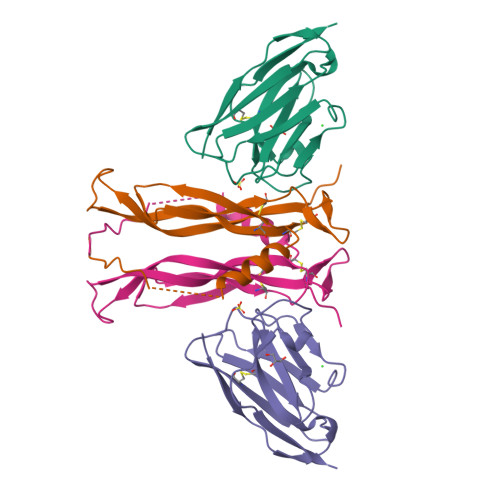
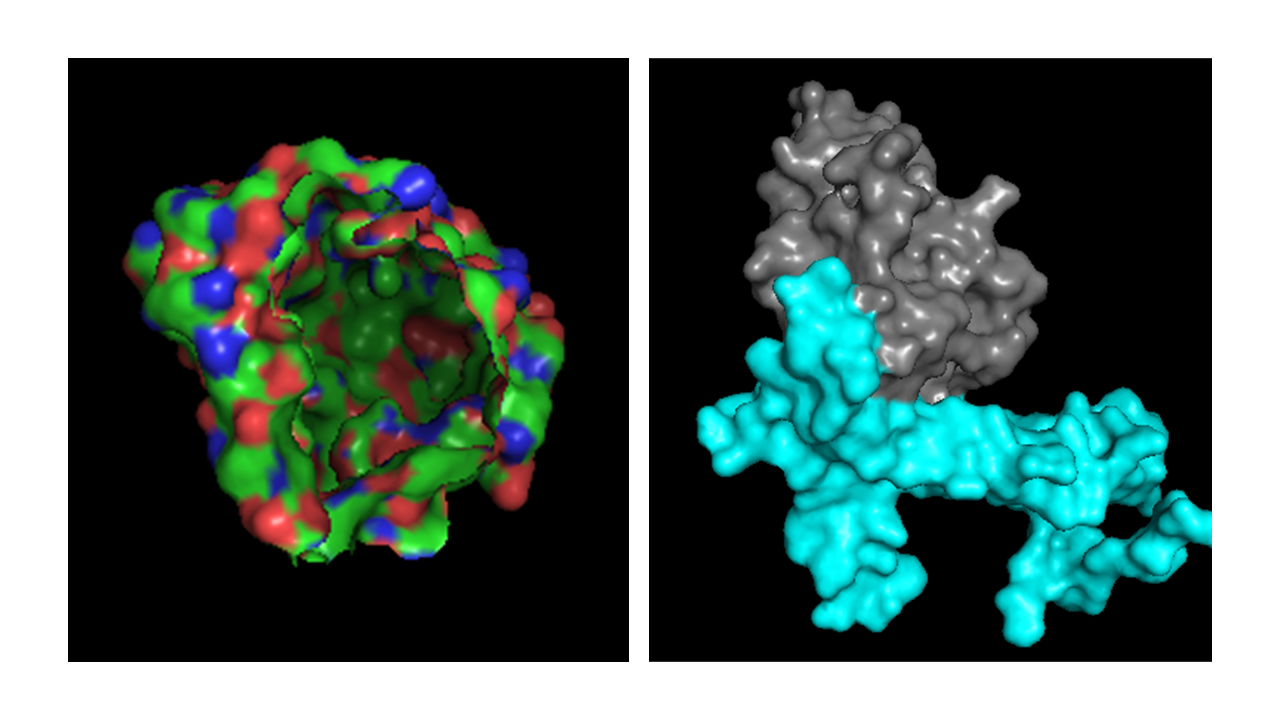
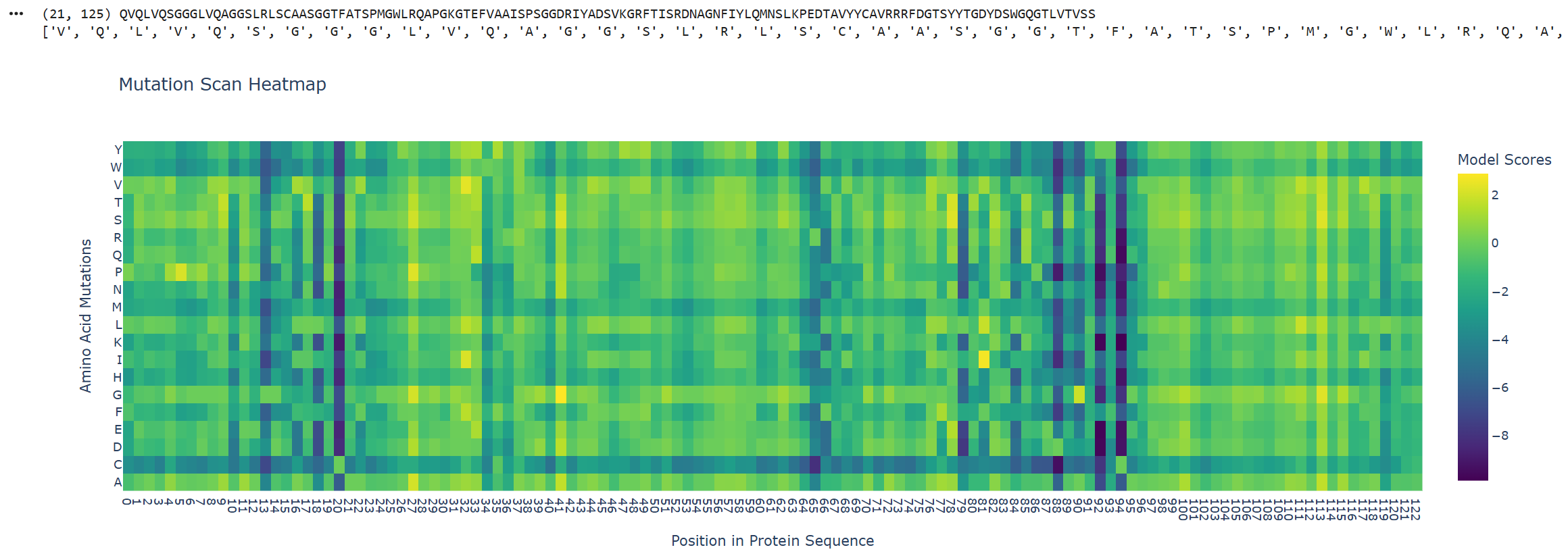
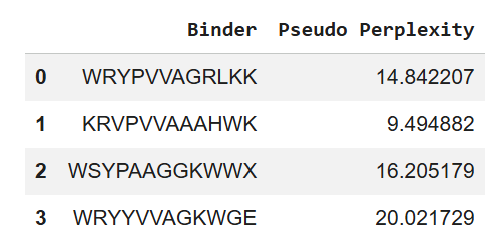
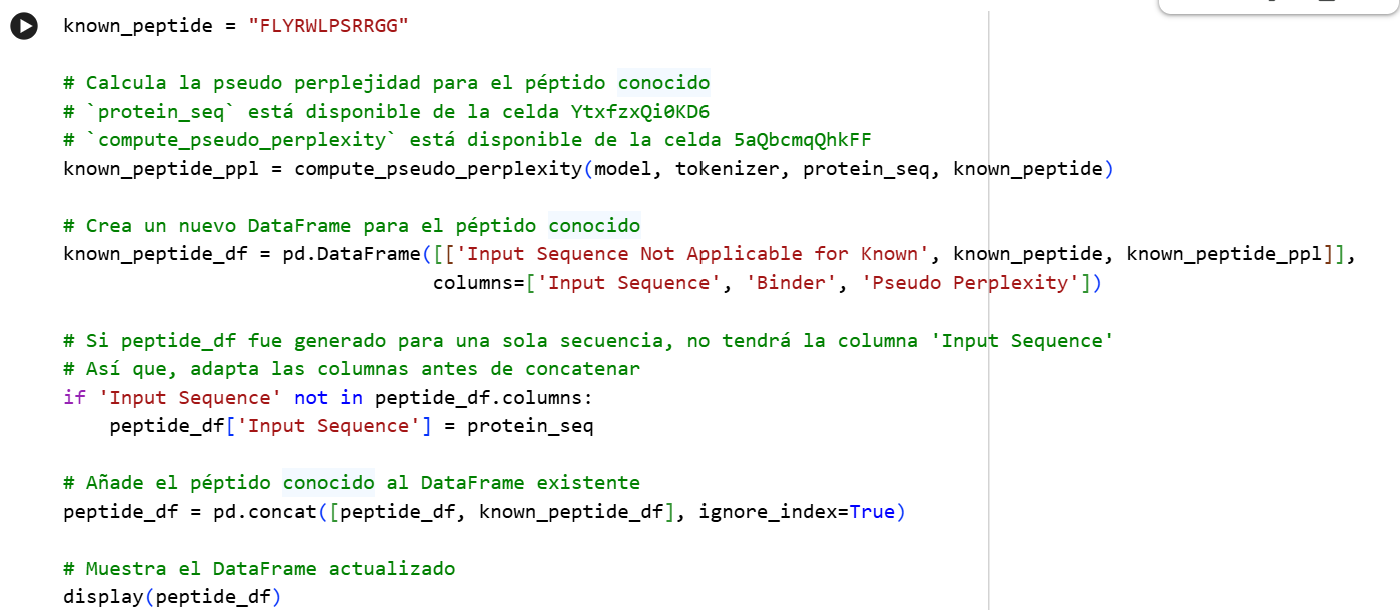
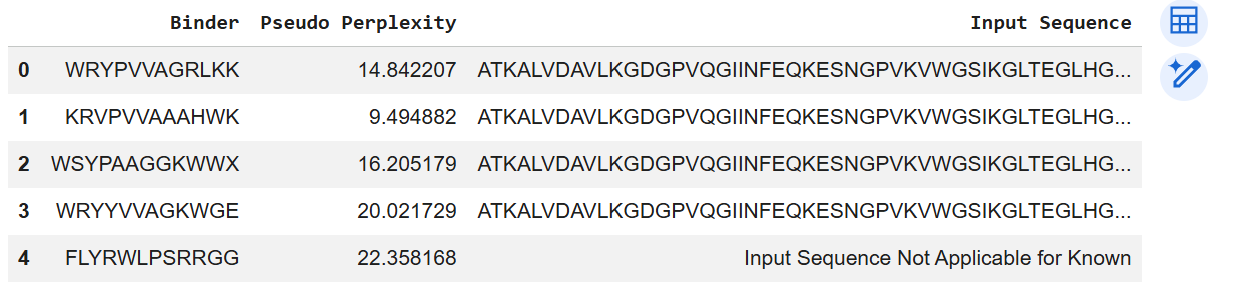
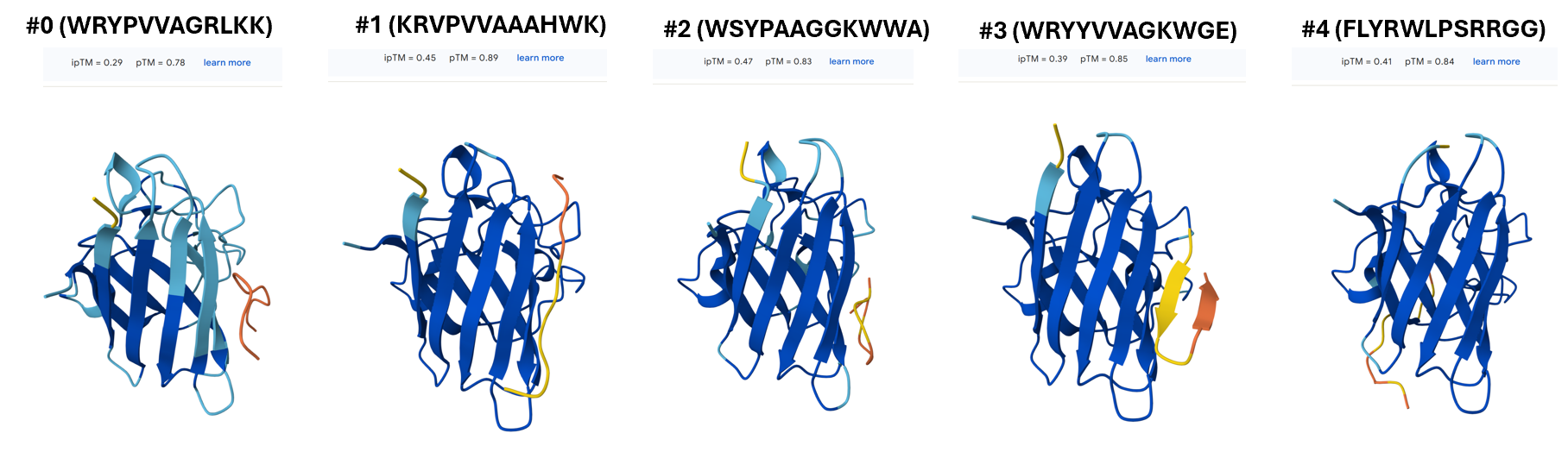
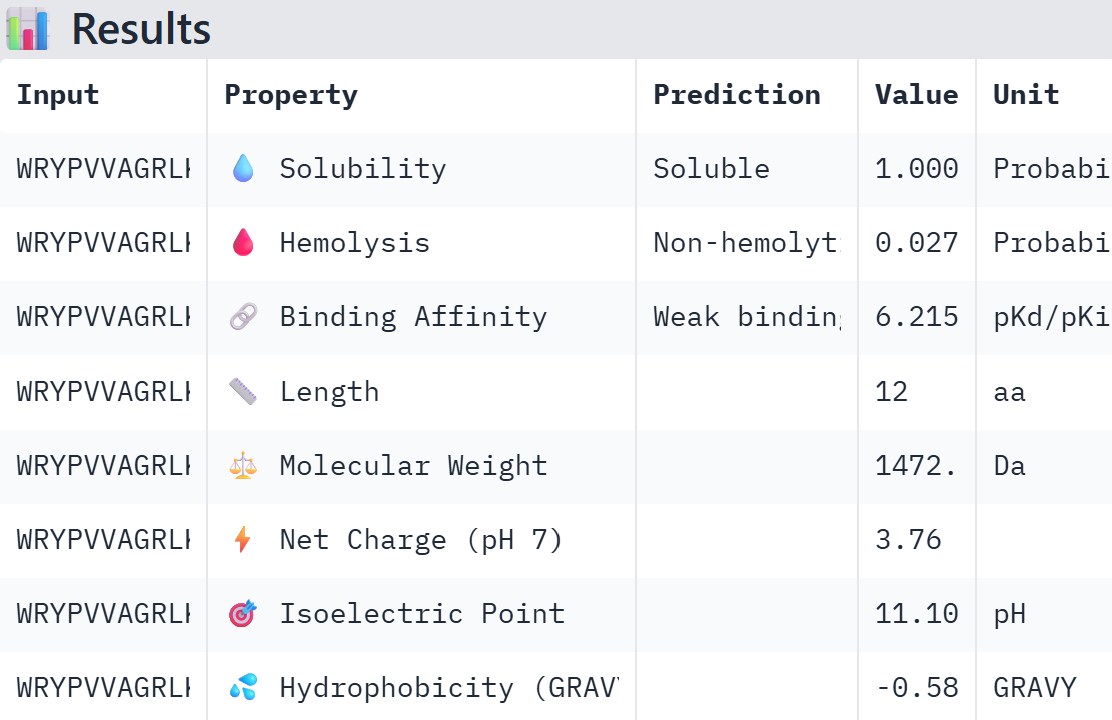
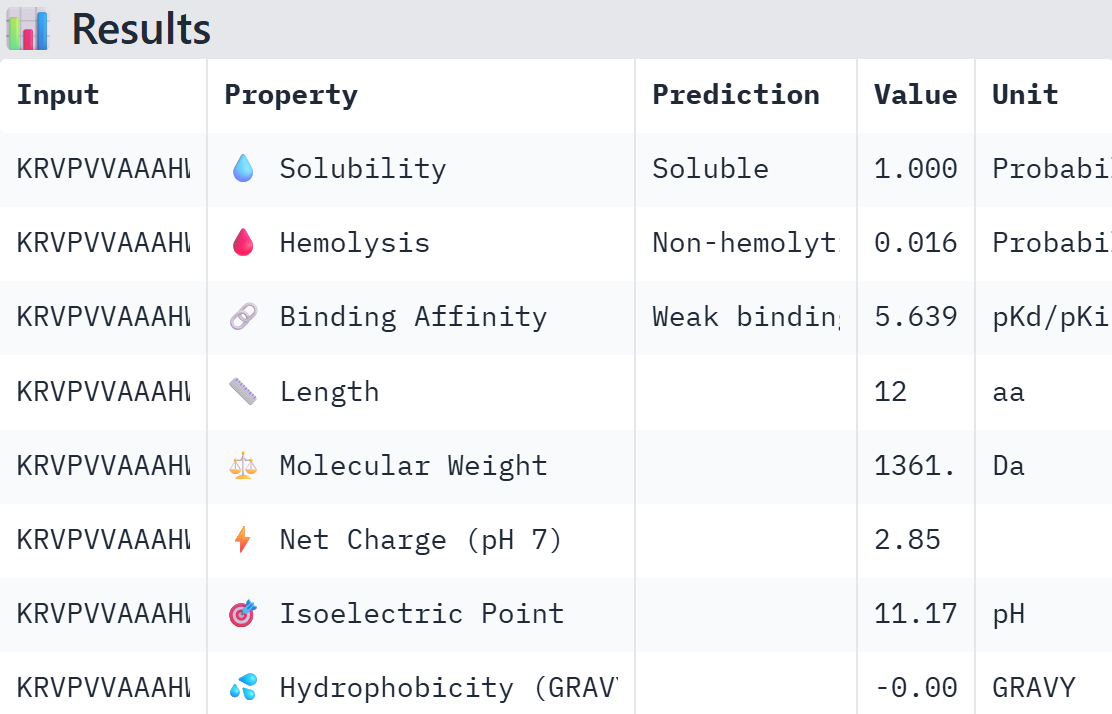
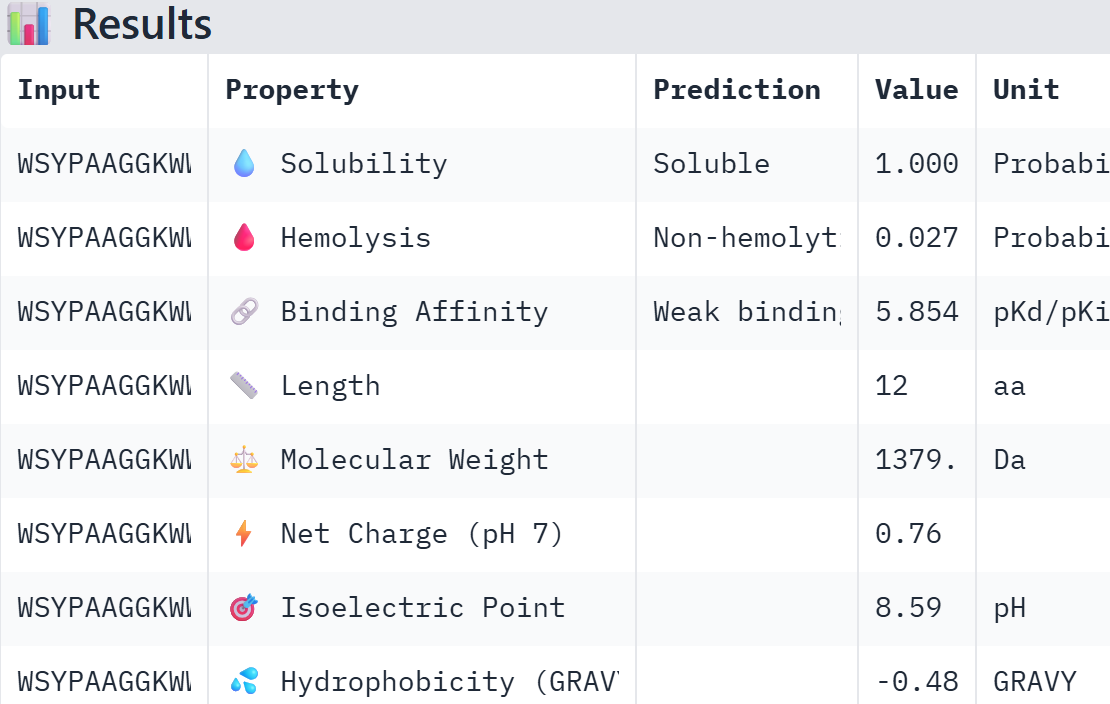
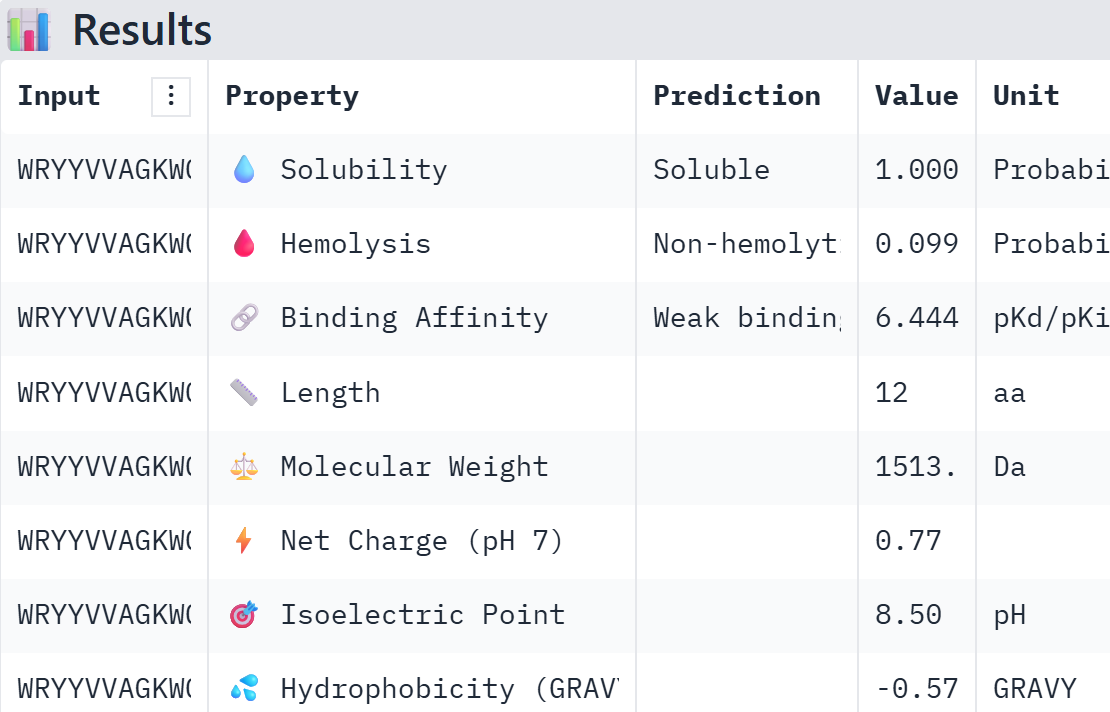
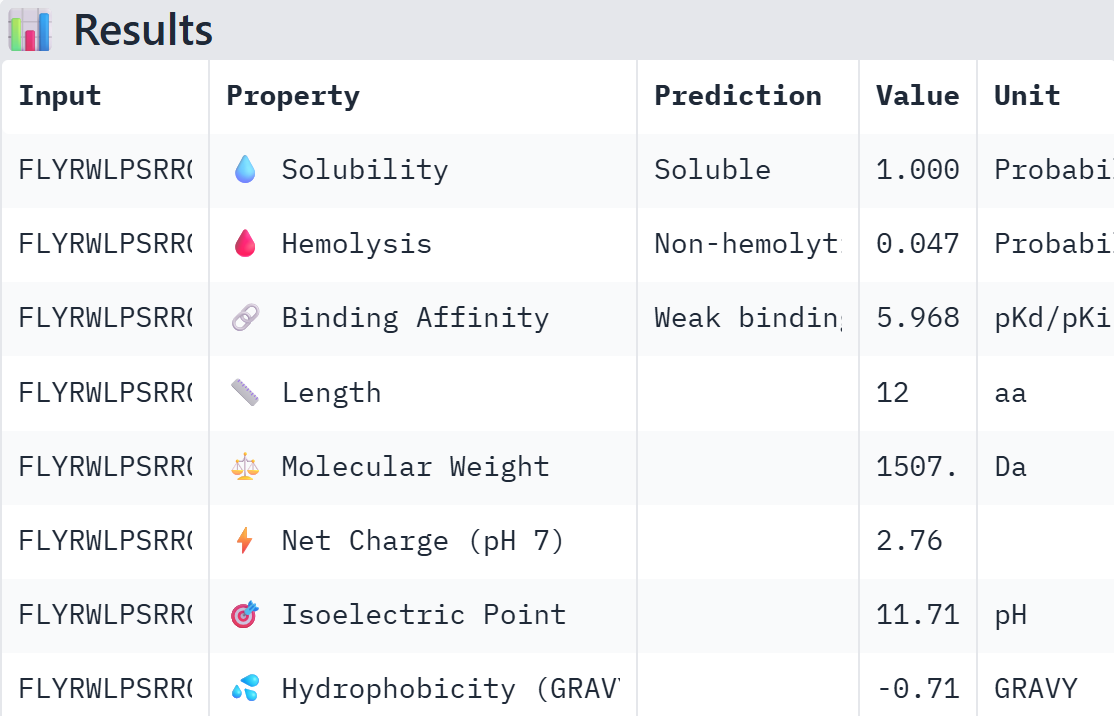
Part A: SOD1 Binder Peptide Design (From Pranam) Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Week 06 HW: Genetic Circuits Part I
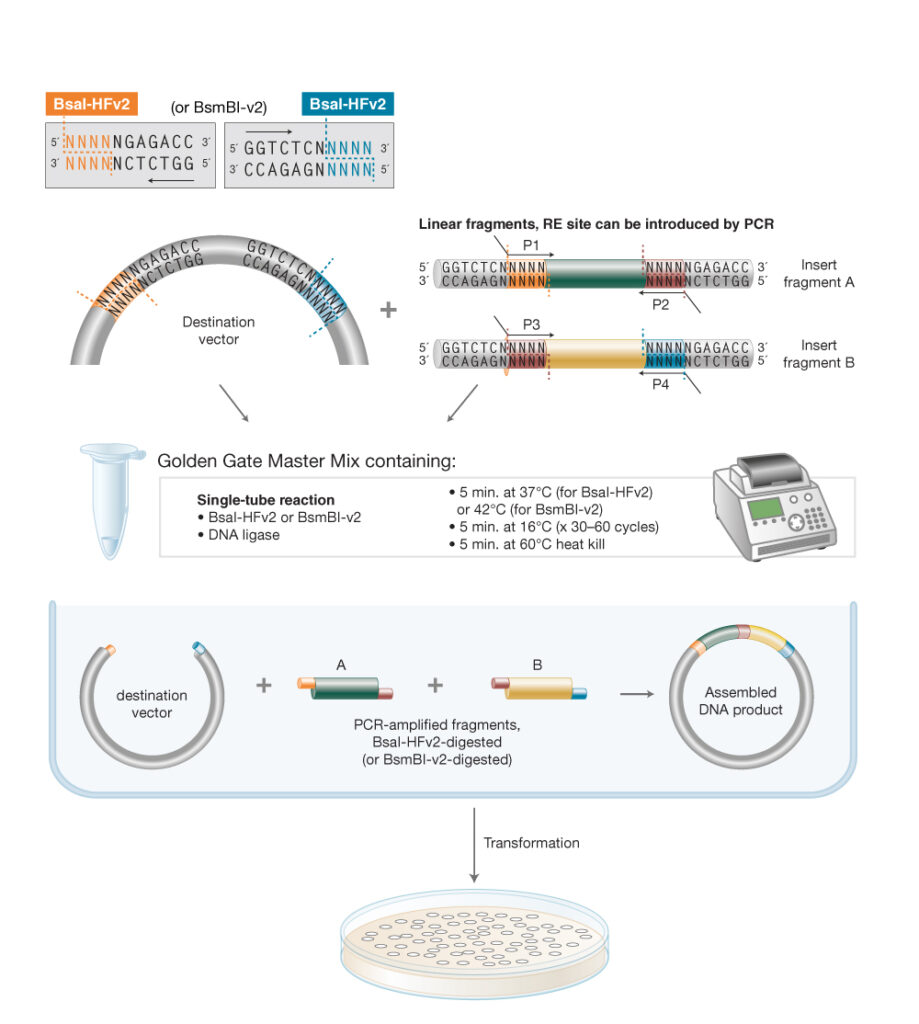
Assignment: DNA Assembly 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? According to New England Biolabs [1]: Phusion DNA polymerase: a DNA polymerase (enzymes that catalyze the synthesis of DNA molecules from dNTPs) that offers high fidelity and speed, with a lower error rate than Taq DNA polymerase.
Week 07 HW: Genetic circuits part II
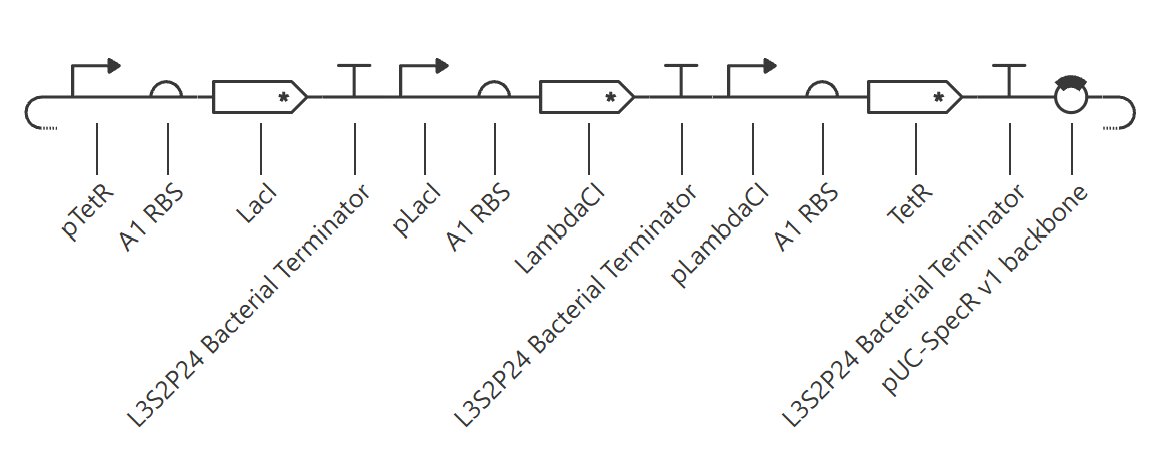
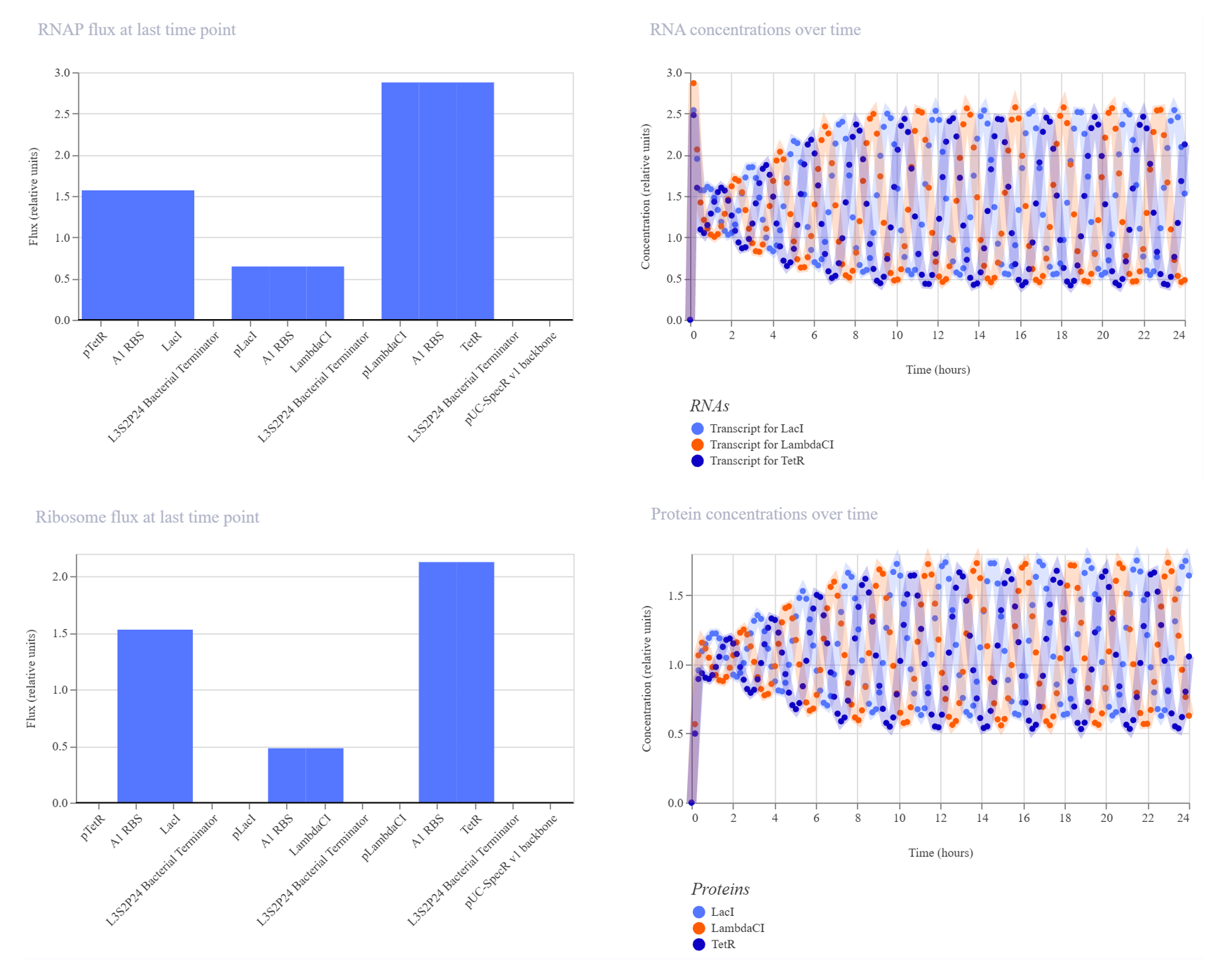
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Boolean functions are limited to discrete on/off states while IANNs are capable of processing analogue signals and, because of that, carry more information. Real world phenomena are analog, inside a cell there is inherent molecular noise, and Boolean circuits are fragile to this, especially at low signal concentrations.
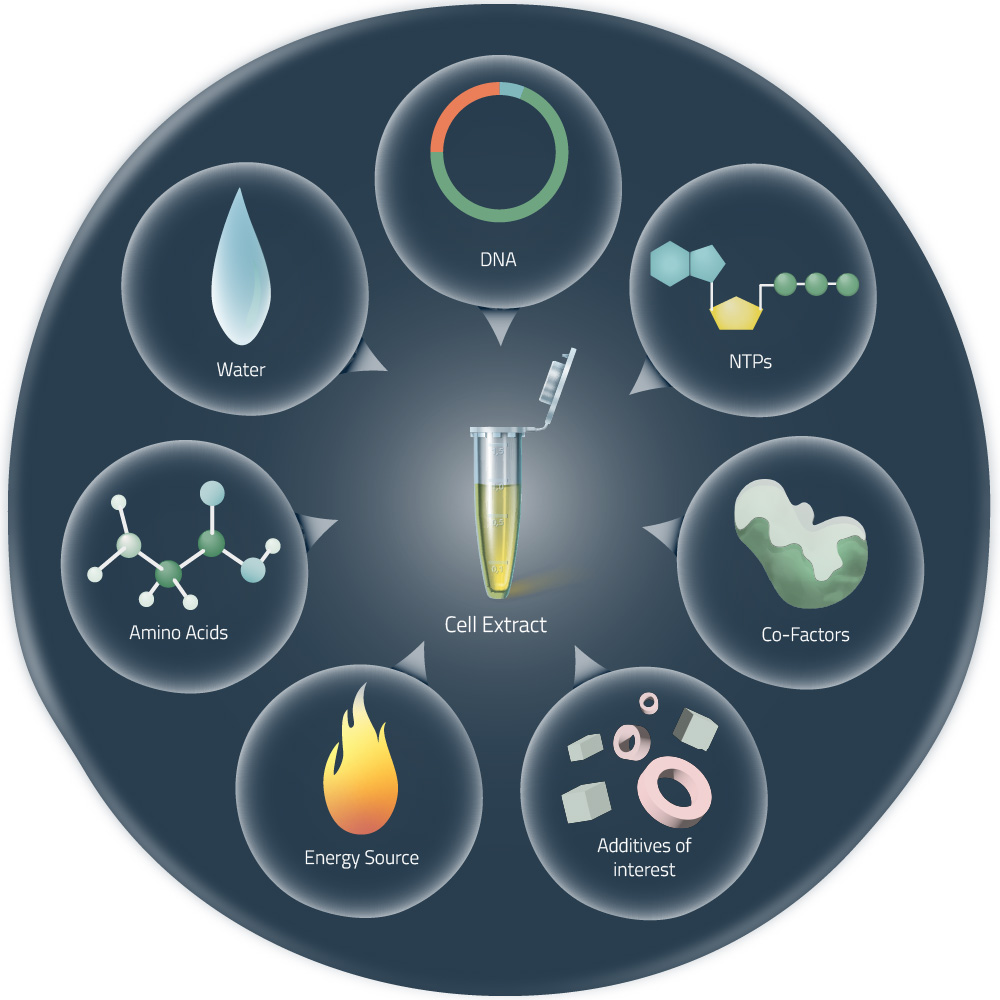
Homework Part A: General and Lecturer-Specific Questions General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Aspect In vivo Cell free Production speed Hours to days (requires growth) Minutes to hours (immediate synthesis) Cell membrane Cell membranes impede scientists from interacting with the components of the reaction or manipulate cellular processes. No cell membranes to get in the way of directly manipulating the reaction components. Toxic proteins If the target protein is toxic to the host cell, the cell may die before it can produce a significant amount of the protein. No living cells to keep alive so it bypasses toxicity issues. Manipulation of Reaction Conditions Cell viability needs to be considerate. Enables optimization (adjust pH, ionic strength, redox potential, metal ion concentrations, or temperature). Interference & purity Host cells need to produce their own proteins to stay alive, interfering with or delaying the production of the target protein. Difficult to separate the target protein from all the other proteins and cellular components. Since CFPS contain the minimal cellular components necessary for protein synthesis it simplifies the extraction and purification process. Non-natural amino acids Restricted to the use of the 20 naturally occurring amino acids. Enable the use of non natural amino acids to produce proteins with novel properties. Storage & Shipping Produced in large batches and shipped cold on ice, which is expensive Can be freeze-dry to make them last longer at room temperature. When is cell-free expression more beneficial than cell production?
Week 10 HW: Imaging and Measurement
For your final project: - Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. - Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. - What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail. —————————————————————————————————————————————————- - Toehold switch activation: Does the switch open specifically in response to LncRNA H19? I would order the toehold switch construct from Twist and it will be expressed at Ginkgo using a PURExpress cell-free reaction with and without the H19 trigger, using sfGFP as reporter. Technology: fluorescence spectroscopy with a plate reader.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design 1. Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each) 1. sfGFP 2. mRFP1 3. mKO2 4. mTurquoise2 5. mScarlet_I 6. Electra2 Fluorescent protein Maturation (min) pKa Brightness Description sfGFP 13.6 54.15 Rapidly-maturing weak dimer mRFP1 60.0 4.5 12.5 Slowly-maturing monomer with low acid sensitivity mKO2 108.0 5.5 39.56 Moderate acid sensitivity mTurquoise2 33.5 3.1 27.9 Rapidly-maturing monomer with very low acid sensitivity mScarlet_I 174.0 5.3 70 Moderate acid sensitivity Electra2 61.48 3. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect. I choose mKO2 because it is acid sensitive and has a slow maturation time.. My hypothesis is that increasing the HEPES-KOH concentration in the 36-hour master mix would help maintain the pH closer to 7.5 throughout the reaction. Which is important because in cell free reactions pH decreases over long incubations due to the accumulation of acidic metabolic byproducts and mKO2 slow maturation time means most of the signal is generated in the later hours when pH tends to drop.