Week 04 HW: Protein Design part I

Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
The average composition of muscle without external fat cover is composed of approximately 70% water, 20% protein, and 9% fat (The exact values vary depending on the animal source) [1]. Therefore 500 g of meat provides 100 g of protein. Since proteins are chains of amino acids, once digested they break down into individual amino acid molecules. We are told the average molecular weight of an amino acid is ~100 Daltons, which means its molar mass is 100 g/mol.
Converting grams of protein to moles of amino acids:
100 g ÷ 100 g/mol = 1 mol of amino acids
Converting moles to number of molecules:
1 mol × 6.022 × 10²³ = ≈ 6 × 10²³ molecules of amino acids
A piece of 500 g of meat contains approximately 6 × 10²³ molecules of amino acids
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Biological identity is not preserved through digestion because the human body breaks down the macromolecules we consume into universal building blocks.
Proteins undergo hydrolysis of their peptide bonds by proteases in the stomach and small intestine, releasing amino acids, the same 20 standard amino acids used by all living organisms. Once absorbed, they enter the bloodstream, they are either incorporated into new human proteins according to the sequence information encoded in human DNA or further metabolized [2].
3. Why are there only 20 natural amino acids?
There are more than 20 amino acids in nature, however, the standard genetic code is restricted to 20 amino acids because it provides a balance between structural diversity and metabolic efficiency.
Chemical coverage: the 2o amino acids set covers the necessary range of hydrophobicity, charge and molecular size required for complex protein folding [2], for example:
- Charge: Positive (Lysine) and Negative (Glutamate).
- Polarity: Hydrophilic (Serine) vs. Hydrophobic (Leucine).
- Specialized Shapes: Small (Glycine), Rigged (Proline), and bulky (Tryptophan).
Frozen Accident: This theory, proposed by Francis Crick, states that “the code is universal because at the present time any change would be lethal, or at least very strongly selected against.” Any attempt by an organism to ‘recode’ or add a new amino acid today would trigger a proteome-wide failure, as it would disrupt the sequence of every existing protein simultaneously [3].
Error minimization: Research suggests that our genetic code is ‘one in a million’ in its ability to ensure that a single-point mutation likely results in a chemically similar amino acid, thereby preserving the protein’s overall structure and function [4].
4. Can you make other non-natural amino acids? Design some new amino acids.
Non-natural amino acids are synthesized compounds that differ from the standard set. They can be synthesized either through organic chemistry or incorporated into proteins via engineered orthogonal translation systems. By modifying side-chain chemistry, we can expand the functional diversity of proteins beyond the constraints of the canonical genetic code, enabling novel catalytic, structural, and responsive properties.
All amino acids share the same basic backbone:
- An amino group (–NH₂)
- A carboxyl group (–COOH)
- A hydrogen
- A variable side chain (R group)
- Attached to the same α-carbon
So to design new amino acids, we keep the backbone and modify the R group to give new chemical properties.
Example: Photoresponsive amino acid with the following side chain:
R = –CH₂–C₆H₄–N=N–C₆H₅
This side chain contains an azobenzene group, which can switch between trans and cis conformations when exposed to different wavelengths of light.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
Elevated levels of IL-17 have been observed in patients during the early stages of the disease. This pro-inflammatory cytokine promotes the proliferation, invasion, and implantation of endometriotic cells by triggering the construction of new blood vessel networks. Blocking IL-17 not only reduces inflammation but also interrupts the development of the blood supply these lesions require to survive and persist outside the uterine cavity.
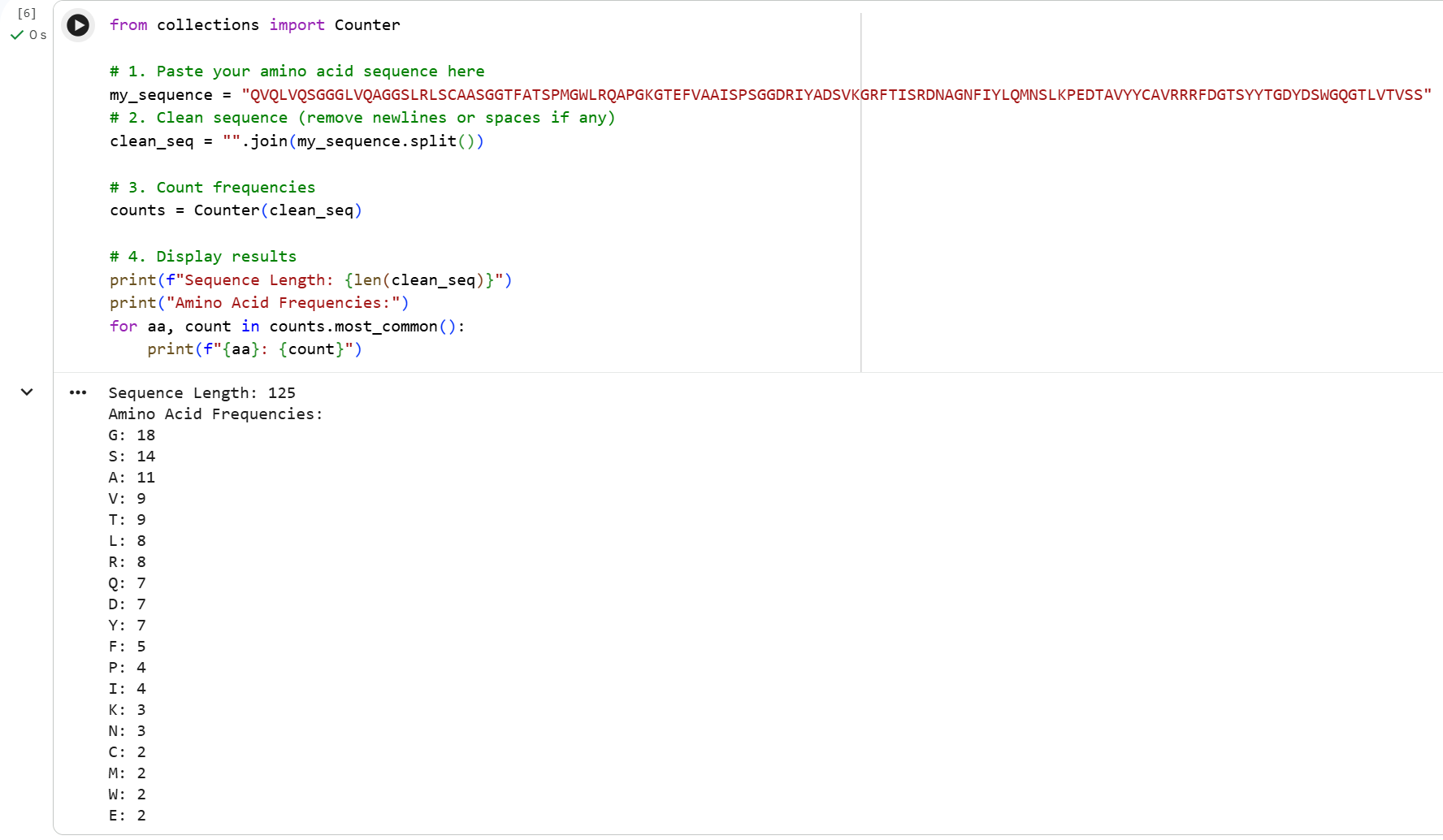
2. Identify the amino acid sequence of your protein.
8B7W_1|Chain A[auth H]|anti-IL-17A-76|Lama glama (9844) QVQLVQSGGGLVQAGGSLRLSCAASGGTFATSPMGWLRQAPGKGTEFVAAISPSGGDRIYADSVKGRFTISRDNAGNFIYLQMNSLKPEDTAVYYCAVRRRFDGTSYYTGDYDSWGQGTLVTVSS
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The most frecuent amino acid in 8B7W_1 is glycine.
How many protein sequence homologs are there for your protein?
250 homologs
Does your protein belong to any protein family?
It´s part of the Single-domain antibody family
3. Identify the structure page of your protein in RCSB
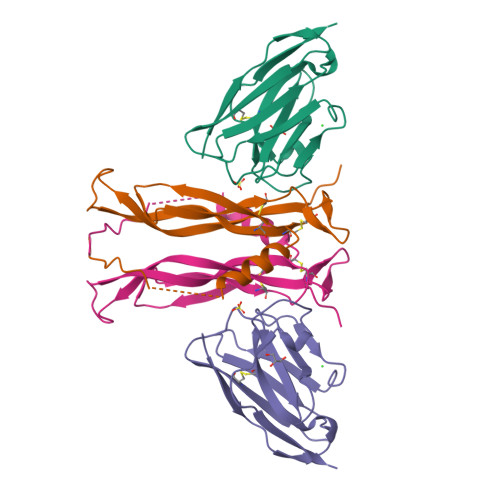
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure was deposited on October 3, 2022 and made publicly available on December 28, 2022. It was solved using X-ray diffraction.
Resolution: 2.85 Å Moderate quality range, close but slightly above the 2.70 Å threshold for a good structure.
Are there any other molecules in the solved structure apart from protein?
Yes, 8B7W is a protein.protein complex containing:
IL-17A: pro-inflammatory cytokine from Homo sapiens Anti-IL-17A-76: nanobody derived from Lama glama (Llama)
Both proteins were expressed in Escherichia coli and the structure contains mutations.
Does your protein belong to any structure classification family?
Classification: IMMUNE SYSTEM/INHIBITOR
4. Open the structure of your protein in any 3D molecule visualization software:
I used PyMol to analyze the three-dimensional structure of 8B7W, a protein complex formed by interleukin-17A (IL-17A) bound to an anti-IL-17A nanobody. Since my main interest lies in the anti IL-17A nanobody, I visualized only this structure referred in PDB as “chain H” hiding “chain B”. This allowed a clearer examination of its structural features, including its secondary structure, residue distribution, and potential interaction regions involved in antigen recognition.
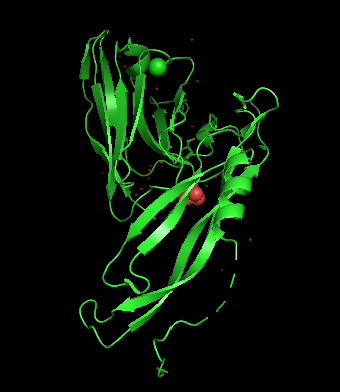
Visualize the protein as:
Cartoon: simplifies the protein structure and highlights the secondary structure elements.
Ribbon: follows the protein backbone and helps visualize the trajectory of the polypeptide chain and how the secondary structure elements are arranged.
Ball and stick: shows atoms as spheres and chemical bonds as sticks. Allows to see individual amino acids, atomic interactions and contacts between residues

Color the protein by secondary structure. Does it have more helices or sheets?
Rojo → α-helices
Amarillo → β-sheets
Verde → loops
The nanobody analyzed shows a structure predominated by β-sheets. This is typical of antibody variable domains that adopt an immunoglobulin fold (many β-sheets and long loops), in this case it plays an important role in forming the antigen-binding interface with IL-17A.
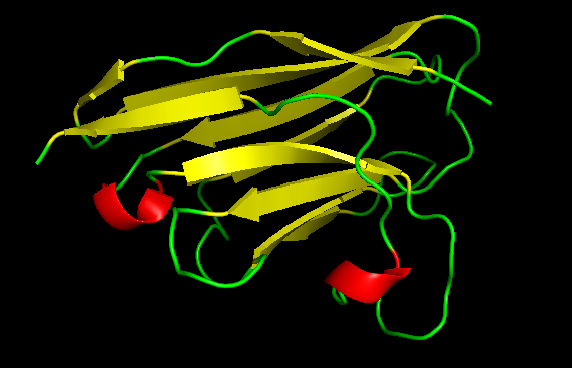
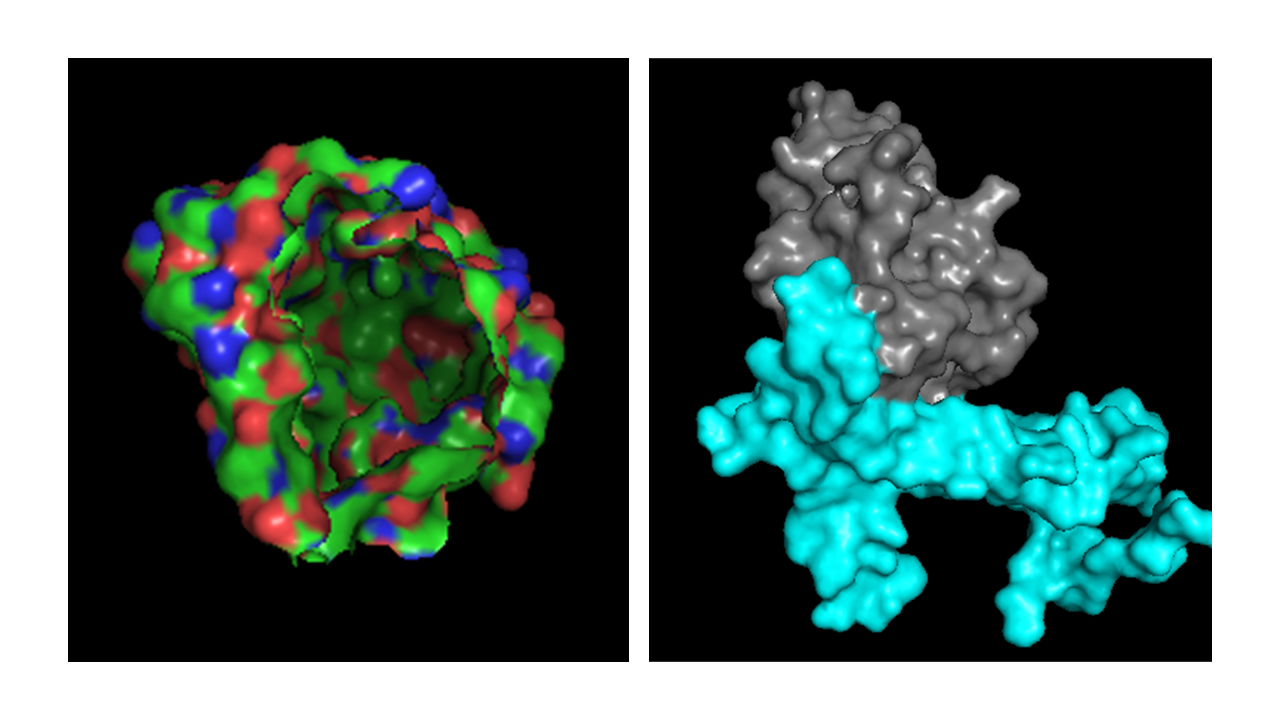
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
When visualizing the surface of the nanobody, a region appears almost like a missing portion of the structure and this happens because the antigen is hidden in the visualization. This is the interface where the nanobody interacts with IL-17A in the complex.
Part C: Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
C1. Protein Language Modeling
1. Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
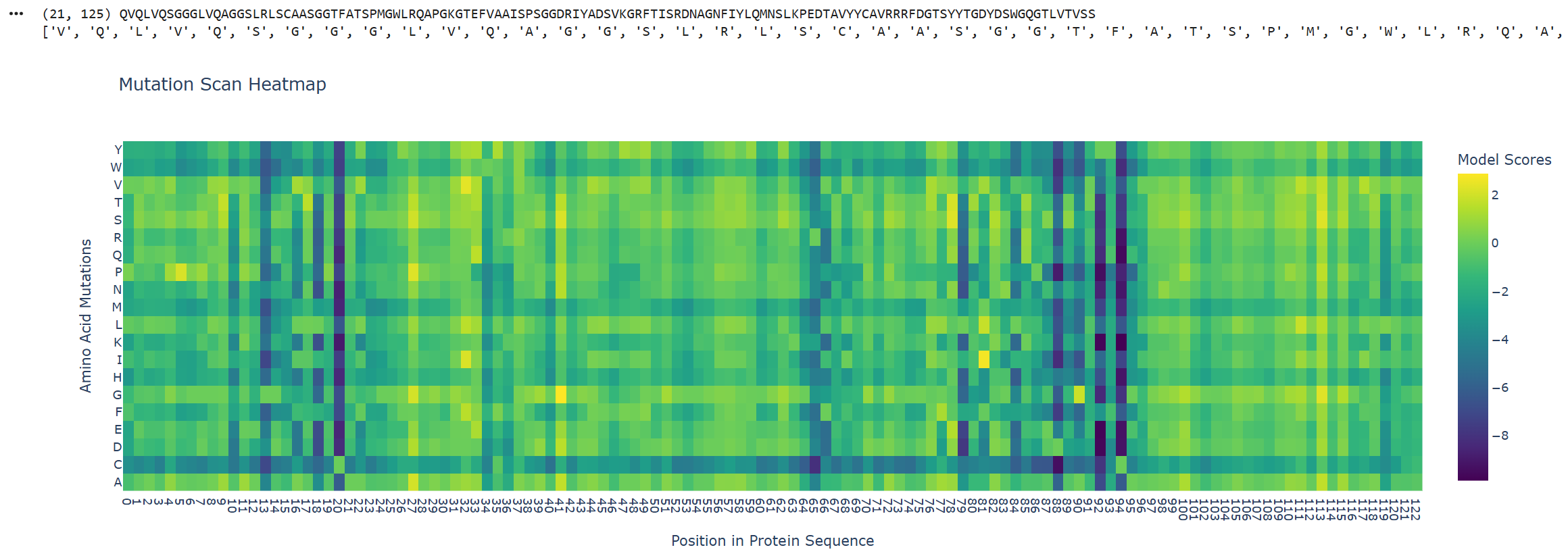
The ESM2 score represents how likely it is evolutionarily that that amino acid will appear in that position, based on millions of protein sequences.
Y-axis: Represents the 20 standard amino acids that could mutate at each position in the sequence.
X-axis: Represents the positions of the amino acids in the input protein sequence.
High score → ESM2 considers this mutation evolutionarily plausible. This amino acid has appeared at this position in similar sequences. The protein likely tolerates this mutation.
Blue (low score) → ESM2 considers this mutation evolutionarily improbable. This amino acid rarely appears at this position. The mutation likely damages the protein.
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Positions 20, 88, 92 and 94 show a vertical dark blue pattern (low score), indicating that these positions dont tolerate mutations. This suggests that the wild-type residues are critical.
Two horizontal lines are also noteworthy due to their dark blue pattern, these correspond to W (Tryptophan) and C (Cysteine). This suggests that introducing these residues in any position is unfavorable. This makes sense because cysteine can form spurious disulfide bonds that disrupt folding, and tryptophan is bulky enough to cause steric clashes in most sequence contexts. With a lighter shade of blue, methionine (M) and histidine (H) also show moderately negative scores across many positions, likely reflecting their more specialized chemical properties and lower natural abundance in protein sequences.
Position 113 shows predominantly positive scores across many substitutions, indicating that this position is highly tolerant to mutation. This suggest that in this position, the identity of the amino acid doesn´t matter much structurally. Probably sits in a flexible loop or solvent-exposed region.
C-terminal region exhibit more yellow/high score regions, suggesting that the C-terminal end of the nanobody is much more tolerant of mutations.
2. Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
b.Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes, I identified a cluster containing different types of protein from bacteria like Mycobacterium Tuberculosis, Pseudomona Aeruginosa y Thermus Thermophilus.
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
I think the data base is limited and has no enough nanobodys sequences because the cluster where my protein is, it´s surrounded by proteins from different animals and even the IL-12. I would have expected to see other VHH sequences from Llama.
C2. Protein Folding
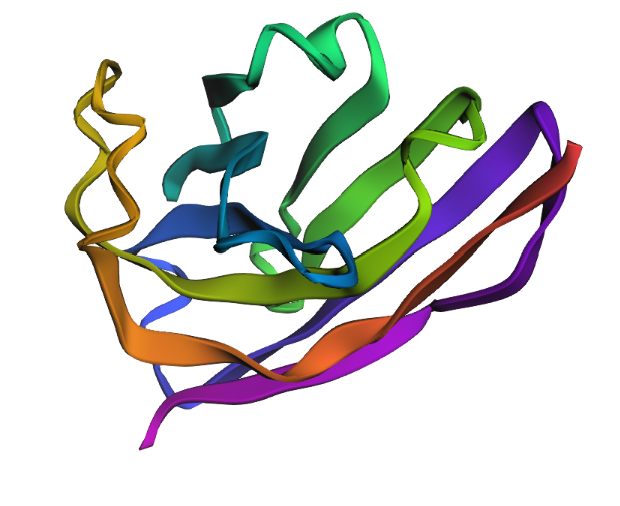
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Yes, It is very similar with the structure I saw in Pymol, The B-Barrel structure and the loops are visible.
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
My protein is resilient to mutations because although I made some mutations that appeared to be unfavorable in the ESM2 Mutation Scan heatmap, I didn´t see drastic changes in the protein´s structure, the B-Barrel is still there as well as the loops.
These are the changes I made:
Position 20: L → E Posición 65: K → C Posición 94: Y → R
There are probably changes but since I can´t compare side to side the structures, to me they appear to be still similar.
Although, if I remove the first ten amino acids, the structure dramatically changes. That is because the first aminoacids in a nanobody are almost the same in their variations.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
8B7W, score=1.5216, fixed_chains=[], designed_chains=[‘H’], model_name=v_48_020 QVQLVQSGGGLVQAGGSLRLSCAASGGTFATSPMGWLRQAPGKGTEFVAAISPSGGDRIYADSVKGRFTISRDNAGNFIYLQMNSLKPEDTAVYYCAVRRRFDGTSYYTGDYDSWGQGTLVTVSS
T=0.1, sample=0, score=0.7640, seq_recovery=0.5760 SLTLSQSGGGTVAAGGSVTLTCSSSGGTFKDQWMGWLRQAPGKPVEFVAAISPSGSTTLYSDKVKGRFTISKDSSGQTVTLTMNDLKPEDTATYYCAVRTSSNGSPMDPSNYQPDGSGQKLTVLP
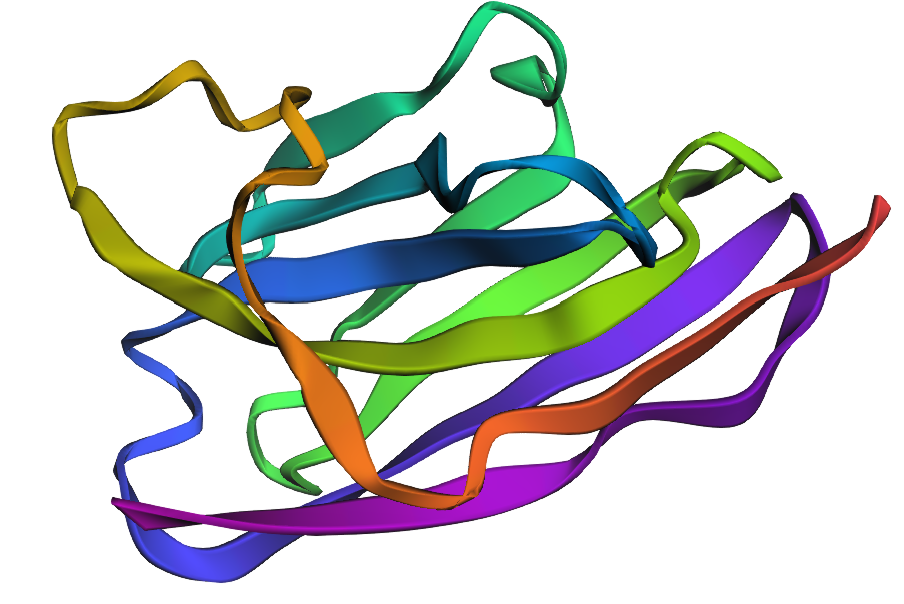
2. Input this sequence into ESMFold and compare the predicted structure to your original.
The predicted structure is similar but not completely the same, it appears to have the same structure but is folding in a slightly different way.
Part D. Group Brainstorm on Bacteriophage Engineering
Choose one or two main goals from the list that you think you can address computationally
Goal: Higher toxicity of lysis protein
Sub-goal: Eliminate the dependence of the L protein on the host chaperone DnaJ (E. coli) to accelerate and enhance bacterial lysis.
Context
- The L protein of bacteriophage MS2 lyses the bacterium Escherichia coli. The authors identify that this process depends on the hostile chaperone DnaJ, specifically through an interaction with its C-terminal domain.
- Using mutants, the study demonstrates that the P330Q mutation in DnaJ blocks the lytic capacity of the L protein at certain temperatures.
- In the absence of interaction with DnaJ, the N-terminal domain of L interferes with it´s ability to bind to it´s unknown target.
- The discovery of variants called Lodj mutants revealed that the N-terminal domain of the L protein is dispensable and is responsible for generating this chaperone dependence.
Tools/approaches
ESM2 for Deep Mutational Scanning (DMS): we want to identify residues in the N-terminal domain that can be removed or mutated without disrupting folding or making the protein unstable while mantain critical residues that are essential for lytic function.
ESMFold for structural prediction: predict the structure of the designed variants.
ProteinMPNN for inverse folding: once we find a stable structure for the L protein we will use ProteinMPNN to generate new sequences that might fold more efficiently.
Why these tools?
Since the N-terminal domain of L is intrinsically disordered and difficult to fold without DnaJ, ESM2 can help identify favorable mutations to improve stability.
ESMFold would help for validating whether a drastic mutation causes the rest of the protein to collapse.
ProteinMPNN can help redesign the sequence of the lysis protein so that it folds more efficiently.
Pitfalls
Bacterial cells could lyse too early, not allowing the production of sufficient phage particules and resulting in very low final phage titers, this could lead to resistance to the phages.
The interaction between the N-terminal domain and DnaJ conferes the L protein with some stability, if we aim to eliminate this dependency, there´s the posibility that the redesigned protein could become unstable and degrade before reaching the membrane.
--- title: Pipeline´s schematic --- graph TD A[WT L-protein sequence] --> B[ESM2 deep mutational scan] B --> |Identify favorable mutations| C[ESMFold structural prediction] C --> |Validate stability| D[ProteinMPNN inverse folding] D --> |New sequences independent from DnaJ and with more efficient folding| E[AF2-Multimer co-fold with DnaJ] E --> |Verify loss of interaction| F[Candidate variants for lab]
AI Prompts:
- What could be the reason there are horizontal dark blue lines in W and C, assigning low score to that residues? M and H also have low score but a bit higher than the others, why could be the chemical reason behind this?
Bibliography
[1] “Meat Composition - an overview | ScienceDirect Topics.” Accessed: Mar. 02, 2026. [Online]. Available: https://www.sciencedirect.com/topics/food-science/meat-composition
[2] D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, 8th ed. New York, NY, USA: W.H. Freeman/Macmillan Learning, 2021, ch. 18, pp. 695–750.
[3] G. K. Philip and S. J. Freeland, “Did evolution select a nonrandom ‘alphabet’ of amino acids?,” Astrobiology, vol. 11, no. 3, pp. 235–240, Apr. 2011, doi: 10.1089/ast.2010.0567.
[4] F. H. C. Crick, “The origin of the genetic code,” J. Mol. Biol., vol. 38, no. 3, pp. 367–379, Dec. 1968, doi: 10.1016/0022-2836(68)90392-6.
[5] S. J. Freeland and L. D. Hurst, “The Genetic Code Is One in a Million,” J. Mol. Evol., vol. 47, no. 3, pp. 238–248, Sep. 1998, doi: 10.1007/PL00006381.
[6] “Understanding Non-natural Amino Acids: A Guide for Researchers.” Accessed: Mar. 03, 2026. [Online]. Available: https://www.nbinno.com/article/pharmaceutical-intermediates/understanding-non-natural-amino-acids-a-guide-for-researchers