Week 5: Protein Design - part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1. Then decide which ones are worth advancing toward therapy.
A. Part 1: Generate Binders with PepMLM
SOD1 SEQUENCE
SOD1 SEQUENCE with A4V mutation
After processing 4 peptides with 12 amino acids in the mutational sequence, we got:
The pseudo perplexity range explains that the lower the range, the higher the confidence of the model. This means that the peptide with 15.42 will be less natural, while the peptide with 10.32 is a more natural and similar peptide to the sequence. Adding the SOD-1 binding sequence marks a difference arrises, have a pseudo perplexisty of 20.63, a very high number, which means that
B. Part 2: Evaluate Binders with AlphaFold3
I took the peptides generated in PepMLM and bound them to Alphafolds using the mutant SOD1 sequence. The results show that the protein sequence is highly confident in the result it generated, indicating that the model has high confidence in the predicted structure. While the iPTM shows numbers under 0.6, which means there is low confidence in the interaction between the peptide and protein. Also, the parts in which the peptide actually binds a little bit to the protein correspond to the beginning of the sequence, which appears to be a more flexible region of the protein.
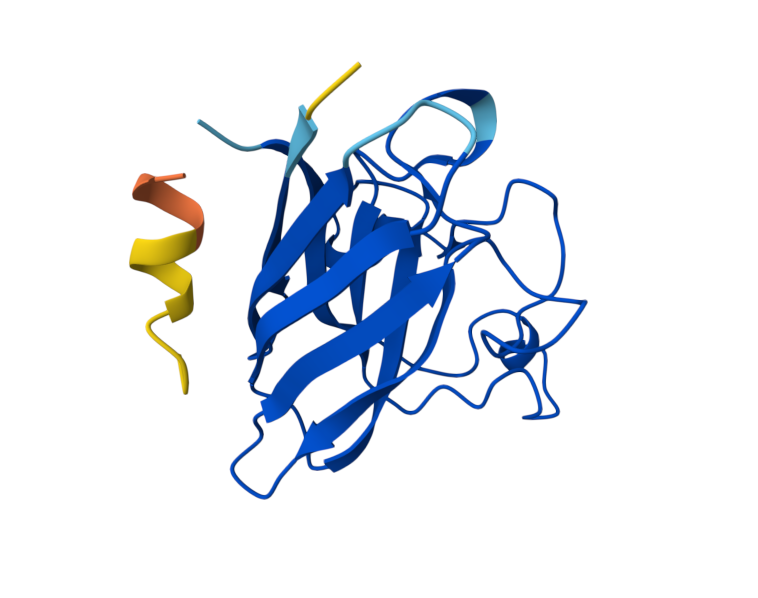
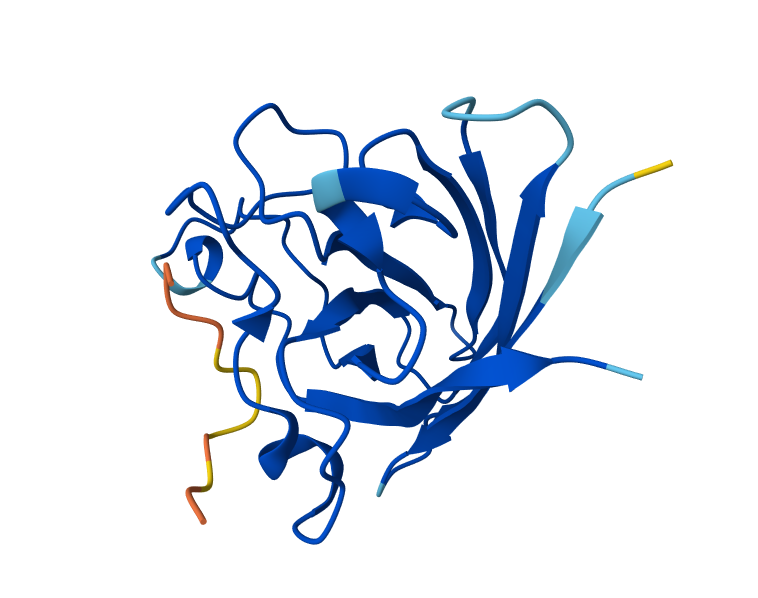
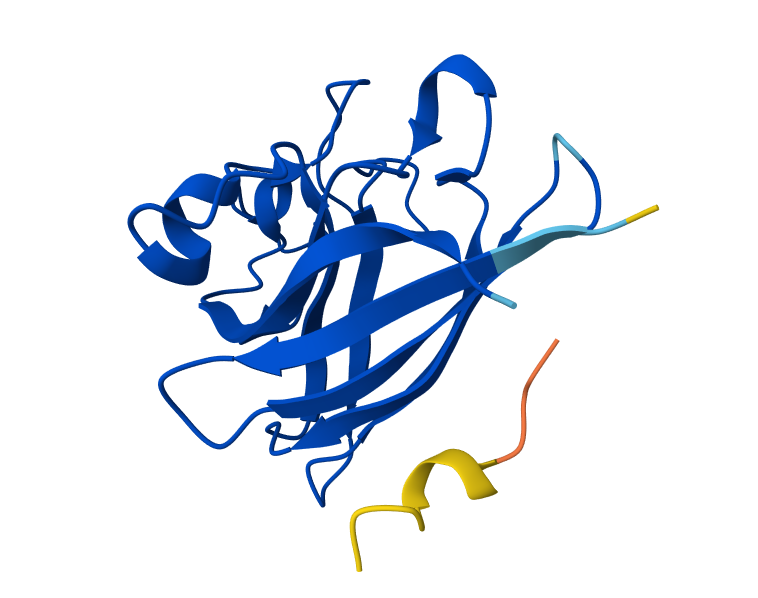
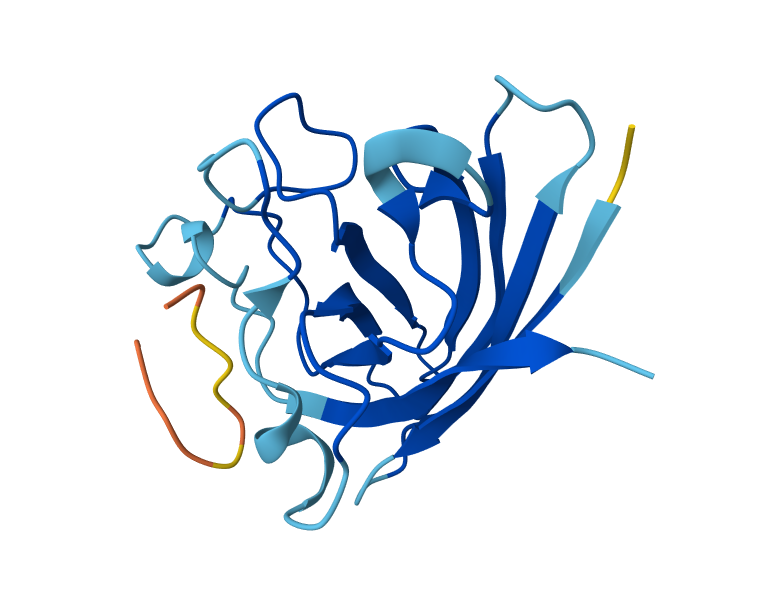
C. Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
It seems that the best candidate as a therapeutic peptide is Peptide 3: KRYPAVALAWWE. Although other candidates show very good qualities and achieve similar results in terms of solubility, hemolysis, molecular weight, and net charge, they do not present a strong binding score. In this case, Peptide 3 shows the highest predicted binding affinity among the candidates.
If we compare these results with the iPTM values predicted by Alphafold, we can observe that the confidence of interaction between peptides and the protein is generally low. For Peptide 3 in particular, the iPTM value lies in the middle of the observed range, suggesting (inside of the low values) moderate structural confidence in the predicted interaction.
Additionally, when designing peptides for therapeutic purposes, several properties must be considered. First, peptides need to be soluble so that they can circulate in the biological fluids without forming aggregates. Second, hemolysis probabilities should remain below 0.2, since higher values indicate that peptides may disrupt red blood cells and release hemoglobin into the bloodstream, which can be toxic. Third, binding affinity is important because it helps to predict whether a peptide will interact strongly with the target protein. Furthermore, molecular weight is preferably small, as smaller peptides are easier to synthesize and diffuse through biological environments. Finally, a moderate positive net charge is often favorable, because it can promote electrostatic interactions with negatively charged regions on protein surfaces, potentially stabilizing the peptide-protein interaction.
D. Part 4: Generate Optimized Peptides with moPPIt
I chose to run the peptide at the nearest residues of the mutation because the flexibility around these spaces is beneficial to peptide-protein binding.
To consider the values of analysis: 💧 Solubility: 1.0 (good) 🩸 Hemolysis : 1.0 (good) 🔗 Binding Affinity: the higher the better 🧩 Motif: 1.0 (good)
Therefore, Peptide 3: YYQKTCLVKKEH reflects that it is the best candidate for binding to the mutant SOD1. It presents balanced and consistent results in every aspect: hemolysis, solubility, affinity, and motif. Although the solubility is slightly lower compared to Peptide 0, it still falls in the favorable range, suggesting that the peptide can remain stable and soluble in physiological conditions. Also, it presents a high affinity and motif, meaning that it can perform a strong and specific interaction with the selected residues of the protein.
Compared to PepMLM peptides, the Moppit results show a good affinity and motif, which did not appear in the PepMLM peptides. I think Moppit has a higher affinity and better chances to bind with the protein because it has developed results with a specific target of residues in a specific region, while PepML gives a general result based on stable and more plausible sequences without focusing on any particular binding site.
- How would you evaluate these peptides before advancing them to clinical studies?
I would first run a few more computational tests to have consistent results in stability and strength of the peptide-protein bond. This would be run by docking and molecular dynamics simulations. Afterward, it will be necessary to do some in vitro experiments to test if the solubility, hemolysis, binding, affinity, motif, and results keep being consistent and similar to the computational simulations. Finally, in vivo models would be run to assess safety, stability, and pharmacokinetic properties to see if the peptide meets the requirements for clinical studies.
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.