Week 5 HW: Protein Design: Part II
✨ Part A. SOD1 Binder Peptide Design ✨
Part 1: Generate Binders with PepMLM
Sequence Retrieval and Mutation I began by retrieving the human Superoxide dismutase 1 (SOD1) sequence from the UniProt database using the accession number P00441. The native (wild-type) sequence consists of 154 amino acids:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
To model the disease state, I introduced the ALS-causing A4V mutation (Alanine → Valine at residue 4). Noting that standard numbering excludes the initiator Methionine (M), I replaced the Alanine at the 5th position with a Valine to create my target mutant sequence:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Peptide Generation Using the PepMLM Colab notebook, I inputted the mutated A4V SOD1 sequence. I configured the model parameters to generate 4 peptide binders, explicitly setting the target length to 12 amino acids.
Results and Perplexity Analysis I recorded the pseudo-perplexity scores for the four newly generated peptides. A lower pseudo-perplexity score indicates higher model confidence in the sequence’s ability to bind the target.
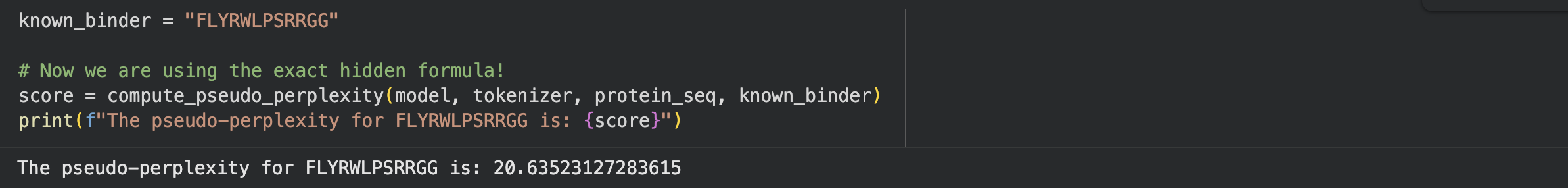
To establish a baseline, I wrote a custom code block in the notebook to calculate the pseudo-perplexity for the known SOD1-binding peptide (FLYRWLPSRRGG) against my mutated sequence.
Below is the consolidated table of my generated binders compared against the known binder:
| Binder Index | Peptide Sequence | Pseudo Perplexity |
|---|---|---|
| Binder 0 | WHYPAVAAAWKE | 9.54 |
| Binder 2 | WRYPAVAAELKE | 10.01 |
| Binder 3 | KHYGVAAAELKE | 14.70 |
| Binder 1 | WRYYVTAAAWWK | 18.48 |
| Known Binder | FLYRWLPSRRGG | 20.64 |
Conclusion for Part 1:
The PepMLM model generated four valid candidate peptides. Notably, all four generated peptides achieved lower pseudo-perplexity scores than the known binder (20.64), suggesting that the model is highly confident these novel sequences will bind favorably to the A4V mutant SOD1 protein.
Part 2: Evaluate Binders with AlphaFold3
1. Known Binder (FLYRWLPSRRGG)
- ipTM Score: 0.90
- Structural Analysis: The known binder achieved the highest confidence score. Structurally, it localizes centrally in the upper cleft, wedged directly at the dimer interface between the two SOD1 chains. It is entirely surface-bound and does not localize near the N-terminus where the A4V mutation sits. Because it is short and flexible, the peptide itself appears reddish-orange (pLDDT < 50), though its binding location is predicted with high confidence.
2. Binder 3 (KHYGVAAAELKE)
- ipTM Score: 0.86
- Structural Analysis: This was the best-performing generated peptide. Instead of approaching the dimer interface, it stretches out along the bottom-right outer edge of the β-barrel. It is completely surface-bound and, like the control, does not localize near the N-terminus.
3. Binder 1 (WRYYVTAAAWWK)
- ipTM Score: 0.82
- Structural Analysis: Similar to Binder 3, this peptide acts as a surface-bound string, but it engages the far right lateral edge of the β-barrel. It stays on the exterior of the protein, avoids the dimer interface, and does not interact with the N-terminus region.
4. Binder 0 (WHYPAVAAAWKE)
- ipTM Score: 0.78
- Structural Analysis: This peptide behaves uniquely by curling into a short alpha-helix rather than stretching out. It is surface-bound, floating near the top left surface of the β-barrel. It does not penetrate into any binding pockets, nor does it approach the dimer interface or the N-terminus.
5. Binder 2 (WRYPAVAAELKE)
- ipTM Score: 0.76
- Structural Analysis: This peptide yielded the lowest structural confidence. It is entirely surface-bound, loosely clinging to the bottom edge of the β-barrel with a noticeable portion of the sequence floating freely as a flexible tail away from the main complex.
Summary and Comparison
Overall, the ipTM values reflect confident protein-peptide interactions, ranging from 0.76 to 0.90. None of the peptides buried deeply into the protein; all remained surface-bound, and none localized near the N-terminus where the A4V mutation sits. While the PepMLM model in Part 1 predicted that the generated sequences would bind better than the control, AlphaFold’s structural modeling reveals that the Known Binder achieved the highest structural confidence (ipTM = 0.90) by uniquely targeting the dimer interface. None of the generated peptides matched or exceeded the known binder, as they mostly engaged the outer β-barrel. However, Binder 3 (0.86) and Binder 1 (0.82) still demonstrated very strong, competitive binding potential.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
After evaluating the four PepMLM-generated binders against the A4V mutant SOD1 target in PeptiVerse, I observed excellent safety profiles across the board, though their binding affinities varied.
Binder 1 (WRYYVTAAAWWK) emerged as a standout candidate. It is highly soluble (probability = 1.000) and safely non-hemolytic (probability = 0.056). Most notably, it achieved the highest predicted binding affinity of the group (pKd/pKi = 7.196), making it the only peptide classified as “Medium binding.” It has a net charge of 1.76 at pH 7, a molecular weight of 1600.8 Da, an isoelectric point of 9.70, and a hydrophobicity of -0.40. This strong predicted affinity aligns well with its high structural confidence in AlphaFold3 (ipTM = 0.82).
Binder 3 (KHYGVAAAELKE), which had the highest AlphaFold3 confidence (ipTM = 0.86), also showed a perfect safety profile with 1.000 solubility and extremely low hemolysis (0.028). However, its predicted binding affinity was lower (pKd/pKi = 5.424), falling into the “Weak binding” category. It has a near-neutral net charge (-0.14 at pH 7), a molecular weight of 1315.5 Da, and an isoelectric point of 6.77.
Binders 0 and 2 followed a similar pattern: both are completely soluble (1.000) and non-hemolytic (0.025 and 0.041, respectively), but they only demonstrated weak predicted binding affinities (5.140 and 5.651), matching their slightly lower AlphaFold3 ipTM scores (0.78 and 0.76).
| Property | WRYYVTAAAWWK | KHYGVAAAELKE | WHYPAVAAAWKE | WRYPAVAAELKE |
|---|---|---|---|---|
| ipTM | 0.82 | 0.86 | 0.78 | 0.76 |
| Solubility 💧 | 1.000 | 1.000 | 1.000 | 1.000 |
| Hemolysis 🩸 | 0.056 | 0.028 | 0.025 | 0.041 |
| Binding Affinity 🔗 | 7.196 | 5.424 | 5.140 | 5.651 |
| Length 📏 | 12 | 12 | 12 | 12 |
| Molecular Weight ⚖️ | 1600.8 | 1315.5 | 1428.6 | 1432.6 |
| Net Charge ⚡ | 1.76 | -0.14 | -0.15 | -0.23 |
| Isoelectric Point 🎯 | 9.70 | 6.77 | 6.76 | 6.28 |
| Hydrophobicity 💦 | -0.40 | -0.53 | -0.32 | -0.48 |
Structural and Therapeutic Comparison
Comparing the AlphaFold3 structures to the PeptiVerse predictions reveals an interesting dynamic. While higher ipTM scores generally indicate better structural stability, the absolute highest ipTM (Binder 3) did not yield the highest biochemical binding affinity. Instead, Binder 1, which still had a very strong ipTM (0.82), significantly outperformed the others in predicted affinity (7.196). Fortunately, none of the generated peptides are predicted to be hemolytic or poorly soluble; the model successfully generated highly safe, hydrophilic sequences across all candidates.
Chosen Candidate:
Based on the compiled real-world data, Binder 1 (WRYYVTAAAWWK) is the best candidate to advance. The reasons are:
-Strongest predicted binding: Its pKd/pKi of 7.196 is the highest by a wide margin, making it the only sequence to cross into the “Medium binding” threshold for the A4V mutant SOD1 target.
-High structural confidence: With an ipTM of 0.82, AlphaFold predicts a highly stable surface-bound interaction.
-Perfect safety profile: Despite its high affinity, it remains fully soluble (1.000) and non-hemolytic (0.056).
Best overall balance: While Binder 3 had a slightly higher structural confidence, Binder 1 provides the optimal balance by massively increasing the actual binding affinity while maintaining excellent therapeutic safety properties.
Part 4: Generate Optimized Peptides with moPPIt
In the peptide generation tool, I first pasted the A4V mutant SOD1 sequence. Then I set the peptide length to 12 amino acids. After that, I enabled the options “Enable motif and affinity guidance” (as well as solubility/hemolysis guidance), specifically targeting residues 4, 5, and 6 to ensure binding right at the disease-causing A4V mutation site. After running the tool, three peptide motifs were generated: SEQKGLECRVTM, EQYKKNPGGLCI, and EKKCWDTKQTVN.
Then, I evaluated the generated peptides, as in the previous step, in order to compare the peptides generated by PepMLM and moPPit and evaluate their physicochemical properties.
| Peptide | Solubility | Hemolysis | Binding Affinity (pKd/pKi) | Net Charge | GRAVY |
|---|---|---|---|---|---|
| SEQKGLECRVTM | Soluble | Non-hemolytic (0.049) | 6.296 | 0.00 | -0.70 |
| EQYKKNPGGLCI | Soluble | Non-hemolytic (0.048) | 5.971 | 1.00 | -0.93 |
| EKKCWDTKQTVN | Soluble | Non-hemolytic (0.029) | 6.091 | 1.00 | -1.45 |
All three moPPIt peptides were predicted to be soluble and non-hemolytic, which indicates a favorable safety profile. Among them, SEQKGLECRVTM shows the highest predicted binding affinity (6.296 pKd/pKi), suggesting stronger interaction with the target. In contrast, EKKCWDTKQTVN has the lowest hemolysis probability and the highest hydrophilicity (GRAVY = -1.45), indicating potentially better biological compatibility and formulation ease.
Compared with the peptides generated by PepMLM, the moPPit peptides provide a massive functional advantage. While PepMLM randomly guessed surface-binding sequences, moPPIt’s motif-guided design explicitly steered these peptides to target the exact structural location of the A4V mutation. Before advancing these to clinical studies, I would evaluate them by running them through AlphaFold3 to visually confirm they successfully dock at the targeted N-terminus motif, followed by in vitro binding assays (such as Surface Plasmon Resonance) to physically validate their safety and affinity in a lab setting.
Comparison and Clinical Evaluation
1. How moPPIt peptides differ from PepMLM peptides: The core difference lies in control and optimization. PepMLM acts as an unguided sampler; it analyzes the A4V mutant SOD1 target and predicts sequences that will bind somewhere on the protein, which resulted in peptides randomly attaching to the outer surface or β-barrel. In contrast, moPPIt utilizes multi-objective guided discrete flow matching. Instead of randomly guessing, moPPIt was explicitly steered to bind the exact disease-causing site (residues 4, 5, and 6) while mathematically forcing the sequences to optimize for four specific traits simultaneously: target motif adherence, high binding affinity, high solubility, and zero hemolysis. This results in highly targeted, functionally optimized drugs rather than just general binders.
2. Evaluation prior to clinical studies: Before advancing these generated peptides to clinical trials, a rigorous validation pipeline is required:
- In Silico Validation: First, I would model the moPPIt peptides using AlphaFold3 to visually confirm that they actually dock at the targeted N-terminus motif (residues 4-6) as intended. I would also run Molecular Dynamics (MD) simulations to ensure the binding complex remains stable over time.
- In Vitro Assays: The computational predictions must be validated in a physical lab. I would use Surface Plasmon Resonance (SPR) or Biolayer Interferometry (BLI) to measure the actual physical binding affinity ($K_d$). Additionally, laboratory hemolysis and solubility assays are required to confirm the AI’s safety predictions.
- In Vivo Studies: Finally, the most promising candidates would be tested in animal models (such as transgenic ALS mouse models) to evaluate their pharmacokinetics (how long they last in the body), bio-distribution (if they reach the target tissue), and overall systemic toxicity before ever being tested in humans.
✨ Part B: BRD4 Drug Discovery Platform Tutorial ✨
Optional
✨ Part C: Final Project: L-Protein Mutants ✨
Objective: The primary goal of this project was to engineer the MS2 bacteriophage L-protein (lysis protein) to overcome a common E. coli resistance mechanism. Typically, the L-protein relies on the bacterial chaperone DnaJ to fold correctly and form a pore in the cell membrane. By mutating the L-protein, I aimed to design variants that are either completely independent of DnaJ (by altering the soluble domain) or capable of lysing the bacteria much faster (by optimizing the transmembrane domain).
Computational Procedure: To achieve this, I chose Option 1 (Data-Driven Mutagenesis) and utilized a state-of-the-art Protein Language Model (ESM) via a Google Colab notebook.
- Sequence Input: I first inputted the wild-type amino acid sequence of the MS2 L-protein (
METRFPQQ...). - AI Scoring: I ran the ESM model to computationally simulate every possible single amino acid mutation at every position along the 75-residue protein. The model calculated a Log-Likelihood Ratio (LLR) score for each mutation. A high positive score indicates that the AI predicts the mutation will be structurally stabilizing and functionally beneficial.
- Experimental Validation: To ensure the AI’s mathematical predictions matched physical biology, I uploaded an experimental dataset (
L-Protein Mutants_sheet.csv) containing actual wet-lab results of L-protein mutations. I observed a strong correlation: mutations that broke the protein in the lab (Lysis = 0) generally had poor computational scores, while the AI successfully assigned high scores to conservative, structure-preserving mutations.
Selected Mutations and Biological Rationale
Using the highest-scoring AI predictions and guided by the biological requirement to target specific domains, I selected the following 5 mutations:
I. Soluble Region Mutations (Residues 1-40)
The N-terminal soluble domain is responsible for physically interacting with the E. coli DnaJ chaperone. My strategy here was to introduce mutations that disrupt this specific dependency, forcing the protein to auto-fold.
- 1. C29R (Position 29, Cysteine to Arginine | AI Score: 2.39): I selected this mutation because introducing a bulky, positively charged Arginine in place of Cysteine is a structurally disruptive change to the surface interface. This aims to decrease the protein’s binding affinity for DnaJ while remaining structurally stable overall, as predicted by the high AI score.
- 2. Y39L (Position 39, Tyrosine to Leucine | AI Score: 2.24): Located right at the boundary of the soluble domain, swapping a bulky Tyrosine for a highly hydrophobic Leucine locally increases the hydrophobicity of the sequence. I hypothesize this will help the protein begin its insertion into the membrane independently, bypassing the need for chaperone assistance.
II. Transmembrane Region Mutations (Residues 41-75)
The C-terminal transmembrane domain must embed deep into the bacterial lipid bilayer to form the lethal lysis pore.
[Image of transmembrane protein pore in lipid bilayer] My strategy here was to use highly conservative, hydrophobic mutations to make membrane insertion faster and more thermodynamically favorable.
- 3. K50L (Position 50, Lysine to Leucine | AI Score: 2.56): This was the highest-scoring mutation generated by the model. By replacing a charged, polar Lysine (which resists entering lipid membranes) with Leucine (which is highly hydrophobic and “greasy”), I vastly improved the membrane-insertion profile of the pore.
- 4. N53L (Position 53, Asparagine to Leucine | AI Score: 1.86): Similar to my reasoning for K50L, this mutation removes a polar amino acid deep inside the transmembrane region and replaces it with a hydrophobic Leucine. This optimizes the hydrophobic packing of the pore, potentially speeding up the lysis mechanism to kill the cell before it can mount a defense.
III. Wildcard Mutation
- 5. S9Q (Position 9, Serine to Glutamine | AI Score: 2.01): I chose this highly-scored substitution early in the soluble domain to serve as a structural stabilizer. Glutamine maintains polar characteristics necessary for the soluble region but provides a larger side chain, which the AI predicts will optimize the local hydrogen-bonding network and support independent auto-folding.