Week 1 HW: Principles and Practices

1. First, describe a biological engineering application or tool you want to develop and why.
Daptomycin is the one of the novel lipopeptide antibiotics, that has high clinical relevance, owning to is effectiveness against multidrug resistant (MDR) bacteria, such as vancomycin resistant enterococci (VRE) and methicillin-resistant Staphylococcus aureus (MRSA), establishing its rule as a last resort antibiotic, one of the drawbacks of daptomycin is its expensive price tag, which puts a load on the patient treated and the health system due to the laborious work required with it, I want to decrease the cost for daptomycin by removing some of the genes required for its manufacturing, and using protein engineering to compensate for the removed genes by increasing affinity of one substrate over the other, and finally, design a de-novo enzyme for the lipid tail addition process to daptomycin.
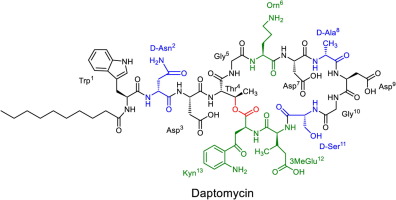
To further clarify my idea, let’s explore together, the structure of daptomycin and how does bacteria naturally produce it, daptomycin is a 13-peptide long molecule, with some amino acids being non proteinogenic (not one of the common 20 amino acids used by the ribosome to create proteins), with a lipid tail attached to the first amino acid, tryptophan, this lipid tail is naturally 13 carbons long, which a problem we will touch upon later on, you might currently be wondering but how does bacteria incorporate non-proteinogenic amino acids using normal protein synthesis pathways? Well, the answer is: by not using normal protein synthesis pathway. https://www.sciencedirect.com/science/article/pii/S0968089616303856?via%3Dihub
Instead of using ribosomes, bacteria use, in this case, a bunch of proteins called non-ribosomal peptide symthetases (NRPSs), which are huge proteins that can use not the only the 20 amino acids, it also has the ability to incorporate 500 different substrates!
They work in modules where each one is responsible for exactly one substrate in the synthesis, each module consists of domains, with the three basic ones being: adenyltation (A), thiolation (T) & condensation (C), the A domain is responsible for the recognition and activation step of the molecule, the thiolation is responsible for anchoring the substrate to the module and passing it between different domains, and the condensation domain is responsible for moving the peptide from one module to the next. (https://doi.org/10.1002/anie.201609079)
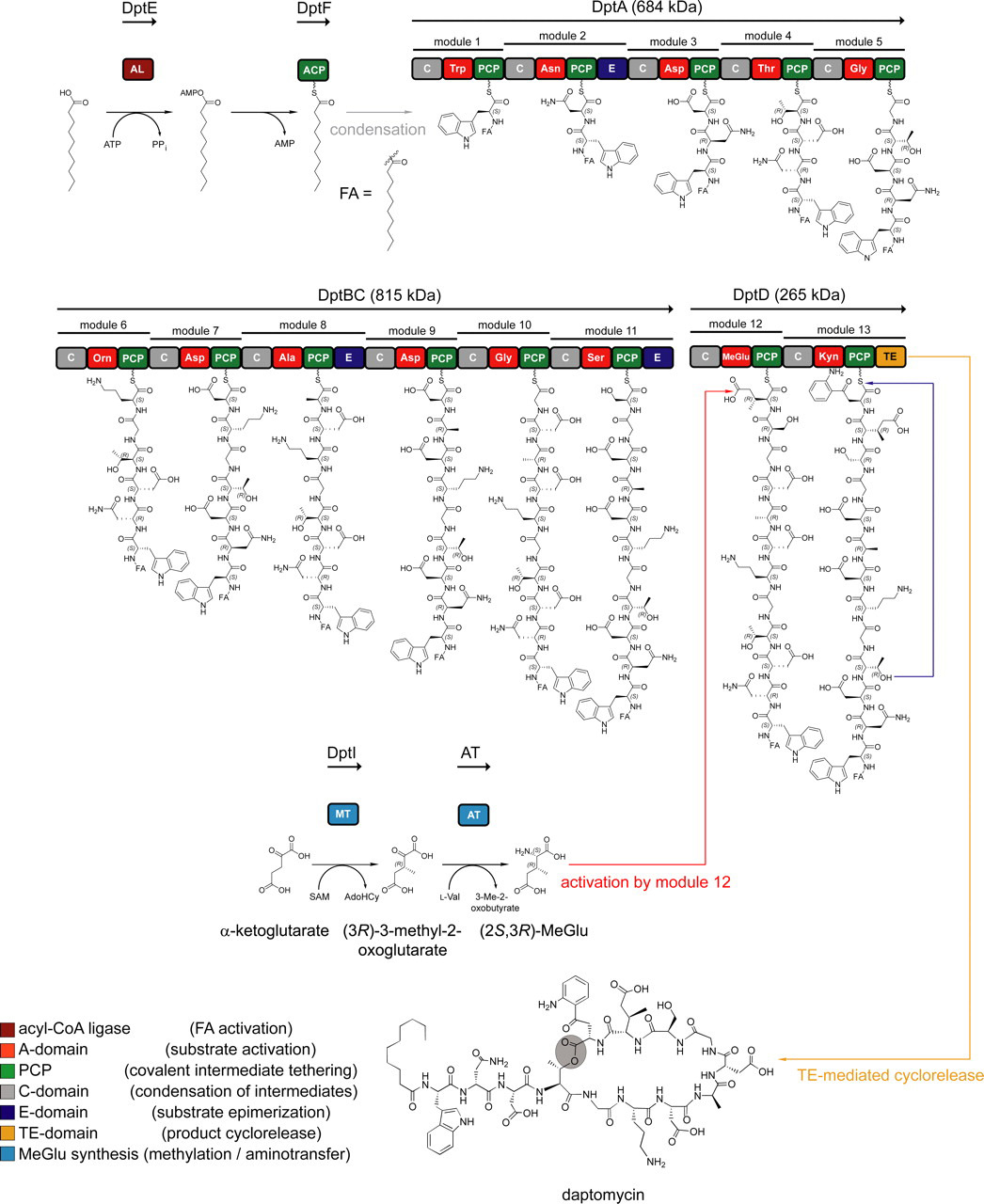
here is an illustration of daptomycin’s gene cluster https://www.jbc.org/article/S0021-9258(19)89079-8/fulltext
The first two modules in daptomycin synthesis (DptE & F) add a lipid tail to tryptophan, which is a must for its anti-bacterial activity, as mentioned above these modules are quite problematic, why is that the case?
Because the lipid tail that gives daptomycin its activity with a tolerable toxicity is C10, this module however can incorporate C11, 12 & 13 because they’re naturally found in the cell of S. roseporous, the bacteria that synthesises daptomycin, the molecules that have a tail rather the C10 are basically discarded as waster after purification using chromatography, in addition to that, decanoic acid (C10) is toxic to the bacterium, which requires very careful addition of this chemical, with exact amounts & rate, slowing down the industrial rate of daptomycin production, additionally, because of daptomycin antibacterial activity, the bacteria has to regularly get the drug outside, through the use of special pumps coded in the ABC transporter pump genes.
Back to the proposed project, how does it address these problems?
I want to remove the DptE & F modules entirely, and engineer the A site of DptA to recognize tryptophan directly, to direct the bacteria to create only the nucleus for daptomycin, which has little to no antibacterial activity, which would allow for the deletion of ABC pump genes too, I would then to create an enzyme responsible for the acylation of nucleus, but this enzyme would have higher affinity for decanoic acid, which would decrease the waste, effectively increasing the amount of drug produced per batch.
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm).
Break big goals down into two or more specific sub-goals.
Goal A: ensure safe biological & ecological handling:
- A I: make sure all the personnel working with these bacteria are qualified and certified to deal with biologics
- A II: ensure the bacterial waste is discarder in an ecologically safe way that doesn’t harm nature
Goal B: promote access to more affordable antibiotics:
- B I: Ensure cost savings translate to lower patient prices, especially in low- and middle-income countries (LMICs)
- B II: Prevent monopolistic control of the improved production process
- B III: Establish responsible sharing practices for engineered strains and protocols
“Claude and ChatGPT were used for help in the following sections and the answers were thoroughly reviewed by me”
3. Question 3: Three Governance Actions
GOVERNANCE ACTION 1: Biosafety Certification for Engineered Production Strains
- Purpose: Currently, antibiotic production facilities follow Good Manufacturing Practices (GMP), but there’s no specific certification for engineered bacteria. Standards vary widely between countries. I propose creating a tiered certification system based on modification complexity - basic modifications get basic oversight.
- Design:
- Actors: National regulators (FDA, EMA), facility operators, independent auditors
- Requirements: Annual inspections, genetic sequencing and tracking of production strains, validated waste treatment, worker training certification, emergency response plans
- Implementation: 3-year phased rollout, starting with new facilities then existing ones
- Assumptions:
- Certification costs won’t make production economically unviable
- Facilities won’t relocate to avoid compliance
- BSL-1 is adequate with proper controls
- Risks:
- Failure: Under-resourced agencies can’t enforce standards, creating safety theater
- Success: Over-regulation drives production offshore to countries with weaker oversight, increasing costs and defeating the affordability goal
GOVERNANCE ACTION 2: Technology Transfer Partnership for LMIC Manufacturing
- Purpose: Antibiotic manufacturing is concentrated in China and India. Technology transfer is ad-hoc. I propose an international consortium (modeled on WHO’s 2021 mRNA vaccine hubs) providing financial and technical support for daptomycin production in LMICs with high MDR burden.
- Design:
- Actors: WHO, national governments, pharmaceutical companies, universities, philanthropic organizations (Gates Foundation, Wellcome Trust)
- Requirements: Facility construction grants, 2-3% royalty caps for LMIC licensing, 6-month technical training fellowships, quality assurance support, price commitments (<$50/treatment)
- Implementation: 3-5 country pilot over 5 years, $100M funding
- Assumptions:
- Sufficient local infrastructure and workforce
- Local demand justifies production scale
- Quality maintainable across distributed manufacturing
- Political stability in partner regions
- Pharma participation despite lower margins
- Risks:
- Failure: Facilities built but can’t sustain operations due to funding gaps or quality issues
- Success: Displaces existing pharmaceutical jobs, creates aid dependency, prices don’t drop if distribution remains inefficient
GOVERNANCE ACTION 3: Standardized Kill-Switches and Biocontainment
- Purpose: Engineered production strains rely only on physical containment. I propose developing and requiring standardized genetic kill-switches and auxotrophy markers for NRPS-engineered antibiotic producers.
- Note: Kill-switch technology exists (deadman circuits, CRISPR systems, synthetic auxotrophy), but a 2022 study found no actual biocontainment standards - this proposes standardizing existing research.
- Design:
- Actors: Academic researchers, biotech companies, standards organizations (BioBricks Foundation), journal editors
- Requirements: Multiple orthogonal kill-switches (temperature-sensitive, chemical dependency), auxotrophy for rare metabolites, open-source plasmid toolkit, journal publication requirements, ISO industry standards
- Implementation: 2-year research phase, then adoption through publication requirements and industry guidelines
- Assumptions:
- Kill-switches remain stable over many generations
- Auxotrophic dependencies can’t be environmentally rescued
- Academic and industry adoption
- Journal enforcement authority
- Minimal impact on production efficiency
- Risks:
- Failure: Kill-switches prove unstable, adding complexity without real safety benefit, slowing research
- Success: False security leads to lax physical containment, doesn’t work for all applications, sophisticated actors could bypass
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
| Does the option: | Option 1 | Option 2 | Option 3 |
|---|---|---|---|
| Enhance Biosecurity | |||
| • By preventing incidents | 2 | 3 | 1 |
| • By helping respond | 1 | 2 | N/A |
| Foster Lab Safety | |||
| • By preventing incident | 1 | 2 | 1 |
| • By helping respond | 1 | 3 | N/A |
| Protect the environment | |||
| • By preventing incident | 2 | 2 | 1 |
| • By helping respond | 1 | 3 | N/A |
| Other considerations | |||
| • Minimizing costs and burdens | 2 | 2 | 1 |
| • Feasibility? | 1 | 3 | 2 |
| • Not impede research | 2 | 1 | 2 |
| • Promote constructive applications | 3 | 1 | 2 |
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why.
Primary Priority: Option 3 (Kill-Switch Standards) - Immediate Implementation I would prioritize developing and standardizing technical biocontainment measures first because:
- Prevention over response: Technical safeguards provide the most robust protection against environmental release and potential dual-use (scored 1s across prevention categories)
- Scalable: Once developed, these tools can be applied globally regardless of regulatory capacity
- Enables other goals: Biocontained strains make both regulation (Option 1) and technology transfer (Option 2) safer and more politically feasible
Secondary Priority: Option 1 (Biosafety Certification) - 3-year rollout After biocontainment tools are available, implement certification requirements because:
- Complements technical safeguards: Adds institutional oversight layer
- High feasibility: Regulatory pathways already exist
- Creates accountability: Ensures standards are maintained over time
Selective Implementation: Option 2 (Tech Transfer) - Targeted pilot Rather than broad implementation, I’d recommend:
- Start with 2-3 pilot countries with strong existing pharmaceutical infrastructure
- Link funding to adoption of Options 1 & 3 (biocontainment + certification)
- Focus on price transparency mechanisms rather than direct manufacturing subsidies
- mechanism and has uncertain feasibility
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate of DNA polymerase I is $1:105$, which translates to a wrong base added every hundred thousand bases, the human genome is made of approximately 6.4 Gbp, which would lead to abou 120,000 errors in every cell replication cycle (https://www.nature.com/scitable/topicpage/dna-replication-and-causes-of-mutation-409/) Biology deals with this by several means, first of all, DNA polymerases actually have proofreading capability, that allows them to remove bases that were added by mistake, secondly, biology provides several means DNA repairing mechanisms, such as the mutS repair system & mismatch repairing enzymes, which decreases the mutation to an astonishing $10{-8}$ mutations/bp/generation! (https://book.bionumbers.org/what-is-the-mutation-rate-during-genome-replication/)
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
On average, a human protein is 476 amino acids, (https://bionumbers.hms.harvard.edu/bionumber.aspx?id=101652) and there is 64 different codon combinations, three of which are stop codons, which leaves us with 61 codes and 20 amino acids, that means on average an amino acid has approximately 3 different combinations; therefore, there is $3^{476}$ (approximately $1.29 \times 10^{277}$)different ways to code for the same protein. I believe one of the reasons this doesn’t work in practice, is because genomes tend to have codon bias, where one has a faster translation rate than the other, causing it to be more common, or for the other to stall the translation; in addition, since biology is just a bunch of random shapes bumping into each other (shoutout to anyone who watches phy the neutrophil) perhaps some of these shapes would stall the translation process
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently? The most common way for oligo synthesis is solid-phase synthesis (https://www.mt.com/sg/en/home/applications/L1_AutoChem_Applications/L2_ReactionAnalysis/oligonucleotide-synthesis.html & https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/oligonucleotide-synthesis)
Why is it difficult to make oligos longer than 200nt via direct synthesis? during acid deprotection, depurination side reactions limit the quality and yields of oligonucleotides above 100 nucleotides, efforts has been made in this area and new detritylation process can help with creating 150 nucleotides with high yield.
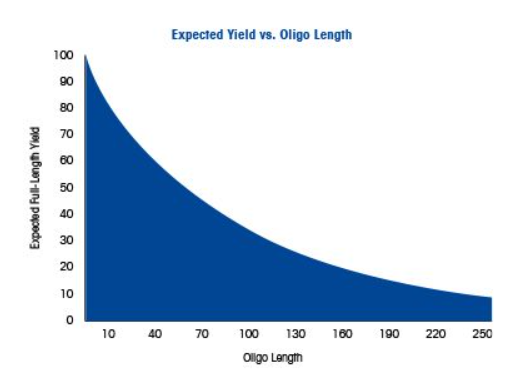
Why can’t you make a 2000bp gene via direct oligo synthesis? because the yield would be so low due to instability and error accumulation.
Homework Questions from George Church:
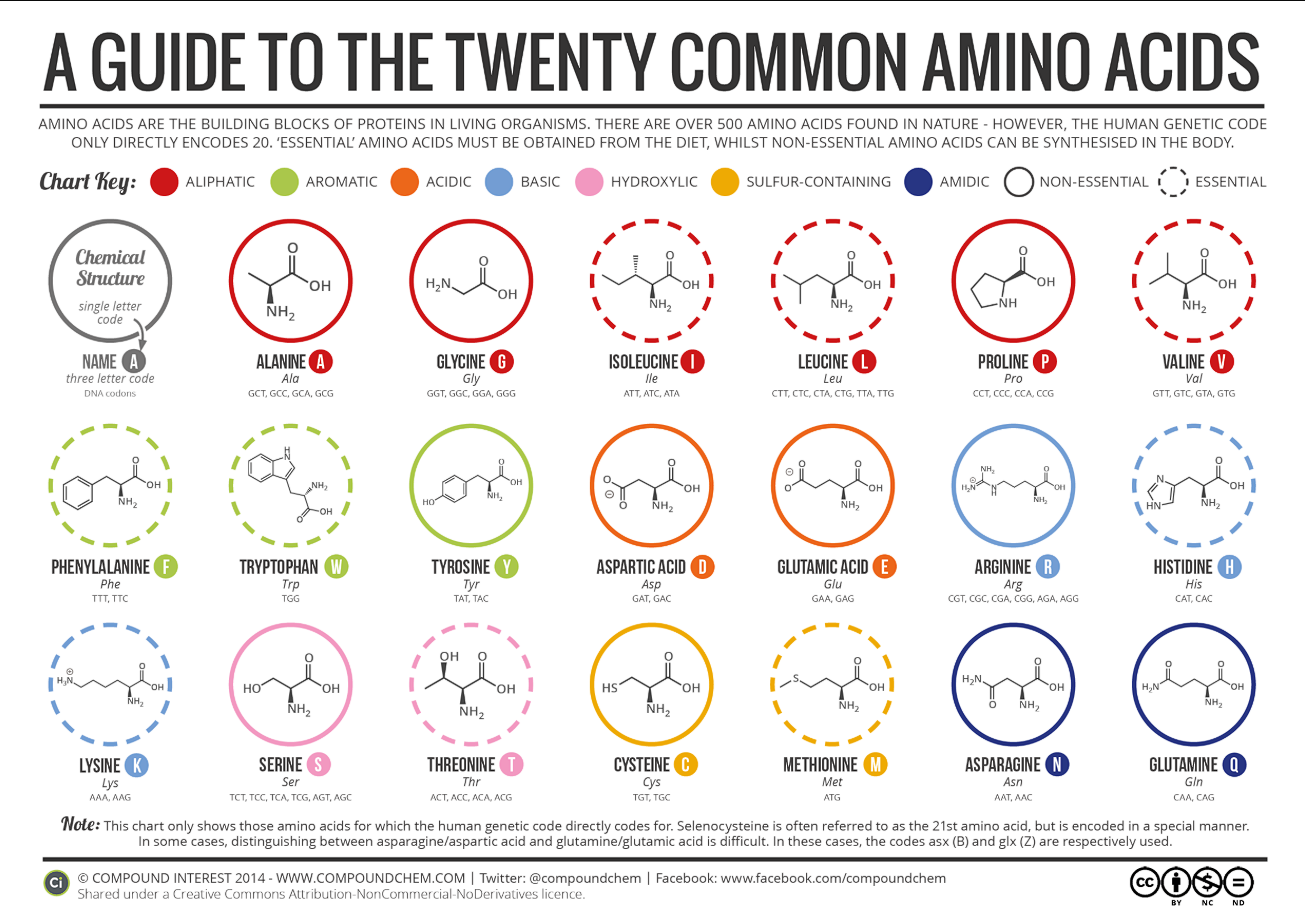
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”? The 10 essential amino acids are: histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine, and arginine. These amino acids must be obtained from nutrition, because the body can’t normally synthesize them. This renders the lysine contingency pointless, because dinosaurs, as animals, lack the ability to synthesis lysine anyways.