Week 4 HW: Protein Design Part 1
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Hey, so I’m a vegetarian, so I don’t take a single gram of meat if I can avoid it.
However, 500 grams of meat is approximately 3.011e+26 Daltons. If an average amino acid is 100 Daltons, we’ll end up with 3.011e+24 amino acid molecules. That’s assuming that all of the meat is protein, which it isn’t. Meat is approximately 20% protein, so 20% of 500 is 100g protein, or 3.01e+24Why do humans eat beef but do not become cows, eat fish but do not become fish?
Because we’re not modifying the human DNA by eating fish or beef. Our DNA makes us who we are. What we eat is digested as macromolecules that become generic molecular building blocksWhy are there only 20 natural amino acids?
Evolutionarily, 20 amino acids may be a near-optimal solution balancing chemical diversity, folding reliability, metabolic cost, and coding efficiency. There is enough redundancy and error minimization that similar codons encode chemically similar amino acids. These amino acids cover the functional space of protein physiochemical dimensions (hydrophobicity, etc)Can you make other non-natural amino acids? Design some new amino acids.
Yes. Non-natural amino acids (nnAAs) can be designed and incorporated into proteins. For example, a photo-switchable amino acid can have light-driven switching which can help Light-controlled enzyme activationWhere did amino acids come from before enzymes that make them, and before life started?
Amino acids likely formed through:Lightning-driven atmospheric chemistry
Strecker-type reactions in aqueous environments
Hydrothermal vent redox chemistry
UV photochemistry
Delivery from meteorites
They are a natural product of carbon, nitrogen, hydrogen, and oxygen chemistry under energetic planetary conditions.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Can you discover additional helices in proteins?
Yes we can both identify and design new helices using structural, computational, and synthetic approaches. We can discovery by mining structural databases, through computational protein design and through non-natural amino acids.Why are most molecular helices right-handed?
Most molecular helices in biology are right-handed because life is built from chiral building blocks of one dominant handedness, and that stereochemistry biases the lowest-energy helical geometry.Why do β-sheets tend to aggregate?
β-sheets tend to aggregate because their hydrogen-bonding pattern and geometry make intermolecular extension energetically favorable and difficult to cap. In other words, the structure is inherently “open-ended.”What is the driving force for β-sheet aggregation?
The driving force for β-sheet aggregation is free-energy minimization dominated by backbone hydrogen bonding and hydrophobic collapse, with cooperativity that makes extension energetically downhill once nucleated.Why do many amyloid diseases form β-sheets?
Many amyloid diseases involve β-sheets because the β-sheet architecture is the lowest-energy, self-templating structure accessible to partially unfolded polypeptides. When proteins misfold, β-structure is the most thermodynamically and kinetically available aggregation solution.Can you use amyloid β-sheets as materials?
Yes. Amyloid β-sheets are exceptionally robust, nanoscale materials. The cross-β architecture is one of the strongest naturally occurring protein material motifs. Amyloid fibrils are mechanically stiff, chemically stable, and self-assembling, which are all desirable properties for materials engineering.Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
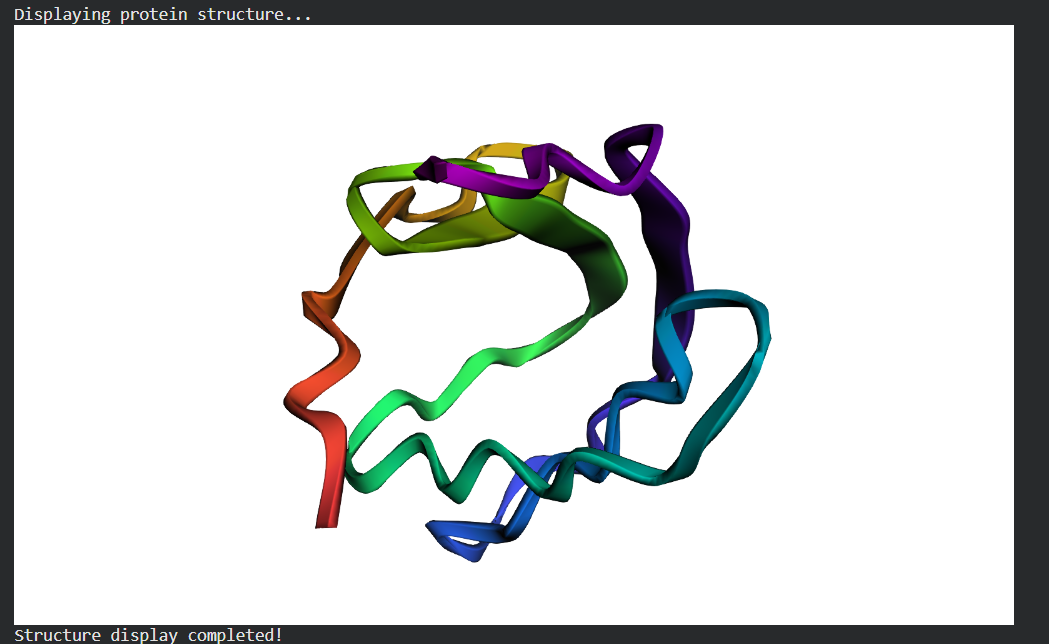
I picked histone-based chromatin https://pdb101.rcsb.org/motm/314, which is the molecule of the month on PDB and arrived at archeal histone https://www.uniprot.org/uniprotkb/A0A060HNU7/entry#sequences. I thought their biological assembly looked cool - it has nice symmetry because they act as spools for DNA. I also found it interesting that Histones were long thought to be unique to the eukaryotic lineage. But recent studies have shown that histones exist in most archaeal and some bacterial cells, shedding light on DNA packaging mechanisms while also raising new questions about the evolution of histones.Identify the amino acid sequence of your protein.
MSSGPEFGLAAMYRIMKKSGAERVSDDAADELRKVLEEVAERIAKQAAELSMHAGRKTIKPEDIRLASKNVIRLHow long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
It’s 74 amino acids long. The most common amino acid is: A, which appears 11 times.How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
The BLAST tool found 250 results in UniProtKBDoes your protein belong to any protein family?
The sequence similarity shows it belongs to the archaeal histone HMF familyIdentify the structure page of your protein in RCSB
There isn’t an exact match. The closest similarity search results in CRYSTAL STRUCTURE OF THE HISTONE HMFA FROM METHANOTHERMUS FERVIDUS https://www.rcsb.org/3d-view/1B67When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Yes, it’s very well resolved for the similar protein in RCSB
Are there any other molecules in the solved structure apart from protein?
NoDoes your protein belong to any structure classification family?
No it doesn’t.Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Cartoon: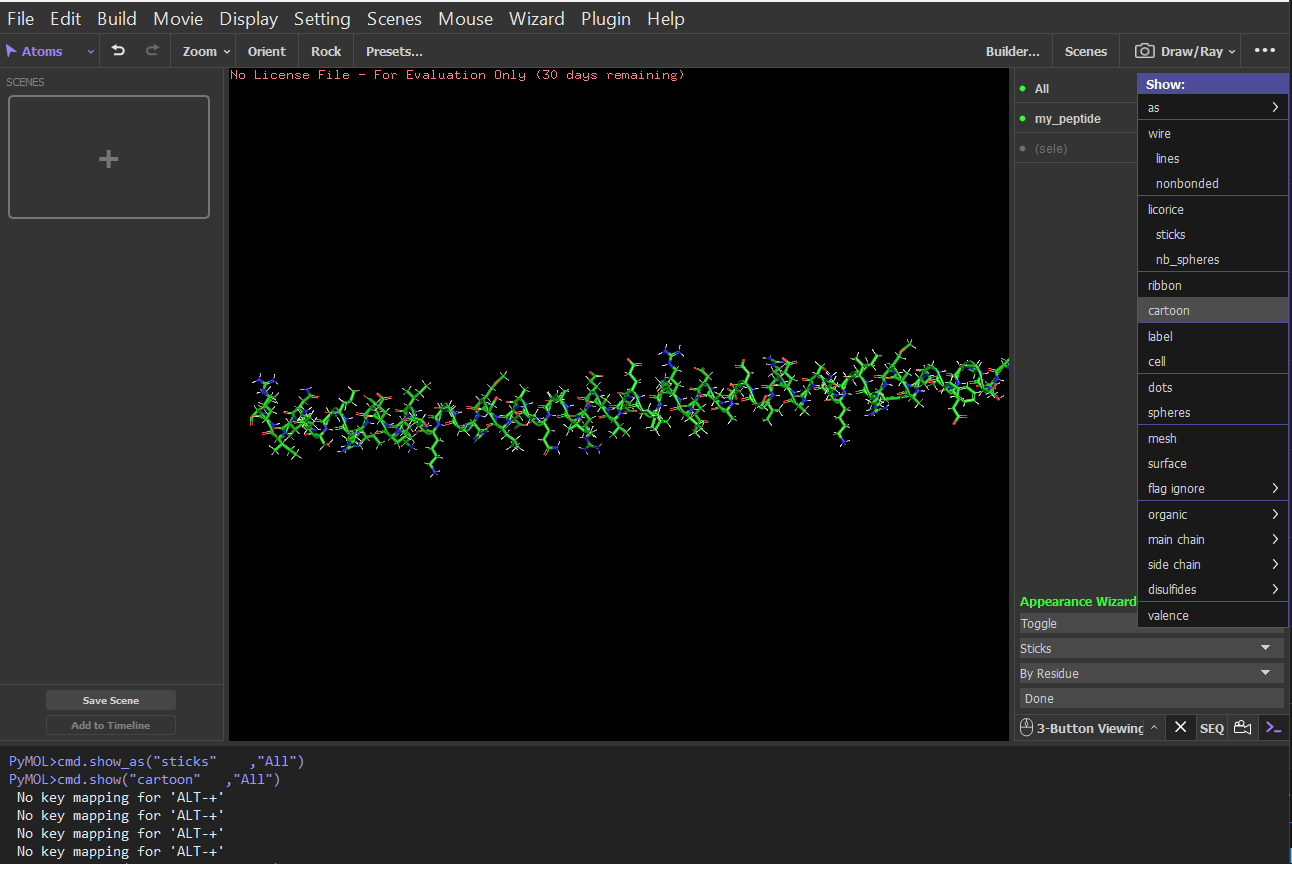 Ribbon:
Ribbon: 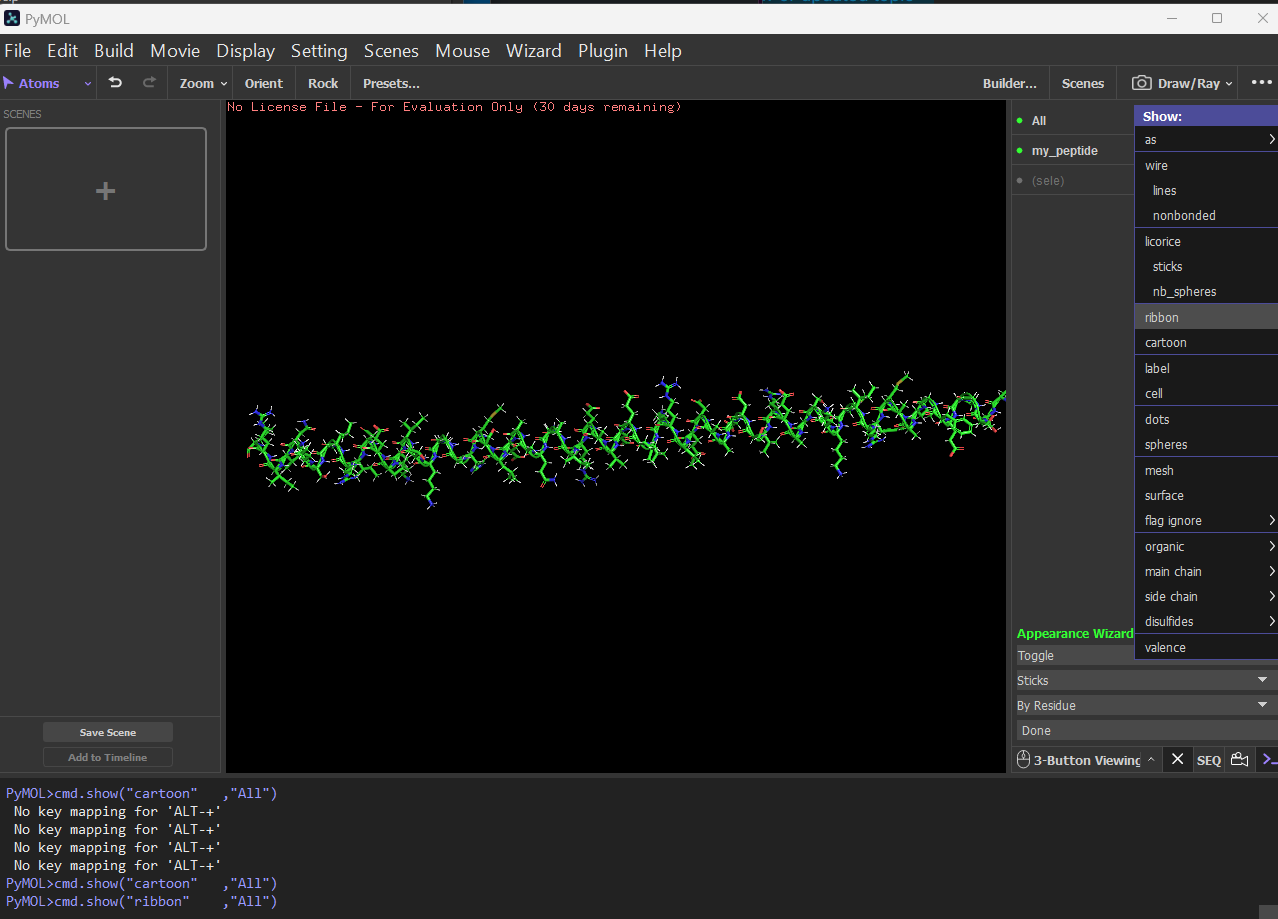 Sticks:
Sticks: 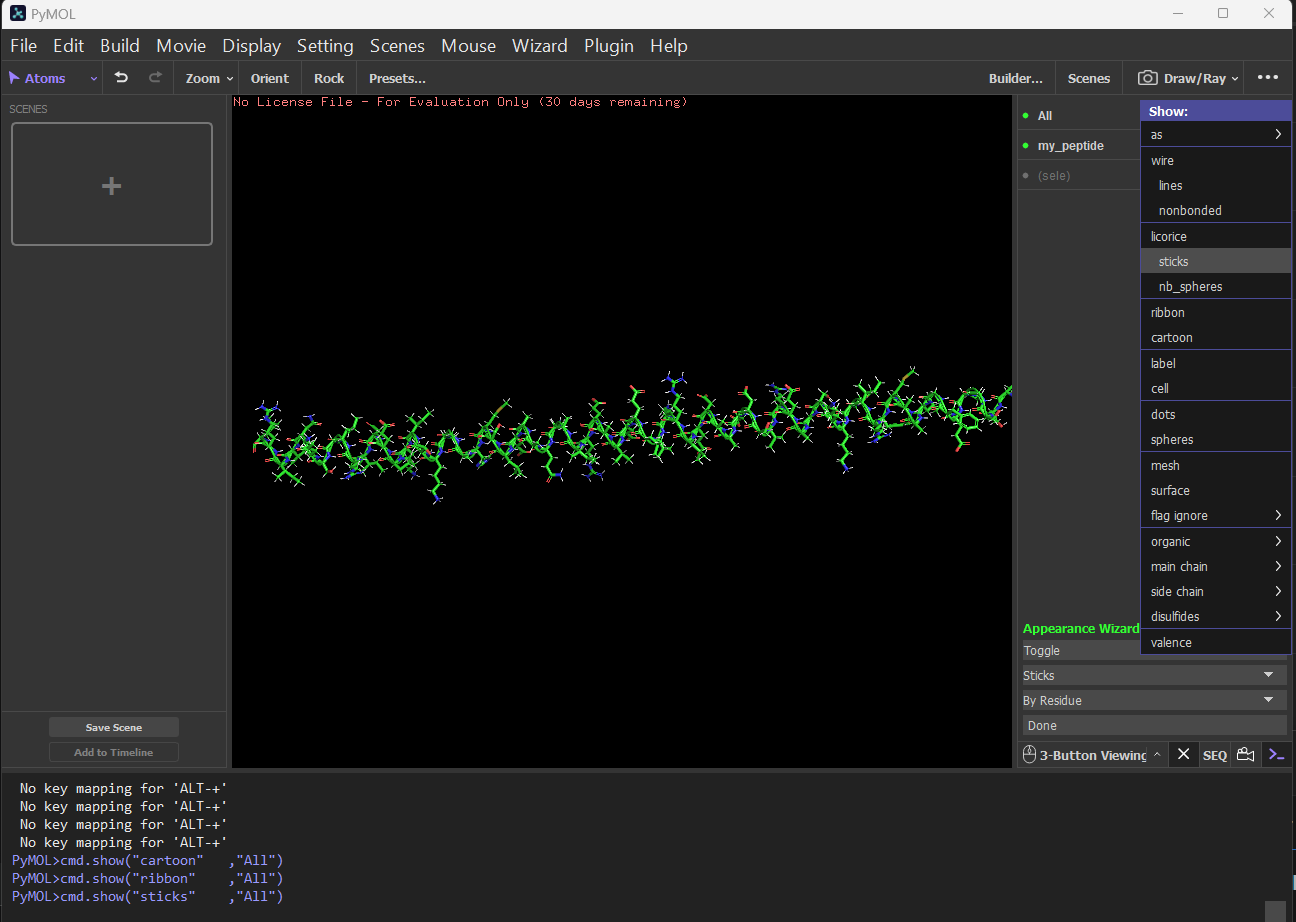
Color the protein by secondary structure. Does it have more helices or sheets?
It has more helices and no sheets. It has loops. \Figure: Helix, Sheets, and Loops Sticks:
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
| Property | Hydrophobic (Orange) | Hydrophilic (Marine) |
|---|---|---|
| Common Residues | Leu, Ile, Val, Phe | Ser, Thr, Asp, Glu, Lys |
| Typical Location | Buried in the core | Exposed on the surface |
| Role | Stability & Folding (Hydrophobic effect) | Solubilization & Ligand Binding |
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
There are no true internal binding pockets in this structure. structure is still a single, extended α-helix (likely a coiled or amphipathic helix), not a folded globular domain. A binding pocket requires:
- Tertiary packing
- Enclosed or partially enclosed cavity volume
- Multiple secondary structure elements forming a concave region
A single helix is essentially a cylinder with protruding side chains. Cylinders do not form pockets unless:
- Multiple helices pack together (helix bundle)
- The helix bends sharply
- Oligomerization creates an interface groove
Part C:
- Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
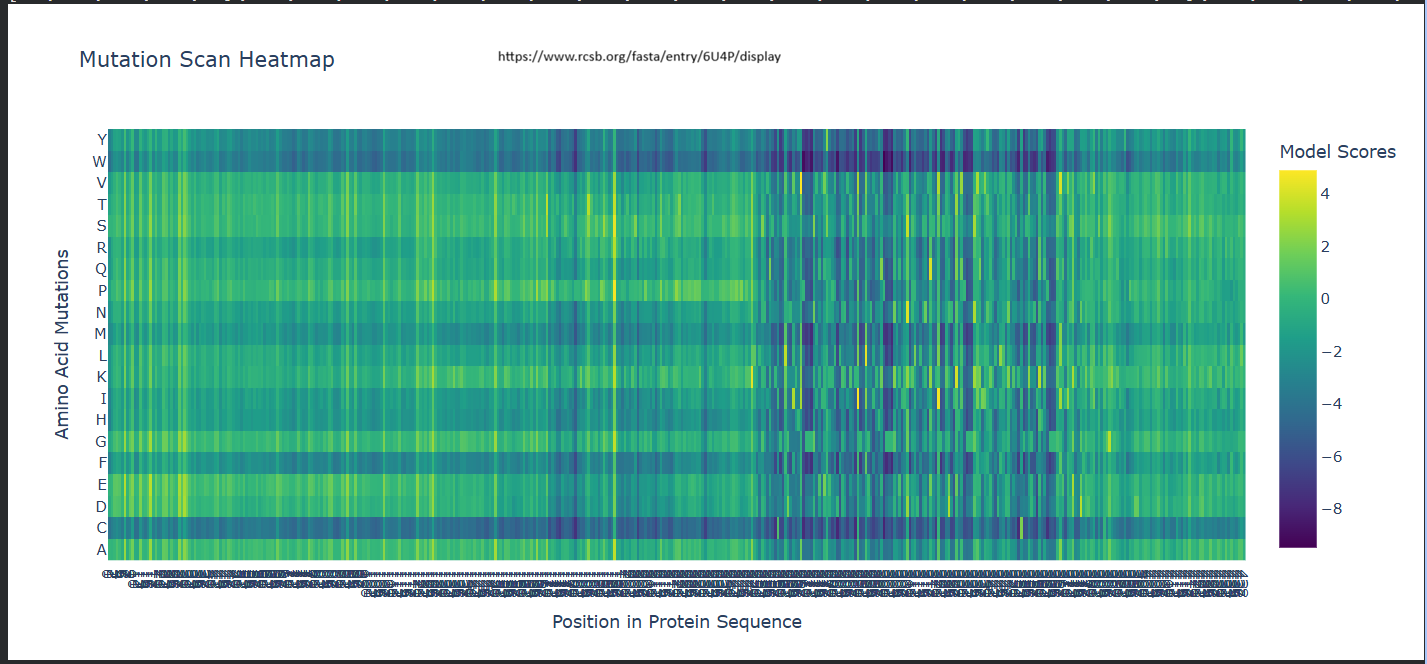
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
It seems like for position 333, Wildtype residue G, It is highly likey for the amino acid to be
-– Scores for Position 331 (Wild-type Residue: P) -–
Amino Acid (Y) | Score (Z)
-————————
A | 0.2499
C | -4.0639
D | -2.3773
E | -1.1166
F | -2.0289
G | -1.5281
H | -1.6049
I | 0.9267
K | 0.0000
L | 0.5773
M | -0.5974
N | -1.6772
P | -0.3847
Q | 1.5133
R | -1.1004
S | -0.9229
T | -0.0714
V | 2.6539
W | -3.7854
Y | -2.9304
My first question was why isn’t the Wild-Type (P) exactly 0?
I understood that when the code calculates these scores, it masks (hides) the residue at position 331 and asks the model: ‘Given the rest of the protein, what should be here?’
The Model’s Preference: The model thinks Lysine (K) fits that spot slightly better than the original Proline (P). Since the ‘Relative’ score is log(P_mutation) - log(P_wildtype), and the model happened to predict Lysine with a probability identical to what it expected for a wild-type residue in that context, it came out to 0.
The Penalty for P: The -0.3847 for Proline means that when the model looks at the surrounding sequence, it’s actually a bit ‘surprised’ to see a Proline there compared to its top choices (like Valine or Lysine).
The Baseline: The true ‘baseline’ is always the Wild-Type residue’s original probability. Any amino acid with a positive score is something the model likes more than the original, and anything negative is something it likes less.
Positive Scores (e.g., V: 2.6539): These are favored mutations. A positive score means the model thinks Valine is actually more likely to be found at this position than the original Proline. This could suggest the mutation might increase stability or is common in similar protein sequences evolutionarily.
Negative Scores (e.g., C: -4.0639): These are disfavored mutations. A negative score indicates the model predicts this change is unlikely or potentially destabilizing. The more negative the number, the more ‘surprised’ the model would be to see that amino acid there.
-–
Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
Done in code.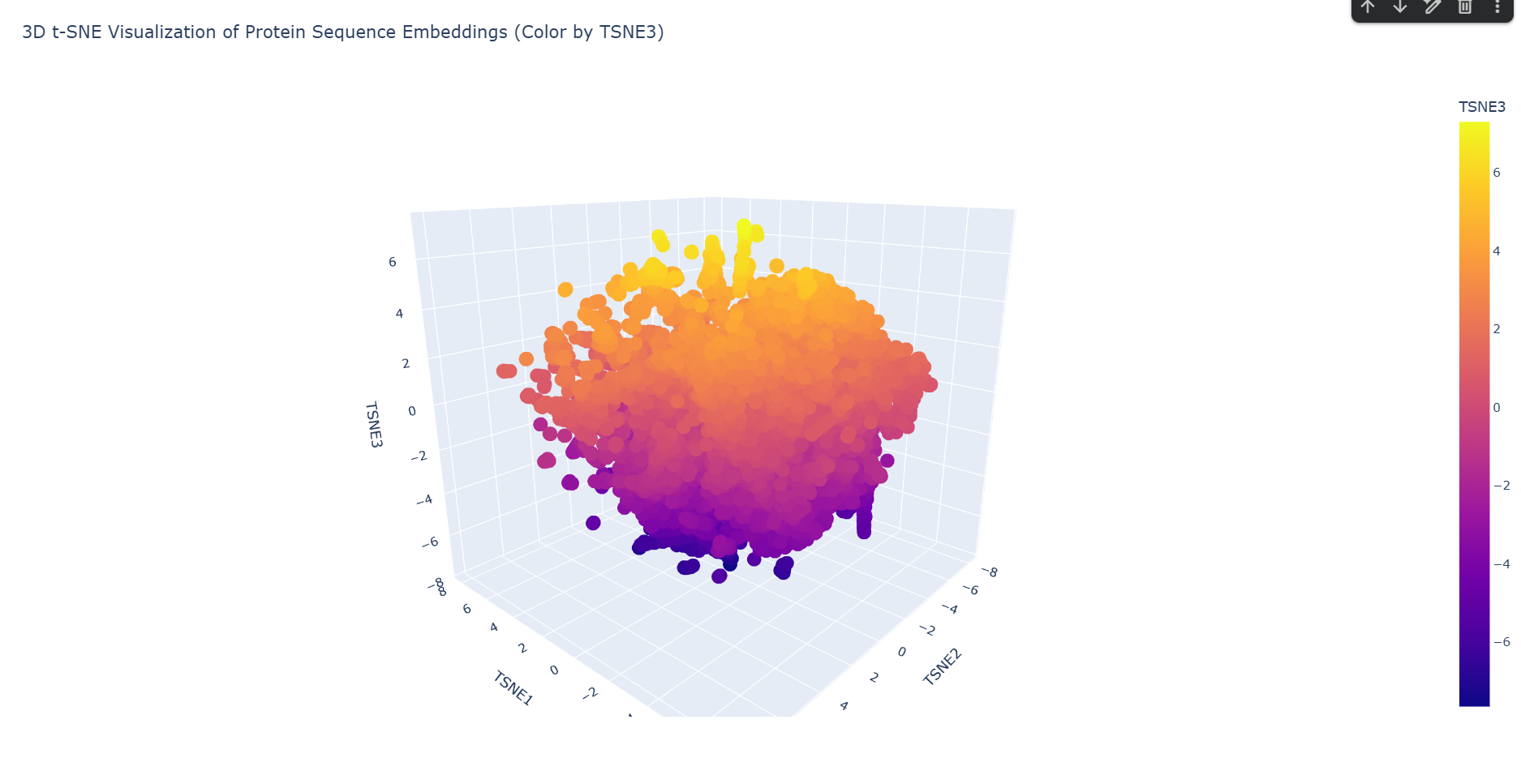 b. Analyze the different formed neighborhoods: do they approximate similar proteins?
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes. Based on the structural neighborhood report, Cluster 6 is the most ‘pure’ (defined as the highest percentage of its dominant fold after ‘automated matches’ are set aside).Here are the top 5 most common structural descriptions in that neighborhood:
- T-cell antigen receptor (Dominant Motif)
- Transglutaminase, two C-terminal domains
- Fibronectin type-III domai ncontaining protein 3a
- Neogenin
- Chondroitinase AC
This neighborhood primarily clusters proteins with Immunoglobulin-like folds and large multi-domain enzymatic proteins, showing that the latent space successfully groups these complex beta-sheet rich structures together.
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
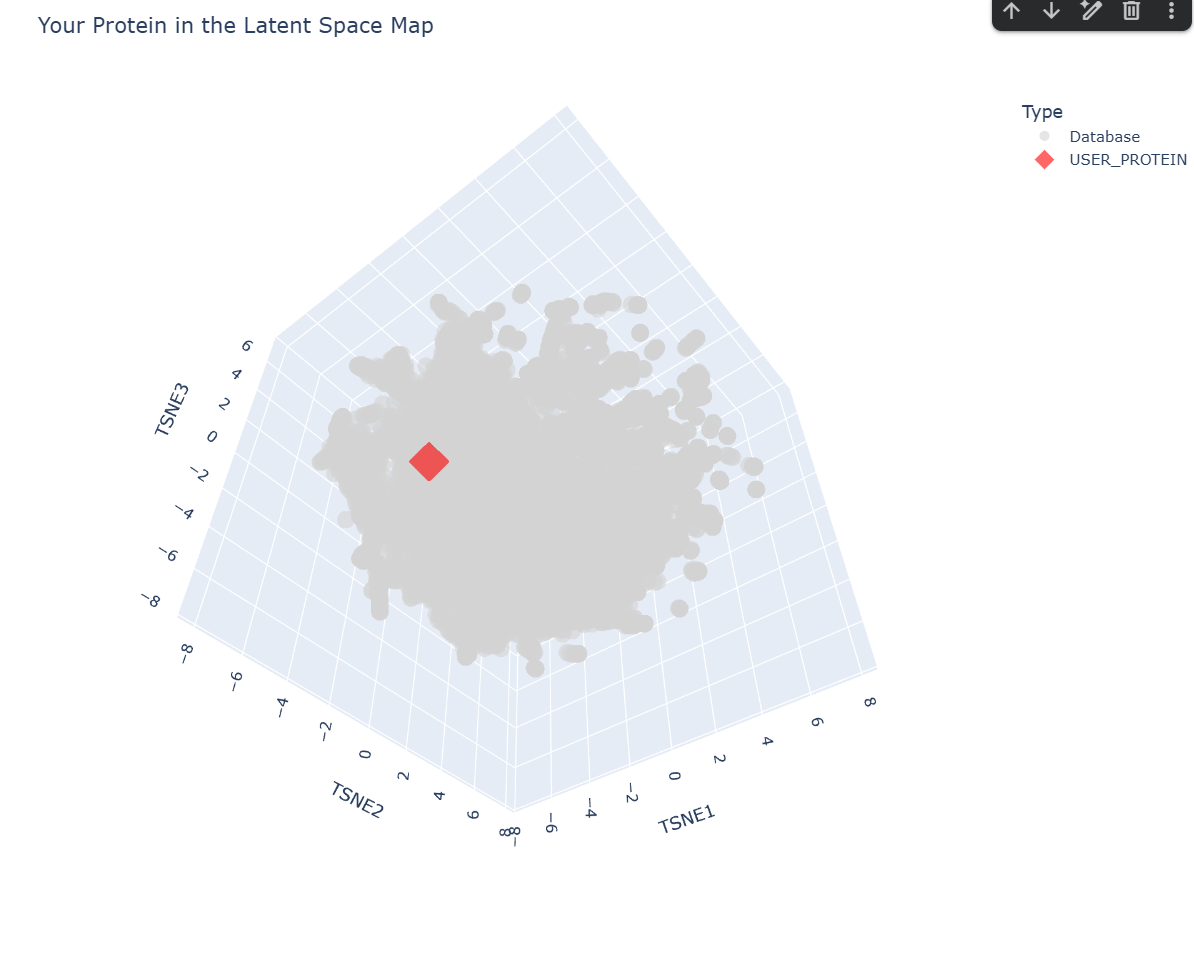
-– Top 10 Nearest Structural Neighbors to myProtein -–
1. Distance: 0.3048 | Description: d2ffta1 g.88.1.1 (A:2-84) Thylakoid soluble phosphoprotein TSP9 {Spinach (Spinacia oleracea) [TaxId: 3562]}
2. Distance: 0.5750 | Description: d2b5lc_ d.384.1.1 (C:) SV5-V core {Simian virus 5 [TaxId: 11207]}
3. Distance: 0.6194 | Description: d4lmsa_ d.184.1.0 (A:) automated matches {Chroomonas sp. [TaxId: 3029]}
4. Distance: 0.6318 | Description: d1xg0a_ d.184.1.1 (A:) Phycoerythrin 545 alpha-subunits {Cryptophyte (Rhodomonas sp. CS24) [TaxId: 79257]}
5. Distance: 0.8230 | Description: d1pbyc_ a.137.9.1 (C:) Quinohemoprotein amine dehydrogenase C chain {Paracoccus denitrificans [TaxId: 266]}
6. Distance: 0.8820 | Description: d4it7a_ d.17.1.0 (A:) automated matches {Ascaris lumbricoides [TaxId: 6252]}
7. Distance: 0.9125 | Description: d5ez2a_ b.60.1.0 (A:) automated matches {Sander vitreus [TaxId: 283036]}
8. Distance: 0.9148 | Description: d4m0wa2 d.3.1.23 (A:63-319) automated matches {SARS coronavirus [TaxId: 227859]}
9. Distance: 0.9433 | Description: d3nwaa_ e.76.1.1 (A:) Glycoprotein B {Human herpesvirus 1 [TaxId: 10298]}
10. Distance: 0.9935 | Description: d2a73a6 g.94.1.1 (A:578-643) Complement C3 linker domain {Human (Homo sapiens) [TaxId: 9606]}
Based on the ESM-2 latent space analysis, my protein’s closest structural neighbor is TSP9 (Thylakoid soluble phosphoprotein) from Spinach.
Analysis of Neighbors:
- TSP9 (Distance: 0.3048): This is a very close match in the embedding space. TSP9 is often associated with photosynthesis and has highly flexible regions.
- Viral and Algal Proteins: The presence of SV5-V core and Phycoerythrin in the top neighbors suggests your protein might share motifs with proteins involved in large complex assemblies or pigment binding.
- Structural Diversity: The neighbors include a mix of alpha-helical and linker domains (like the Complement C3 linker). This suggests your protein might have a modular or relatively disordered/flexible structure, which is common for proteins that show up near ’linker’ or ‘phosphoprotein’ domains in the latent space.
C2. Protein Folding
Folding a protein
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
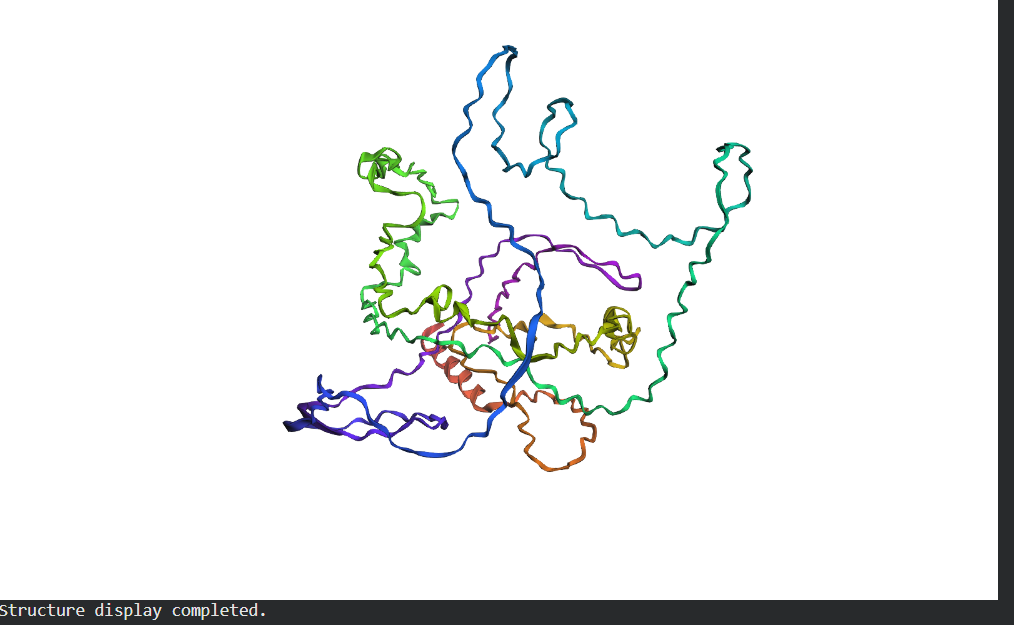
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Folded Protein Small Change change: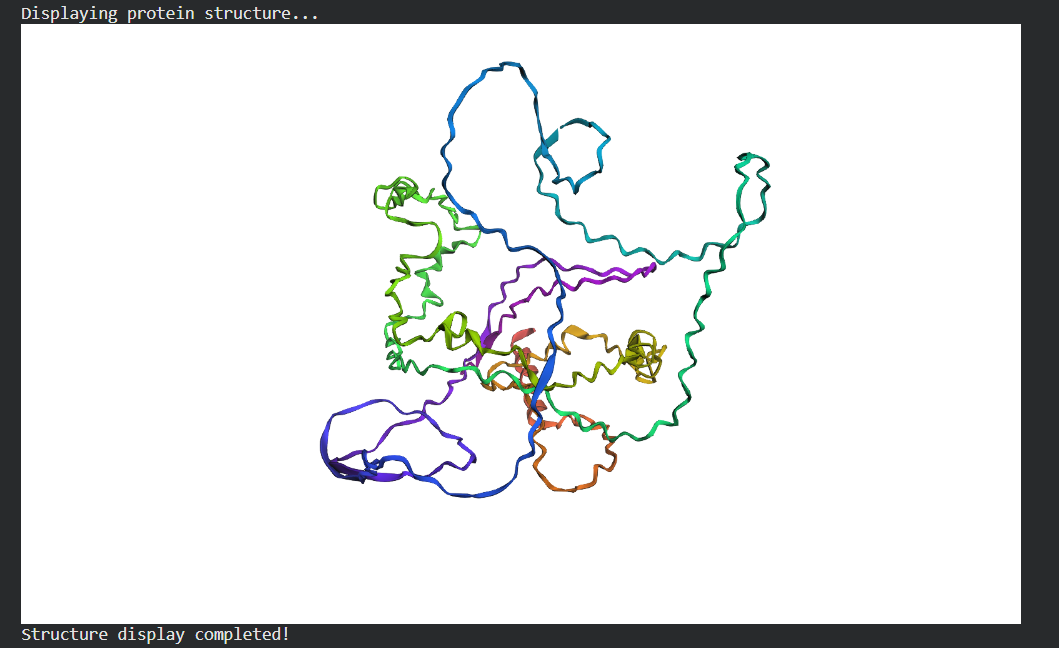
Folded Protein Big Change change:
## C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Generating sequences...
>10KR, score=2.4363, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020
QIVYKPVDLSKVTSKCGSLGNIHHKPGGGQMEVKSEKLDFKDRVQSKIGSLDNITHVPGGGNKKIETHKLT
>T=0.1, sample=0, score=1.3713, seq_recovery=0.2535
TVVVTPVVTVVTTVTTGAVGTVVVDPGGGEEVVTVEGVVVTGPVVVEVGVHGVTVTTPAGGTVVTVVVTET
New Sequence:TVVVTPVVTVVTTVTTGAVGTVVVDPGGGEEVVTVEGVVVTGPVVVEVGVHGVTVTTPAGGTVVTVVVTET
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
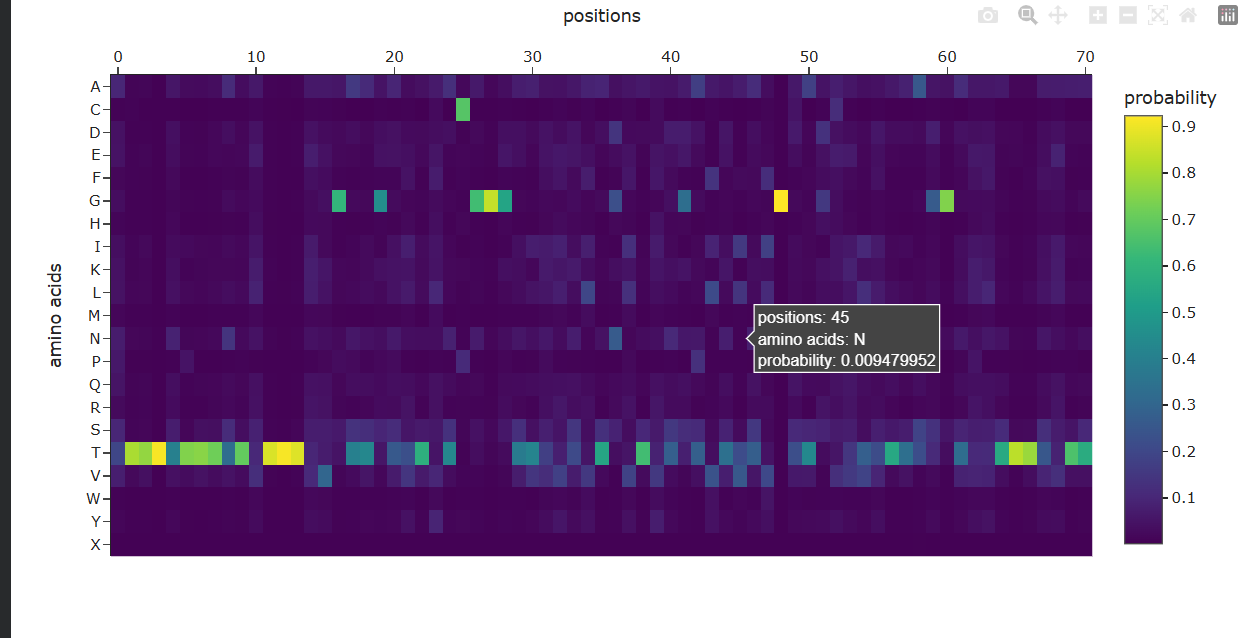
Input this sequence into ESMFold and compare the predicted structure to your original.