Week 04 HW: Protein Design Part 1
Part A. Conceptual Questions
Answer any NINE of the following:
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assume that the mass of other components like fat, collagen etc. are negligible compared to proteins in meat.
Average wight of 1 molecule of amino acid = 100 Dalton = 1.7 * $10^{-24}$ g
Weight of piece of meat = 5 * $10^{2}$ g
Therefore, number of amino acid molecules = (5 * $10^{2}$) / 1.7 * $10^{-24}$ = 2.94 * $10^{26}$
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
All the biomolecules found in foods get broken down into their constituent molecules via different enzymes and get resynthesized into the required biomolecules in various metabolic cycles as per the requirement of the body. This is vastly different from the synthesis of proteins through genetic translation. However, if we can encode bovine genes into the human genome, there is a possibility that some of the proteins synthesised via translation resemble that of the cow rather than human proteins. However, this is not enough for a human to become a cow because amino acids alone do not maketh species!
3. Why are there only 20 natural amino acids? Can you make other non-natural amino acids?
To answer the second part of the question, I would say yes. It is indeed possible to make non-natural amino acids.
The first part however, is interesting. This paper lists out several reasons so as to why only 20 proteogenic amino acids exists. Some of the interesting takeaways I found are:
- Some amino acids are highly “expensive to produce”. So, if two amino acids are (almost) similar in properties, life would favour the one with less productin cost. Case in point: Leucine requires 1 ATP, but Isoleucine requires 11.
- Some of the possible side chains, especially the aromatic ones, would make it completely insoluble in water- which is detrimental for reactions. Therefore, any amino acid would have to be at least as soluble, if not more than the least soluble amino aicd (at pH 7), Tyrosine.
- Some other side chains like esters and anhydrides are easily hydrolysed; ketones and aldehydes are suseptible to oxidation, reduction, and nucleophilic attacks; and carbon-carbon double and triple bonds are more reacctive than their single bond counterparts. Therefore, amino acids with those side chains are best avoided.
- Secondary structure that does not form bonds with other amino acids; molten globules (non-polar parts hidden inside) will have flexible side chains that msut be frozen into fixed positions, costing energy; and aggregated clumps of amino acids, especially beta sheets, are useless and oftentimes even toxic (amyloids). Therefore, these can’t be overly favoured.
- Incorporating other elements beside C, H, O, N also comes with a high energy costs. S containing amino acids (methionine and cysteine) are energetically expensive than other amino acids of the similar size. Therefore, evolution didn’t favour the ones with other elements.
4. Design some new amino acids.

5. Where did amino acids come from before enzymes that make them, and before life started?
The RNA world hypothesis can explain this to an extent. If we consider the first catalysing molecules to be RNAs, then it could very well have been possible for them to catalyse the synthesis of amino acids. Clay particles arranging themeselves in such a geometry that they would catalyse the synthesis is also a proposed hypothesis. Since these are biotic synthesis, only one of the racemic form would have been favoured. If abiotic synthesis of amino acds is considered, then the Miller-Urey experiment proved that high energy discharge in the form of lightining can lead to the formation of amino acids. Some other ways for abiotic snthesis would be; undersea volcanic eruptions; and meteorite impact etc. Although in these cases, equal proportion of L and D forms are likely to be formed.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
I would expect it to be left-handed helix. Since D-amino acids are the mirror images of L-amino acids, the steric and geometric parameters would flip, leading to the left-handed helix.
7. Can you discover additional helices in proteins?
Apart from $\alpha$ helices, $3_{10}$ helices, and $\pi$ helices, polyproline helices, collagen helices are known to exist. So, yes, it is possible to discover additonal helices in proteins.
8. Why are most molecular helices right-handed?
Due to the potential for steiric hindarance, biologically, either the biomolecules can all be either right-handed or left-handed helices. It is not possible for some molecules to be right-handed and some others to be left-handed. The right-handedness of proteins is due to the existence of L-amino acids, and the right-handedness of nucleic acids is due to D-sugars. Computation simulations show that L-amino acids consistently fold into stable structures than their D coutnerparts in ambient conditions. The same can be inferred for the stability of nucleic acids, although left-handed stuctures are known to exist under specific conditions.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
- Extensive hydrogen bonding
- Hydrophobic side chains intergigitated with hydrophilic ones
- Flat planarity as opposed to curved nature of helices
- Strucutral complementarity of the edges, that favour inter-strucutral bond formation.
10. Why do many amyloid diseases form β-sheets?
Skipped.
11. Can you use amyloid β-sheets as materials?
Skipped.
12. Design a β-sheet motif that forms a well-ordered structure.
Skipped.
Part B. Protein Analysis and Visualization
Pick any protein with a 3D structure and answer:
1. Briefly describe the protein you selected and why you selected it.
Proline-Betaine Transporter is the protein I have selected, and it is because it acts as an osmoprotectant for bacteria under high salt concentrations.
2. Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid?
Sequence: MLKRKKVKPI TLRDVTIIDD GKLRKAITAA SLGNAMEWFD FGVYGFVAYA LGKVFFPGAD PSVQMVAALA TFSVPFLIRP LGGLFFGMLG DKYGRQKILA ITIVIMSIST FCIGLIPSYD TIGIWAPILL LICKMAQGFS VGGEYTGASI FVAEYSPDRK RGFMGSWLDF GSIAGFVLGA GVVVLISTIV GEANFLDWGW RIPFFIALPL GIIGLYLRHA LEETPAFQQH VDKLEQGDRE GLQDGPKVSF KEIATKYWRS LLTCIGLVIA TNVTYYMLLT YMPSYLSHNL HYSEDHGVLI IIAIMIGMLF VQPVMGLLSD RFGRRPFVLL GSVALFVLAI PAFILINSNV IGLIFAGLLM LAVILNCFTG VMASTLPAMF PTHIRYSALA AAFNISVLVA GLTPTLAAWL VESSQNLMMP AYYLMVVAVV GLITGVTMKE TANRPLKGAT PAASDIQEAK EILVEHYDNI EQKIDDIDHE IADLQAKRTR LVQQHPRIDE
Length: 500
Most frequent amino acid: Leucine (L)
3. How many protein sequence homologs are there for your protein? (Hint: Use Uniprot’s BLAST tool)
Lorem Ipsum
4. Does your protein belong to any protein family?
It is a transport protein.
5. Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure?
Yes, the structure was solved. It is a good quality structure too.
6. Are there any other molecules in the solved structure apart from protein?
None
7. Does your protein belong to any structure classification family?
I don’t think it does.
8. Open the structure in PyMol (or similar).
- Visualize as “cartoon”, “ribbon”, “ball and stick”.
- Color by secondary structure. Does it have more helices or sheets?
- Color by residue type. Distribution of hydrophobic vs hydrophilic residues?
- Visualize surface. Does it have binding pockets?
It has only helices.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
- Generate deep mutational scan with ESM2. Can you explain any particular pattern?
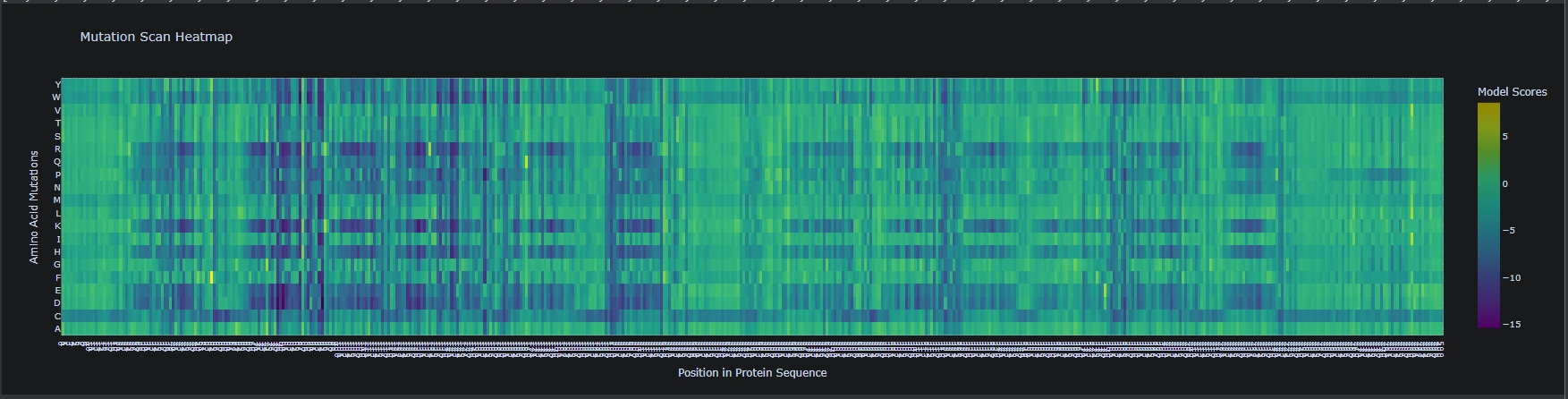
- (Bonus) Compare predictions to experimental scans.
Skipped - Latent space analysis: Place your protein in the map and explain its position.
Skipped
C2. Protein Folding
- Fold your protein with ESMFold. Do predicted coordinates match original?
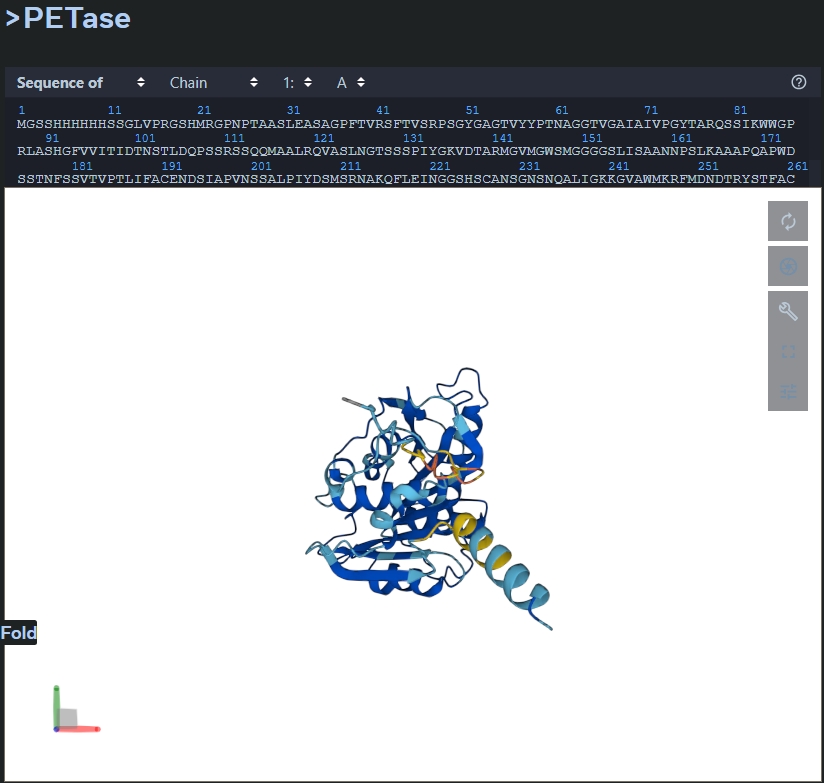
The predictied coordinates do not match the original. - Try mutations and large sequence changes. Is structure resilient?
The structure is not resilient as the conformation changes. The mutation carried out was: 5-10 H were replaced with A, and 242-247 were replaced with H.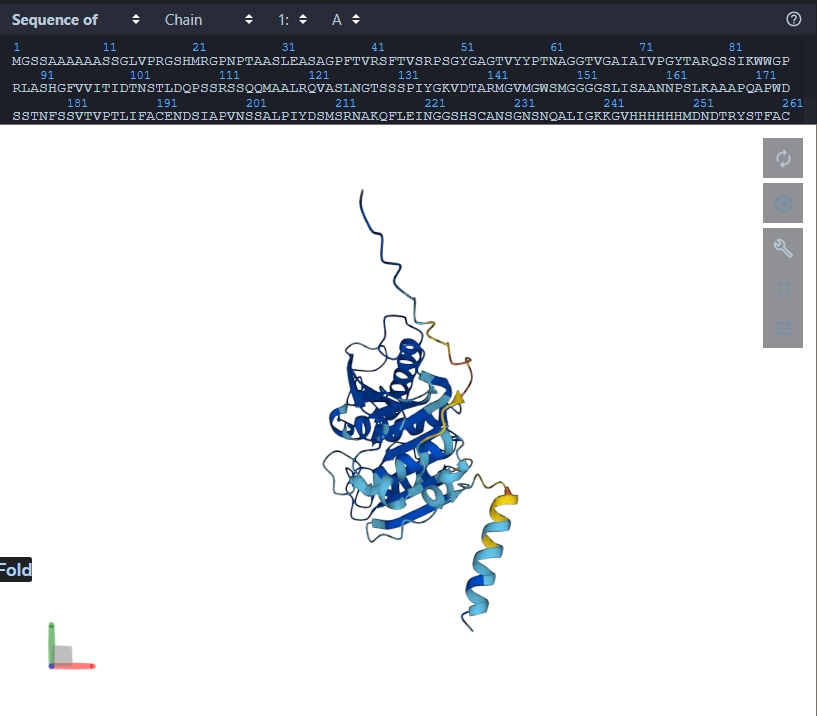
C3. Protein Generation
- Inverse-folding with ProteinMPNN. Compare predicted vs original sequence.
The predicted sequence is 50.33% similar to the orignial sequence according to this python program - Input sequence into ESMFold and compare predicted structure.
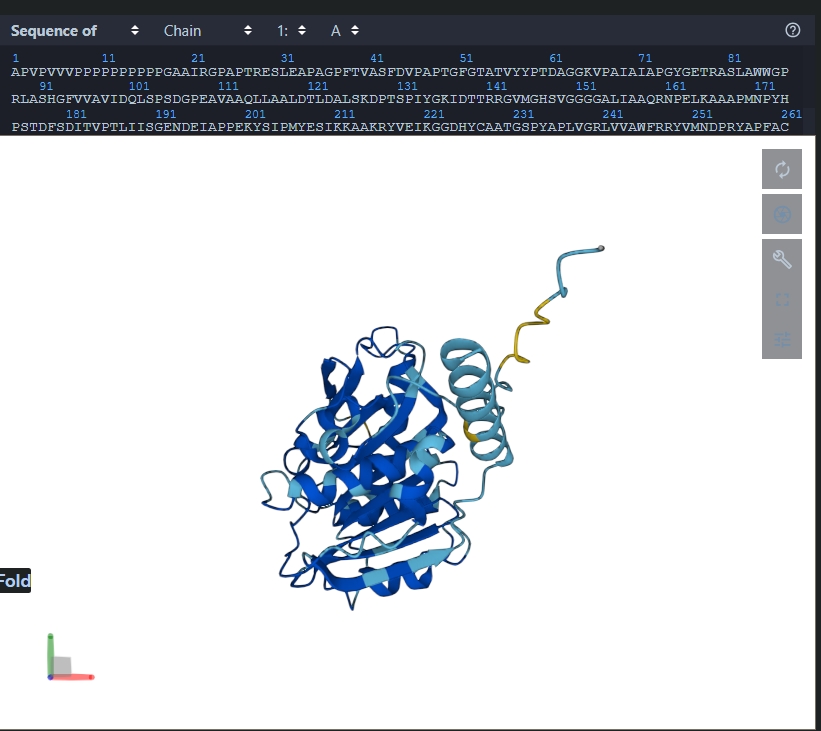
Part D. Group Brainstorm on Bacteriophage Engineering
1. Choose one or two main goals (e.g., stabilize lysis protein, disrupt interaction with E. coli DnaJ).
My group consisted of 4 people: @lorem ipsum, @lorem ipsum, @lorem ipsum, and @lorem ipsum. After brainstorming, we decided to focus on stabilizing the lysis protein.
2. Write a 1-page proposal describing:
- Tools/approaches you propose using
- Why those tools might help
- Potential pitfalls
- Include schematic of pipeline
Proposal:
By: 2026a-nourelden-rihan, 2026a-ritika-saha, 2026a-rahul-yaji, 2026a-keerthana-gunaretnam
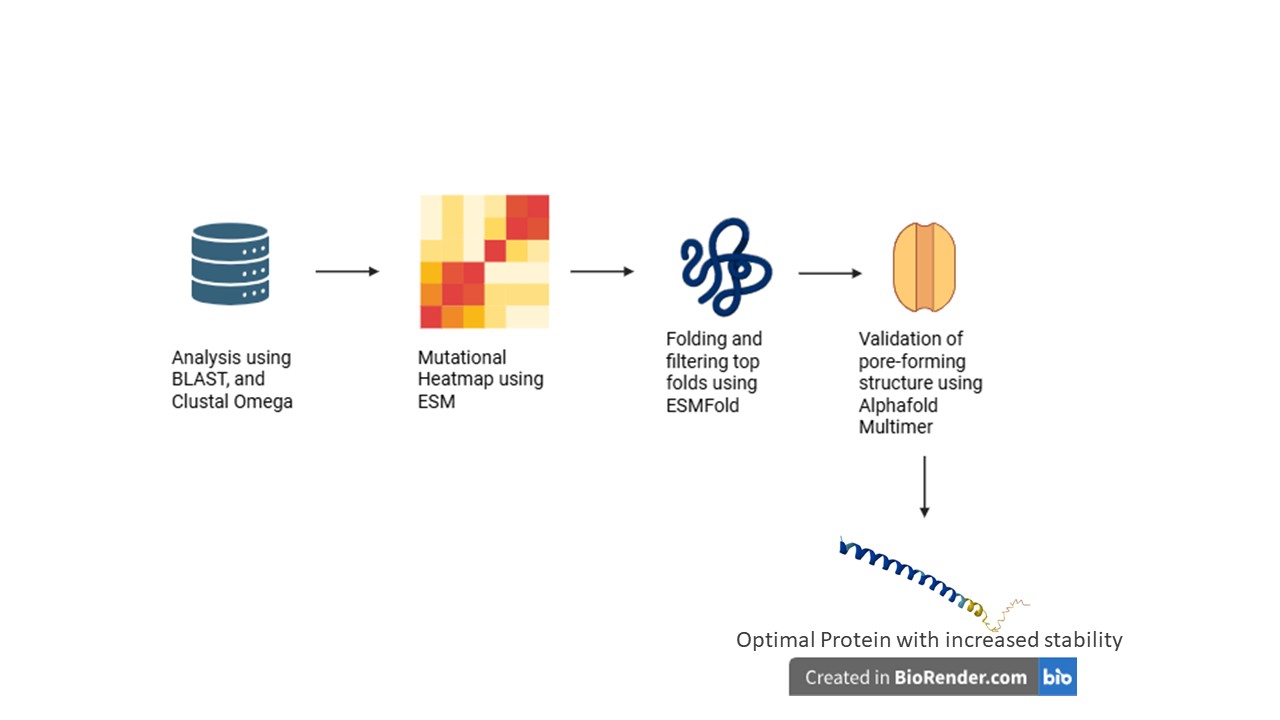
We decided to focus on the main area of increasing the stability of the MS2 phage lysis protein L, with a possible secondary goal of reducing the dependency on host DnaJ, while still maintaining the lysis action.
The tools AlphaFold, Clustal Omega, BLAST, ESM, and ESMFold were discussed.
BLAST can pull out homologous lysis proteins from the databases.
Clustal Omega can create MSAs to identify essential L48-S49 residues, and the pore-forming regions that must not be mutated.
ESM can create mutation heatmaps, which can guide the use of ESMFold to obtain highest score foldings in mutatable regions.
AlphaFold Multimer predicts whether the subunits of our protein can successfully create a pore in the host membrane, and also to check whether N-terminus can break the interaction with DnaJ.
We also identified a few pitfalls, with majors ones dealing with limited training datasets, that may not be properly aligned towards creating a transmembrane lysis protein.
Some other pitfalls include the lack of proper annotations for amurins; the possibility of an over-stable protein to form non-functional aggregates; and the vulnerability of modified protein to host proteases.
Schematic: