Week 05 HW: Protein Design Part 2
Part 1: Generate Binders with PepMLM
- Retrieve sequence and introduce mutation: (Pasted the sequence from UniPort, deleted M at 1st position, changed A to V at 4th position.)
ATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
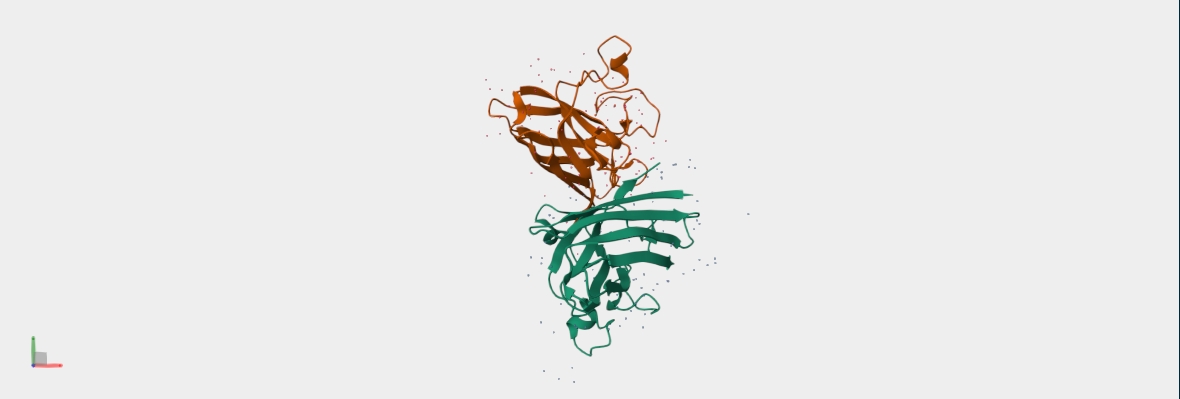
Structure of the native sequence- predicted vs actual:
- Generate 4 peptides using PepMLM Colab:
| index | Binder | Pseudo Perplexity |
|---|---|---|
| 1 | WRSPAVAVAHWE | 7.76721411356481 |
| 2 | WRVGWVGVELKE | 24.2058244561383 |
| 3 | WRSPAAXIEHKX | 11.243453670563373 |
| 4 | WRVYAAXIEWGK | 20.449723821548965 |
- Known binder: FLYRWLPSRRGG
- Perplexity score: 22.5252
A note about perplexity score: A key evaluation metric for language models that measures how well a probability model predicts a sample. Lower the score, higher the confidence of the model that the output satisfies the criteria.
Part 2: Evaluate Binders with AlphaFold3
Peptide Binding location ipTM score WRSPAVAVAHWE None 0.28 WRVGWVGVELKE None 0.35 WRSPAAXIEHKX None 0.33 WRVYAAXIEWGK None 0.34
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Peptide Comparison Results:
| Input Sequence | Solubility | Hemolysis (Prob.) | Binding Affinity | Length (aa) | Mol. Weight (Da) | Net Charge (pH 7) | Isoelectric Point (pH) | Hydrophobicity (GRAVY) |
|---|---|---|---|---|---|---|---|---|
| WRSPAVAVAHWE | 1.0 | 0.044 (Non-hemolytic) | 5.361 (Weak) | 12 | 1408.6 | -0.14 | 6.76 | -0.13 |
| WRVGWVGVELKE | 1.0 | 0.117 (Non-hemolytic) | 7.089 (Medium) | 12 | 1457.7 | -0.23 | 6.28 | -0.13 |
| WRSPAAXIEHKX | 1.0 | 0.011 (Non-hemolytic) | 4.645 (Weak) | 12 | 1158.5 | 0.85 | 8.76 | -0.86 |
| WRVYAAXIEWGK | 1.0 | 0.043 (Non-hemolytic) | 6.724 (Weak) | 12 | 1360.7 | 0.76 | 8.59 | -0.26 |
| (Known) FLYRWLPSRRGG | 1.0 | 0.047 (Non-hemolytic) | 5.962 (Weak) | 12 | 1507.7 | 2.76 | 11.71 | -0.71 |
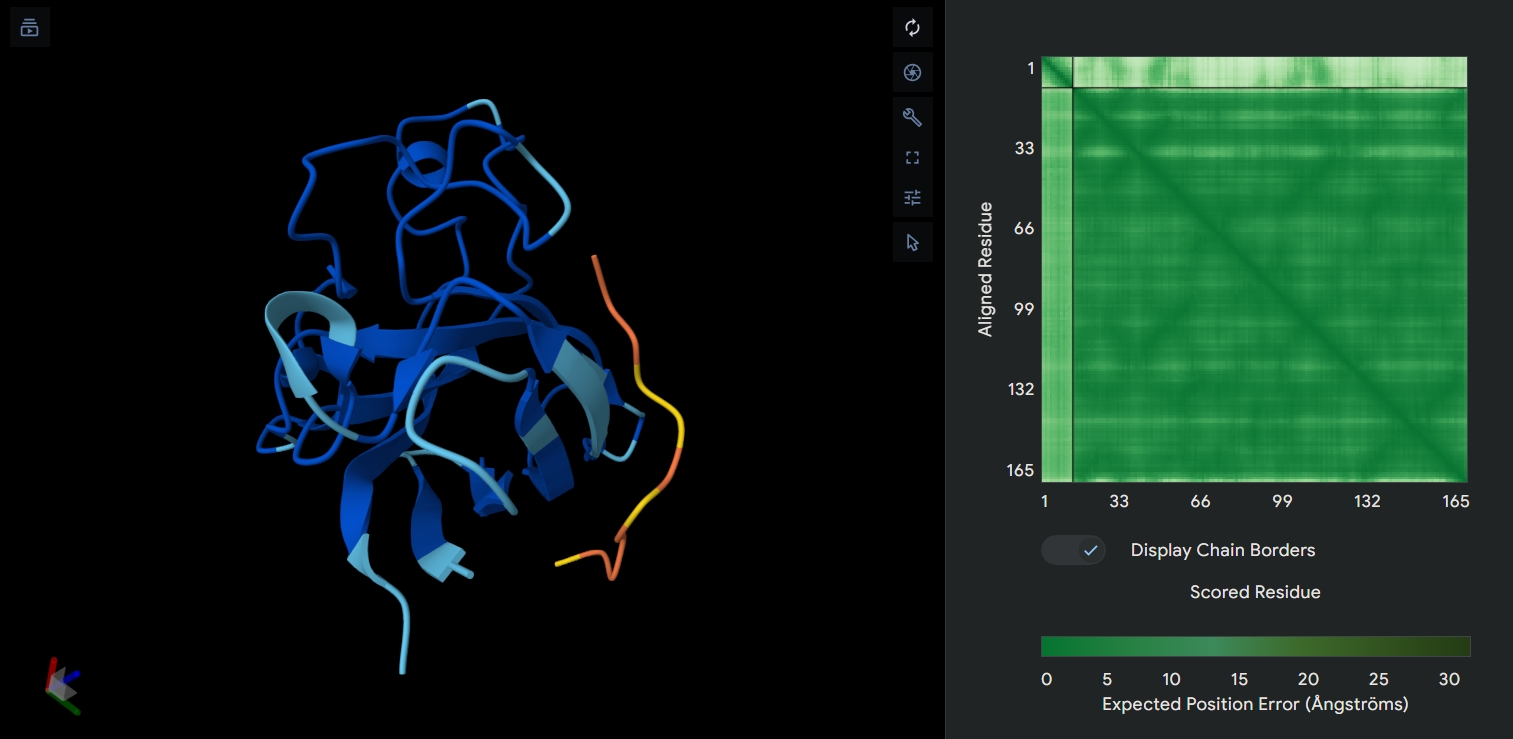
- The best peptide I would chose for wet lab validation would be WRVGWVGVELKE due to its relatively high binding affinity.
Part 4: Generate Optimized Peptides with moPPIt
Parameters:
Binder Hemolysis Solubility Affinity Motif SVKTKCCTTYQS 0.96447 0.916667 6.5756 0.890471 DDTKKCSCIQTH 0.974932 0.916667 6.31426 0.914592 ENGETFQCTKKV 0.970342 0.833333 6.04386 0.934673 KKSKKAFVCCVC 0.963174 0.666667 8.17171 0.613892
For the very long execution time, and the computational resources this program took, the only significant advantage it has (in this particular context) over PepMLM is the motif score, since there was no option to check for the motif specificity in the Peptiverse. All the other properties of the PepMLM generated sequences (predicted using Petptiverse) and those of the moPPIt peptides are comparable.
Part B: BRD4 Drug Discovery Platform Tutorial (Optional)
- Skipped
Part C: Group Project: L-Protein Mutants
- I chose the third option- Generating random mutations in the Lysis protein while avoiding the loss of function or non sense codons.The Python script was generated solely by the Google Gemini 2.5 Flash, that is in-built in Google Colab. The prompt was:
Develop a Python program in Google Colab that processes an amino acid sequence and generates mutated versions of it based on experimental data. The program should perform the following steps:
Prompt the user to enter an amino acid sequence.
Load mutation data from a publicly accessible Google Sheet URL (https://docs.google.com/spreadsheets/d/11WzDDNkQDEiqbUSGV0ZCqITGctyNFpD7xnPlhsj2BhE/edit?gid=0#gid=0).
The data contains information about amino acid changes and their associated ‘Lysis’ activity.
Filter the mutation data to include only ‘active’ mutations (where ‘Lysis’ is not 0). Extract the ‘Original_Residue’, ‘Position’, and ‘Mutated_Residue’ from the relevant columns (e.g., ‘Amino Acid Change’ and ‘Amino Acid Position’ or a ‘Mutation’ column like ‘X###Y’).
Create a helper function to format amino acid sequences by inserting a space after every 5 amino acids for better readability.
Implement a function generate_random_mutation_combinations(sequence, mutation_df, num_mutations) that takes an original amino acid sequence, the filtered active mutations DataFrame, and the desired number of mutations as input.
This function should:
- Identify all valid mutation sites where the original residue in the sequence matches an original residue in the mutation_df.
- Ensure that the num_mutations are applied to unique positions in the sequence. If there are fewer available unique mutation positions than num_mutations, it should apply all available unique mutations.
Randomly select mutations from the available options for the chosen unique positions.
Return the new mutated sequence and print the applied mutations.
Generate Multiple Mutated Sequences: Prompt the user for the number of mutated sequences they wish to generate. For each requested sequence:
Call the generate_random_mutation_combinations function.
Display the generated sequence with a clear heading (e.g., ‘Sequence 1:’, ‘Sequence 2:’, etc.).
Print both the original and the mutated sequences, using the formatting function defined in step 5.
In a separate code block, display each generated mutated sequence individually using display() so that each sequence is easily copyable by the user.
The generated mutational sequences were:
0. METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT (Original)- METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAFFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- METRFPQQSQQTLAATNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- METRFPQQSQQTPASTNRRRPFKHGGYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
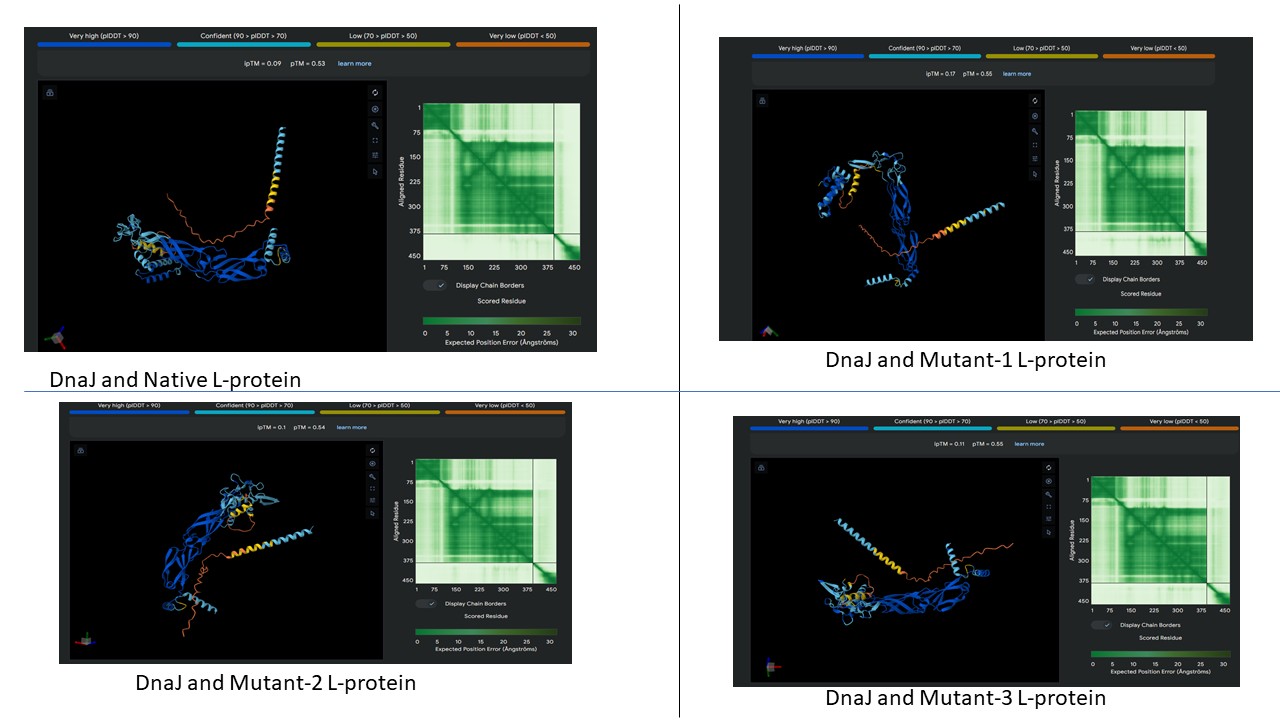
AF2 Multimer was used to co-fold the mutant Lysis protein (METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAFFLSKFTNQLLLSLLEAVIRTVTTLQQLLT) and DnaJ:

Cofolding was not performed for the other two sequences as my laptop started getting stuck while running the program.
The plDDT score indicates that the model is not confident about the folding of the input random mutated L protein. Overall, it suggests that the random mutation approach is very time consuming to obtain leads.
Later, cofolding was performed using Alphafold server, and the results obtained are shown below: